

Abstract
Several multicomponent assembly processes have been developed for the synthesis of intermediates that may be elaborated by a variety of cyclizations to generate a diverse array of highly functionalized heterocycles from readily-available starting materials. The overall approach enables the efficient preparation of libraries of small molecules derived from fused, privileged scaffolds.
INTRODUCTION
One of the first challenges in drug discovery programs is identifying small molecules that exhibit activity in biological screening assays that are relevant to treating human health disorders. Once such compounds are identified, they are structurally modified toward optimizing important parameters such as potency, specificity, toxicity, and bioavailability. This approach has also proven to be effective for the development of small molecule probes that can be used for functional and mechanistic studies of biological systems. In this context, the preparation of collections of compounds containing privileged structures, which are molecular scaffolds possessing a recognized ability to elicit biological activity across a range of targets,1,2 has proven to be a successful paradigm for identifying small molecules with useful pharmacological profiles. When contemplating new strategies for preparing novel compound libraries, it is thus important to develop efficient approaches that enable access to functionalized heterocyclic frameworks that incorporate privileged scaffolds and may be readily diversified.
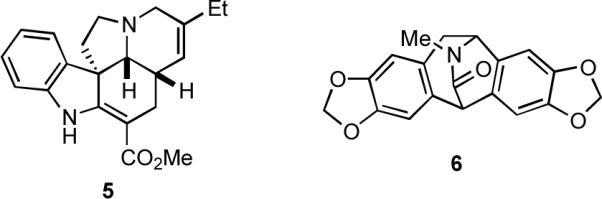
During work directed toward the total synthesis of the complex indole alkaloid (±)-tetrahydroalstonine (4), we invented a powerful Mannich-type multicomponent assembly process (MCAP) to generate 2, which underwent a hetero-Diels-Alder cycloaddition to deliver the pentacyclic core 3 that was then elaborated into the natural product via a remarkably short sequence of reactions (Scheme 1).3 The discovery of this facile method for constructing 2 laid the foundation for the development of the vinylogous Mannich reaction, which has proven an effective construct for alkaloid synthesis.4 We also realized that related MCAPs could be used as key steps to prepare other alkaloid natural products and applied this strategy to the concise syntheses of (±)-pseudotabersonine (5)5 and (±)-roelactamine (6) (Figure 1).6a
Scheme 1.

Figure 1.
Beyond the obvious application to the synthesis of alkaloids, it was apparent that we could implement such MCAPs as key steps in the assembly of intermediates that could be quickly elaborated into scaffolds of medicinal relevance via cyclizations preprogrammed by the functionality resident in the inputs 7–10 (Scheme 2).6 In general terms, our methodology allows rapid access to a diverse array of compounds comprising an aryl aminomethyl subunit 11, a structural motif common to a range of biologically active compounds, including those comprising heterocyclic privileged structures. We herein report the details of some recent applications of this approach to the diversity oriented synthesis (DOS) of skeletally distinct, unnatural products containing fused tetrahydroisoquinolines, benzodiazepines, 2-aryl piperidines, norbenzomorphans, and isoindolinones.7
Scheme 2.

RESULTS AND DISCUSSION
The tetrahydroisoquinoline ring system, which possesses a constrained aryl aminomethyl subunit, is present in a variety of complex natural products and pharmaceuticals that exhibit a wide range of biological properties, including antihypertensive,8 anticancer,9 and antimalarial10 activities. Starting with dihydroisoquinoline (12) or the readily-available 7-bromodihyroisoquinoline (13)11 as a preformed imine input in our MCAPs, we developed several novel MCAP/cyclization sequences to access a diverse array of unique heterocyclic ring systems incorporating the tetrahydroisoquinoline core (Scheme 3). Accordingly, treatment of imine 13 with silyl ketene acetal 1412 and chloroacetyl chloride in the presence of a catalytic amount of TMSOTf provided the phenyl ester 15 in 89% yield via a Mannich-type process.
Scheme 3.

Subsequent heating of 15 with benzylamine and Hünig's base furnished the fused tricycle 16 in 61% yield. The phenyl ester moiety resident in 15 was necessary to facilitate a one-pot displacement/cyclization process, as the corresponding methyl ester required more forcing conditions for lactam formation, which led to lower yields. In an interesting variant of this Mannich-type process, activation of 12 with TMSOTf,13 followed by addition of lithium phenylacetylide and N-acylation with o-azidobenzoyl chloride14 gave an intermediate amide that underwent a facile Huisgen cycloaddition to form the 1,2,3-triazole-fused 1,5-benzodiazepine-2-one 17 in one-pot and in 82% yield.
Compounds containing a piperazinone ring fused to a tetrahydroisoquinoline, as in 19, exhibit potent anthelmintic activity.15 We envisioned an MCAP that would allow the facile synthesis of triazolo piperazinone derivatives of these compounds (Scheme 3). Activation of 13 with TMSOTf, followed by addition of 1-hexynylzinc chloride gave an intermediate N-silyl amine that underwent reaction with chloroacetyl chloride to provide amide 18 in 91% yield. Subsequent displacement of the chloride ion with azide ion, followed by dipolar cycloaddition provided the novel triazolo piperazinone 19 in a two-step process.16
Aryl aminomethyl subunits bearing an α-aminonitrile moiety are synthetically versatile intermediates that we envisioned might be exploited for the construction of a number of fused heterocyclic ring systems.17 α-Aminonitriles can be readily accessed via the venerable Strecker reaction,18 wherein an intermediate imine or iminium ion that is formed upon condensation of a carbonyl input and an amine is trapped with cyanide ion. Following our sequential MCAP/cyclization strategy,5,6 the Strecker reaction was employed to assemble α-aminonitrile 21 from known benzaldehyde 20.19 Heating 21 resulted in a facile intramolecular Huisgen cycloaddition to afford the triazolo benzodiazepine scaffold 22 (Scheme 4). It was essential to perform this cycloaddition at low concentration and to maintain the temperature below 60 °C in order to avoid the deleterious elimination of HCN from compound 22. It is noteworthy that benzodiazepines are the archetypal privileged structures,1,2 and compounds containing 1,2,3-triazolo-1,4-benzodiazepines are known to bind to the benzodiazepine receptor.20
Scheme 4.

The α-aryl, α-aminonitrile subunit embedded within 22 was then exploited as a functional handle to access a variety of novel heterocyclic systems incorporating a benzodiazepine ring. To this end, the secondary amine 22 was first acylated with several different acid chlorides to furnish the amides 23–26 in high yields (Scheme 5). Deprotonation of chloroacetamide 23 at the acidic nitrile α-position, followed by the intramolecular displacement of chloride ion afforded the β-lactam 27 in 78% yield. The β-lactam substructure is prevalent within pharmaceutical agents and other biologically important molecules that elicit a diverse range of activities.21 Treatment of o-fluorobenzamide 24 with NaH effected an unusual cyclization involving a nucleophilic aromatic substitution by a nitrile anion to form the fused isoindolinone 28 in 76% yield. This sequence represents a novel approach to isoindolinones, which are themselves privileged structures.22 Indeed, 1,4-benzodiazepine fused isoindolinones are of medicinal interest as potassium channel antagonists.23
Scheme 5.

Inspired by the work of McEwen and Cooney,24 the anion derived from amide 25 was allowed to react with vinyltriphenylphosphonium bromide (Schweizer's reagent) to produce the pyrrolo benzodiazepine 29 in 91% yield. This transformation capitalizes on both the acidity of the proton adjacent to the nitrile group and the stability of cyanide ion as a leaving group. Notably, this reaction represents the first example of pyrrole formation from a heteroaromatic-substituted amide, further extending the scope of the original report. The Wittig reaction and aromatization steps were complete within ninety minutes at ambient temperature, a stark contrast to the extended reaction times and high temperatures originally reported for acyclic systems.24 Some pyrrolo-1,4-benzodiazepines have been identified as HIV-1 reverse transcriptase inhibitors.25
The nitrile functionality offers additional possibilities for diversification as a dipolarophile. For example, o-azidobenzamide 26 underwent an intramolecular dipolar cycloaddition upon heating to afford hexacycle 30, the structure of which incorporates two azole fused benzodiazepine units. Garanti and co-workers reported a similar approach to tetrazole fused 1,4-benzodiazepines,26 structures which are known benzodiazepine receptor binders.27
2-Aryl-substituted piperidines are found in a number of biologically active molecules.2a,28 Compounds containing this heterocyclic scaffold can be rapidly obtained through an MCAP/cyclization sequence to provide 2-arylpiperidines that are well suited to subsequent diversification reactions. When the imine formed upon condensation of allylamine and 2-bromobenzaldehyde (31) was treated with 4-pentenoyl chloride (32) and vinyl ether 33 in the presence of catalytic amounts of TMSOTf, the β-amido aldehyde 34 was obtained in 70% yield (Scheme 6). Aldehyde 34 then underwent a facile condensation and intramolecular nitrone cycloaddition upon heating with N-methylhydroxylamine to furnish the cis-fused octahydroisoxazolo[4,3-c]pyridine 35 in excellent yield as a single diastereomer. The 4-pentenoyl unit, which both activated the imine toward nucleophilic addition and masked the amino group, was readily cleaved with iodine in a mixture of THF and aqueous HCl to give the secondary amine 36.29
Scheme 6.

We then explored several methods by which the 2-bromophenyl substituent could be used in subsequent transformations to give novel heterocyclic scaffolds (Scheme 7). When 37, which was formed in an analogous fashion to 35,6a was treated with zinc in aqueous HCl at 0 °C, the N–O bond was cleaved to afford the amino alcohol 38. Palladium-catalyzed, intramolecular N-arylation furnished the tetrahydroquinoline 39 in 70% yield; this represents a novel entry to this tricyclic scaffold. The combined presence of the piperidine nitrogen atom in 36 and the pendant 2-bromophenyl substituent presented a number of interesting opportunities for ring forming reactions that provide access to some unique, tetracyclic scaffolds. For example, the phenylacetamide 40, which was obtained by acylation of 36, underwent tandem enolate arylation/allylation to give the 4,4-disubstituted dihydroisoquinolin-3-one 41 with high diastereoselectivity (>95:5) upon sequential reaction with excess LDA in the presence of DMPU followed by the addition of allyl bromide.30 We previously reported an analogous reaction using methyl iodide as the alkylating agent, which proceeded with similar selectivity to give predominantly one diastereomer, for which the relative stereochemistry was determined by X-ray crystallography.7c Presumably, in both cases the high degree of diastereoselectivity arises as a result of the preferential approach of the alkylating agent to the more sterically accessible convex face of the intermediate enolate. Protonation of the intermediate enolate was also highly diastereoselective, but the product underwent facile epimerization to afford a 2:1 mixture of diastereomers.
Scheme 7.

The free secondary amino group in 36 was also utilized to enable one-pot syntheses of several fused heterocyclic ring systems. For example, treatment of 36 with phenylisocyanate provided an intermediate urea, which underwent a palladium-catalyzed N-arylation in the presence of palladium acetate and BINAP to deliver the dihydroquinazolin-2-one 43. Ferraccioli and co-workers described a similar transformation that was reported to be general for (2-bromophenyl)methylamines.31 However, considerable optimization of the catalyst system was required in our hands, and we found that it was essential to ensure complete formation of the intermediate urea 42 before heating. Failure to do so resulted in incomplete cyclization, a phenomenon we attributed to catalyst poisoning by unreacted 36 at elevated temperatures. We also developed a related palladium-catalyzed cyclization of thioureas as illustrated by the initial conversion of 36 into 44 by reaction with an isothiocyanate, followed by an intramolecular S-arylation to furnish the 2-imino-1,3-benzothiazinane 45.32 This one-pot process for generating and cyclizing thioureas tethered to aryl bromides is an unprecedented tandem process that required the use of Pd[(t-Bu)3P]2 to achieve high conversion.
Varying the nucleophilic input in the MCAP by using an allyl zinc reagent provided a convenient means of accessing novel 2-aryl tetrahydropyridines that could be elaborated in a number of useful transformations. For example, sequential treatment of 2-bromo-6-chlorobenzaldehyde (46) with allylamine, CbzCl, and allylzinc bromide gave an 89% yield of diene 47. When 47 was treated with Grubbs II catalyst, it underwent a facile RCM reaction to afford 48 in 93% yield (Scheme 8).
Scheme 8.

The tetrahydropyridine 48 proved to be a versatile intermediate that could be readily elaborated into several, skeletally-diverse heterocycles of biological interest. Among these, norbenzomorphans are known to exhibit a variety of important neurological activities, including acetylcholinesterase (AChE) inhibition33 and codeine-like analgesic activity.34 We discovered that 48 could be readily converted to the norbenzomorphan core 49 in a single step by an intramolecular Heck cyclization (Scheme 9). Further derivatization of 49 was conveniently achieved by exploiting the electron-rich enecarbamate as a dienophile in the Povarov cyclization.35 In initial experiments, we found that treating 49 with p-toluidine (50), ethyl glyoxylate, and a catalytic amount of squaric acid gave a mixture (1.2:1) of diastereomeric cycloadducts.36 When Sc(OTf)3 was used as the acid catalyst, the reaction was both cleaner and higher yielding. Although this mixture of cycloadducts (1.2:1) could be easily separated by chromatography, both isomers converged to a single quinoline-fused norbenzomorphan 51 upon DDQ oxidation.
Scheme 9.

The isoindolinone ring system is a structural subunit common to both natural products and pharmacologically important compounds.22,37–39 It therefore occurred to us that the Parham cyclization40 might be applied to compounds such as 48 to create novel isoindolinones. In the event, addition of n-BuLi to 48 at −100 °C, followed by warming to −78 °C and cyclization of the intermediate aryllithium reagent onto the carbamate carbonyl provided the isoindolinone 52 in 60% yield.
Compounds containing the 1,2,3,4-tetrahydrobenzo[h][1,6]naphthyridine motif elicit a diverse range of biological properties, including selective 5-HT4 antagonist activity.41 Accordingly, we envisioned that the tetrahydropyridine 48 could serve as a precursor to this fused tricycle by a sequence of olefin isomerization and a Povarov reaction. Although methods for isomerizing tetrahydropyridines to enecarbamates are known,42 we found that conventional heating of 48 in the presence of 10% Pd/C required lengthy reaction times (24 h) and gave irreproducible yields. However, when the reaction was promoted with microwave heating, enecarbamate 53 was quickly obtained in 77% yield. Notably, no dehydrohalogenation was observed under these conditions. The enecarbamate 53 underwent a facile Povarov cyclization when exposed to 2-bromoaniline (54), ethyl glyoxylate, and 20 mol % Sc(OTf)3 to give a mixture (2:1) of diastereomeric tetrahydroquinolines that were oxidized with DDQ to deliver 55 in 52% overall yield.
In summary, we have extended our sequential MCAP/cyclization approach for DOS to the efficient preparation of several heterocyclic scaffolds containing the tetrahydroisoquinoline, benzodiazepine and 2-aryl piperidine substructures. Implementation of innovative reaction sequences subsequent to the assembly step enabled the facile formation of additional fused heterocyclic ring systems to give novel molecules of potential medicinal interest. The further development and application of these and related tactics to DOS and to the rapid synthesis of diverse compound libraries is an ongoing area of research within our laboratories, and new findings will be reported in due course.
REPRESENTATIVE EXPERIMENTAL PROCEDURES
Methanol (MeOH), acetonitrile (MeCN), and N,N-dimethylformamide (DMF) were dried by filtration through two columns of activated molecular sieves. Tetrahydrofuran (THF) and toluene were passed through two columns of activated neutral alumina prior to use. Triethylamine (Et3N), N,N-diisopropylamine, N,N-diisopropylethylamine, benzene, dichloromethane (CH2Cl2), pyridine, N,N'-dimethylpropylene urea (DMPU), phenyl isocyanate, and 1,4-dioxane were freshly distilled over CaH2. Trimethylsilyl trifluoromethanesulfonate (TMSOTf) was distilled over P2O5. Thionyl chloride (SOCl2) was distilled from triphenylphosphite. Zinc granules were activated by stirring with aqueous HCl (1.0 M) for 10 min, then filtered, rinsed with H2O, MeOH, then Et2O, and dried under high vacuum (ca. 0.5 mmHg) before use. Zinc chloride was fused under high vacuum (ca. 0.5 mmHg) prior to use. All other reagents and solvents were reagent grade and were purchased and used as received unless otherwise noted. Reactions were performed under nitrogen or argon atmosphere in round bottom flasks sealed under rubber septa with magnetic stirring, unless otherwise noted. Water sensitive reactions were performed with flame-or oven-dried glassware, stir bars and steel needles. Reaction temperatures are reported as the temperatures of the bath surrounding the vessel. Sensitive reagents and solvents were transferred using plastic or oven-dried glass syringes and steel needles using standard techniques.
Proton nuclear magnetic resonance (1H NMR) and carbon nuclear magnetic resonance (13C NMR) spectra were acquired in CDCl3 unless otherwise noted. Chemical shifts are reported in parts per million (ppm, δ), downfield from tetramethylsilane (TMS, δ = 0.00 ppm) and are referenced to residual solvent. Coupling constants (J) are reported in hertz (Hz) and the resonance multiplicity abbreviations used are: s, singlet; d, doublet; t, triplet; q, quartet; dd, doublet of doublets; ddd, doublet of doublet of doublets; dt, doublet of triplets; td, triplet of doublets; dq, doublet of quartets; m, multiplet; comp, overlapping multiplets of magnetically non-equivalent protons. The abbreviations br and app stand for broad and apparent, respectively. Infrared (IR) spectra were obtained with a Thermo Scientific Nicolet IR-100 FT-IR series spectrometer as thin films on sodium chloride plates. Melting points were determined using a Thomas-Hoover Uni-melt capillary melting point apparatus. Thin-layer chromatography (TLC) was performed on EMD 60 F254 glass-backed pre-coated silica gel plates and was visualized using one or more of the following methods: UV light (254 nm) and staining with iodine (I2), basic potassium permanganate (KMnO4) or acidic p-anisaldehyde (PAA). Flash chromatography was performed using glass columns and with Silicycle SiliaFlash F60 (40–63 μm) silica gel eluting with the solvents indicated according to the procedure of Still.43
Phenyl 2-(7-bromo-2-(2-chloroacetyl)-1,2,3,4-tetrahydroisoquinolin-1-yl)acetate (15)
Trimethylsilyl trifluoromethanesulfonate (27.1 mg, 22 μL, 0.12 mmol) was added to a solution of imine 13 in THF (4.8 mL) at −78 °C. To this mixture was added silyl ketene acetal 1412 (743 mg, 3.57 mmol) followed by chloroacetyl chloride (162 mg, 114 μL, 1.43 mmol) and the reaction was stirred at −78 °C for 22 h. The reaction was partitioned between CH2Cl2 (20 mL) and H2O (20 mL), and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 15 mL), and the combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The resultant yellow oil was purified by flash column chromatography eluting with hexanes/EtOAc (7 : 3) to give 446 mg (89%) of 15 as a white powder: mp 139–141 °C; 1H NMR (600 MHz) (rotamers) δ 7.47 (d, J = 1.8 Hz, 0.63 H), 7.42–7.36 (comp, 3.45 H), 7.27–7.28 (m, 0.30 H), 7.23–7.21 (m, 0.67 H), 7.11–7.09 (comp, 1.27 H), 7.06–7.05 (comp, 1.71 H), 6.07 (t, J = 6.6 Hz, 0.64 H), 5.48 (dd, J = 9.6, 4.8 Hz, 0.36 H), 4.73 (dd, J = 13.2, 6.0 Hz, 0.38 H), 4.54 (d, J = 12.6 Hz, 0.37 H), 4.19–4.09 (comp, 1.69 H), 3.93 (ddd, J = 9.0, 4.8, 3.6 Hz, 0.66 H), 3.73 (ddd, J = 13.2, 10.2, 4.2 Hz, 0.65 H), 3.24 (dd, J = 16.2, 9.6 Hz, 0.38 H), 3.19 (ddd, J = 13.2, 12.0, 4.2 Hz, 0.38 H), 3.10–2.94 (comp, 2.73 H), 2.88 (app dt, J = 7.8, 4.2 Hz, 0.66 H), 2.77–2.74 (m, 0.37 H); 13C NMR (150 MHz) (rotamers) δ 169.3, 168.6, 166.2, 165.8, 150.5, 150.1, 137.3, 137.0, 133.0, 132.4, 131.3, 131.0, 130.7, 130.5, 130.0, 129.7, 129.5, 129.2, 126.4, 126.0, 121.6, 121.3, 120.5, 120.2, 53.4, 50.1, 41.7, 41.2, 41.1, 40.6, 35.9, 28.6, 27.1; IR (neat) 2946, 1751, 1654, 1485, 1430, 1193, 1136, 816, 750, 690 cm−1; mass spectrum (CI) m/z 422.0156 [C19H1879Br35ClNO3 (M+1) requires 422.0080].
11-Bromo-3-(3,4-dimethoxyphenethyl)-1,3,4,7,8,12b-hexahydro-[1,4]diazepino[7,1-a]isoquinoline-2, 5-dione(16)
A solution of amide 15 (37 mg, 0.092 mmol), benzylamine (11 mg, 12 μL, 0.11 mmol), and N,N-diisopropylethylamine (15 mg, 20 μL, 0.11 mmol) in MeCN (2.2 mL) was heated at 75 °C for 24 h. The reaction was cooled to room temperature and partitioned between CH2Cl2 (15 mL) and saturated aqueous NaHCO3 (15 mL), and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 15 mL), and the combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The resultant yellow oil was purified by flash column chromatography eluting with toluene/EtOAc (1 : 1) to provide 21 mg (61%) of 16 as a white solid: mp 60–62 °C; 1H NMR (500 MHz) δ 7.42 (d, J = 1.5 Hz, 1 H), 7.38 (dd, J = 8.5, 1.5 Hz, 1 H), 7.34–7.27 (comp, 3 H), 7.24–7.23 (comp, 2 H), 7.06 (d, J = 8.5 Hz, 1 H), 5.09 (dd, J = 11.5, 3.5 Hz, 1 H), 4.72 (d, J = 14.5 Hz, 1 H), 4.67 (d, J = 14.5 Hz, 1 H), 4.31 (ddd, J = 9.0, 5.0, 4.5 Hz, 1 H), 4.22 (d, J = 16.5 Hz, 1 H), 4.04 (d, J = 16.5 Hz, 1 H), 3.26 (dd, J = 16.5, 4.0 Hz, 1 H), 3.20 (ddd, J = 13.0, 10.5, 4.0 Hz, 1 H), 3.12 (dd, J = 16.0, 11.5 Hz, 1 H), 2.93 (ddd, J = 16.5, 10.5, 5.5 Hz, 1 H), 2.74 (app dt, J = 16.5, 4.0 Hz, 1 H); 13C NMR (125 MHz) δ 169.0, 167.2, 137.2, 136.3, 134.3, 130.8, 130.7, 128.8, 128.6, 128.2, 127.9, 120.5, 54.5, 52.9, 51.8, 41.6, 41.4, 27.9; IR (neat) 2925, 1664, 1636, 1485, 1436, 912, 731, 699 cm−1; mass spectrum (CI) m/z 399.0710 [C20H2079BrN2O2 (M+1) requires 399.0630].
20-Bromo-4,5,6,14-tetraazapentacyclo[12.8.0.02,6.07,12.017,22]docosa-2,4,7(12),8,10,17,19,21-octaen-13-one (17)
A solution of o-azidobenzoic acid (112 mg, 0.69 mmol) in thionyl chloride (1.63 g, 1.0 mL, 13.7 mmol) was heated under reflux for 3 h. The cooled solution was concentrated under reduced pressure, and the residue was azeotroped with anhydrous benzene (3 × 4 mL) to provide crude o-azidobenzoyl chloride as a yellow oil.14 In a separate flask, trimethylsilyl trifluoromethanesulfonate (112 mg, 91 μL, 0.50 mmol) was added dropwise to a solution of dihydroisoquinoline (12) (60 mg, 0.46 mmol) in THF (3.6 mL) at −23 °C. In a separate flask, n-BuLi (0.22 mL, 0.59 mmol, 2.75 M in hexanes) was added dropwise to a solution of phenylacetylene (65 mg, 70 μL, 0.64 mmol) in THF (1.8 mL) and the brown solution was stirred at room temperature for 10 min.44 The phenyl acetylide was then added dropwise to the solution of activated imine at −78 °C, and the reaction was stirred at −78 °C for 5 h, whereupon o-azidobenzoyl chloride (0.69 mmol) in THF (2.0 mL) was added dropwise at −78 °C. The reaction was warmed to room temperature and stirred for 20 h. The reaction mixture was partitioned between saturated aqueous NaHCO3 (20 mL) and CH2Cl2 (20 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (2 × 20 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The resultant yellow oil was purified by flash chromatography eluting with pentane/EtOAc (3 : 2) to give 142 mg (82%) of 16 as a yellow solid: mp 154 °C (dec.); 1H NMR (600 MHz) δ 8.14-8.12 (comp, 2 H) 7.77 (ddd, J = 8.1, 7.4, 1.5 Hz, 1 H), 7.63 (ddd, J = 7.8, 7.4, 1.3 Hz, 1 H), 7.12–7.06 (comp, 2 H), 7.02–6.99 (comp, 3 H), 6.94–6.92 (comp, 2 H), 6.88–6.84 (comp, 2 H), 5.95 (s, 1 H), 4.28 (ddd, J = 12.8, 6.5, 4.9 Hz, 1 H), 3.72 (ddd, J = 12.8, 8.3, 4.3 Hz, 1 H), 2.93–2.81 (comp, 2 H); 13C NMR (150 MHz) δ 165.9, 143.8, 135.8, 134.1, 133.0, 132.9, 131.8, 129.5, 129.3, 128.7, 128.6, 128.4, 128.2, 127.9, 127.8, 127.7, 127.6, 126.7, 123.2, 50.6, 39.4, 28.5; IR (neat) 3442, 3060, 2963, 2910, 2862, 1643, 1604, 1484, 1408, 1260, 1084, 1020, 795, 750, 698 cm−1; mass spectrum (CI) m/z 379.1558[C24H19N4O (M+1) requires 379.1481].
1-(7-Bromo-1-(hex-1-ynyl)-3,4-dihydroisoquinolin-2(1H)-yl)-2-chloroethanone (18)
Trimethylsilyl trifluoromethanesulfonate (344 mg, 0.28 mL, 1.57 mmol) was added slowly to a solution of dihydroisoquinoline 13 (300 mg, 1.43 mmol) in THF (11 mL) at −23 °C, and the reaction was then cooled to −78 °C. In a separate flask, n-BuLi (0.9 mL, 1.86 mmol, 2.2 M in hexanes) was added dropwise to a solution of 1-hexyne (164 mg, 0.23 mL, 2.00 mmol) in THF (6 mL) at 0 °C, and the clear colorless solution was stirred for 5 min at 0 °C. ZnCl2 (2.3 mL, 2.3 mmol, 1 M in THF) was added dropwise at 0 °C, and the solution was warmed to room temperature and stirred for 15 min.45 The solution of freshly prepared organozinc reagent was added dropwise to the solution of activated imine at −78 °C, and the reaction was stirred for 5 h at −78 °C, whereupon chloroacetyl chloride (284 mg, 0.20 mL, 2.57 mmol) was added slowly at −78 °C. The bath was removed, and the reaction was warmed to room temperature and stirred for 10 min. The reaction mixture was partitioned between saturated aqueous NaHCO3 (20 mL) and CH2Cl2 (20 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (2 × 20 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The resultant yellow gum was purified by flash chromatography eluting with pentane/EtOAc (8 : 2) to give 479 mg (91%) of 18 as a pale yellow oil: 1H NMR (600 MHz) (rotamers) δ 7.44 (d, J = 1.2 Hz, 1 H), 7.33–7.32 (m, 1 H), 7.03–7.00 (m, 1 H), 6.24 (s, 0.58 H), 5.64 (s, 0.33 H), 4.56–4.50 (m, 0.40 H), 4.32–4.30 (m, 0.37 H), 4.16 (d, J = 12.0 Hz, 1 H), 4.13–4.10 (m, 0.72 H), 3.94–3.91 (m, 0.62 H), 3.77–3.73 (m, 0.63 H), 3.27 (m, 0.37 H), 3.05–2.98 (m, 0.67 H), 2.85–2.78 (comp, 1.44 H), 2.17–2.16 (comp, 2 H), 1.46–1.43 (comp, 2 H), 1.38–1.32 (comp, 2 H), 0.92–0.87 (comp, 3 H); 13C NMR (150 MHz) (rotamers) δ 165.4, 164.9, 136.6, 135.7, 132.8, 131.5, 130.8, 130.5, 130.4, 130.1, 120.2, 120.1, 86.4, 84.8, 77.8, 48.1, 44.8, 41.3, 41.1, 40.8, 37.2, 30.5, 30.4, 28.5, 27.6, 21.9, 18.4, 13.6; IR (neat) 2957, 2933, 2871, 1657, 1431, 1188, 929, 814, 796, 647 cm −1; mass spectrum (CI) m/z 368.0415 [C17H2079Br35ClNO (M+1) requires 368.0339].
4-Bromo-16-butyl-10,13,14,15-tetraazatetracyclo[8.7.0.02,7.03,17]heptadeca-2,4,6,14,16-pentaen-11-one (19)
A mixture of amide 18 (50 mg, 0.14 mmol) and sodium azide (10 mg, 0.15 mmol) in anhydrous DMF (0.6 mL) was stirred at room temperature for 2 h. The reaction was partitioned between H2O (5 mL) and toluene (5 mL). The layers were separated, and the aqueous layer was extracted with toluene (1 × 5 mL). The combined organic layers were washed with H2O (5 × 5 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude yellow azide thus obtained was dissolved in anhydrous DMF (2.7 mL), and the solution was heated at 100 °C for 48 h. The reaction was partitioned between H2O (10 mL) and toluene (10 mL). The layers were separated, and the aqueous layer was extracted with toluene (1 × 10 mL). The organic layer was washed with H2O (5 × 10 mL), dried (Na2SO4) and concentrated under reduced pressure. The resultant brown oil was purified by flash column chromatography eluting with pentane/EtOAc (3 : 7) to give 43 mg (85%) of 19 as a white solid: mp 135–136 °C; 1H NMR (600 MHz) δ 7.43 (dd, J = 8.4, 2.4 Hz, 1 H), 7.14 (d, J = 8.4 Hz, 1 H), 6.95 (d, J = 2.4 Hz, 1 H), 5.80 (s, 1 H), 5.20 (d, J = 18.0 Hz, 1 H), 4.83 (d, J = 18.0 Hz, 1 H), 4.53 (ddd, J = 13.2, 7.2, 6.0 Hz, 1 H), 3.52–3.47 (m, 1 H), 3.22 (ddd, J = 16.2, 7.2, 6.6 Hz, 1 H), 2.96 (ddd, J = 13.2, 7.2, 6.6 Hz, 1 H), 2.84 (app dt, J = 15.0, 7.2 Hz, 1 H), 2.68 (app dt, J = 15.0, 7.2 Hz, 1 H), 1.85–1.80 (comp, 2 H), 1.47–1.41 (comp, 2 H), 0.96 (t, J = 7.2 Hz, 3 H); 13C NMR (150 MHz) δ 162.2, 144.6, 136.1, 133.7, 132.0, 130.9, 127.3, 124.5, 120.6, 52.1, 48.9, 42.1, 30.9, 26.6, 25.2, 22.6, 13.8; IR (neat) 2956, 2870, 1667, 1481, 1429, 1258, 828, 734 cm−1; mass spectrum (CI) m/z 375.0821 [C17H2079BrN4O (M+1) requires 375.0742].
2-(2-(Benzo[d][1,3]dioxol-5-ylmethyl)phenyl)-2-(prop-2-ynylamino)acetonitrile (21)
Aqueous HCl (4.28 mL, 1.0 M, 4.28 mmol) was added dropwise to a solution of o-azidobenzaldehyde 20 (600 mg, 4.08 mmol),19 propargylamine (236 mg, 274 μL, 4.28 mmol) and sodium cyanide (210 mg, 4.28 mmol) in MeOH (8.6 mL) and the reaction stirred at room temperature for 2.5 h. The reaction was diluted with H2O (50 mL) and the pH raised to 10 with aqueous NaOH (ca. 300 μL, 1.0 M). The resulting mixture was extracted with EtOAc (3 × 70 mL) and the combined organic layers were dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with hexanes/Et2O (4 : 1 → 3 : 2) to give 673 mg of amine 21 (78%) as an orange oil: 1H NMR (500 MHz) δ 7.53 (dd, J = 7.6, 1.5 Hz, 1 H), 7.45 (ddd, J = 8.1, 7.6, 1.5 Hz, 1 H), 7.22 (dd, J = 8.1, 1.5 Hz, 1 H), 7.20 (app td J = 7.6, 1.5 Hz, 1 H), 5.11 (d, J = 7.5 Hz, 1 H), 3.62 (m, 2 H), 2.34 (t, J = 2.5 Hz, 1 H) 2.00 (m, 1 H); 13CNMR (100 MHz) δ 138.1, 130.8, 129.4, 125.4, 125.2, 118.7, 117.9, 79.6, 73.2, 48.6, 36.5; IR (neat) 3295, 2132, 1586, 1491, 1452, 1297, 1106 cm−1; mass spectrum (CI) m/z 212.0940 [C11H10N5 (M+1) requires 212.0936].
5,6-Dihydro-4H-benzo[f][1,2,3]triazolo[1,5-a][1,4]diazepine-6-carbonitrile (22)
A solution of amine 21 (667 mg, 3.02 mmol) in toluene (158 mL) was stirred at 60 °C for 34 h. The cooled reaction was concentrated under reduced pressure, and the residue was purified by flash column chromatography eluting with toluene/EtOAc (1 : 1 → 0 : 1) to give 589 mg of amine 22 (88%) as a colorless solid: mp 133 °C (dec.) (colorless needles from hexanes/CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 7.94 (d, J = 7.9 Hz, 1 H), 7.90 (s, 1 H), 7.75–7.66 (comp, 2 H), 7.61 (app t, J = 7.2 Hz, 1 H), 5.49 (d, J = 5.1 Hz, 1 H), 4.20 (ddd, J = 6.3, 5.1, 4.9 Hz, 1 H), 4.10 (dd, J = 14.6, 4.9 Hz, 1 H), 3.73 (dd, J = 14.6, 6.3 Hz, 1 H); 13C NMR (75 MHz, d6-DMSO) δ 135.4, 135.2, 132.2, 131.0, 130.1, 129.7, 127.0, 123.4, 119.2, 48.9, 36.7; IR (neat) 3312, 2920, 2851, 1495, 1469, 1230, 1136, 1095 cm−1; mass spectrum (CI) m/z 212.0940 [C11H10N5 (M+1) requires 212.0936].
5-(2-Fluorobenzoyl)-5,6-dihydro-4H-benzo[f][1,2,3]triazolo[1,5-a][1,4]diazepine-6-carbonitrile (24)
2-Fluorobenzoyl chloride (90 mg, 68 μL, 0.57 mmol) was added to a solution of amine 22 (60 mg, 0.28 mmol) and pyridine (67 mg, 69 μL, 0.85 mmol) in anhydrous MeCN (1.5 mL) at 0 °C, and the reaction was stirred at room temperature for 1 h. The reaction was diluted with CH2Cl2 (20 mL) and the mixture was washed with aqueous HCl (10 mL, 1.0 M) and saturated aqueous NaHCO3 (10 mL). The organic layer was dried (MgSO4) and concentrated under reduced pressure, and the residue was purified by flash column chromatography eluting with hexanes/EtOAc (4 : 6) to give 93 mg (98%) of amide 24 as a colorless solid: mp 217–219 °C (colorless prisms from hexanes/EtOAc); 1H NMR (500 MHz, DMSO-d6, 100 °C) δ 8.01 (dd, J = 7.8, 1.2 Hz, 1 H), 7.96 (s, 1 H), 7.93–7.80 (m, 1 H), 7.83 (app td, J = 7.8, 1.4 Hz, 1 H), 7.67 (app td, J = 7.8, 1.2 Hz, 1 H), 7.66–7.60 (m, 1 H), 7.54 (app td, J = 7.4, 1.8 Hz, 1 H), 7.41–7.32 (comp, 2 H), 6.90–6.52 (m, 1 H), 5.28–4.89 (m, 1 H), 4.33 (d, J = 15.3 Hz, 1 H); 13C NMR (125 MHz, d6-DMSO, 100 °C) δ 164.5, 157.6 (JC-F = 347.6 Hz), 134.7, 132.8, 132.3 (JC-F = 8.3 Hz), 131.8, 131.3, 131.0, 129.5, 128.6, 124.7 (JC-F = 3.5 Hz), 123.3, 123.3, 121.5 (JC-F = 16.7 Hz), 115.8 (JC-F = 21.0 Hz), 115.3, 47.1, 38.2; IR (neat) 3063, 2923, 1652, 1614, 1450, 1455, 1394, 1325, 1236, 1093 cm−1; mass spectrum (ESI) m/z 356.0918 [C18H12N5OFNa (M+Na) requires 356.0918].
5-Nicotinoyl-5,6-dihydro-4H-benzo[f][1,2,3]triazolo[1,5-a][1,4]diazepine-6-carbonitrile (25)
A mixture of amine 22 (110 mg, 0.52 mmol), nicotinoyl chloride hydrochloride (185 mg, 1.0 mmol) and pyridine (164 mg, 168 μL, 2.1 mmol) in anhydrous CH2Cl2 (3.0 mL) was stirred at rt for 2 h. Saturated aqueous NaHCO3 (5 mL) was added and the reaction was stirred at rt for 20 min. The reaction was then diluted with CH2Cl2 (30 mL) and washed with saturated aqueous NaHCO3 (20 mL). The organic layer was dried (MgSO4) and concentrated under reduced pressure, and the residue was purified by flash chromatography eluting with CH2Cl2/MeOH (24 : 1 → 19 : 1 ) to give 153 mg (92%) of amide 25 as a colorless solid: mp 187–189 °C (colorless microcrystals from i-PrOH); 1H NMR (400 MHz) δ 8.88–8.76, (comp, 2 H), 8.09 (d, J = 8.2 Hz, 1 H), 7.95 (app dt, J = 7.9, 2.0 Hz, 1 H), 7.90–7.74 (comp, 2 H), 7.70–7.46 (comp, 2 H), 7.51 (dd, J = 7.9, 5.0 Hz, 1 H), 6.49 (br s, 1 H), 5.10 (br s, 1 H), 4.34 (br s, 1 H); 13C NMR (100 MHz) δ 168.0, 152.7, 148.4, 135.9, 135.5, 133.8, 132.9, 131.5, 130.6, 130.2, 129.2, 124.7, 124.1, 123.3, 115.1, 47.7, 40.9; IR (neat) 2941, 2361, 1651, 1589, 1501, 1391, 1326, 1238, 1102 cm−1; mass spectrum (CI) m/z 317.1154 [C17H13N6O (M+1) requires 317.1151].
11-Oxo-11,13a-dihydro-9H-benzo[f]isoindolino[1,2-d][1,2,3]triazolo[1,5-a][1,4]diazepine-13a-carbonitrile (28)
A solution of amide 24 (28 mg, 0.084 mmol) in anhydrous DMF (0.7 mL) was added to a suspension of sodium hydride (3.7 mg, 0.092 mmol, 60% dispersion in mineral oil) in DMF (0.7 mL) and the reaction was stirred at room temperature for 2 h. The solution was diluted with toluene (30 mL) and the mixture was washed with H2O (3 × 15 mL) and saturated aqueous NaHCO3 (10 mL). The organic layer was dried (MgSO4) and concentrated under reduced pressure, and the residue was purified by flash column chromatography eluting with hexanes/EtOAc (4 : 6) to give 20 mg (76%) of isoindolinone 28 as a colorless solid: mp 245–247 °C; 1H NMR (400 MHz) δ 8.10 (dd, J = 8.0, 1.2, Hz, 1 H), 8.02 (d, J = 7.6 Hz, 1 H), 7.99 (s, 1 H), 7.87 (app t, J = 7.5 Hz, 1 H), 7.82–7.72 (comp, 3 H), 7.50 (app td, J = 7.8, 1.2 Hz, 1 H), 7.20 (dd, J = 7.8, 1.4 Hz, 1 H), 5.47 (d, J = 15.1 Hz, 1 H), 4.25 (d, J = 15.1 Hz, 1 H); 13C NMR (100 MHz) δ 165.9, 139.2, 135.8, 134.1, 133.7, 132.5, 131.6, 131.3, 130.7, 130.3, 127.0, 126.1, 126.0, 125.4, 125.3, 115.9, 61.0, 35.1; IR (neat) 3084, 2924, 2855, 1714, 1493, 1468, 1358, 1238, 1153, 1104 cm−1; mass spectrum (ESI) m/z 314.1038 [C18H12N5O (M+1) requires 314.1036].
11-(Pyridin-3-yl)-9H-benzo[f]pyrrolo[1,2-d][1,2,3]triazolo[1,5-a][1,4]diazepine (29)
A solution of amide 25 (78 mg, 0.25 mmol) in DMF (2.8 mL) was added dropwise over 2 min to sodium hydride (10.8 mg, 0.27 mmol), and the mixture was stirred at room temperature for 45 min. A solution of triphenylvinylphosphonium bromide (105 mg, 0.28 mmol) in DMF (1.8 mL) was added, and the reaction was stirred at room temperature for 1.5 h. The reaction was diluted with toluene (30 mL) and washed with H2O (3 × 15 mL) and saturated aqueous NaHCO3 (10 mL). The organic layer was dried (MgSO4) and concentrated under reduced pressure, and the residue was purified by flash chromatography eluting with toluene/EtOAc/MeOH (50 : 50 : 1) to give 67 mg (91%) of pyrrole 29 as a pale yellow solid: mp 182–184 °C (pale yellow needles from hexanes/EtOAc); 1H NMR (400 MHz) δ 8.72–8.67 (m, 1 H), 8.67–8.62 (m, 1 H), 8.13–8.06 (m, 1 H), 7.79–7.74 (comp, 2 H), 7.72 (app dt, J = 7.8, 2.0 Hz, 1 H), 7.56–7.48 (comp, 2 H), 7.45 (dd, J = 7.8, 4.9 Hz, 1 H), 6.60 (d, J = 3.9 Hz, 1 H), 6.39 (d, J = 3.9 Hz, 1 H), 5.12 (br s, 2 H); 13C NMR (100 MHz) δ 149.7, 148.9, 136.2, 134.2, 132.7, 132.0, 131.9, 130.9, 129.7, 129.5, 128.6, 128.1, 124.4, 124.0, 123.8, 111.5, 110.7, 37.5; IR (neat) 3031, 2924, 2854, 1567, 1482, 1454, 1421, 1335, 1253, 1231 cm−1; mass spectrum (ESI) m/z 300.1245 [C18H14N5 (M+1) requires 300.1244].
N-Allyl-N-(1-(2-bromophenyl)-3-oxopropyl)pent-4-enamide (34)
Vinyl TBS ether (33) was prepared according to the procedure of Kawakamie and coworkers.46 4-Pentenoyl chloride (32) was prepared according to the procedure of Rosenblum and coworkers and purified by fractional distillation under nitrogen prior to use. 47 Allylamine (1.9 g, 2.5 mL, 33 mmol), MgSO4 (8.1 g, 67 mmol) and 2-bromobenzaldehyde (4.15 g, 22.4 mmol) were combined in CH2Cl2 (44 mL) and the mixture was stirred for 24 h. The mixture was filtered and the filtrate was concentrated in vacuo to give the allylimine, which was used without further purification. Vinyl TBS ether (33) (10.6 g, 67 mmol) and CH2Cl2 (29 mL) were added and the solution was cooled to 0 °C, followed by dropwise addition of 4-pentenoyl chloride (32) (2.9 g, 2.7 mL, 24 mmol). After 5 min, TMSOTf (0.50 g, 0.41 mL, 2.2 mmol) was added dropwise and the mixture was warmed to room temperature and stirred for 40 h. The mixture was concentrated under reduced pressure and the residue was partitioned between CH2Cl2 (70 mL) and saturated aqueous NaHCO3 (70 mL), and the mixture was stirred rapidly for 1 h. The phases were separated, and the aqueous layer was extracted with CH2Cl2 (2 × 40 mL). The combined organic layers were dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with hexanes/EtOAc/MeOH (75 : 25 : 1 → 50 : 50 : 1 → 25 : 75 : 1) to give 5.50 g (70%) of the aldehyde 34 as a viscous, golden oil: 1H NMR (600 MHz) (9 : 1 rotamer mixture, data given for the major rotamer) δ 9.76 (dd, J = 2.7, 2.1 Hz, 1 H) 7.64–7.58 (m, 1 H), 7.37–7.30 (comp, 2 H), 7.20 (ddd, J = 8.1, 6.6, 2.4 Hz ,1 H), 6.26 (dd, J = 8.8, 6.3 Hz, 1 H), 5.90–5.79 (m, 1 H), 5.54–5.44 (m, 1 H), 5.08–4.94 (comp, 4 H), 3.72 (ddt, J = 17.4, 5.1, 1.7 Hz, 1 H), 3.64 (dd, J = 17.4, 5.9 Hz, 1 H), 3.18 (ddd, J = 15.6, 8.8, 2.7 Hz, 1 H), 2.99 (ddd, J = 15.6, 6.3, 2.1 Hz, 1 H), 2.48–2.34 (comp, 4 H); 13C NMR (150 MHz) (9 : 1 rotamer mixture, data given for the major rotamer): δ 199.6, 172.9, 137.4, 137.2, 133.9, 133.6, 129.8, 129.4, 127.6, 125.5, 117.2, 115.2, 53.2, 47.6, 46.4, 33.0, 29.2; IR (neat) 2979, 1723, 1643, 1470, 1415, 1025 cm−1; mass spectrum (ESI) m/z 350.0750 [C17H21NO2 79Br (M+1) requires 350.0750].
1-((3aR,6S,7aS)-6-(2-Bromophenyl)-1-methyltetrahydroisoxazolo[4,3-c]pyridin-5(1H,3H,6H)-yl)-pent-4-en-1-one (35)
N-Methylhydroxylamine hydrochloride (1.25 g, 15.0 mmol), aldehyde 34 (3.50 g, 9.99 mmol) and Et3N (3.0 g, 4.2 mL, 30 mmol) were combined in toluene (116 mL) and the mixture was heated under reflux for 90 min. After cooling to room temperature, H2O (60 mL) was added and the layers were separated. The aqueous layer was saturated with NaCl then extracted with CH2Cl2 (2 × 30 mL). The combined organic layers were dried (MgSO4) and concentrated under reduced pressure and the residue was purified by column chromatography eluting with hexanes/EtOAc/MeOH (75 : 25 : 1 → 50 : 50 : 1 → 0 : 95 : 5) to afford 3.63 g (96%) of the isoxazolidine 35 as a highly viscous, pale yellow gum that crystallized on standing: mp 70–71 °C; 1H NMR (600 MHz) (rotamers) δ 7.55 (dd, J = 8.1, 1.1 Hz, 0.75 H), 7.51 (d, J = 7.7Hz, 0.25 H), 7.35–7.30 (m, 0.75 H), 7.25 (dd, J = 8.5, 1.7 Hz, 0.75 H), δ 7.25–7.20 (m, 0.25 H), 7.18–7.13 (m, 0.75 H), 7.11 (dd, J = 7.8, 1.2 Hz, 0.25 H), 7.08–7.04 (m, 0.25 H), 5.86–5.77 (m, 0.25 H), 5.73–5.63 (m, 0.75 H), 5.23 (dd, J = 13.7, 4.8 Hz, 0.25 H), 5.07–5.00 (comp, 1 H), 5.00–4.92 (comp, 1 H), 4.91–4.84 (comp, 1.5 H), 4.20–4.10 (comp, 1 H), 3.97 (dd, J = 13.7, 5.4 Hz, 0.25 H), 3.60–3.53 (comp, 1.25 H), 3.07–2.88 (comp, 2.75 H), 2.70 (s, 3 H), 2.55–2.47 (m, 0.25 H), 2.47–2.39 (m, 0.25 H), 2.39–2.32 (m, 1.5 H), 2.32–2.20 (comp, 1.5 H), 2.16–2.07 (m, 0.75 H), 1.86–1.77 (m, 0.75 H), 1.77–1.67 (comp, 1 H); 13C NMR (150 MHz) (rotamers) δ 173.2, 171.2, 142.7, 142.5, 137.4, 137.2, 133.2, 129.2, 128.7, 128.3, 127.9, 126.4, 125.3, 121.8, 121.1, 115.3, 115.1, 68.3, 68.0, 64.6, 55.7, 54.6, 43.9, 43.7, 42.8, 39.6, 33.3, 33.1, 32.6, 31.8, 29.0, 28.9; IR (neat) 2956, 2876, 1650, 1418, 1240, 1026, 915, 756 cm−1; mass spectrum (ESI) m/z 379.1017 [C18H24N2O2 79Br (M+1) requires 379.1016].
(3aR,6S,7aS)-6-(2-Bromophenyl)-1-methyloctahydroisoxazolo[4,3-c]pyridine (36)
Iodine (6.67 g, 26.3 mmol) was added to a stirred solution of the amide 35 (2.00 g, 5.27 mmol) in THF (41 mL) and 1.5 M aqueous HCl (15 mL) at 0 °C. The stirred mixture was warmed to room temperature and after 60 min, saturated aqueous Na2S2O3 was added until the solution became colorless. The mixture was then diluted with H2O (120 mL) and washed with Et2O (3 × 120 mL). The pH of the aqueous layer was adjusted to 12 by addition of 1 M aqueous NaOH and the solution was then saturated with NaCl and extracted with CH2Cl2 (4 × 100 mL). The combined CH2Cl2 extracts were dried (MgSO4) and concentrated under reduced pressure and the residue was purified by flash column chromatography eluting with EtOAc/MeOH (100 : 1 → 90 : 10) to give 1.40 g (89%) of the amine 36 as a brown solid: mp 106–108 °C (pale yellow needles from ethyl acetate/hexanes); 1H NMR (400 MHz): δ 7.60–7.49 (comp, 2 H), 7.34–7.27 (m, 1 H), 7.11 (td, J = 7.7, 1.6 Hz, 1 H), 4.24 (dd, J = 9.2, 7.2 Hz, 1 H), 3.96 (dd, J = 9.8, 2.2 Hz, 1 H), 4.02–3.94 (m, 1 H), 3.29 (d, J = 12.5 Hz, 1 H), 3.26–3.16 (comp, 2 H), 3.07–2.95 (m, 1 H), 2.68 (s, 3 H), 2.12–2.02 (m, 1 H), 1.68–1.50 (comp, 2 H); 13C NMR (100 MHz): 142.7, 132.7, 128.7, 127.9, 127.9, 123.2, 68.0, 64.4, 57.7, 44.6, 44.6, 37.8, 34.8; IR (neat) 3313, 2950, 1469, 1438, 1023 cm−1; mass spectrum (ESI) m/z 297.0597 [C13H18NO2 79Br (M+1) requires 297.0602].
1-((2S,4S,5R)-2-(2-Bromophenyl)-5-(hydroxymethyl)-4-(methylamino)piperidin-1-yl)ethanone (38)
Zinc dust (3.9 g, 60 mmol) was added at 0 °C to a stirred solution of isoxazolidine 376a (1.00 g, 2.95 mmol) in 10% aqueous HCl (45 mL). After 1 h, the mixture was filtered through Celite rinsing with 10% aqueous HCl (20 mL). The pH of the combined filtrate and rinsings was adjusted to 12 by the addition of 30% aqueous NH4OH and the mixture was extracted with CH2Cl2 (4 × 50 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure and the residue was purified by flash column chromatography eluting with CH2Cl2/MeOH (100 : 1 → 1 : 1) to give 837 mg (83%) of the amino alcohol 38 as a clear, colorless gum: 1H NMR (500 MHz, DMSO-d6, 130 °C) δ 7.53–7.57 (m, 1 H), 7.32 (dd, J = 7.9, 1.2 Hz, 1 H), 7.32–7.27 (m, 1 H), 7.14 (ddd, J = 7.9, 6.0, 2.7 Hz, 1 H), 5.21 (app t, J = 7.7 Hz, 1 H), 4.16 (dd, J = 13.7, 6.1 Hz, 1 H), 3.59 (dd, J = 11.0, 5.4 Hz, 1 H), 3.55 (dd, J = 11.0, 6.4 Hz, 1 H), 3.03 (br s, 2 H), 2.84 (ddd, J = 9.1, 6.1, 3.3 Hz, 1 H), 2.18–2.12 (comp, 2 H), 2.16 (s, 3 H), 1.95–1.90 (m, 1 H), 1.94 (ddd, J = 13.9, 8.8, 7.7 Hz, 1 H), 1.92 (s, 3 H); 13C NMR (125 MHz, DMSO-d6, 130 °C) δ 168.9, 142.5, 132.0, 127.6, 127.1, 126.0, 120.4, 59.9, 54.6, 54.5, 41.5, 39.2, 33.1, 31.0, 20.6; IR (neat) 3323, 2923, 1633, 1420, 1024 cm−1; mass spectrum (ESI) m/z 341.0858 [C15H22N2O2 79Br (M+1) requires 341.0859].
1-[(1S,9S,10R)-10-(Hydroxymethyl)-8-methyl-8,12-diazatricyclo[7.3.1.02,7]trideca-2(7),3,5-trien-12-yl]ethan-1-one (39)
Pd(OAc)2 (6.0 mg, 0.027 mmol) and (±)-BINAP (20 mg, 0.032 mmol) were combined in toluene (5 mL) and heated at 40 °C until all solid had dissolved. The solution was cooled to room temperature over 10 min, then combined with amine 38 (92 mg, 0.27 mmol) and Cs2CO3 (177 mg, 0.543 mmol). The mixture was heated under reflux for 14 h, then cooled to room temperature and filtered through celite, rinsing with toluene (5 mL). The combined filtrate and rinsings were concentrated under reduced pressure and the residue was purified by flash column chromatography eluting with CHCl3/MeOH (95 : 5) to afford 49 mg (70%) of the tetrahydroquinoline 39 as a pale yellow gum: 1H NMR (600 MHz) (rotamers) δ 7.22–7.15 (comp, 1.6 H), 7.01 (dd, J = 7.6, 1.5 Hz, 0.4 H), 6.62–6.55 (comp, 2 H), 5.91 (br s, 0.6 H), 4.93 (br s, 0.4 H), 4.36 (dd, J = 13.3, 4.8 Hz, 0.4 H), 3.83 (d, J = 1.8 Hz, 0.4 H), 3.72 (d, J = 2.1 Hz, 0.6 H), 3.66 (dd, J = 10.4, 7.6 Hz, 0.6 H), 3.60–3.49 (comp, 2 H), 3.14 (s, 1.2 H), 3.08 (s, 1.8 H), 2.72 (br s, 1 H), 2.69 (app t, J = 12.8 Hz, 0.6 H), 2.34 (s, 1.2 H), 2.11 (app t, J = 13.3 Hz, 0.4 H), 2.05–1.81 (comp, 3 H), 2.04 (s, 1.8 H); 13C NMR (150 MHz) (rotamers) δ 168.7, 168.1, 146.5, 146.4, 129.9, 129.7, 129.2, 128.9, 120.5, 119.3, 115.6, 115.3, 109.7, 109.5, 62.7, 62.3, 54.4, 53.6, 46.6, 45.8, 45.7, 42.0, 40.0, 39.8, 36.4, 29.3, 28.4, 22.0, 22.0; IR (neat) 3392, 2931, 1614, 1503, 1436, 1045 cm−1; mass spectrum (CI) m/z 260.1522 [C15H20N2O2 (M·+) requires 260.1525].
1-((3aR,6S,7aS)-6-(2-Bromophenyl)-1-methyltetrahydroisoxazolo[4,3-c]pyridin-5(1H,3H,6H)-yl)-2-phenylethanone (40)
Phenylacetyl chloride (64 mg, 55 μL, 0.41 mmol) was added dropwise to a solution of amine 36 (100 mg, 0.336 mmol) and Et3N (47 mg, 65 μL, 0.46 mmol) in CH2Cl2 (2 mL). After 1 h, the mixture was diluted with CH2Cl2 (10 mL) and partitioned with saturated aqueous NaHCO3 (10 mL). The phases were separated and the aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The combined organic extracts were dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with hexanes/EtOAc (1 : 1 → 1 : 3) to afford 117 mg (84%) of the amide 40 as a white foam: 1H NMR (400 MHz) (rotamers) δ 7.57 (d, J = 8.0 Hz, 0.7 H), 7.51 (d, J = 7.8 Hz, 0.3 H), 7.35–7.03 (comp, 7.7 H), 7.00 (d, J = 6.6 Hz, 0.3 H), 5.24 (dd, J = 13.5, 4.7 Hz, 0.3 H), 5.04 (dd, J = 12.5, 5.1 Hz, 0.7 H), 5.01–4.93 (m, 0.7 H), 4.20–4.08 (m, 0.7 H), 4.05–3.92 (comp, 0.6 H), 3.75 (s, 0.6 H), 3.60–3.53 (m, 0.7 H), 3.48–3.39 (comp, 0.6 H), 3.36 (d, J = 15.1 Hz, 0.7 H), 3.28 (d, J = 15.1 Hz, 0.7 H), 3.10–2.82 (comp, 2.4 H), 2.75–2.53 (m, 0.3 H), 2.66 (s, 2 H), 2.64 (s, 1 H), 2.44–2.27 (comp, 1 H), 1.77–1.57 (comp, 1 H); 13C NMR (100 MHz) (rotamers) δ 171.8, 169.9, 142.5, 142.4, 134.6, 134.5, 133.4, 133.2, 129.4, 129.0, 128.8, 128.7, 128.5, 128.4, 127.8, 127.1, 126.8, 126.4, 125.3, 121.9, 121.1, 68.3, 67.9, 64.5, 55.8, 54.7, 44.3, 43.7, 43.5, 42.5, 42.1, 40.6, 39.9, 32.8, 31.8; IR (neat) 2955, 2874, 1646, 1414, 1026 cm−1; mass spectrum (ESI) m/z 415.1016 [C22H25N2O2 79Br (M+1) requires 415.1016].
(1S,8R,12R,16S)-8-Allyl-15-methyl-8-phenyl-14-oxa-10,15-diazatetracyclo[8.7.0.02,7.012,16]heptadeca-2(7),3,5-trien-9-one (41)
LDA was prepared by addition of n-BuLi (3.90 mL, 9.0 mmol, 2.3 M in hexanes) to a solution of diisopropylamine (1.09 g, 0.78 mL, 10.8 mmol) in THF (9.0 mL) at 0 °C. After 30 minutes, the solution was warmed to room temperature. A portion of the LDA solution so obtained (0.65 M in THF/hexanes, 2.2 mL, 1.4 mmol) was added dropwise to a solution of amide 40 (100 mg, 0.241 mmol) and DMPU (0.37 g, 0.35 mL, 2.9 mmol) in THF (3 mL) at −78 °C. The solution was then warmed to 0 °C and stirred for 1 h. The mixture was recooled to −100 °C, and a solution of allyl bromide (0.35 g, 0.25 mL, 2.9 mmol) in THF (2 mL) was added dropwise. The mixture was warmed to −78 °C and held at this temperature for 20 min, before warming to 0 °C. Toluene (5 mL) and saturated aqueous NH4Cl (5 mL) were added, and the mixture was concentrated under reduced pressure to remove the THF. The residue was partitioned between H2O (5 mL) and toluene (5 mL), and the layers were separated. The toluene layer was washed with H2O (4 × 10 mL), dried (MgSO4) and concentrated under reduced pressure. The residue was purified by column chromatography eluting with pentane/ EtOAc/MeOH (25 : 75 : 1 → 0 : 100 : 1) to afford 60 mg (66%) of the dihydroisoquinolin-3-one 41 as a yellow gum: 1H NMR (400 MHz) δ 7.36–7.27 (m, 2 H), 7.26–7.12 (comp, 7 H), 5.50–5.38 (m, 1 H), 5.04 (dd, J = 17.1, 1.5 Hz, 1 H), 5.03–4.94 (m, 1 H), 4.90 (dd, J = 10.3, 1.5 Hz, 1 H), 4.45 (dd, J = 12.6, 2.2 Hz, 1 H), 4.08–4.00 (m, 1 H), 3.77 (dd, J = 14.0, 6.8 Hz, 1 H), 3.39–3.28 (m, 1 H), 3.14–2.97 (comp, 3 H), 2.88 (dd, J = 14.0, 7.2 Hz, 1 H), 2.58 (s, 3 H), 2.26–2.16 (m, 1 H), 1.47 (app q, J = 12.6 Hz, 1 H); 13C NMR (75 MHz) δ 170.4, 145.0, 136.0, 134.4, 134.2, 129.0, 128.5, 127.8, 127.3, 127.2, 127.1, 125.4, 118.3, 67.4, 64.0, 57.4, 53.8, 44.5, 43.4, 40.6, 38.1, 37.9; IR (neat) 2953, 2680, 1643, 1443, 1243 cm−1; mass spectrum (CI) m/z 374.1994 [C24H26N2O2 (M+1) requires 374.1994].
(1S,12R,16S)-15-Methyl-8-phenyl-14-oxa-8,10,15-triazatetracyclo[8.7.0.02,7.012,16]heptadeca-2(7),3,5-trien-9-one (43)
Pd(OAc)2 (2.4 mg, 0.011 mmol) and (±)-BINAP (7.8 mg, 0.013 mmol) were combined in toluene (0.8 mL) and heated at 40 °C until all solid had dissolved. The mixture was cooled to room temperature, then combined with amine 36 (30 mg, 0.10 mmol) and Cs2CO3 (67 mg, 0.21 mmol) in a screw-cap vial. The mixture was cooled to 0 °C before dropwise addition of a solution of phenyl isocyanate (24 mg, 0.20 mmol) in toluene (0.2 mL). The mixture was warmed to room temperature and stirred for 30 min, before heating at 120 °C for 13 h. The mixture was filtered through Celite and concentrated under reduced pressure and the residue was purified by flash column chromatography eluting with EtOAc/MeOH (100 : 1 → 90 : 10) to give 31 mg (92%) of the dihydroquinazolin-2-one 43 as a yellow glass: 1H NMR (400 MHz) δ 7.50 (t, J = 7.6 Hz, 2 H), 7.41 (t, J = 7.6 Hz, 1 H), 7.30–7.25 (m, 2 H), 7.10 (dd, J = 7.3, 1.6 Hz, 1 H), 7.04 (td, J = 8.0, 1.6 Hz, 1 H), 6.98 (td, J = 7.3, 1.1 Hz, 1 H), 6.21 (dd, J = 8.0, 1.1 Hz, 1 H), 4.62 (dd, J = 14.3, 2.0 Hz, 1 H), 4.56 (dd, J = 12.5, 2.3 Hz, 1 H), 4.25 (app t, J = 8.6 Hz, 1 H), 3.78 (app t, J = 8.6 Hz, 1 H), 3.39–3.23 (comp, 2 H), 3.13–3.02 (m, 1 H), 2.69 (s, 3 H), 2 .29–2.18 (m, 1 H), 1.99 (app q, J = 12.5 Hz, 1 H); 13C NMR (75 MHz) δ 153.9, 139.1, 138.3, 2 × 129.8, 128.2, 128.1, 125.1, 122.3, 121.4, 115.1, 67.8, 64.1, 55.0, 44.3, 41.5, 38.5, 35.6; IR (neat) 2952, 1659, 1465, 1289, 1266 cm−1; mass spectrum (ESI) m/z 336.1706 [C20H22N3O2 (M+1) requires 336.1707].
Allyl-[1-(2-bromo-6-chloro-phenyl)-but-3-enyl]carbamic acid benzyl ester (47)
A mixture of allylamine (971 mg, 1.27 mL, 17.0 mmol), 2-bromo-6-chlorobenzaldehyde (46) (1.87 g, 8.51 mmol) and 4 Å molecular sieves (2.0 g) was stirred in CH2Cl2 (20 mL) for 12 h at room temperature. The sieves were removed by filtration through Celite, and the filtrate was concentrated under reduced pressure to afford 2.20 g (ca. 100%) of crude imine, which was used directly in the next step. Benzyl chloroformate (1.60 g, 1.34 mL, 9.39 mmol) was added to a solution of imine in THF (17 mL) and was heated at 60 °C for 1 h. The reaction was then cooled to −78 °C, and a freshly prepared solution of allylzinc bromide48 (ca. 13.1 mmol) in THF (10 mL) was added and the reaction stirred for 2 h. The cooling bath was removed, and the reaction was allowed to warm to 0 °C and quenched with saturated aqueous NH4Cl (~10 mL). The mixture was partitioned between water (100 mL) and Et2O (100 mL), and the layers were separated. The aqueous layer was extracted with Et2O (3 × 50 mL) and the combined organic extracts were washed with saturated aqueous NaHCO3 (100 mL) and brine (50 mL), dried (MgSO4), filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography eluting with hexanes/EtOAc (100 : 0 → 95 : 5 → 90 : 10 → 85 : 15) to give 3.31 g (89%) of 47 as a pale yellow oil: 1H NMR (400 MHz) δ 7.60–7.40 (m, 1 H), 7.40–7.10 (comp, 6 H), 7.00 (t, J = 10.8 Hz, 1 H), 5.94–5.82 (m, 1 H), 5.80–5.66 (comp, 2 H), 5.35–4.80 (comp, 6 H), 4.32 (br d, J = 22.4 Hz, 1 H), 4.05 (dd, J = 23.0, 6.80 Hz, 1 H), 3.05–2.94 (m, 1 H), 2.89–2.81 (m, 1 H); 13C NMR (75 MHz) δ 155.9, 137.3, 136.4, 135.5, 133.4, 132.5, 130.1, 128.7, 128.2, 127.9, 127.7, 117.9, 115.1, 67.1, 59.3, 47.6, 35.0; IR (neat) 3078, 2978, 1715, 1448, 1401, 1254 cm−1; mass spectrum (ESI) m/z 434.0518 [C21H22NO235Cl79Br (M+1) requires 434.0517].
Benzyl 6-(2-bromo-6-chlorophenyl)-5,6-tetrahydropyridine-1(2H)-carboxylate (48)
Grubbs 2nd generation catalyst (49 mg, 58 μmol) was added to a solution of 47 (500 mg, 1.15 mmol) in CH2Cl2 (25 mL). After stirring for 14.5 h at room temperature, the reaction was concentrated under reduced pressure. The residue was dissolved in 15% EtOAc/hexanes and filtered through a plug of silica gel to remove the catalyst. The filter plug was rinsed with 15% EtOAc/hexanes (3 × 20 mL) and the combined washings and filtrate were concentrated under reduced pressure to give the crude product, which was purified via flash column chromatography eluting with hexanes/EtOAc (100 : 0 → 95 : 5) to give 434 mg (93%) of 48 as a colorless oil: 1H NMR (400 MHz) δ 7.42 (d, J = 7.8 Hz, 1 H), 7.28–7.05 (comp, 6 H), 6.97 (t, J = 8.4 Hz, 1 H), 6.16–6.03 (comp, 2 H), 5.43 (t, J = 8.0 Hz, 1 H), 5.03 (d, J = 11.6 Hz, 1 H), 4.92–4.84 (m, 1 H), 4.47–4.43 (m, 1 H), 4.00 (d, J = 16.8 Hz, 1 H), 2.49 (app d, J = 8.0 Hz, 2 H); 13C NMR (75 MHz) δ 155.9, 139.9, 136.6, 133.5, 132.7, 130.4, 128.5, 128.4, 128.3, 128.0, 127.4, 126.3, 123.8, 67.5, 55.1, 53.7, 42.7, 27.7; IR (neat) 3044, 2944, 2851, 1695, 1415, 1328, 1228 cm−1; mass spectrum (ESI) m/z 428.0023 (M+Na) requires 428.0023].
10-Chloro-1,10b-dihydropyrido[2,1-a]isoindol-6(4H)-one (52)
n-BuLi (0.16 mL, 0.33 mmol, 2.04 M hexanes) was added to a solution of dry THF (2.0 mL) and 48 (135 mg, 0.33 mmol) at −100 °C and for 5 min. The reaction was warmed to −78 °C for 10 min then quenched with a solution of MeOH and saturated aqueous NH4Cl (1:1; 2 mL). After warming to room temperature, the mixture was concentrated under reduced pressure and the residue purified via radial plc eluting with hexanes/EtOAc (100 : 0 → 90 : 10 → 80 : 20 → 70 : 30) to give 41 mg (60%) of 52 as a yellow oil: 1H NMR (400 MHz) δ 7.78 (d, J = Hz, 1 H), 7.50 (d, J = 8.0 Hz, 1 H), 7.43 (t, J = 7.8 Hz, 1 H), 5.94–5.85 (m, 2 H), 4.70–4.64 (m, 1 H), 4.54 J = 10.8, 4.8 Hz, 1 H), 3.88–3.86 (m, 1 H), 3.19–3.12 (m, 1 H), 2.02–1.92 (m, 1 H); 13C NMR (75 MHz) δ 166.1, 143.5, 135.1, 131.9, 130.0, 129.4, 128.7, 127.9, 127.2, 123.6, 123.3, 122.4; IR (neat) 3038, 2851, 1688, 1468, 1421, 1274 cm−1; mass spectrum (ESI) m/z 220.0524 [C12H11NO35Cl (M+1) requires 220.0524].
2-(2-Bromo-6-chlorophenyl)-3,4-dihydro-2H-pyridine-1-carboxylic acid benzyl ester (53)
A microwave vial containing tetrahydropyridine 48 (225 mg, 0.55 mmol) in dry THF (2.0 mL) and Et3N mL) was degassed with Ar for 20 min with stirring. The vial was sealed and heated in the microwave (120 °C, 300 W) for 50 min with vigorous stirring. The reaction was cooled to room temperature and concentrated under reduced pressure. EtOAc (30 mL) was added and the mixture was filtered through Celite and rinsed with EtOAc (2 × 10 mL). The combined filtrate and washings were concentrated under reduced pressure, and the residue was purified via radial plc eluting with hexanes/EtOAc (100 : 0 → 95 : to give 173 mg (77%) of 53 as a colorless oil: 1H NMR (400 MHz) δ 7.50–7.15 (comp, 6 H), 7.02–6.86 (comp, 3 H), 5.63–5.53 (m, 1 H), 5.25–4.85 (comp, 3 H), 2.20–1.98 (comp, 4 H); 13C NMR (125 MHz) (rotamers) δ 153.4, 152.7, 139.0, 138.5, 136.1, 135.4, 133.6, 132.7, 131.8, 130.8, 129.4, 127.8, 126.5, 124.8, 120.5, 107.1, 67.4, 57.2, 54.9, 26.6, 19.8; IR (neat) 3073, 2942, 2846, 1712, 1657, 1403, 1320, 1073, 922 cm−1; mass spectrum (ESI) m/z 406.0206 [C19H18NO235Cl79Br (M+1) requires 406.0209].
ACKNOWLEDGMENTS
We thank the National Institutes of Health (GM 24539 and GM 86192) and the Robert A. Welch Foundation (F-0652) for their generous support of this work. We are also grateful to Dr. Richard (Materia, Inc.) for providing metathesis catalysts used in these studies.
Footnotes
This paper is dedicated to my longtime friend Al Padwa, a true innovator in developing novel entries to diverse classes of heterocyclic compounds, on the occasion of his 75th birthday.
REFERENCES
- 1.For the origin of the term “privileged scaffolds”, see: Evans BE, Rittle KE, Bock MG, DiPardo RM, Freidinger RM, Whitter WL, Lundell GF, Veber DF, Anderson PS, Chang RSL, Lotti VJ, Cerino DJ, Chen TB, Kling PJ, Kunkel KA, Springer JP, Hirshfield J. J. Med. Chem. 1988;31:2235. doi: 10.1021/jm00120a002..
- 2.For reviews on privileged scaffolds, see: Horton DA, Bourne GT, Smythe ML. Chem. Rev. 2003;103:893. doi: 10.1021/cr020033s.; DeSimone RW, Currie KS, Mitchell SA, Darrow JW, Pippin DA. Comb. Chem. High Throughput Screening. 2004;7:473. doi: 10.2174/1386207043328544.; Constantino L, Barlocco D. Curr. Med. Chem. 2006;13:65.; Welsch ME, Snyder SA, Stockwell BR. Curr. Opin. Chem. Biol. 2010;14:347. doi: 10.1016/j.cbpa.2010.02.018..
- 3.Martin SF, Benage B, Hunter JE. J. Am. Chem. Soc. 1988;110:5925. [Google Scholar]
- 4.Martin SF. Acc. Chem. Res. 2002;35:895. doi: 10.1021/ar950230w. [DOI] [PubMed] [Google Scholar]
- 5.Cheng B, Sunderhaus JD, Martin SF. Org. Lett. 2010;12:3622. doi: 10.1021/ol101356u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Sunderhaus JD, Dockendorff C, Martin SF. Org. Lett. 2007;9:4223. doi: 10.1021/ol7018357. [DOI] [PubMed] [Google Scholar]; (b) Sunderhaus JD, Dockendorff C, Martin SF. Tetrahedron. 2009;65:6454. doi: 10.1016/j.tet.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sunderhaus JD, Martin SF. Chem. Eur. J. 2009;15:1300. doi: 10.1002/chem.200802140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For preliminary accounts of some of this work, see: Donald JR, Martin SF. Org. Lett. 2011;13:852. doi: 10.1021/ol1028404.; Sahn JJ, Su JY, Martin SF. Org. Lett. 2011;13:2590. doi: 10.1021/ol200709h.; Hardy S, Martin SF. Org. Lett. 2011;13:3102. doi: 10.1021/ol201010s.; Granger BA, Kaneda K, Martin SF. Org. Lett. 2011;13:4542. doi: 10.1021/ol201739u..
- 8.Gavras I, Vlahakos D, Melby JC, Gavras H. J. Clin. Pharm. 1984;24:343. doi: 10.1002/j.1552-4604.1984.tb02786.x. [DOI] [PubMed] [Google Scholar]
- 9.Asaoka T, Yazawa K, Mikami Y, Takahashi K. J. Antibiot. 1982;35:1708. doi: 10.7164/antibiotics.35.1708. [DOI] [PubMed] [Google Scholar]
- 10.Francois G, Bringmann G, Phillipson JD, Boyd MR, Assi LA, Schneider C, Timperman G. PCT Int. Appl. WO 9521616 A1 19951717 [Google Scholar]
- 11.(a) Worrall DE. Org. Syn. 1929;9:66. [Google Scholar]; (b) Schumacher RW, Davidson BS. Tetrahedron. 1999;55:935. [Google Scholar]; (c) Mjalli AMM, Gohimmukkulah DR, Tyagi S. PCT Int. Appl. WO 2006093823 [Google Scholar]
- 12.Slougui N, Rousseau G. Synth. Commun. 1987;17:1. [Google Scholar]
- 13.Kawate T, Nakagawa M, Yamazaki H, Hirayama M, Hino T. Chem. Pharm. Bull. 1993;41:287. [Google Scholar]
- 14.Cledera P, Avendaño C, Menéndez JC. Tetrahedron. 1998;54:12349. [Google Scholar]
- 15.Dong Y, Chollet J, Vargas M, Mansour NR, Bickle Q, Alnouti Y, Huang J, Keiser J, Vennerstrom JL. Bioorg. Med. Chem. Lett. 2010;20:2481. doi: 10.1016/j.bmcl.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 16.This procedure can be telescoped in a one pot procedure, although in preliminary experiments we found the yields were less reproducible.
- 17.For recent reviews on the use of α-amino nitriles in the generation of molecular diversity, see: Opatz T. Synthesis. 2009;12:1941.; González-Vera JA, García-López MT, Herranz R. Mini-Rev. Org. Chem. 2008;5:209.; Enders D, Shilvock JP. Chem. Soc. Rev. 2000;29:359..
- 18.For a recent review, see: Galatsis P. In: Name Reactions for Functional Group Transformations. Li JJ, Corey EJ, editors. Wiley; Hoboken: 2007. p. 477..
- 19.Pelkey ET, Gribble GW. Tetrahedron Lett. 1997;38:5603. [Google Scholar]
- 20.Bertelli L, Biagi G, Giorgi I, Livi O, Manera C, Scartoni V, Martini C, Giannaccini G, Trincavelli L, Barili PL. Farmaco. 1998;53:305. doi: 10.1016/s0014-827x(98)00025-1. [DOI] [PubMed] [Google Scholar]
- 21.(a) del Pozo C, Macías A, López-Oritz F, Maestro MA, Alonso E, González J. Eur. J. Org. Chem. 2004:535. [Google Scholar]; (b) Mehta PD, Sengar NPS, Pathak AK. Eur. J. Med. Chem. 2010;45:5541. doi: 10.1016/j.ejmech.2010.09.035. [DOI] [PubMed] [Google Scholar]
- 22.Salcedo A, Neuville L, Zhu J. J. Org. Chem. 2008;73:3600. doi: 10.1021/jo800266y. [DOI] [PubMed] [Google Scholar]
- 23.(a) Baldwin JJ, Claremon DA, Elliott JM, Liverton N, Remy DC, Selnick HG, Merck & Co. Inc. World Patent WO 9514471 (A1) 1995; (b) Lynch JJ, Jr., Salata JJ, Merck & Co. Inc. World Patent WO 9800405 (A1) 1998
- 24.(a) Cooney JV, McEwen WE. J. Org. Chem. 1981;46:2570. [Google Scholar]; (b) Cooney JV, Beaver BD, McEwen WE. J. Heterocycl. Chem. 1985;22:635. [Google Scholar]
- 25.De Lucca GV, Otto MJ. Bioorg. Med. Chem. Lett. 1992;2:1639. [Google Scholar]
- 26.Broggini G, Garanti L, Molenti G, Zecchi G. Heterocycles. 1999;51:1295. [Google Scholar]
- 27.Boulouard M, Gillard AC, Guillaumat PO, Daoust M, Legrand E, Quermonne MA, Rault S. Pharm. Pharmacol. Commun. 1998;4:43. [Google Scholar]
- 28.Xiao D, Palani A, Wang C, Tsui H-C, Shih N-Y, Reichard GA. European Patent EP 1,747,221. 2005
- 29.For the original report of this method to cleave 4-pentenamides, see: Madsen R, Roberts C, Fraser-Reid B. J. Org. Chem. 1995;60:7920..
- 30.For the only other known example of a tandem enolate arylation/alkylation process, see: Goehring RR, Sachdeva YP, Pisipati JS, Sleevi MC, Wolfe JF. J. Am. Chem. Soc. 1985;107:435..
- 31.Ferraccioli R, Carenzi D. Synthesis. 2003;9:1383. [Google Scholar]
- 32.For a similar reaction, using aryl iodides in conjunction with pre-formed thioureas, see: Orain D, Blumstein A-C, Tasdelen E, Haessig S. Synlett. 2008;16:2433..
- 33.Jones S, Johnson J, Ives J, Hedberg K, Dunaiskis A, Chapin D, Nielsen J, Liston D, Chen YL. J. Med. Chem. 1994;37:1996. doi: 10.1021/jm00039a013. [DOI] [PubMed] [Google Scholar]
- 34.Jacobson AE, Mokotoff M. J. Med. Chem. 1970;13:7. doi: 10.1021/jm00295a002. [DOI] [PubMed] [Google Scholar]
- 35.(a) Povarov LS. Russ. Chem. Rev. 1967;36:656. [Google Scholar]; (b) Kouznetsov VV. Tetrahedron. 2009;65:2721. [Google Scholar]
- 36.Xia C, Heng L, Ma D. Tetrahedron Lett. 2002;43:9405. [Google Scholar]
- 37.Minagawa K, Kouzuki S, Kamigauchi T. J. Antibiot. 2002;55:165. doi: 10.7164/antibiotics.55.165. [DOI] [PubMed] [Google Scholar]
- 38.Luci DK, Lawson EC, Ghosh E, Kinny WA, Smith CE, Qi J, Wang Y, Minor LK, Maryanoff BE. Tetrahedron Lett. 2009;50:4958. [Google Scholar]
- 39.Curtin ML, Frey RR, Heyman RH, Sarris KA, Steinman DH, Holmes JH, Bousquet PJ, Cunha GA, Moskey MD, Ahmed AA, Pease LJ, Glaser KB, Stewart KD, Davidsen SK, Michaelides MR. Bioorg. Med. Chem. Lett. 2004;14:4505. doi: 10.1016/j.bmcl.2004.06.041. [DOI] [PubMed] [Google Scholar]
- 40.Parham WE, Bradsher CK. Acc. Chem. Res. 1982;15:300. [Google Scholar]
- 41.Hinschberger A, Butt S, Lelong B, Boulouard M, Dumuis A, Francois D, Bureau R, Pfeiffer B, Renard P, Rault S. J. Med. Chem. 2003;46:138. doi: 10.1021/jm020954v. [DOI] [PubMed] [Google Scholar]
- 42.For example, see: Painter FF, Wanner KT. Tetrahedron. 1994;50:3113..
- 43.Still WC, Kahn M, Mitra A. J. Org. Chem. 1978;43:2923. [Google Scholar]
- 44.Sperger CA, Wanner KT. Tetrahedron. 2009;65:5824. [Google Scholar]
- 45.King AO, Negishi E, Villani FJ, Jr., Silveira A., Jr. J. Org. Chem. 1978;43:358. [Google Scholar]
- 46.Kawakamie Y, Aoki T, Yamashita Y. Polym. Bull. 1987;18:473. [Google Scholar]
- 47.Rosenblum SB, Huynh T, Afonso A, Davis HR., Jr. Tetrahedron. 2000;56:5735. [Google Scholar]
- 48.Negishi E, Boardman LD, Vahid S, Bagheri A, Stoll T, Tour JM, Rand CL. J. Am. Chem. Soc. 1988;110:5383. [Google Scholar]