

Abstract
Microscopic eukaryotes are abundant, diverse, and fill critical ecological roles across every ecosystem on earth, yet there is a well-recognized gap in our understanding of their global biodiversity. Fundamental advances in DNA sequencing and bioinformatics now allow accurate en masse biodiversity assessments of microscopic eukaryotes from environmental samples. Despite a promising outlook, the field of eukaryotic marker gene surveys faces significant challenges: how to generate data that is most useful to the community, especially in the face of evolving sequencing technology and bioinformatics pipelines, and how to incorporate an expanding number of target genes.
Microscopic Eukaryotes: Global dominance, scant knowledge
Microscopic eukaryotic taxa are abundant and diverse, playing a globally important role in the functioning of ecosystems [1, 2] and host-associated habitats [3]. Here, we consider taxa generally represented by individuals <1mm in size; the term “microscopic eukaryotes” thus encompasses meiofaunal metazoa (e.g. Nematoda, Platyhelminthes, Gastrotricha and Kinorhyncha), fungi, microbial eukaryotes including deep protist lineages (Alveolata, Rhizaria, Amoebozoa, Algal taxa in the Chlorophyta and Rhodophyta, etc.), and eggs and juvenile stages of some larger metazoan species. These ubiquitous eukaryote groups play key roles as decomposers, predators, producers and parasites, yet we know little about their biology, ecology, and diversity. Analyses of eukaryotic community structure often reveal divergent lineages [4–6] and long lists of previously undiscovered sequences [7, 8]. Nematodes, for instance, account for 80–90% of all metazoa on Earth, yet less than 4% of the estimated >1 million species are formally known and described [9]. This discrepancy between known and estimated diversity is common for all microscopic eukaryote groups, and generally stems from the difficulty of applying traditional approaches in species identification to high-throughput sequence data. Traditional approaches, though well-validated, do not scale to the large numbers of sequences now being collected [6, 9–12].
In many ways, the problems faced in the study of microscopic eukaryotes mirror those facing studies of archaea and bacteria. The exploration of archaeal and bacterial diversity long ago adopted a molecular taxonomy [13] —early uses of high-throughput sequencing allowed the characterization of microbial taxa in environmental samples ranging from the world’s oceans [14, 15] to our own bodies [16, 17]. These approaches clearly illuminate a path for the exploration of eukaryotic diversity, but also highlight the pitfalls we will need to address along the way. While advances in the study of archaeal and bacterial diversity provide valuable knowledge and infrastructure for high-throughput analyses of eukaryotes, eukaryotes also have four unique features. First, for many groups of microscopic eukaryotes, we have access to biologically informative morphology and a substantial body of existing taxonomic resources (expertise, keys, specimen vouchers). Therefore we can (and should) collect and employ morphological metadata as a valuable component of marker gene surveys, especially when it is desirable to compare results to historical or fossil specimens from which DNA cannot be extracted. Second, the increased complexity of eukaryotic genomes is correlated with an increased number and variability of the traditional target loci for molecular taxonomy (rRNA; Box 1) [18]. While the ribosomal locus varies in copy number (1–15) and length heterogeneity in archaea and bacteria [19] the variation can be much more extensive in eukaryotes (extending to tens of thousands of copies in some taxa [18, 20]). This issue severely complicates both the clustering of sequences into Operational Taxonomic Units (OTUs) and the use of read counts for estimating species abundances (Box 3). Third, most eukaryotes have mitochondrial genomes. In multicellular animals, the mitochondrial genome evolves rapidly (especially in the non-coding regions), offering higher resolution for detecting more recent evolutionary forces—mitochondria might thus provide a basis for large-scale analyses of gene flow. Finally, many groups of eukaryotes appear to evolve with a smaller contribution of horizontal gene transfer (metazoa, fungi; [21]). Consequently, the evolutionary framework inferred from a single locus can better reflect the history of these eukaryotic genomes as a whole.
Box 1. Intragenomic rRNA variation in Eukaryotes.
Ribosomal RNA in eukaryotes is encoded by 18S, 5.8S and 28S subunit genes, organized in tandemly repeated arrays within a genome. The number of gene copies can vary dramatically across taxa, with eukaryotic species exhibiting hundreds to many thousands of ribosomal arrays [18, 20]; these are sometimes found at a single locus but are also known to exist in multiple distinct loci [20]. Concerted evolution results in high levels of identity among intraspecific repeats but higher divergence across interspecific gene copies [69]. However, The number of rRNA copies can vary dramatically even within species [70, 71], confounding the ability to correlate the number of reads generated in a marker gene survey with the number of individuals in a sample. While the phenomenon of concerted evolution [69] predicts that new mutations are rapidly propagated across the rRNA gene copies within a species, it is clear that intragenomic ribosomal variation is extensive in some cases [72] and some of these variants might represent pseudogenes [73]. While such variation can be incorporated into the appropriate OTU by clustering approaches, levels of rRNA diversity are significantly different across taxa [31], and significant empirical data will be required to understand the pattern and consequences of intra-genomic variation across diverse eukaryotes [74].
Box 3. Major challenges for Eukaryotic Marker Gene Surveys.
A major challenge for marker gene surveys is the accurate identification of biological taxa across multiple samples. While overall per-nucleotide error rates can be lower in high-throughput than Sanger sequencing [86], larger numbers of sequences mean larger numbers of sequences containing errors. Although it is possible to predict and identify the source of errors in high-throughput datasets, such errors result from the interplay of multiple factors—position in sequence, presence of homopolymers, and physical location of DNA on sequencing platforms [31, 86, 87]. Regardless of sequencing method, PCR-derived artifacts (e.g. chimeras) can artificially increase estimates of diversity [31].
A second challenge concerns our ability to quantify the absolute abundance of individuals based on sequence read counts. This relationship is complicated by the extreme variation in the number of rRNA gene copies per nucleus among species and the number of nuclei per individual. A practical solution has been to compare environmental communities only in terms of relative taxon abundance (normalizing sequence reads per OTU). Early proof-of-concept control experiments in nematodes revealed a strong consistency in rRNA patterns but highlighted the difficulty of correlating OTUs with biological species and defining absolute abundances [10, 88, 89]; while technical replicates agreed well with one another, the rRNA read number per individual was highly variable—even within a single specimen. OTUs corresponding to reference Sanger sequences comprise the majority of the reads from any given species but many other variants exist, some differing from known references by >3 bp [88]. When we consider that some well-described nematode species differ at a single nucleotide in the 18S rRNA gene [88], it is clear that this intragenomic variation can confound efforts to differentially identify sequencing errors and the “rare biosphere” in environmental datasets. Although a rare biosphere [14] certainly exists, any observations of low abundance taxa need to be critically evaluated. Rare species can be important, especially for ecosystem responses, and these low-abundance taxa tend to be diverse (e.g. [90]).
Emerging insight from Environmental Data
Following earlier 16S rRNA (reference GenBank accession X80721.1 for E. coli) investigations of archaeal and bacterial communities [14, 22], high-throughput marker gene approaches were developed for different groups of microscopic eukaryotes using the 18S nuclear small subunit rRNA gene (nSSU; reference GenBank accession X03680.1 for C. elegans), focusing on protists [11, 12, 23–26] and meiofauna [9, 10, 27]. Similar to 16S investigations, these early 18S studies uncovered concordant patterns of high eukaryotic richness and an extended rare biosphere [11, 28]. Although the field is not yet mature, environmental datasets are already yielding novel molecular taxonomic insights into the magnitude and composition of the eukaryotic biosphere in a range of habitats. However, bioinformatic analyses of eukaryotic control communities indicate that additional work is needed in order to recover actual taxon richness [29–31].
Both 454 and Illumina sequencing datasets have suggested that the composition of marine meiofaunal [30] and protist [5, 32] communities differ significantly from estimates derived from morphological taxonomy; in these sequence datasets, the unexpected prominence of turbellarian flatworms and monothalamous foraminiferans has highlighted biases stemming from sample preservation methods in traditional taxonomic approaches. The isolation of deep alveolate lineages further suggests that divergent taxa identified in high-throughput datasets likely lack the characteristic morphological features typified by closely related clades [5]. Marker gene surveys thus facilitate objective comparisons of taxonomic richness at higher-level ranks, on a scale that is not possible using morphology alone. Accordingly, the cost-effective, high-throughput comparative power of marker gene surveys is ideally suited to (amongst other applications) biomonitoring [33–35], phylogeography [36] and exploring the relationships between biological diversity and ecosystem functioning using a “systems ecology” approach [37].
Analyzing High-Throughput Data
Over the last few years, high-throughput sequencing techniques have been informed by rapid progress in sequencing technology, bioinformatics tools and analytical pipelines. Here we present an overview of the analytical considerations for high-throughput studies. Following sample collection, extraction of environmental DNA, PCR, and sequencing (Figure 1), large datasets can be processed using a number of existing tools (Table 1).
Figure 1.
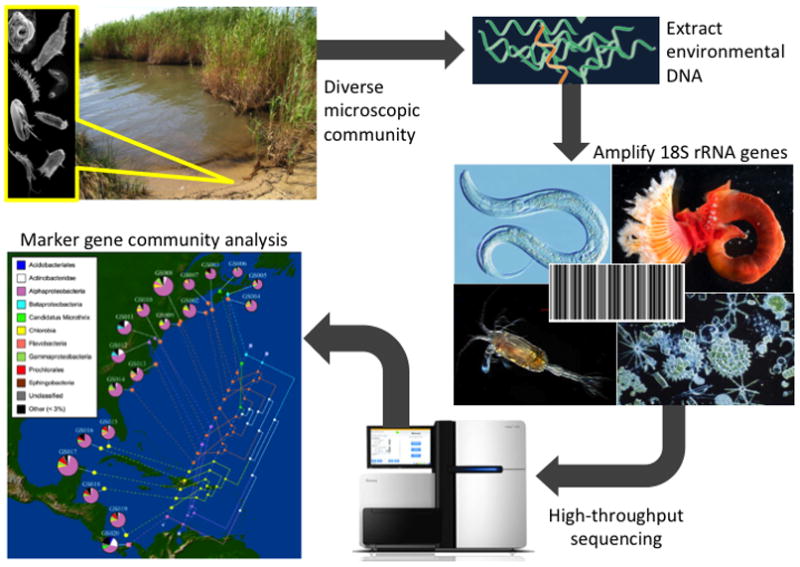
Typical standardized workflow (from environment to sequences) for high-throughput marker gene studies. Soils and sediments are typically frozen upon collection (−80°C to preserve RNA) and brought back to the lab for bulk extraction of environmental DNA. Marker genes (e.g. rRNA) are amplified from genomic extracts using barcoded, conserved primer pairs. Following high-throughput sequencing (typically conducted on 454 or Illumina platforms), datasets are processed and clustered into Operational Taxonomic Units (OTUs) under a range of pairwise identity cutoffs. OTUs are subsequently used to conduct alpha and beta diversity analyses, summarize community taxonomy, and interpret assemblages in a phylogenetic context. (Depiction of community analysis modified from Parks et al. [56])
Table 1.
Online Resources and popular software tools for eukaryotic marker gene surveys
| Resource | Capabilities | Interface | Website | Reference |
|---|---|---|---|---|
| AmpliconNoise | Denoising | Command line executables | http://code.google.com/p/ampliconnoise/ | [38] |
| Denoiser | Denoising | QIIME | http://www.qiime.org | [51] |
| QIIME | Data processing, OTU picking, taxonomy assignment, ecological analyses | Command Line, Amazon Cloud | http://www.qiime.org | [51] |
| OCTUPUS | Data processing, OTU picking, taxonomy assignment | Pipeline of perl scirpts | http://octupus.sourceforge.net/ | [30] |
| VAMPS | Data processing, OTU picking, taxonomy assignment, ecological analyses | Web-based tools | http://vamps.mbl.edu | NA |
| Galaxy | Data processing, OTU | Web, GUI | http://main.g2.bx.psu.edu | [55] |
| clustering, Ecological Analyses | ||||
| CANGS | Data processing, taxonomy assignment, ecological analyses | Pipeline of perl scirpts | http://i122server.vu-wien.ac.at/pop/software.html | [96] |
| RDP | Data processing, OTU picking, taxonomy assignment, ecological analyses | Web-based tools | http://pyro.cme.msu.edu/ | [45] |
| CLOTU | Data processing, OTU picking, taxonomy assignment | Web-based pipeline | http://www.bioportal.uio.no | [97] |
| Mothur | Data processing, OTU picking, taxonomy assignment, chimera checking, ecological analyses | Command line executables | http://www.mothur.org/ | [42] |
| ESPRIT | OTU picking | Command line executables | http://www.biotech.ufl.edu/people/sun/esprit.html | [44, 98] |
| ChimeraSlayer | Chimera checking | Command line executables | http://microbiomeutil.sourceforge.net/ | [48] |
| Perseus | Chimera checking | Command line executables | http://code.google.com/p/ampliconnoise/ | [38] |
| UCHIME | Chimera checking | Command line executable | http://www.drive5.com/uchime/ | [49] |
| MEGAN4 | Taxonomic assingment (BLAST-Based), exploring trees | GUI | http://ab.inf.uni-tuebingen.de/software/megan/ | [99] |
| EPA | Tree-insertion | Web, GUI | http://i12k-exelixis3.informatik.tu-muenchen.de/raxml | [40] |
| pplacer | Tree-insertion | Command line executables | http://matsen.fhcrc.org/software.html | [39] |
| guppy | Edge PCoA | Command line executables | http://matsen.fhcrc.org/software.html | [58] |
| TopiaryExplorer | Taxon labeling and visualization in tree topologies | GUI | http://topiaryexplorer.sourceforge.net | [67] |
| GenGIS | Data Vizualization | GUI with python console | http://kiwi.cs.dal.ca/GenGIS/Main_Page | [56] |
| VisTrails | Data Visualization | GUI (XML based) | http://www.vistrails.org | [57] |
| MG-RAST | Metagenomic analysis, Data Storage | Web | http://metagenomics.anl.gov | [62] |
| Dryad | Data Repository | Web | http://datadryad.org | [100] |
| SILVA | Curated Reference Database, Sequence alignment | Web | http://www.arb-silva.de | [50] |
| MetaBar | Metadata entry and tracking system | Graphical web tool | http://www.megx.net/metabar/ | [101] |
Denoising
Denoising (correcting pyrosequencing errors), using tools such as AmpliconNoise [38] or Denoiser [29] is often encouraged as a pre-processing step for 454 datasets, but this step requires considerable computational resources. Although denoising reduces sequencing error, existing algorithms employ read abundance information [38] and consequently, can also function to remove valuable biological signals (e.g. intragenomic rRNA variants, Box 1) and rare species from eukaryotic datasets. It seems likely that sequence ‘noise’ might only present concerns for specific biological questions such as species counts, although further studies comparing the affect of denoising on a wide range of environmental datasets and applicable to different sequencing platforms are needed.
Processing Raw Reads
Primers, sequencing adaptors, and barcode tags are typically removed from raw sequencing reads, with the relevant metadata (sample site, primer name) inserted into FASTA headers or sequence mapping files. Short and noisy reads are discarded completely, and low-quality base calls are trimmed from longer sequences (typically the tail end of raw reads where sequencing quality deteriorates). A multitude of tools can be utilized for these basic processing steps (Table 1).
OTU Picking
OTU picking tools fall into three general categories: de novo OTU picking, where reads are clustered only against one another; closed-reference OTU picking, where reads are searched against a database and clusters are defined by the best database match for each read, with nonmatching reads discarded; and open-reference OTU picking, where clusters are similarly defined by the best database matches, but nonmatching reads are instead retained and clustered de novo. It is important to note that high-throughput analyses do not necessarily depend on defining OTUs; direct phylogenetic placement methods [39, 40] can handle unclustered, processed sequence reads (Figure 3). However, the sheer volume of raw sequence data means that some degree of filtering or clustering is usually necessary for minimizing downstream computational demands. In this sense, OTU picking can be viewed simply as another processing step—not an attempt to define biological species from sequence data per se.
Figure 3.

OTU reference sequences can be placed into an evolutionary context using tools such as pplacer [39] or the Evolutionary Placement Algorithm (EPA [40]), which place short reads into a guide tree framework constructed from full-length reference sequences. For each pre-aligned OTU or sequencing read, likelihood scores are calculated for all possible positions in the tree, and the sequence is subsequently inserted at the node exhibiting the best score.
Pre-clustering reads for computational efficiency
Pre-clustering of raw reads is commonly employed in order to reduce the compute time of OTU picking. Most OTU picking approaches require all-by-all comparisons (each sequence compared to every other sequence), with total computational time being proportional to the square of the number of input sequences—thus, algorithms useful for small datasets quickly become infeasible with larger datasets. The most common pre-filtering method collapses only those sequences that are 100% identical prior to OTU picking. A similar filter involves collapsing sequences that have identical prefixes or suffixes (nested sequences beginning at the 5′ or 3′ end of the gene, respectively) meeting a user-specified maximum length parameter. In all cases, abundance information (defined by unique ID tags for filtered reads) is retained for each representative sequence.
De novo OTU picking
De novo algorithms are the most ubiquitous, primarily because they do not require a specific external database and inherently retain all input sequence reads, and therefore cannot introduce biases stemming from database incompleteness. There are many existing algorithms for de novo OTU picking including uclust [41], OCTUPUS [30], mother [42], cd-hit [43] and ESPRIT-Tree [44] (Table 1). A useful strategy in de novo OTU picking (and the de novo step of open-reference OTU picking) is to pre-sort sequences by their abundance. For algorithms that sort through the sequences in order (e.g., uclust), the most abundant sequences (less likely to represent errors) are the reads initially used to form OTU clusters.
Closed-reference OTU picking
When using closed reference OTU picking, a minimal threshold is usually applied for accepting database hits as matches, and sequences that do not match this threshold are discarded as failures. Although closed-reference OTU picking can exclude many good reads, it can also be considered a strict quality filter (and screen for contaminants) since all observed sequences must be similar to previously known sequences. Closed-reference OTU picking has primarily been applied to bacterial and archaeal studies that can take advantage of large data repositories and multiple curated databases (e.g. the Ribsomal Database Project (RDP) [45], Greengenes [46]). This method is more difficult for eukaryotic studies given the limited number of eukaryotic database resources (although the SILVA database [51] can be harnessed) and the patchy coverage across diverse taxa in terms of published rRNA sequences.
BLAST and uclust are both commonly used for closed-reference OTU picking, with assignments differing in the use of local (BLAST) versus global (uclust) alignments. For this reason uclust is more conservative: for a sequence to match at 97% identity the uclust match must span the full length of the read, whereas a BLAST-assigned sequence can exhibit 97% identity, but only cover, for example, only 50% of the total read length. Minimum aligned percentages can be applied to circumvent this issue with BLAST, although uclust is considerably faster for OTU picking. However, global alignments can be problematic when the sequence termini are especially noisy, as is the case with Sanger sequencing (where both ends tend to be lower-quality) and high-throughput platforms (where quality drops off, sometimes dramatically, towards the 3′ end of the sequence).
Despite discarding valid reads that are not represented in the reference database, closed-reference OTU picking exhibits several benefits over de novo clustering. First, reads from multiple hypervariable gene regions (or genomic loci [47]) can be compared in a single analysis if clustered against full-length reference sequences. In de novo clustering, independent, non- overlapping amplicons would instead cluster independently of each other. Closed-reference approaches also facilitate direct comparisons and meta-analysis across studies utilizing the same collection of reference sequences, as new unique datasets are incrementally added into a centralized database resource. Closed-reference OTU picking additionally results in stable OTU identifiers, so the “same” OTU can be pinpointed in different studies. Finally, this method is easily parallelized, and therefore more amenable to increasingly large sequence outputs such as those generated on Illumina platforms. Because no new clusters are created in the process, it is possible to split an input collection of reads into tens or hundreds of smaller input collections, process all of the sub-inputs in parallel, and subsequently collate the results.
Open-reference OTU picking
Open-reference OTU picking is a combination of de novo and closed-reference OTU picking strategies, and any pair of methods could theoretically be combined for tailored workflows. However, users should be wary of the differences in how OTUs are defined if combining strategies such as BLAST and de novo uclust for OTU picking. The open-reference approach begins the same way as closed-reference OTU picking, but sequences that fail to hit a reference sequence are retained through de novo clustering at the end of the process. Although the initial reference-based OTU picking can be run in parallel (as in closed-reference OTU picking), the de novo process must consider all remaining sequences simultaneously. For eukaryotic studies (and in general), open-reference approaches are preferable over closed-reference OTU picking, since environmental datasets typically contain a signification proportion of taxa with no close relatives in public sequence repositories. De novo clustering thus retains unknown diversity and supplies high-throughput database resources with new divergent lineages.
Identifying Chimeras
New algorithms are emerging for the identification of chimeras (Box 4) in high-throughput datasets, including ChimeraSlayer [48], Perseus [38], and UCHIME [49]. Both Perseus and UCHIME take advantage of the intuitive assumption that chimeras (hybrid, daughter sequences) should be less frequent than parental sequences that have undergone at least one more round of PCR amplification. Control experiments show that Perseus and UCHIME (tools used without a reference database) both outperform ChimeraSlayer (which compares taxonomic lineage information from reference databases), at least in 16S datasets [38, 49]. Detecting hybrid sequences between closely related taxa currently represents one of the most significant challenges. In practice, no method is 100% effective for preventing or identifying chimeras: chimeras can theoretically be generated at any position along an amplicon, and detection ability will correlate with the length of the chimeric fragment.
Box 4. The perils of chimeras.
Chimeras are in vitro DNA artefacts derived from the mixture of two or more parent molecules in a PCR reaction of a homologous gene region [9, 91], typically formed when incomplete extension occurs in a preliminary round of PCR and the resulting sequence fragment acts as a primer for a different sequence in subsequent thermal cycles. The result is an artificial recombinant molecule with discrete break points (and specific hotspots along an amplicon; [92]), corresponding to the transition between the different parent molecules [38]. Chimeras substantially overinflate richness estimates and suggest the presence of spurious taxa and OTUs, potentially across multiple samples [38]. They are additionally likely to skew our interpretation of the extent of the “rare biosphere” [14], since chimeras usually appear as divergent, low frequency sequences [29, 93].
Sample and taxonomic diversity, the composition and structure of the locus employed, and DNA polymerases and thermal cycling conditions will all interact to create different levels of chimeric amplicons [38, 48]. Identifying optimal “low chimera” PCR protocols for marker loci will require a series of carefully controlled experiments, but general rules include keeping the number of PCR thermal cycles to a minimum and increasing extension times [28]. The latter allows DNA polymerases to reach the 3′ terminus of the target molecule and minimizes chimera formation in longer PCR reactions.
Assigning Taxonomy
Although interesting biological patterns can be investigated in the absence of species names, taxonomic frameworks are useful for relating sequences to what is known about the organisms. A number of different approaches can be used for assigning taxonomy (alone or in conjunction), including BLAST, Global Alignments for Sequence Taxonomy (GAST), probabilistic classifiers, or tree-based assignments. BLAST assignments utilize pairwise alignment scores and have the advantage of being the easiest to use: a local database and single step can identify close relatives from millions of published sequences. BLAST methods can assign taxonomy from public sequence repositories (GenBank, EMBL) or use a smaller database of reference sequences with trusted taxonomy (e.g. curated from SILVA [50]). GAST approaches utilize BLAST searches against reference datasets for hypervariable gene regions, in conjunction with global alignments and RDP classifier scores; this method is presently implemented in the VAMPS pipeline (http://vamps.mbl.edu). Probabilistic approaches such as the RDP classifier instead apply a naïve Bayesian classifier to match 8-base sequence “words” to a reference training set (the RDP [45] or another user-defined database when implemented in QIIME [51]), also returning confidence scores for each taxonomic assignment. Tree-based assignment (Figure 3) provides an alternate (and in theory, more robust) approach: a predefined guide tree containing full-length reference sequences is used place unknown OTU sequences within a known phylogeny. Using likelihood scores, aligned short reads are tested across all nodes in the reference topology and subsequently inserted as a branch at the highest scoring position. Current tools include the RAxML-based Evolutionary Placement Algorithm [40] accessible through a web interface (http://i12k-exelixis3.informatik.tu-muenchen.de/raxml), and the open-source command-line tool pplacer [39] that can be run on local machines.
Ecological analyses
Vague or absent taxonomy does not preclude ecological analyses nor the testing of ecological hypotheses (phylogeograpic patterns, community assemblages)—producing OTU tables and taxonomy tables are thus intermediate steps towards these end goals. A suite of new tools are on the horizon (Figure 4), and many useful ecological analyses are currently incorporated within the QIIME pipeline [51], including community summaries (pie, bar, or area charts detailing taxonomic proportions), heatmaps displaying OTU abundance across sample sites, alpha diversity (including rarefaction, and based on OTU counts, Chao1 estimation, or phylogenetic diversity), phylogenetically-informed beta diversity and ordination (Principal Coordinate Analysis based on UniFrac distances [52] between-sample and jackknifed UPGMA analysis), and OTU network analysis (cytoscape [53]). High-throughput data can also be imported into other ecological workbenches (Primer-E [54], Galaxy [55]) which provide some additional multivariate statistics. Other methods include visualization workbenches (Figure 4B) such as GenGIS [56] and VisTrails [57] that allow graphical explorations of high-throughput sequence data, and Edge Principal Component Analysis (Figure 4C) for visually identifying key community lineages in tree topologies [58].
Figure 4.

High-throughput biodiversity research is an active and rapidly evolving field. Future analytical tools will expand towards a number of exciting, emerging research areas including (A) OTU network analysis, (B) visualization as an exploratory tool (modified from Shapiro et al. [95]), (C) Edge Principal Component Analysis to defining biological lineages that define community assemblages (after Matsen & Evans), and (D) Quantifying the impact of different OTU picking strategies on cluster formation.
Comparative Meta-analyses
As publicly available high-throughput data accumulate, we will soon gain an unprecedented view of microscopic eukaryote species on a truly global scale. Unfortunately, our ability to carry out such comparative eukaryotic meta-analyses is presently hindered by methodological heterogeneity, data accessibility, and underdeveloped computational infrastructure. The differential use of primer sets or gene regions (varying according to taxon and investigator preference) presents a substantial long-term challenge. Conserved primers targeting informative 18S regions [9] will provide a broad taxonomic view of eukaryote communities (although with consistent amplification biases) and provide useful data for biodiversity research, while other primers (e.g. ITS for fungi [59]) can deliver species-level data and address certain specific biological questions.
Future meta-analyses will ideally harness a robust subset of biologically meaningful OTUs, potentially made accessible through reference-based OTU picking. This evokes an urgent question: how do we compare the output of different OTU pickers and ensure the deposition of ‘real’ OTUs (e.g. species analogues) in reference databases? The goal of clustering is not to maximize or minimize the final OTU count, yet current approaches do not allow researchers to visualize and quantify how OTU picking affects the biological interpretation of sequence data [60]. New tools (Figure 4D) or tree-based algorithms (employing phylogenetic species concepts) could be applied to dynamically define species “clouds” from high-throughput rRNA sequence reads.
Data storage and access presents another significant barrier towards the effective use of published sequences. The fluctuating status and accessibility issues of the NCBI Short Read Archive (SRA) makes this resource less than suitable for meeting long-term community needs. Repositories such as Dryad (http://datadryad.org) and CAMERA [61] exist as alternatives but are limited by file size (10GB limit for Dryad) or are targeted towards a different purpose (microbial metagenomics for CAMERA). MG-RAST [62] accepts amplicon datasets in addition to its primary focus on metagenomes, and can be used as a resource for sequence deposition. Increasing data volumes also present an increasing need for data standards: for both sequences and metadata, standard workflows and documentation can provide a solid foundation to drive innovation and promote information discovery. The Genomic Standards Consortium [63] (http://gensc.org) is working to coordinate efforts such as MIMARKS standards (Minimum Information about a marker gene sequence [64]), promoting the collection of rich, contextual metadata to ensure the long term utility of published datasets and encourage comparative studies.
The need for robust guide trees and reference databases
Limited eukaryote reference databases and inconsistent taxonomic levels currently hinder the development of robust computational pipelines for marker gene data (e.g. reference-based OTU picking and confident taxonomy assignments [60]), and limit use of tree-based methods and deeper sequencing technologies with shorter sequence reads (such as those derived from the Illumina platforms). Microscopic eukaryotic taxa have been historically underrepresented in public repositories, with some divergent phyla represented by a single published rRNA sequence (e.g., the phylum Loricifera contains one reference sequence in SILVA release 106 [50]). The accuracy of BLAST-derived taxonomy depends on the database coverage for a given taxonomic group: for well-sampled groups (Arthropods, Annelids), it is possible to obtain genus-level accuracy, whereas only phylum-level accuracy (at best) might be possible for neglected ‘minor’ phyla (Loricifera, Gnathostomulida, deep protist lineages). Divergent lineages, common in poorly characterized environmental samples, often return few or no ‘good’ matches (>90%) from public repositories. Underpopulated databases also prevent the use of tools requiring taxonomic lineage information (e.g. RDP classifier [65]) and decrease the accuracy of probabilistic tools [66]. Databases for microscopic eukaryotes covering alternate loci are smaller (28S rRNA [50]) or virtually nonexistent (mtDNA), deterring the use of additional genes in high-throughput studies. Ultimately, a much larger collection of full-length eukaryotic reference sequences (or whole genomes) will be necessary for identifying erroneous reads, and providing a strong link between sequence data and morphology.
In theory, tree-insertion methods (pplacer [39], EPA [40]; Figure 3) will circumvent many of the issues that confound BLAST assignments, and these tools have recently become more broadly accessible with their incorporation into the QIIME pipeline [45]. The availability of software such as TopiaryExplorer [67] will greatly aid the visualization and interpretation of OTUs within phylogenetic trees. No high-quality, densely sampled guide tree exists for the eukaryotic domain, although recent efforts have substantially improved the backbone of deep splits across major taxa [68] (a eukaryotic guide tree currently exists for the SILVA reference database within the ARB software suite, but this tree also suffers from the above-mentioned taxon sampling issues, and represents only an approximated phylogenetic topology). In the future, tree-based tools will be a critical component of high-throughput analyses: providing an additional line of evidence to supplement BLAST hits (particularly where reference sequences are unclassified or misnamed), helping to identify divergent lineages (long branch taxa with no close reference sequences), and aiding the development of phylogenetic species concepts to delineate OTUs as putative species.
Future outlook and challenges
Although we are making substantial progress with high-throughput eukaryotic studies, many challenges lie ahead. A strong emphasis on morphological and environmental data collection, guide trees and reference sequence databases, and open-access repositories for high-throughput datasets is urgently needed. Large-scale sequencing methods offer substantial promise for basic and applied biodiversity research, yet the wider adoption of these approaches will likely hinge on the ease-of-use and accuracy of analytical tools and pipelines. Traditional ecologists and biologists typically have limited computational backgrounds, yet computer scientists rarely design cutting-edge tools with this fact in mind. Similarly, computational pipelines are not always subjected to rigorous benchmarking, unit testing, or proof-of-concept validation in relation to real-world biology. Encouraging an ongoing dialogue between computer scientists (who want to develop clever algorithms) and biologists (who want knowledge of ecology and taxonomy) will enable interdisciplinary collaborations that promote the development of easy-to-use, reliable, and well-documented software.
Conclusions
The promise and accessibility of high-throughput sequencing is now poised to attract increasing numbers of non-computationally trained researchers. With ongoing declines in the price of sequencing, deep sequencing will inevitably represent the most cost-effective approach for elucidating ecological and functional roles of complex communities. However, exploiting the data will require the continued refinement of bioinformatics pipelines and database resources, which will in turn require an ongoing and reciprocal collaboration between computational and biological scientists (Figure 5). A key challenge for high-throughput studies is to move beyond descriptions of differences between ecosystems, towards capturing whole ecosystem function. For any given sample, marker gene surveys will define community assemblages, metagenomics will reveal the genomic potential of a community, and metatranscriptomics, metaproteomics and metabolomics will provide a snapshot in time of community function. Combined with environmental information, these complementary approaches will enable us to define the driving forces that govern species distributions and drive ecological assemblages on scales ranging from microscopic to global.
Figure 5.
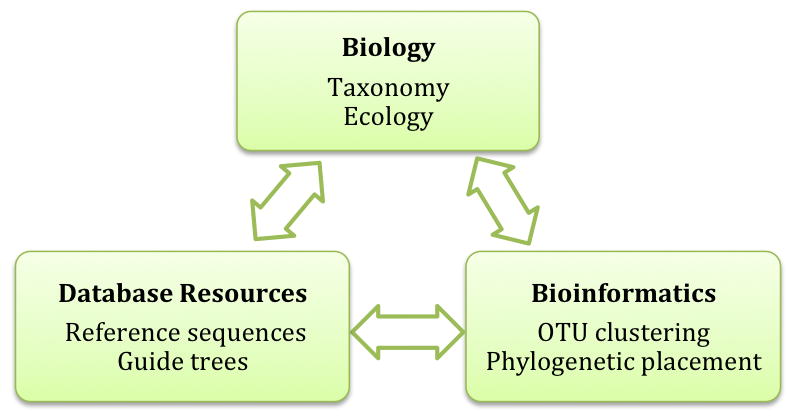
For future success, biodiversity research must adhere to a trifecta of biology, bioinformatics and database resources; none of these foci can exist in isolation, and each area must serve to inform the others. Biological questions drive high-throughput studies, and so computational pipelines and cyberinfrastructure need to functionally inform our knowledge of ecosystem processes. Likewise, computational resources must be complementary, whereby bioinformatic outputs are effectively databased, and evolving database resources produce continuing refinements in analytical pipelines. Seamless integration between these sectors will be crucial for enabling comparative metadata analyses and untangling complex ecological patterns – for example, mining published datasets for co-occurring species, or linking specific OTUs with environmental parameters (pH, salinity, temperature, etc.).
Figure 2.

High-throughput studies follow a common workflow that begins with raw sequence data and sample metadata (primer barcodes and environmental data). Raw data is filtered and processed, with the option of denoising (a step currently applicable only to 454 data) before Operational Taxonomic Units (OTUs) are picked through reference-based or de novo approaches. OTU picking can include pre-clustering steps such as Single Linkage Preclustering (SLP, [94]), prefix-suffix filtering or collapsing of identical sequences to reduce compute time (all methods available within the QIIME pipeline [51]); the recommended and default OTU picking workflow in QIIME currently involves sorting sequences by abundance, collapsing identical reads, picking OTUs de novo with uclust, and subsequently inflating the ‘identical reads’ to recapture abundance information about the initial sequences). Taxonomy is next assigned to OTU reference sequences, followed by construction of an OTU abundance matrix and a phylogenetic tree; when working with a closed reference-based OTU picking protocol it is not necessary to make taxonomic assignments or build a phylogenetic tree as these can be obtained directly from the reference data set. These outputs can be subsequently utilized for ecological diversity analyses and visualization approaches.
Box 2. Biases in physical and genomic sampling.
A typical assessment of eukaryotic diversity derived from environmental DNA comprises field, lab and bioinformatics components (methodologically similar to archaeal and bacterial approaches; Figure 1), each accompanied by specific challenges; a recent review by Creer et al. [9] provides a comprehensive outline of the workflow and methodological considerations involved with eukaryotic studies].
Replicated sampling schemes must effectively capture eukaryotic community diversity, given that species diversity and population densities can vary (spatially and temporally) by several orders of magnitudes. Although aquatic organisms from pelagic habitats can simply be concentrated from their environment [11, 28], interstitial eukaryotes are accompanied by a solid matrix (soil or sediment) that precludes direct environmental DNA extractions large enough to capture the diversity of rare taxa. Approaches for extraction have been tried and tested in unconsolidated marine [9, 30] and estuarine [33] sediments, but comprehensively separating the eukaryotic specimens from terrestrial soils, including muds, clays and large amounts of organic matter and inhibitors, poses additional challenges. Biases in taxon representation should be assumed whenever such extraction protocols are adopted. A further consideration for whole-sediment extractions is the potential existence of extracellular DNA or transient fauna [75, 76].
Following the separation of organisms from soil or sediment, bulk environmental DNA is extracted and specific gene targets are amplified via polymerase chain reaction (PCR) using appropriately selected, barcoded [77, 78] degenerate primers. The goal of marker gene surveys is to appraise as broad a taxonomic breadth as possible but both DNA extraction and PCR amplification are key steps that introduce biases [11, 31, 75]. To minimize such biases, different DNA extraction approaches can be compared [9], the use of PCR-primer cocktails implemented [79, 80] and the conservation of degenerate primer binding sites assessed using rRNA databases [45, 46, 50] and primer design tools.
While both physical and genomic sampling involve many steps known to introduce biases, certain steps can be taken to reduce such discrepancies. For example, applying multiple methods to physically extract organisms and using different combinations of primer sets (and genetic loci) to minimize the potential exclusion of taxa. To date, there has been little effort towards quantifying the impact of sampling protocols in eukaryotes (although sampling bias has been exhaustively assessed in archaea and bacteria [81–83], and parasitology studies [84, 85]; a robust understanding of these biases will be critical for our interpretation of community assemblages and informing practical applications for high-throughput techniques.
Outstanding questions for high-throughput marker gene studies include:
How diverse are communities of microscopic eukaryotes?
How geographically structured are these communities?
Are there taxonomic or life-history biases for true cosmopolitan species?
To what degree do environmental factors, bacteria and archaea, and eukaryotic communities interact to drive biotic assemblages?
Acknowledgments
The authors would like to thank the anonymous reviewers for their insightful comments that significantly helped to improve an earlier version of the manuscript. Development of this manuscript was made possible by a Catalysis Meeting award (HB and WKT) from the National Evolutionary Synthesis Center. HB and WKT supported through NSF (DEB-1058458). SC supported by a Natural Environment Research Council (NERC) New Investigator Grant (NE/E001505/1), a Post Genomic and Proteomics Grant (NE/F001266/1) and a Molecular Genetics Facility Grant (MGF-167). DP acknowledges funding from USDA/CSREES – TSTAR (grants 2006-04347 and 2008-34135-19505), NSF (DEB-0450537) and the CR-USA Foundation. RK and JGC supported in part by the National Institutes of Health, Bill and Melinda Gates Foundation, the Crohns and Colitis Foundation of America, the Sloan Indoor Environment program, and the Howard Hughes Medical Institute.
Glossary Box Terms
- Marker gene surveys
high-throughput environmental sequencing utilizing homologous genetic loci (e.g. 16S, 18S rRNA) amplified via conserved primer sets
- Metagenomics
high-throughput, random sequencing of genomic DNA from environmental isolates
- Metatranscriptomics
high-throughput sequencing of expressed gene transcripts (mRNA) from environmental isolates
- Meiofauna
a loose term to define metazoan species with a body size <1mm, although this size fraction often varies across studies
- OTU
Operational Taxonomic Unit, typically defined from high-throughput sequence data that is filtered for quality and subsequently clustered under pairwise identity cutoffs
- 454
Common term for the Roche GS platforms that use bead emulsion methods and typically return ~1.2 million sequences per full plate run (reads currently averaging 350–450 bp)
- Illumina
Company producing the newest Hi-Seq and MiSeq platforms, which uses bridge amplification to produce 1.6 billion sequences per 8-lane Hi-Seq flow cell (current max length for paired-end reads is 300 bp)
- Pyrosequencing
General term referring to light-based high-throughput sequencing techniques (e.g. 454)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Danovaro R, et al. Exponential decline of deep-sea ecosystem functioning linked to benthic biodiversity loss. Current Biology. 2008;18:1–18. doi: 10.1016/j.cub.2007.11.056. [DOI] [PubMed] [Google Scholar]
- 2.Wardle DA. The influence of biotic interactions on soil biodiversity. Ecology Letters. 2006;9:870–886. doi: 10.1111/j.1461-0248.2006.00931.x. [DOI] [PubMed] [Google Scholar]
- 3.Wegner Parfrey L, et al. Microbial eukaryotes in the human microbiome: ecology, evolution, and future directions. Frontiers in Microbiology. 2011;2:1–16. doi: 10.3389/fmicb.2011.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Behnke A, et al. Microeukaryote community patterns along an O2/H2S Gradient in a supersulfidic Anoxic Fjord (Framvaren, Norway) Applied and environmental microbiology. 2006;72:3626–3636. doi: 10.1128/AEM.72.5.3626-3636.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Groisillier A, et al. Genetic diversity and habitats of two enigmatic marine alveolate lineages. Aquatic Microbial Ecology. 2006;42:277–291. [Google Scholar]
- 6.Richards T, Bass D. Molecular screening of free-living microbial eukaryotes: diversity and distribution using a meta-analysis. Current Opinion in Microbiology. 2005;8:240–252. doi: 10.1016/j.mib.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 7.Rosling A, et al. Archaeorhizomycetes: Unearthing an ancient class of ubiqutous soil fungi. Science. 2011;333:876–879. doi: 10.1126/science.1206958. [DOI] [PubMed] [Google Scholar]
- 8.Jones MDM, et al. Discovery of novel intermediate forms redefines the fungal tree of life. Nature. 2011;474:200–203. doi: 10.1038/nature09984. [DOI] [PubMed] [Google Scholar]
- 9.Creer S, et al. Ultrasequencing of the meiofaunal biosphere: practice, pitfalls, and promises. Molecular Ecology. 2010;19:4–20. doi: 10.1111/j.1365-294X.2009.04473.x. [DOI] [PubMed] [Google Scholar]
- 10.Porazinska DL, et al. Evaulating high-throughput sequencing as a method for metagenomic analysis of nematode diversity. Molecular Ecology Resources. 2009;9:1439–1450. doi: 10.1111/j.1755-0998.2009.02611.x. [DOI] [PubMed] [Google Scholar]
- 11.Stoeck T, et al. Multiple marker parallel tag enviornmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Molecular Ecology. 2010;19:21–31. doi: 10.1111/j.1365-294X.2009.04480.x. [DOI] [PubMed] [Google Scholar]
- 12.Stoeck T, et al. Massively parallel tag sequencing reveals the complexity of anaerobic marine protistan communities. BMC Biology. 2009;7:72. doi: 10.1186/1741-7007-7-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pace NR. A molecular view of microbial diversity and the biosphere. Science. 1997;276:734–740. doi: 10.1126/science.276.5313.734. [DOI] [PubMed] [Google Scholar]
- 14.Sogin ML, et al. Microbial diversity in the deep sea and the unexplored “rare biosphere”. Proceedings of the National Academy of Science USA. 2006;103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amaral-Zettler L, et al. A global census of marine microbes. In: McIntyre AD, editor. Life in the World’s Oceans: Diversity, Distribution and Abundance. Blackwell Publishing; 2010. pp. 223–245. [Google Scholar]
- 16.Fierer N, et al. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proceedings of the National Academy of Science USA. 2008;105:17994–17999. doi: 10.1073/pnas.0807920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turnbaugh PJ, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prokopowich CD, et al. The correlation between rDNA copy number and genome size in eukaryotes. Genome. 2003;46:48–50. doi: 10.1139/g02-103. [DOI] [PubMed] [Google Scholar]
- 19.Pei AY, et al. Diversity of 16S rRNA genes within individual prokaryotic genomes. Applied and environmental microbiology. 2010;76:3886–3897. doi: 10.1128/AEM.02953-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu F, et al. Mapping of picoeukaryotes in marine ecosystems with a quantitative PCR of the 18S rRNA gene. FEMS Microbiology Ecology. 2005;52:79–92. doi: 10.1016/j.femsec.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 21.Andersson JO. Lateral gene transfer in eukaryotes. Celluar and molecular life sciences. 2005;62:1182–1197. doi: 10.1007/s00018-005-4539-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huber JA, et al. Microbial population structures in the deep marine biosphere. Science. 2007;318:97–100. doi: 10.1126/science.1146689. [DOI] [PubMed] [Google Scholar]
- 23.Amaral-Zettler L, et al. A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small-subunit ribosomal RNA genes. PLoS One. 2009;4:e6372. doi: 10.1371/journal.pone.0006372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Medinger R, et al. Diversity in a hidden world: potential an limitation of next-generation sequencing for surveys of molecular diversity of eukaryotic microorganisms. Molecular Ecology. 2010;19:32–40. doi: 10.1111/j.1365-294X.2009.04478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Orsi W, et al. Protistan microbial observatory in the Cariaco Basin, Caribbean. II. Habitat specialization. The ISME Journal. 2011;5:1357–1373. doi: 10.1038/ismej.2011.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edgcomb V, et al. Protistan microbial observatory in the Cariaco Basin, Caribbean. I. Pyrosequencing vs Sanger insights into species richness. The ISME Journal. 2011;5:1344–1356. doi: 10.1038/ismej.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bik HM, et al. Metagenetic community analysis of microbial eukaryotes illuminates biogeographic patterns in deep-sea and shallow water sediments. Molecular Ecology. 2011 doi: 10.1111/j.1365–1294X.2011.05297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nolte V, et al. Contrasting seasonal niche separation between rare and abundant taxa conceals the extent of protist diversity. Molecular Ecology. 2010;19:2908–2915. doi: 10.1111/j.1365-294X.2010.04669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reeder J, Knight R. Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nature Methods. 2010;7:668–669. doi: 10.1038/nmeth0910-668b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fonseca VG, et al. Second-generation environmental sequencing unmasks marine metazoan biodiversity. Nature Communications. 2010;1:98. doi: 10.1038/ncomms1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Behnke A, et al. Depicting more accurate pictures of protistan community complexity using pyrosequencing of hypervariable SSU rRNA gene regions. Environmental Microbiology. 2011;13:340–349. doi: 10.1111/j.1462-2920.2010.02332.x. [DOI] [PubMed] [Google Scholar]
- 32.Lecroq B, et al. Ultra-deep sequencing of foraminiferal microbarcodes unveils hidden richness of early monothalamous lineages in deep-sea sediments. Proceedings of the National Academy of Science USA. 2011 doi: 10.1073/pnas.1018426108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chariton A, et al. Ecological assessment of estuarine sediments by pyrosequencing eukaryotic ribosomal DNA. Frontiers in Ecology and the Environment. 2010;8:233–238. [Google Scholar]
- 34.Hajibabaei M, et al. Environmental Barcoding: A next-generation sequencing approach for biomonitoring applications using river benthos. PLoS One. 2011;6:e17497. doi: 10.1371/journal.pone.0017497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pfrender ME, et al. Assessing macroinvertebrate biodiversity in freshwater ecosystems: advances and challenges in DNA-based approaches. Quarterly Review of Biology. 2010;85:319–340. doi: 10.1086/655118. [DOI] [PubMed] [Google Scholar]
- 36.Emerson B, et al. Phylogeny, phylogeography, phylobetadiversity and the molecular analysis of biological communities. Philosophical Transactions of the Royal Society B. 2011;366:2391–2402. doi: 10.1098/rstb.2011.0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Purdy KJ, et al. Systems Biology for Ecology: From molecules to ecosystems. Advances in Ecological Research. 2010;43:87–149. [Google Scholar]
- 38.Quince C, et al. Removing noise from pyrosequenced amplicons. BMC Bioinformatics. 2011;12:38. doi: 10.1186/1471-2105-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsen FA, et al. pplacer: linear time maximum-likelihood Bayesian phyogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics. 2010;11:538. doi: 10.1186/1471-2105-11-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berger SA, Stamatakis A. Aligning short reads to reference alignments and trees. Bioinformatics. 2011;27:2068–2075. doi: 10.1093/bioinformatics/btr320. [DOI] [PubMed] [Google Scholar]
- 41.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 42.Schloss PD, et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and environmental microbiology. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li W, Godzik A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 44.Cai Y, Sun Y. ESPRIT-Tree: hierarchial clustring analysis of millions of 16S rRNA pyrosequences in quasilinear computational time. Nucleic Acids Research. 2011;39:e95. doi: 10.1093/nar/gkr349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cole JR, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Research. 2009;37:D141–145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DeSantis T, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and environmental microbiology. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caporaso JG, et al. Host-associated and free-living phage communities differ profoundly in phylogenetic composition. PLoS One. 2011;6:e16900. doi: 10.1371/journal.pone.0016900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haas BJ, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Research. 2011;21:494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edgar RC, et al. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pruesse E, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Research. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lozupone C, et al. Unifrac: an effective distance metric for microbial community comparison. The ISME Journal. 2011;5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cline MS, et al. Integration of biological networks and gene expression data using Cytoscape. Nature Protocols. 2007;2:2366–2383. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clarke K, Gorley R. PRIMER v6: User Manual/Tutorial. 2006. [Google Scholar]
- 55.Blankenberg D, et al. Galaxy: A web-based genome analysis tool for experimentalists. Current Protocols in Molecular Biology. 2010;Chapter 19 doi: 10.1002/0471142727.mb1910s89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parks DH, et al. GenGIS: A geospatial information system for genomic data. Genome Research. 2009;19:1896–1904. doi: 10.1101/gr.095612.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Callahan SP, et al. VisTrails: Visualization meets Data Management. Proceedings of ACM SIGMOD; June 27–29, 2006; ACM Press; 2006. pp. 745–747. [Google Scholar]
- 58.Matsen FA, Evans S. Edge principal components and squash clustering: using the special structure of phylogenetic placement data for sample comparison. 2011 doi: 10.1371/journal.pone.0056859. arXiv:1107.5095v1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bellemain E, et al. ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. BMC Microbiology. 2010;10:189. doi: 10.1186/1471-2180-10-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Christen R. Global sequencing: A review of current molecular data and new methods available to assess microbial diversity. Microbes and Environments. 2008;23:253–268. doi: 10.1264/jsme2.me08525. [DOI] [PubMed] [Google Scholar]
- 61.Seshadri R, et al. CAMERA: a community resource for metagenomics. Plos Biology. 2007;5:e75. doi: 10.1371/journal.pbio.0050075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meyer F, et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Field D, et al. The Genomic Standards Consortium. Plos Biology. 2011;9:e1001088. doi: 10.1371/journal.pbio.1001088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yilmaz P, et al. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nature Biotechnology. 2011;29:415–420. doi: 10.1038/nbt.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Q, et al. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Werner JJ, et al. Impact of training sets on classification of high-throughput bacerial 16S rRNA gene surveys. The ISME Journal. 2011 doi: 10.1038/ismej.2011.1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pirrung M, et al. TopiaryExplorer: Visualizing large phylogenetic trees with environmental metadata. Bioinformatics. 2011 doi: 10.1093/bioinformatics/btr1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wegener Parfrey L, et al. Broadly sampled multigene analyses yield a well-resolved eukaryotic tree of life. Systematic Biology. 2010;59:518–533. doi: 10.1093/sysbio/syq037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dover G. Molecular drive: a cohesive mode of species evolution. Nature. 1982;299:111–117. doi: 10.1038/299111a0. [DOI] [PubMed] [Google Scholar]
- 70.Lyckegaard EM, Clark AG. Evolution of ribosomal RNA gene copy number on the sex chromosomes of Drosophila melanogaster. Molecular Biology and Evolution. 1991;8:458–474. doi: 10.1093/oxfordjournals.molbev.a040664. [DOI] [PubMed] [Google Scholar]
- 71.Averbeck KT, Eickbush TH. Monitoring the mode and tempo of concerted evolution in Drosophila melanogaster rDNA locus. Genetics. 2005;171:1837–1846. doi: 10.1534/genetics.105.047670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.James SA, et al. Repetitive sequence variation and dynamics in the ribosomal DNA array of Saccharomyces cerevisiae as revealed by whole-genome resequencing. Genome Research. 2009;19:626–635. doi: 10.1101/gr.084517.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Santos SR, et al. Molecular characterization of nuclear small subunit (18S)-rDNA pseudogenes in a symbiotic dinoflagellate (Symbiodinium, Dinophyta) Journal of Eukaryotic Microbiology. 2003;50:417–421. doi: 10.1111/j.1550-7408.2003.tb00264.x. [DOI] [PubMed] [Google Scholar]
- 74.Schlöetterer C, Tautz D. Chromosomal homogeneity of Drosophila ribosomal DNA arrays suggests intrachromosomal exchanges drive concerted evolution. Current Biology. 1994;4:777–783. doi: 10.1016/s0960-9822(00)00175-5. [DOI] [PubMed] [Google Scholar]
- 75.Pawlowski J, et al. Eukaryotic richness in the abyss: Insights from Pyrotag sequencing. PLoS One. 2011;6:e18169. doi: 10.1371/journal.pone.0018169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dell’Anno A, Danovaro R. Extracellular DNA plays a key role in deep-sea ecosystem functioning. Science. 2005;309:2179. doi: 10.1126/science.1117475. [DOI] [PubMed] [Google Scholar]
- 77.Binladen J, et al. The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing. PLoS One. 2007;2:e197. doi: 10.1371/journal.pone.0000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hamady M, et al. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nature Methods. 2008;5:235–237. doi: 10.1038/nmeth.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ivanova NV, et al. Universal primer cocktails for fish DNA barcoding. Molecular Ecology. 2007;7:544–548. [Google Scholar]
- 80.Matzen da Silva J, et al. Systematic and evolutionary insights derived from mtDNA COI barcode diversity in the Decapoda (Crustacea: Malacostraca) PLoS One. 2011;6:e19449. doi: 10.1371/journal.pone.0019449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Feinstein LM, et al. Assessment of bias associated with incomplete extraction of microbial DNA from soil. Applied and environmental microbiology. 2009;75:5428–5433. doi: 10.1128/AEM.00120-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Engelbrektson A, et al. Experimental factors affecting PCR-based estimates of microbial species richness and evenness. The ISME Journal. 2010;4:642–647. doi: 10.1038/ismej.2009.153. [DOI] [PubMed] [Google Scholar]
- 83.Hong S, et al. Polymerase chain reaction primers miss half of rRNA microbial diversity. The ISME Journal. 2009;3:1365–1373. doi: 10.1038/ismej.2009.89. [DOI] [PubMed] [Google Scholar]
- 84.Valkiunas G, et al. A comparative analysis of microscopy and PCR-based detection methods for blood parasites. Journal of Parasitology. 2008;94:1395–1401. doi: 10.1645/GE-1570.1. [DOI] [PubMed] [Google Scholar]
- 85.Desquesnes M, Dávila AMR. Applications of PCR-based tools for detection and identification of animal trypanosomes: a review and perspectives. Veterinary Parasitology. 2002;109:213–231. doi: 10.1016/s0304-4017(02)00270-4. [DOI] [PubMed] [Google Scholar]
- 86.Huse SM, et al. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biology. 2007;8:R143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gilles A, et al. Accuracy and quality assessment of 454 GS-FLX Titanium pyrosequencing. BMC Genomics. 2011;12:245. doi: 10.1186/1471-2164-12-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Porazinska DL, et al. Linking operational clustered taxonomic units (OCTUs) from parallel ultra sequencing (PUS) to nematode species. Zootaxa. 2010;2427:55–63. [Google Scholar]
- 89.Porazinska DL, et al. Reproducibility of read numbers in high-throughput sequencing analysis of nematode community composition and structure. Molecular Ecology Resources. 2009;10:666–676. doi: 10.1111/j.1755-0998.2009.02819.x. [DOI] [PubMed] [Google Scholar]
- 90.Pester M, et al. A “rare biosphere” microorganism contributes to sulfate reduction in a peatland. The ISME Journal. 2010;4:1591–1602. doi: 10.1038/ismej.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Creer S. Second-generation seequencing derived insights into the temporal biodiversity dynamics of freshwater protists. Molecular Ecology. 2010;19:2829–2831. doi: 10.1111/j.1365-294X.2010.04670.x. [DOI] [PubMed] [Google Scholar]
- 92.Smyth RP, et al. Reducing chimera formation during PCR amplification to ensure accurate genotyping. Gene. 2010;469:45–51. doi: 10.1016/j.gene.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 93.Quince C, et al. Accurate determination of microbial diversity from 454 pyrosequencing data. Nature Methods. 2009;6:639–641. doi: 10.1038/nmeth.1361. [DOI] [PubMed] [Google Scholar]
- 94.Huse SM, et al. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environmental Microbiology. 2010;12:1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shapiro LH, et al. Molecular phylogeny of Banza (Orthoptera: Tettigoniidae), the endemic katydids of the Hawaiian Archipelago. Molecular Phylogenetics and Evolution. 2006;41:53–63. doi: 10.1016/j.ympev.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 96.Pandey R, et al. CANGS: a user-friendly utility for processing and analyzing 454 GS-FLX data in biodiversity studies. BMC Research Notes. 2010;12:182. doi: 10.1186/1756-0500-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kumar S, et al. CLOTU: An online pipeline for processing and clustering of 454 amplicon reads into OTUs followed by taxonomic annotation. BMC Bioinformatics. 2011;12:182. doi: 10.1186/1471-2105-12-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sun Y, et al. ESPRIT: estimating species richness uing large collections of 16S rRNA pyrosequences. Nucleic Acids Research. 2009;37:e76. doi: 10.1093/nar/gkp285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huson DH, et al. Integrative analysis of environmental sequences using MEGAN4. Genome Research. 2011;21:1552–1560. doi: 10.1101/gr.120618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Greenberg J. Theoretical considerations of lifecycle modeling: An analysis of the Dryad repository demonstrating automatic metadata propagation, inheritance, and value system adoption. Cataloging & Classification Quarterly. 2009;47:380–402. [Google Scholar]
- 101.Hankeln W, et al. MetaBar - a tool for consistent contextual data acquisition and standards compliant submission. BMC Bioinformatics. 2010;11:358. doi: 10.1186/1471-2105-11-358. [DOI] [PMC free article] [PubMed] [Google Scholar]