

Abstract
Three new O-prenylated flavonoids, amyrisins A – C (1 –2), were isolated from the leaves and twigs of Amyris madrensis, along with the known compound, polygamain (4). The structures of 1 – 3 were elucidated based on the analysis of spectroscopic data. Amyrisins B (2) and C (3) showed moderate cytotoxicity against PC-3 and DU 145 prostate cancer cells with IC50 values of 17.5 and 23 μM, respectively while amyrisin A (1) did not show any cytotoxicity at the highest concentration tested, 50 μM. Polygamain (4) exhibited potent antiproliferative and microtubule depolymerizing activities.
A focus of our laboratory is the identification of new cytotoxic compounds with potential antitumor activity from plants that thrive in the harsh environment of South Texas.1–3 Over 360 species of plants have been collected and the fresh plant material extracted. One of the plants collected, Amyris madrensis S. Watson, the mountain torchwood, is a perennial shrub belonging to the Rutaceae family that is distributed throughout South Texas and Northern Mexico. The aerial parts of A. madrensis have occasionally been used in folk medicine in Mexico and two coumarins were previously identified from the stems and leaves of A. madrensis.4 The supercritical CO2 extract from the leaves and stems of A. madrensis was toxic to prostate cancer cells. The extract inhibited the growth of PC-3 and DU 145 prostate cancer cells with IC50 values of 6.0 and 7.3 μg/mL, respectively. Additionally, mechanistic assays showed that the crude extract caused cellular microtubule loss similar to the effects of vinblastine. In this study, we report the identification of three new O-prenylated flavonoids, named amyrisins A (1), B (2), and C (3), along with the microtubule destabilizer polygamain (4) from this extract.
The stems and leaves of A. madrensis were extracted using supercritical CO2. The extract was subjected to silica gel column chromatography followed by reversed-phase HPLC to yield amyrisins A – C (1 – 3) and polygamain (4).
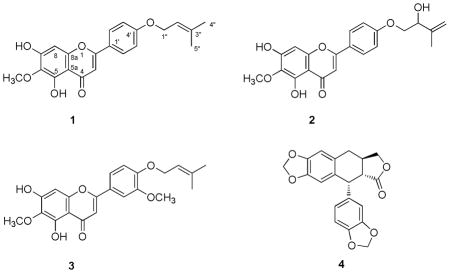
Amyrisin A (1) was obtained as a yellow powder and the molecular formula C21H20O6 was determined by HRMS at m/z 369.1396 [M+H]+ (calcd 369.1388). The proton and carbon NMR spectra suggested a flavonoid skeleton for 1. The 1H NMR spectrum (Table 1) showed a pair of aromatic signals at δ 7.03 (2H, d, J = 9.0 Hz) and 7.83 (2H, d, J = 9.0 Hz), which were assigned to H-3′,5′ and H-2′,6′, suggesting oxygenation at C-4′ for this flavone. Two singlet proton signals at δ 6.58 and 6.60 were ascribed to H-3 and H-8, respectively, based on the HMBC correlation between H-3/C-1′, C-2, C-4, C-10 and H-8/C-6, C-7, C-9, C-10. A downfield signal at δ 13.12 was characteristic for an OH-5 group. A prenyloxy unit could be deduced from the methylene signal at δ 4.60 (2H, d, J = 6.5 Hz), an olefinic signal at δ 5.51 (t, J = 6.5 Hz), and two methyl signals at δ 1.78 (3H, s) and 1.83 (3H, s). Additional signals belonging to a methoxy group at δ 4.05 (3H, s) and a hydroxy group at δ 6.49 (br) were observed. The HMBC correlations between the OH-5 (13.12 ppm) and C-10 (105.9 ppm), C-5 (152.3), and C-6 (130.5 ppm), allowed assignments of the C-5, C-6, and C-10 signals. The methoxy substituent was determined at C-6 by the HMBC correlation between the methoxy group protons and C-6. The location of isoprenyloxy at C-4′ was evidenced by the HMBC correlation between H-1″ and C-4′. Thus, the structure of 1 was determined as 5,7-dihydroxy-6-methoxy-2-(4-((3-methylbut-2-en-1-yl)oxy)phenyl)-4H-chromen-4-one.
Table 1.
1H and 13C NMR Data of Amyrisins A–C (1 – 3) in CDCl3
| Position | 1 | 2a | 3a | |||
|---|---|---|---|---|---|---|
| 13C | 1H | 13C | 1H | 13C | 1H | |
| 2 | 164.4 | 164.1 | 164.1 | |||
| 3 | 103.9 | 6.58 s | 103.9 | 6.58 s | 103.9 | 6.96 s |
| 4 | 183.1 | 182.7 | 6.60 s | 182.7 | ||
| 5 | 152.3 | 152.3 | nd | |||
| 6 | 130.5 | 130.4 | 130.1 | |||
| 7 | 155.0 | 155.1 | 155.0 | |||
| 8 | 93.4 | 6.60 s | 93.4 | 6.60 s | 93.4 | 6.98 s |
| 9 | 153.3 | 153.8 | 153.1 | |||
| 10 | 105.9 | 105.7 | 105.8 | |||
| 1′ | 123.6 | 124.5 | 123.5 | |||
| 2′ | 128.2 | 7.83 d, 9.0 | 128.3 | 7.84 d, 9.0 | 108.8 | 7.22, d, 2.1 |
| 3′ | 115.4 | 7.03 d, 9.0 | 115.1 | 7.05 d, 9.0 | 149.6 | |
| 4′ | 162.1 | 161.7 | 151.6 | |||
| 5′ | 115.4 | 7.03 d, 9.0 | 115.1 | 7.05 d, 9.0 | 112.7 | 6.97 d, 8.6 |
| 6′ | 128.2 | 7.83 d, 9.0 | 128.3 | 7.84 d, 9.0 | 120.1 | 7.49 dd, 8.5, 2.1 |
| 1″ | 65.3 | 4.60 d, 6.5 | 71.9 | 4.14 dd, 9.5, 3.2 4.04 t, 9.2 |
66.1 | 4.67 d, 6.6 |
| 2″ | 119.1 | 5.51 t, 6.5 | 74.1 | 4.53 m | 119.3 | 5.52 t, 6.6 |
| 3″ | 139.2 | 143.3 | 138.7 | |||
| 4″ | 18.4 | 1.78 s, 3H | 113.3 | 5.19 s 5.06 s |
18.0 | 1.77 s |
| 5″ | 26.0 | 1.83 s, 3H | 18.7 | 1.86 s, 3H | 25.5 | 1.80 s |
| OCH3-6 | 61.0 | 4.05 s, 3H | 60.7 | 4.05 s, 3H | 61.0 | 4.05 s |
| OCH3-3′ | 56.3 | 3.96 s | ||||
| OH-5 | 13.12 s | 13.01 s | 13.09 s | |||
| OH-7 | 6.49 brs | 6.49 s | ||||
13C NMR data were obtained from the HSQC and HMBC spectra due to the small quantity of material available, nd = not detected.
Amyrisin B (2) was obtained as yellow powder and the molecular formula, C21H20O7,wasdeduced by HRMS at m/z 385.1299 [M+H]+ (calcd 385.1287) and the NMR data. The 1H and 13C NMR data (Table 1) for 2 were identical to that of amyrisin A (1) except for the signals observed for the isoprenyloxy group present. The latter group in 2 was determined to be 2-hydroxy-isopentenyloxy by the proton NMR data at δ 4.14 (dd, J = 9.5, 3.2 Hz, H-1″), 4.04 (t, J = 9.2 Hz, H-1″), 4.53 (m, H-2″), 5.19 (s, H-4″),5.06 (s, H-4″), and 1.86 (s, H-5″), and the 13C NMR data at δ 71.9 (C-1″), 74.1 (C-2″), 143.3 (C-3″), 113.3 (C-4″), 18.7 (C-5″). The HMBC correlations between H-1″ (both 4.14 and 4.04) and C-4′ (δ 161.7) indicated the prenyloxy group was at C-4′. Thus, the structure of 2 was determined to be 5,7-dihydroxy-2-(4-((2-hydroxy-3-methylbut-3-en-1-yl)oxy)phenyl)-6-methoxy-4H-chromen -4-one. The limited quantity of 2 obtained precluded determination of the absolute configuration of the hydroxyl group at C-2″.
Amyrisin C (3) was also obtained as yellow powder. A molecular formula of C22H22O7 was deduced by HRMS at m/z 399.1447 [M+H]+ (calcd 399.1444) and the NMR data. The 1H NMR (Table 1) showed signals at δ 6.96 (s, H-3), 6.98 (s, H-8), 13.09 (s, OH-5), and 4.05 (s, OCH3-6), indicating that 3 has the same A and C rings as 1. Substitution at C-3′, C-4′ of the B ring was evidenced by proton signals at δ 7.22 (d, J = 2.1 Hz, H-2′), 6.97 (d, J = 8.6 Hz, H-5′), and 7.49 (dd, J = 8.5, 2.1 Hz, H-6′). Additional signals for a 3-methyl-2-butene-1-ol substituent at δ 4.67 (d, J = 6.6, Hz, H-1″), 5.52 (t, J = 6.6, Hz, H-2″), 1.80 (s, H-4″), and 1.77 (s, H-5″) and for a methoxy group at δ 3.96 were observed. The HMBC correlation between δ 3.96 and δ 149.6 (C-3′) indicated this methoxy group to be located at C-3′. Thus, 3 was determined to be 5,7-dihydroxy-6-methoxy-2-(3-methoxy-4-((3-methylbut-2-en-1-yl)oxy)phenyl)-4H-chromen-4-one.
The structure of polygamain (4) was determined by 2 D NMR data and comparison with the data previously published.5,6
The cytotoxic and microtubule disrupting activity of the compounds were evaluated. Compounds 2 and 3 exhibited moderate cytotoxicity against PC-3 cells with IC50 values of 17.5 ± 4.5 and 23.0 ± 5.3 μM, respectively. In contrast, 1 did not cause any cytotoxicity even at concentrations up to 50 μM. The known lignan, polygamain(4), was the most potent of this series with an IC50 value of 70.6 ± 2.6 nM in PC-3 cells. Compounds 1 – 3 were evaluated for their effects on cellular microtubules, but no disruption of microtubules was observed, suggesting that the cytotoxicity exhibited by these compounds was not microtubule mediated. Polygamain (4) was found to be a potent microtubule depolymerizer with effects similar to podophyllotoxin and combretastatin A-4.7
Further assays were conducted with 3 in an attempt to determine its cytotoxic mechanism of action. PC-3 cells were treated with a 50 μM concentration of 3 for 18 h and cell cycle distribution determined using flow cytometry. The results showed that 3 had no effects on the cell cycle distribution, eliminating many common mechanisms of cytotoxicity, which inhibit normal cell cycle progression and cause cells to accumulate in specific phases of the cell cycle.
Experimental Section
General Experimental Procedures
Specific rotation was recorded on a Rudolph Autopol IV polarimeter. UV spectra were obtained online by Waters 996 PDA Detector. HRESIMS were measured using an Agilent Technologies 6224 TOF LC/MS mass system. NMR spectra were recorded on a Bruker Avance 600 MHz or 500 MHz instrument. All spectra were measured and reported in ppm by using the residual solvent (CDCl3) as an internal standard. HRMS were measured using Agilent Technologies 6224 TOF LC/MS system. TLC was performed on aluminum sheets (silica gel 60 F254, Merck KGaA, Germany). HPLC was performed on a Waters Breeze HPLC system and a Phenomenex Luna C18(2) 250 × 21.2 mm column was used. LC/MS was recorded on a Waters Alliance 2695 HPLC equipped with Micromass Quattro triple quadrupole mass spectrometer using ESI model.
Plant Material
Leaves and twigs of Amyris madrensis were obtained from the San Antonio Botanical Gardens in San Antonio, Texas in July 2007. The samples were harvested, transported to the laboratory, the leaves and stems removed and then they were frozen and lyophilized. Voucher specimens (SLM188) were deposited in our herbarium and authenticated byPaul Cox, Superintendent of the San Antonio Botanical Gardens.
Extraction and Isolation
The lyophilized plant material was ground to a powder (166 g) and then extracted using supercritical fluid CO2 at 500 bar and 50 °C to yield 5.80 g of extract. A portion of the extract (3.92 g) was dissolved in 150 mL of hexanes and the soluble material was removed. The hexane- insoluble residue (1.04 g) was solubilized in methylene chloride and subjected to silica gel (Biotage 40+S column) flash chromatography and eluted with a gradient of methylene chloride and ethyl acetate. Fraction 5, eluted using 100% methylene chloride, was further separated by silica gel (eluted with hexanes and ethyl acetate 9:1) and reversed phase HPLC (eluted with a gradient of methanol and water) to yield compound 4 (2.0 mg). Fraction 55, which was eluted with methylene chloride and ethyl acetate (8:2), was separated using reversed-phase HPLC to obtain compounds 1 (0.9 mg) and 3 (0.5 mg). Fraction 78, which was eluted with ethyl acetate, was purified by HPLC to yield compound 2 (0.4 mg).
Amyrisin A (1)
Yellow powder, UV λmax (ACN-H2O) 274, 334 nm; 1H and 13C NMR data, see Table 1; HRMS m/z 369.1396 [M+H]+ (calcd 369.1388); ESIMS m/z 369.1 [M+H]+, 301.1 [M+H-isoprenyl]+.
Amyrisin B (2)
Yellow powder, [α]20D + 6.3 (c 0.03, MeOH); 1H and 13C NMR data, see Table 1; UV λmax (ACN-H2O) 274, 335 nm; HRMS m/z 385.1299 [M+H]+ (calcd 385.1287).
Amyrisin C (3)
Yellow powder, UV λmax (ACN-H2O) 275, 342 nm; 1H and 13C NMR data, see Table 1; HRMS m/z 399.1447 [M+H]+ (calcd 399.1444); ESI-MS m/z 399.1 [M+H]+, 331.1 [M+H-isoprenyl]+.
Biological Assays
PC-3 and DU 145 prostate cancer cells were purchased from the American Type Culture Collection (Manassas, VA). PC-3 cells were cultured in RPMI Medium 1640 (Invitrogen, Carlsbad, CA) with 10% FBS and 50 μg/mL gentamicin and DU 145 cells were cultured in Richter’s IMEM (Invitrogen) with 10% FBS and 25 μg/mL gentamicin. The SRB assay8 was used to evaluate the potency of the compounds as previously described.1 Cells plated in 96-well plates at predetermined densities were incubated with a range of concentrations of 1–4 for 48 h. The concentration that caused 50% inhibition of cellular proliferation (IC50) was determined and is an average of two independent experiments conducted in triplicate. The effect of each compound on cellular microtubules was evaluated as previously described.9
Flow cytometry
The effects of amyrisin C on cell cycle distribution were evaluated using flow cytometry. HeLa cells were treated with vehicle (DMSO), 12.5 nM paclitaxel as a positive control, or 50 μM amyrisin C for 18 h and then the cells were harvested, stained with Krishan’s reagent10 and cell cycle distribution analyzed with a FACSCalibur flow cytometer (BD Biosciences).
Acknowledgments
This work was supported by a grant from the Department of Defense Prostate Cancer Program W81XWH-08-1-0395 (SLM). Rachel Hartley was supported by NIDCR grant number T32DE14318, the COSTAR Program. Support from the CTRC Cancer Center Support Grant, P30CA054174, and the Flow Cytometry, Mass Spectrometry and Macromolecular Structure Shared Resources are gratefully acknowledged. The authors thank Paul Cox for his help in plant identification.
Footnotes
Dedicated to Dr. Gordon M. Cragg, formerly of the National Cancer Institute, Frederick, Maryland, for his pioneering work on the development of natural product anticancer agents.
Supporting Information Available: NMR spectra of compounds 1–3. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Risinger AL, Jackson EM, Polin LA, Helms GL, LeBoeuf DA, Joe PA, Hopper-Borge E, Luduena RF, Kruh GD, Mooberry SL. Cancer Res. 2008;68:8881–8888. doi: 10.1158/0008-5472.CAN-08-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peng J, Jackson EM, Babinski DJ, Risinger AL, Helms G, Frantz DE, Mooberry SL. J Nat Prod. 2010;73:1590–1592. doi: 10.1021/np100350s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peng J, Risinger AL, Fest GA, Jackson EM, Helms G, Polin LA, Mooberry SL. J Med Chem. 2011;54:6117–6124. doi: 10.1021/jm200757g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dominguez XA, Cano G, Luna I, Dieck A. Phytochemistry. 1977;16:1096. [Google Scholar]
- 5.Hokanson GC. J Nat Prod. 1979;42:378–384. [Google Scholar]
- 6.Sheriha GM, Abouamer K, Elshtaiwi BZ, Ashour AS, Abed FA, Alhallaq HH. Phytochemistry. 1987;26:3339–3341. [Google Scholar]
- 7.Hartley RM, Peng J, Fest GA, Dakshanamurthy S, Frantz DE, Brown M, Mooberry SL. Mol Pharmacol. 2011 doi: 10.1124/mol.111.075838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 9.Tinley TL, Randall-Hlubek DA, Leal RM, Jackson EM, Cessac JW, Quada JC, Jr, Hemscheidt TK, Mooberry SL. Cancer Res. 2003;63:3211–3220. [PubMed] [Google Scholar]
- 10.Krishan A. J Cell Biol. 1975;66:188–193. doi: 10.1083/jcb.66.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]