

Abstract
Barleria alluaudii and Diospyros maritima were both investigated as part of an ongoing search for synergistic TRAIL (tumor necrosis factor-α-related apoptosis-inducing ligand) sensitizers. As a result of this study, two naphthoquinone epoxides, 2,3-epoxy-2,3-dihydrolapachol (1) and 2,3-epoxy-2,3-dihydro-8-hydroxylapachol (2), both not previously isolated from natural sources, and the known 2-methyl anthraquinone (3) were identified from B. alluaudii. Time-dependent density functional theory (TD-DFT) calculations of electronic circular dichroism (ECD) spectra were utilized to establish the absolute configuration of 1 and 2. Additionally, five known naphthoquinone derivatives, maritinone (4), elliptinone (5), plumbagin (6), (+)-cis-isoshinanolone (7), and ethylidene-6,6′-biplumbagin (8) were isolated from D. maritima. Compounds 1, 2, and 4–6 showed varying levels of synergy with TRAIL. Maritinone (4) and elliptinone (5) showed the highest synergistic effect, with more than a three-fold increase in activity observed with TRAIL than with compound alone.
Tumor necrosis factor-α-related apoptosis-inducing ligand (TRAIL/Apo2L) is a member of the tumor necrosis factor (TNF) family of apoptosis triggering proteins.1 TRAIL promotes recruitment of the adaptor protein FADD (Fas associated death domain) upon binding to death domain-containing transmembrane receptors, death receptors 4 and 5 (DR4, DR5). FADD is responsible for recruiting procaspase-8 and procaspase-10, which results in the formation of a trimerized receptor-ligand complex called DISC (death-inducing signaling complex). The activated initiator caspase-8 then activates effector caspase-3, caspase-6, and caspase-7, which triggers the caspase cascade and subsequently results in apoptosis.2,3 In type I cancer cells, caspase-8 can directly activate downstream effector caspases to promote apoptosis. However, in type II cancer cells, apoptosis proceeds through cross-talk between the extrinsic and intrinsic pathways and involves the participation of the mitochondria.4 TRAIL is particularly important because it selectively induces apoptosis in cancer cells, while showing little to no effect in normal cells.1,5 However, TRAIL resistance has been widely documented,5–9 and there is evidence to suggest that combination chemotherapy regimens may be more effective than traditional cytotoxic mono-chemotherapy.10–12 In this manner, TRAIL activity may be restored by sensitizing tumor cells with certain chemical agents. Therefore, a high-throughput screen was developed to identify compounds that could sensitize tumor cells to the killing effects of TRAIL.5 As a result of this screen, we reported previously eight new clerodane diterpenes isolated from Casearia arguta with varying levels of TRAIL synergy.13 Here we describe the isolation and structure elucidation of two naphthoquinone epoxides from Barleria alluaudii as well as six known quinones from both B. alluaudii and Diospyros maritima, and their TRAIL-sensitizing activity.
RESULTS AND DISCUSSION
Bioassay-guided fractionation of the organic-soluble extracts of B. alluaudii and D. maritima, utilizing normal-phase chromatography, size-exclusion chromatography, and reversed-phase HPLC resulted in the isolation of two naphthoquinone epoxides, 2,3-epoxy-2,3-dihydrolapachol (1) and 2,3-epoxy-2,3-dihydro-8-hydroxylapachol (2), as well as six known compounds (3–8).
The molecular formula for 1, C15H14O3, was derived from NMR data (Table 1) and the HRESIMS ion at m/z 243.1020 ([M + H]+; Δ + 1.65 ppm). 1D- and 2D-NMR studies were utilized to establish the structure of 1. Initial interpretation of the NMR data (Table 1) indicated that 1 contains six quaternary carbons, six methines, one methylene, and two methyls. The NMR data suggested this compound was a substituted naphthoquinone, with a 3-methyl-2-butenyl moiety, and a one-proton singlet (δH 3.83). The identity of 1 was established as 2,3-epoxy-2,3-dihydrolapachol (or 2-(3′-methyl-2′-butenyl)-2,3-epoxy-1,4-naphthoquinone) upon comparison of spectroscopic data with those reported in the literature.14–17 This is the first reported incidence of 1 as a natural product, although it has been produced by total synthesis14–16 and semi-synthetically from 2-(3′-methyl-2′-buteny1)-2,3-epoxy-1,4-naphthalenedione-4,4-dimethoxy ketal.17
Table 1.
NMR data for 1 and 2 in CDCl3.
| position | δC | δH, mult (J, Hz) | δC | δH, mult (J, Hz) | HMBC |
|---|---|---|---|---|---|
| 1 | 2 | ||||
| 1 | 192.0 | 197.3 | |||
| 2 | 63.8 | 63.6 | |||
| 3 | 59.2 | 3.83, s | 59.1 | 3.79, s | 2, 4, 4a, 1′ |
| 4 | 192.3 | 191.1 | |||
| 4a | 132.1 | 132.4 | |||
| 5 | 127.0 | 7.92, ddd (7.1, 2.1, 0.8) | 119.2 | 7.44, dd (7.5, 0.9) | 1, 4, 6, 7, 8, 8a |
| 6 | 134.5 | 7.72, ddd (7.5, 7.1, 2.1) | 137.1 | 7.55, dd (8.4, 7.5) | 4, 4a, 5, 7, 8, 8a |
| 7 | 134.7 | 7.72, ddd (7.5, 7.1, 2.1) | 124.7 | 7.18, dd (8.4, 0.9) | 1, 4a, 5, 6, 8, 8a |
| 8 | 127.7 | 8.00, ddd (7.1, 2.1, 0.8) | 162.1 | ||
| 8a | 132.6 | 114.7 | |||
| 1a′ | 26.4 | 3.02, dd (15.4, 8.1) | 26.0 | 3.04, dd (15.3, 8.1) | 1, 2, 3, 2′, 3′ |
| 1b′ | 2.66, dd (15.4, 6.9) | 2.58, dd (15.3, 6.8) | 1, 2, 3, 2′, 3′ | ||
| 2′ | 115.6 | 5.06, ddq (8.1, 6.9, 1.0) | 115.2 | 5.01, ddq (8.1, 6.8, 1.0) | 2, 3, 3′, 4′, 5′ |
| 3′ | 137.5 | 137.7 | |||
| 4′ | 18.3 | 1.66, brs | 18.2 | 1.63, brs | 2′, 3′, 4′ |
| 5′ | 26.1 | 1.70, brs | 26.1 | 1.68, brs | 2′, 3′, 5′ |
| OH-8 | 11.39, brs | ||||
The molecular formula for 2, C15H14O4, derived from the HRESIMS ion at m/z 259.0966 ([M + H]+; Δ + 0.39 ppm) indicated that 2 contains one oxygen atom more than 1. Initial interpretation of the NMR data (Table 1) indicated that 2 contains seven quaternary carbons, five methines, one methylene, and two methyls. The dispersion of the aromatic signals and the appearance of a broad OH singlet (δH 11.39) suggested the addition of a hydroxy substituent on the aromatic ring. Furthermore, the downfield shift of the phenol proton signal indicated that the proton is hydrogen bonded to the quinone oxygen. HMBC data (H-5/C-8; H-6/C-8; H-7/C-8) indicated the hydroxyl group to be located at C-8. The identity of 2 was established as 2,3-epoxy-2,3-dihydro-8-hydroxylapachol (or 2-(3′-methyl-2′-butenyl)-2,3-epoxy-8-hydroxy-1,4-naphthoquinone) upon comparison of MS and 1H NMR data with that of a reported intermediate in the synthesis of α-caryopterone.18 Compound 2 has also not been reported as a natural product previously.
The absolute configuration of the naturally occurring epoxy naphthoquinone 2 was established by comparison of the observed electronic circular dichroism (ECD) spectrum with time-dependent density functional theory (TD-DFT) calculated ECD spectra.19,20 A conformational search using molecular mechanics calculations yielded 14 possible conformers within a 10 kcal/mol energy threshold. The geometries of 2 were optimized at the B3LYP/6-31G(d) level affording six possible conformers within a 1 kcal/mol energy difference (2a–2f, Figure 1). The relative energies, relative zero point energies, relative Gibbs free energies and their respective conformational distributions are given in Table 2. The ECD calculations for 2a–2f were then conducted with the B3LYP-6-31G(d) basis set using the IEFPCM solvent continuum model with methanol as the solvent. The calculated ECD spectra, weighted based on the Gibbs free energy, of both the 2S,3R and 2R,3S enantiomers are shown in Figure 2, along with the experimental ECD for 2. The weighted ECD spectra for the 2S,3R enantiomer is in good accordance with the experimental ECD. In particular, the positive cotton effect at 380 nm and the negative cotton effect at 330 nm clearly follow the ECD pattern for 2. Comparison between the experimental and calculated ORD values for both possible enantiomers using DFT at three different wavelengths further supports the 2S,3R configuration (Figure S2, Table S3). Therefore, the absolute configuration of 2 was established as (2S,3R)-2,3-epoxy-2,3-dihydro-8-hydroxylapachol.
Figure 1.
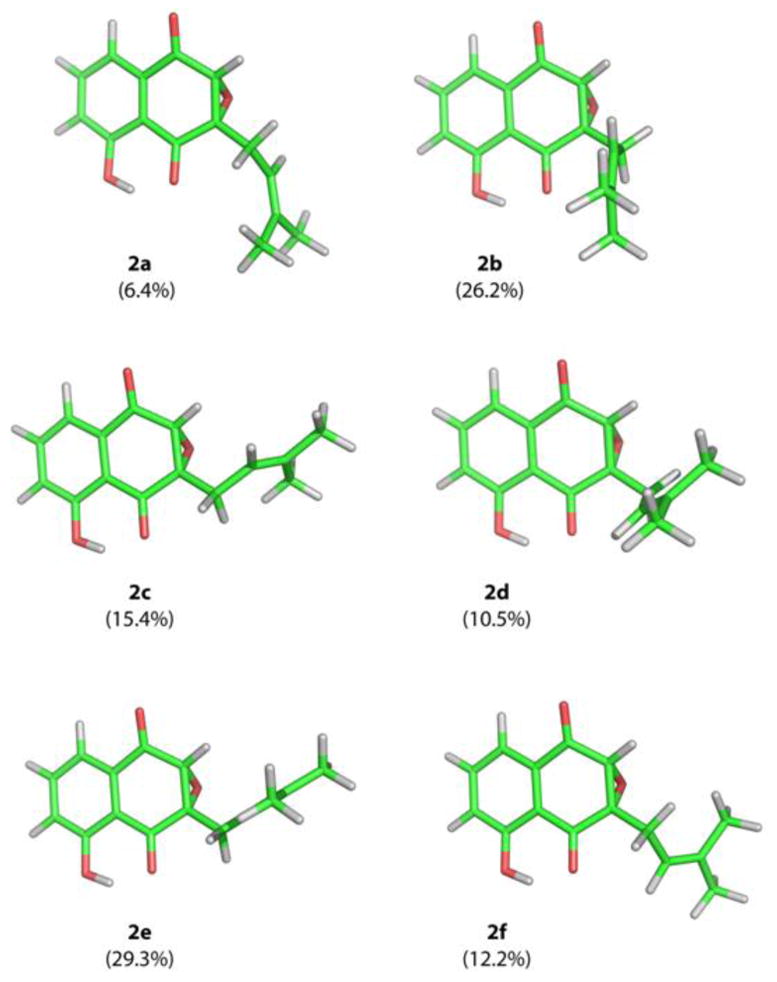
Six lowest energy conformers of (2S,3R)-2,3-epoxy-2,3-dihydro-8-hydroxylapachol (2). Conformer populations were calculated using the Gibbs free energy.
Table 2.
Conformational analysis of 2 using the IEFPCM solvent continuum model (methanol)
| Conformer | ΔEa | PE (%)b | ΔE0a | PE0 (%)b | ΔGa | PG (%)b |
|---|---|---|---|---|---|---|
| 2a | 1.02 | 5.0 | 1.33 | 3.1 | 0.89 | 6.4 |
| 2b | 0.05 | 25.9 | 0.04 | 27.4 | 0.07 | 26.2 |
| 2c | 0.38 | 14.8 | 0.37 | 15.7 | 0.38 | 15.4 |
| 2d | 0.21 | 19.7 | 0.30 | 17.8 | 0.61 | 10.5 |
| 2e | 0 | 28.2 | 0 | 29.3 | 0 | 29.3 |
| 2f | 0.88 | 6.4 | 0.87 | 6.7 | 0.52 | 12.2 |
Relative energy, relative zero point energy, and relative Gibbs free energy at the B3LYP/6-31G(d) level, respectively (kcal/mol).
Conformational distribution calculated by using the respective parameters above at the B3LYP/6-31G(d) level.
Figure 2.
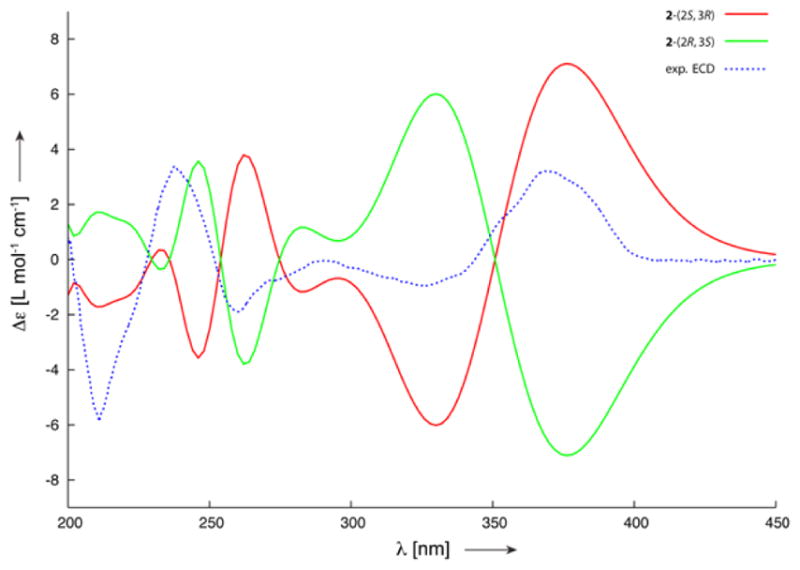
Comparison of experimental ECD values with those calculated for the two possible enantiomers (2S,3R and 2R,3S) of 2. The calculations were performed with DFT at the B3LYP/6-31G(d) level using the IEFPCM solvent continuum model with methanol as the solvent. The calculated ECD values are weighted based on the Gibbs free energy.
The absolute configuration of 1 was established by comparison of experimental and TD-DFT calculated ECD spectra. Following the approach outlined above, geometry optimization of 1 resulted in six possible conformers within a 1 kcal/mol energy difference (1a–1f, Figure S3). A comparison of the observed ECD spectrum for 1 with spectra calculated by TD-DFT for the 2S,3R-1 and 2R,3S-1 enantiomers is seen in Figure 3. The weighted ECD spectra for the 2S,3R enantiomer is in good accordance with the experimental ECD for 1. Interestingly, the optical rotations for compounds 1 and 2 are of opposite sign (1, +92.8; 2, −9.0). Compound 1 was subjected to chiral HPLC analysis to determine whether a mixture of enantiomers was present, and a single peak was observed on six chiral columns (Supporting Information). The ORD values for the 2S,3R-1 and 2R,3S-1 enantiomers were also calculated using DFT at three different wavelengths (Supporting Information, Table S8, Figure S5). The ORD values for 1a–1f are highly divergent, with half being of the opposite sign. Consequently, a small error in the energy calculation could result in an average ORD of the wrong sign. This is in contrast to the calculations for 2a–2f, where only one conformer was the opposite sign. In a similar case, the correct ensemble with an accurate population approximating the full conformational space of the molecule was essential for calculating the chiroptical properties.21 Furthermore, according to McCann and Stephens, the ORD values are not reliable if the absolute value of the difference between calculated and experimental for both enantiomers is less than 74.0.23 Therefore, in the case of compound 1, the conformational averaged [α]D for the two enantiomers is +/− 28.9, and cannot be considered a reliable predictor of absolute configuration. Taken together with the good agreement between the 2S,3R-1 calculated and experimental ECD spectra, biosynthetic principles support 1 having the same 2S,3R absolute configuration, given that 1 and 2 were concurrently isolated from the same organism. Therefore, a 2S,3R configuration is also proposed for 1.
Figure 3.

Comparison of (a) experimental ECD values with (b) those calculated for the two possible enantiomers (2S,3R and 2R,3S) of 1. The calculations were performed with DFT at the B3LYP/6-31G(d) using the IEFPCM solvent continuum model with methanol as the solvent. The calculated ECD values are weighted based on the Gibbs free energy.
Compounds 3–8 were found to be 2-methyl anthraquinone (tectoquinone) (3),24,25 maritinone (4),26,27 elliptinone (5),28,29 plumbagin (6),27,30 (+)-cis-isoshinanolone (7),26,31 and ethylidene-6,6′-biplumbagin (8)32,33 by comparison of their NMR and MS data with those in the literature. The relative configuration of cis-isoshinanolone (7) was supported by the small coupling constant observed between H-3 and H-4 (J = 2.8 Hz); cis and trans isomers show significantly different coupling constants (cis: J = 2.5 Hz; trans: J = 7.5 Hz).26,31 The absolute configuration (3R,4R) was established by comparison of the ECD spectrum measured for 7 with those of the two possible enantiomers.31
ACHN cells are not sensitive to recombinant TRAIL ligand at concentrations up to 10 μg/mL, but they can be sensitized with pre-exposure to certain chemical sensitizers (e.g. the proteasome inhibitor bortezomib).5 The TRAIL activities of compounds 1–8 are summarized in Table 3. Compounds 1, 2, and 4–6 fall into the “synergistic/toxic” category that has been previously described.5 They have some toxicity alone, but synergize with TRAIL to kill cells (greater than an additive effect in combination). Therefore, they are more potent in the presence of TRAIL than in its absence. Maritinone (4) and elliptinone (5) showed the highest degree of TRAIL as their synergistic effects were more than 3-fold greater than the compounds alone. 2,3-Epoxy-2,3-dihydro-8-hydroxylapachol (2) was a more potent cytotoxic agent than 2,3-epoxy-2,3-dihydrolapachol (1), however, 1 showed a higher synergistic effect with TRAIL. Interestingly, 2-methyl-anthraquinone (3), (+)-cis-isoshinanolone (7), and ethylidene-6,6′-biplumbagin (8) were not cytotoxic, despite their structural similarities to the active compounds.
Table 3.
Biological Effects of 1–8 with and without TRAIL
| compound | EC50a w/TRAILb | EC50a w/o TRAIL | synergistic effectc |
|---|---|---|---|
| 2,3-epoxy-2,3-dihydrolapachol (1) | 16.7 | 39.7 | 2.4 |
| 2,3-epoxy-2,3-dihydro-8-hydroxylapachol (2) | 4.7 | 6.4 | 1.4 |
| 2-methyl-anthraquinone (3) | > 50 | > 50 | NA |
| maritinone (4) | 1.5 | 5.8 | 3.8 |
| elliptinone (5) | 3.8 | 13.2 | 3.5 |
| plumbagin (6) | 3.7 | 7.3 | 2.0 |
| (+)-cis-isoshinanolone (7) | > 60 | > 60 | NA |
| ethylidene-6,6′-biplumbagin (8) | > 30 | > 30 | NA |
μM.
40 ng/mL.
Synergistic effect is defined as the ratio of the two half-maximal (EC50) values obtained from cell death curves.
In summary, 2,3-epoxy-2,3-dihydrolapachol (1), 2,3-epoxy-2,3-dihydro-8-hydroxylapachol (2) and 2-methyl anthraquinone (3) were isolated from B. alluaudii. Neither 1 nor 2 have been reported previously from natural sources, and the absolute configuration of 1 and 2 was determined using TD-DFT calculations of ECD spectra. Additionally, five known compounds were isolated from D. maritima (4–8). Compounds 1,2 and 4–6 showed TRAIL sensitization, and compounds 4 and 5 showed more than three times the effect with TRAIL than without. No previous investigations of B. alluaudii, endemic to Madagascar, have been reported in the chemical literature. Compounds 1 and 2 are members of the lapachol family of quinones. β-Lapachol was evaluated by the NCI in the clinic during the 1970s, but was later withdrawn due to high levels of toxicity.34 The closely related β-lapachone is currently in Phase II clinical trials for advanced solid tumors.34,35
Experimental Section
General Experimental Procedures
Optical rotations were measured on a Perkin-Elmer 241 polarimeter. UV spectra were acquired in spectroscopic grade MeOH using a Varian Cary 50 UV-vis spectrophotometer. ECD spectra were recorded on a JASCO J-720 spectropolarimeter or an AVIV 202. NMR data were collected using a Bruker Avance III DRX-600 (1H 600 MHz, 13C 150 MHz) NMR spectrometer (Bruker Biospin) with a 3-mm CPTCI probe, referenced to residual solvent (δH 7.24, δC 77.23 for CDCl3; δH 3.31, δC 49.15 for CD3OD). MS were measured with an Agilent Technologies 6510 Q-TOF LC-MS and an Applied Biosystems, Inc. QSTAR XL hybrid triple-quad time-of-flight (QqTOF) mass spectrometer. Initial purification was performed on Diol SPE cartridges (Applied Separations) and Sephadex LH-20 resin (Amersham Biosciences). All HPLC was performed on a Rainin SD-1/UV-1 system utilizing a Rainin Dynamax C18 column (250 × 10 mm, 5 μm particle size) at 4.5 mL/min.
Plant Material
The stems of Barleria alluaudii Benoist (Acanthaceae) were collected approximately 60 km west of Fort-Dauphin, Toliara, Madagascar (24 59.00 S; 46 33.00 E) by J. Zarucchi, E. Rakotobe, A. Randrianasolo and A. Pool (May 24, 1991). The plant was identified by J. Zarucchi, and a voucher specimen (collection number Q66V600) is maintained at the Missouri Botanical Garden.
The fruits of Diospyros maritima Blume (Ebenaceae) were collected on the Ujung Kulon Reserve, Handeleum Island (Timor) near Java, Indonesia (6 12.00 S; 105 26.00 E) by A. McDonald and Afriastini (May 23, 1992). The plant was identified by A. McDonald, and a voucher specimen (collection number U44Z4517) is maintained at the Field Museum, Chicago, Illinois.
Extraction and Isolation
The stems of Barleria alluaudii (1.192 kg) were extracted successively with CH2Cl2/MeOH (1:1) and MeOH.36 The combined extracts were reduced to dryness in vacuo to give 49.04 g of crude extract. A portion of this extract (203.6 mg) was separated by two Diol SPE cartridges (2 g resin each) and the equivalent fractions were combined to give five total fractions (74A–74E); Fraction A = 9:1 hexanes-CH2Cl2, Fraction B = 20:1 CH2Cl2-EtOAc, Fraction C = EtOAc, Fraction D = 5:1 EtOAc-MeOH, Fraction E = MeOH. Size-exclusion chromatography of the active fraction 74A on Sephadex LH-20 (2.5 × 70 cm) using hexanes-CH2Cl2-MeOH (2:5:1) yielded 11 fractions (90A–90K). Fraction 90D was separated further by Sephadex LH-20 (0.75 × 50 cm) using CH2Cl2-MeOH (1:1) to yield six fractions (110A–110F). Fraction 110D and 110E were purified by HPLC employing an isocratic method of 75% CH3CN-25% H2O (+0.1% AcOH) over 15 min to yield 2,3-epoxy-2,3-dihydro-8-hydroxylapachol (1, 8.5 mg) and 2-methyl-anthraquinone (3, 0.9 mg). Fractions 110B and 110C were purified by HPLC employing a gradient of 60% CH3CN-40% H2O (+0.1% AcOH) to 95% CH3CN over 20 min to yield 2,3-epoxy-2,3-dihydrolapachol (2, 4.1 mg) and 1 (4.4 mg).
The fruits of Diospyros maritima (0.756 kg) were extracted successively with CH2Cl2-MeOH (1:1) and MeOH.36 The combined extracts were reduced to dryness in vacuo to give 24.3 g of crude extract. A portion of this extract (199.3 mg) was separated by two Diol SPE cartridges (2 g resin each) to give five total fractions (75A–75E), as highlighted above. Size-exclusion chromatography of active fraction 75B on Sephadex LH-20 (2.5 × 70 cm) using hexanes-CH2Cl2-MeOH (2:5:1) yielded (+)-cis-isoshinanolone (7, 3.2 mg) and nine other fractions (78A–78G, 78I, and 78J). Fractions 78D and 78E were purified by HPLC employing a gradient of 60% CH3CN-40% H2O (+0.1% AcOH) to 100% CH3CN over 20 min to yield elliptinone (5, 1.1 mg). Size-exclusion chromatography of active fraction 75A on Sephadex LH-20 (2.5 × 70 cm) using hexanes-CH2Cl2-MeOH (2:5:1) yielded plumbagin (6, 42.4 mg) and seven other fractions (89A–89F, and 89H). Fraction 89E was purified by HPLC employing a gradient of 60% CH3CN-40% H2O (+0.1% AcOH) to 90% CH3CN over 20 min to yield maritinone (4, 1.2 mg).
A larger portion of the extract (1.0 g) was separated on a Diol column (2.0 × 5.5 cm, 10 g resin) to give five total fractions (130A–130E), as highlighted above. Size-exclusion chromatography of fraction 130A on Sephadex LH-20 (2.5 × 70 cm) using hexanes-CH2Cl2-MeOH (2:5:1) yielded eight fractions (159A–159H). Fractions 159B–159D were combined and separated further by Sephadex LH-20 (0.75 × 50 cm) using CH2Cl2-MeOH (1:1) to yield six fractions (168A–168F). Fractions 168D and 168E were purified employing the gradient utilized to purify elliptinone to yield ethylidene-6,6′-biplumbagin (8, 0.7 mg).
(2S,3R)-2,3-Epoxy-2,3-dihydrolapachol (1)
[α]25D − 9.0 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 304 (3.22) 263 (3.79) 228 (4.42) nm; 1H NMR and 13C NMR data, see Table 1; HRESIMS m/z 243.1020 [M+H]+ (calcd for C15H15O3, 243.1016).
(2S,3R)-2,3-Epoxy-2,3-dihydro-8-hydroxylapachol (2)
[α]25D + 92.8 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 360 (3.88) 262 (3.66) 233 (4.26) 206 (4.28) nm; 1H NMR and 13C NMR data, see Table 1; HRESIMS m/z 259.0966 [M+H]+ (calcd for C15H15O4, 259.0965).
Conformational Analysis, Geometry Optimization, ECD and ORD calculations
The initial structures of 1 and 2 were built with Discovery Studio 2.5 (Accelrys) and all trial structures were first minimized based on molecular mechanics calculations (CFF force field37). To sample the conformational space, a conformational search was performed (BEST method in Discovery Studio 2.5) based on molecular mechanics calculations (CFF force field). Conformers occurring within a 10 kcal/mol energy window from the global minimum were chosen for geometry optimization and energy calculation using DFT19,20 with the B3LYP functional and the 6-31(d) basis set with the Gaussian09 program.38 Time-dependent DFT (TD-DFT)19,20 with the basis set B3LYP/631G(d) was used to calculate the spin-allowed excitation energies, rotatory (Rn) and oscillator strengths (fn) of the lowest 50 excited states. The spectra were combined after Boltzmann weighting according to their population contribution. The optical rotation dispersion calculations at the three wavelengths 546, 578 and 589 nm were performed with the optimized structures as input coordinates with the same basis set as the geometry optimization and ECD calculations. All the calculations were performed in vacuo with the integral equation formalism variant polarizable continuum model (IEFPCM) as implemented in Gaussian 09 (methanol as solvent).
TRAIL Assay
The biological activity of each extract, chromatographic fraction, or pure compound was monitored using the screening assay described previously.5 Briefly, ACHN cell numbers were assessed after 20–24 h treatment with varying concentrations of extract, fraction, or pure compound in the absence or presence of 40 ng/mL TRAIL. TRAIL has no effect on ACHN cells up to 10 ug/mL. 40 nM bortezomib (final concentration) was used as the positive control.
Supplementary Material
Acknowledgments
The authors thank J. A. McDonald & Afriastini for collection and J. A. McDonald for taxonomic identification of D. maritima (N053469) as part of the collection contract with D.D. Soejarto (UIC) and J. Zarucchi, E. Rakotobe, A. Randrianasolo and A. Poole for collection and J. Zarucchi for taxonomic identification of B. alluaudii (N037953) as part of the collection contract with J. Miller (MO Botanical Garden). The authors also thank D. Newman (NPB) for collection and contract administration, T. McCloud and the Natural Products Support Laboratory for plant extractions, S. Tarasov and M. Dyba of the Biophysics Resource of the Structural Biophysics Laboratory for providing technical assistance with Q-TOF LC-MS experiments and ECD experiments, and M. P. McCoy of Phenomenex for chiral HPLC analysis. This research was supported in part by the Intramural Research Program of NIH, National Cancer Institute, Center for Cancer Research. This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the U.S. Government. C.G. thanks the Max Planck Society, the Deutsche Forschungsgemeinschaft (GRK 782 and FOR 934), and the Fonds der Chemischen Industrie for financial support.
Footnotes
Dedicated to Dr. Gordon M. Cragg, formerly of the National Cancer Institute, Frederick, Maryland, for his pioneering work on the development of natural product anticancer agents.
ASSOCIATED CONTENT:
Supporting Information: 1H and 13C NMR spectra for 1–2, chiral HPLC analysis of 1, Gaussian keywords, calculated excitation energies, oscillator strengths, and rotational strengths for 1–2, important dihedral angles of conformers 1a–1f and 2a–2f, conformational analysis of 1, calculated ORD values for 1–2, and coordinates of conformers 1a–1f and 2a–2f. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mahalingam D, Szegezdi E, Keane M, Jong S, Samali A. Cancer Treat Rev. 2009;35:280–288. doi: 10.1016/j.ctrv.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 2.MacFarlane M, Williams AC. EMBO Rep. 2004;5:674–678. doi: 10.1038/sj.embor.7400191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mahmood Z, Shukla Y. Exp Cell Res. 2010;316:887–899. doi: 10.1016/j.yexcr.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 4.Sayers TJ. Cancer Immunol Immunother. 2011;60:1173–1180. doi: 10.1007/s00262-011-1008-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Booth NL, Sayers TJ, Brooks AD, Thomas CL, Jacobsen K, Goncharova EI, McMahon JB, Henrich CJ. Cancer Immunol Immunother. 2009;58:1229–1244. doi: 10.1007/s00262-008-0637-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang L, Fang B. Cancer Gene Ther. 2005;12:228–237. doi: 10.1038/sj.cgt.7700792. [DOI] [PubMed] [Google Scholar]
- 7.Van Geelen CM, de Vries EG, de Jong S. Drug Resist Updat. 2004;7:345–358. doi: 10.1016/j.drup.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 8.O’Kane HF, Watson CJ, Johnston SR, Petak I, Watson RW, Williamson KE. J Urol. 2006;175:432–438. doi: 10.1016/S0022-5347(05)00160-6. [DOI] [PubMed] [Google Scholar]
- 9.Cheng J, Hylander BL, Baer MR, Chen X, Repasky EA. Mol Cancer Ther. 2006;5:1844–1853. doi: 10.1158/1535-7163.MCT-06-0050. [DOI] [PubMed] [Google Scholar]
- 10.Huang Y, Sheikh MS. Toxicol Appl Pharmacol. 2007;224:284–289. doi: 10.1016/j.taap.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 11.Duiker EW, Mom CH, de Jong S, Willemse PH, Gietema JA, van der Zee AG, de Vries EG. Eur J Cancer. 2006;42:2233–2240. doi: 10.1016/j.ejca.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 12.Buchsbaum DJ, Forero-Torres A, LoBuglio AF. Future Oncol. 2007;3:405–409. doi: 10.2217/14796694.3.4.405. [DOI] [PubMed] [Google Scholar]
- 13.Whitson EL, Thomas CL, Henrich CJ, Sayers TJ, McMahon JB, McKee TC. J Nat Prod. 2010;73:2013–2018. doi: 10.1021/np1004455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Claessens S, Habonimana P, De Kimpe N. Org Biomol Chem. 2010;8:3790–3795. doi: 10.1039/c004580b. [DOI] [PubMed] [Google Scholar]
- 15.Preusch PC, Suttie JW. J Org Chem. 1983;48:2291–2293. [Google Scholar]
- 16.Pluim H, Wynberg H. J Org Chem. 1980;45:2498–2502. [Google Scholar]
- 17.Perry NB, Blunt JW, Munro MH. J Nat Prod. 1991;54:978–985. doi: 10.1021/np50076a009. [DOI] [PubMed] [Google Scholar]
- 18.Matsumoto T, Ichihara A, Yanagiya M, Yuzawa T, Sannai A, Oikawa H, Sakamura S, Eugster CH. Helv Chim Acta. 1985;68:2324–2331. [Google Scholar]
- 19.Di Bari L, Guillarme S, Hermitage S, Jay DA, Pescitelli G, Whiting A. Chirality. 2005;17:323–331. doi: 10.1002/chir.20167. [DOI] [PubMed] [Google Scholar]
- 20.Furche F, Ahlrichs R, Wachsmann C, Weber E, Sobanski A, Vogtle F, Grimme S. J Am Chem Soc. 2000;122:1717–1724. [Google Scholar]
- 21.Sun H, d’Auvergne EJ, Reinscheid UM, Dias LC, Andrade CK, Rocha RO, Griesinger C. Chem Eur J. 17:1811–1817. doi: 10.1002/chem.201002520. [DOI] [PubMed] [Google Scholar]
- 22.Stephens PJ, McCann DM, Cheeseman JR, Frisch MJ. Chirality. 2005;17:S52–S64. doi: 10.1002/chir.20109. [DOI] [PubMed] [Google Scholar]
- 23.McCann DM, Stephens PJ. J Org Chem. 2006;71:6074–6098. doi: 10.1021/jo060755+. [DOI] [PubMed] [Google Scholar]
- 24.Kafuku K, Sebe K. Bull Chem Soc Jpn. 1932;7:114–127. [Google Scholar]
- 25.Rath G, Ndonzao M, Hostettmann K. Int J Pharmacogn. 1995;33:107–114. [Google Scholar]
- 26.Tezuka M, Takahashi C, Kuroyanagi M, Satake M, Yoshihira K, Natori S. Phytochemistry. 1973;12:175–183. [Google Scholar]
- 27.Gu JQ, Graf TN, Lee D, Chai HB, Mi Q, Kardono LB, Setyowati FM, Ismail R, Riswan S, Farnsworth NR, Cordell GA, Pezzuto JM, Swanson SM, Kroll DJ, Falkinham JO, 3rd, Wall ME, Wani MC, Kinghorn AD, Oberlies NH. J Nat Prod. 2004;67:1156–1161. doi: 10.1021/np040027m. [DOI] [PubMed] [Google Scholar]
- 28.Fallas AL, Thomson RH. J Chem Soc C. 1968:2279–2282. [Google Scholar]
- 29.Zakaria MB, Jeffreys JAD, Waterman PG, Zhong SM. Phytochemistry. 1984;23:1481–1484. [Google Scholar]
- 30.d’Astafort D. J Pharm Chim. 1828;14:441. [Google Scholar]
- 31.Bringmann G, Messer K, Saeb W, Peters EM, Peters K. Phytochemistry. 2001;56:387–391. doi: 10.1016/s0031-9422(00)00386-1. [DOI] [PubMed] [Google Scholar]
- 32.Higa M. Chem Pharm Bull. 1988;36:3234–3236. [Google Scholar]
- 33.Higa M, Ogihara K, Yogi S. Chem Pharm Bull. 1998;46:1189–1193. [Google Scholar]
- 34.Cragg GM, Grothaus PG, Newman DJ. Chem Rev. 2009;109:3012–43. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- 35.Ravelo AG, Estevez-Braun A, Chavez-Orellana H, Perez-Sacau E, Mesa-Siverio D. Curr Top Med Chem. 2004;4:241–265. doi: 10.2174/1568026043451500. [DOI] [PubMed] [Google Scholar]
- 36.McCloud TG. Molecules. 2010;15:4526–4563. doi: 10.3390/molecules15074526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lifson S, Warshel A. J Chem Phys. 1968;49:5116–5129. [Google Scholar]
- 38.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Whitson Emily L, Sun Han, Thomas Cheryl L, Henrich Curtis J, Sayers Thomas J, McMahon James B, Griesinger Christian, McKee Tawnya C. Gaussian 09, Revision A.1. Gaussian, Inc; Wallingford, CT: 2009. Synergistic TRAIL sensitizers from Barleria alluaudii and Diospyros maritima. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.