

SUMMARY
Pigment gallstones, which are much less frequent than cholesterol stones, are classified descriptively as ‘black’ or ‘brown’. They are composed mostly of calcium hydrogen bilirubinate, Ca(HUCB)2, which is polymerized and oxidized in ‘black‘ stones but remains unpolymerized in ‘brown’ stones. Black stones form in sterile gallbladder bile but brown stones form secondary to stasis and anaerobic bacterial infection in any part of the biliary tree, including the gallbladder. Other calcium salts coprecipitate in both stone types; crystalline calcium phosphate and/or carbonate in the case of ‘black’ stones and amorphous calcium salts of long chain saturated fatty acids (‘soaps’) in the case of ‘brown’ stones. Cholesterol is present in variable proportions in ‘brown’ more than ‘black’ stones and, in the latter, the bile sterol may be totally absent. The ‘scaffolding’ of both stone types is a mixed mucin glycoprotein matrix secreted by epithelial cells lining the biliary tree. The critical pathophysiological prerequisite for ‘black’ stone formation is ‘hyperbilirubinbilia’ (biliary hypersecretion of bilirubin conjugates). It is due principally to hemolysis, ineffective erythropoiesis, or pathologic enterohepatic cycling of unconjugated bilirubin. Endogenous biliary β-glucuronidase hydrolysis of bilirubin conjugates in gallbladder bile provides HUCB− molecules that precipitate as insoluble salts with ionized Ca. Putatively, reactive oxygen species secreted by an inflamed gallbladder mucosa are responsible for transforming the initial soft yellow precipitates into hard black [Ca(HUCB)2]n polymers. Despite ‘brown’ gallstones being soft and amenable to mechanical removal, chronic anaerobic infection of the biliary tree is often markedly resistant to eradication.
INTRODUCTION
Although cholesterol gallstones can contain as much as 30% calcium bilirubinate and are sometimes called ‘mixed’ stones [1], pigment gallstones are distinct containing calcium bilirubinate, Ca(HUCB)2, as the principal component. They are classified descriptively as ‘black’ stones, which are hard, and ‘brown’ stones, which are soft [2]. True prevalence as opposed to clinical, autopsy or surgical prevalence in free-living populations has not been determined. Black stones are mostly amorphous containing primarily polymerized calcium monoacidic unconjugated bilirubin, Ca(HUCB)2. They often contain crystalline salts of calcium phosphate and/or calcium carbonate in one of its polymorphic forms, calcite, aragonite or valerite and may also contain many metals found in bile [3]. They form in sterile gallbladder bile, and the principal risk factor is ‘hyperbilirubinbilia’ (secretion of excess bilirubin conjugates into bile). This results particularly from hemolysis from any cause, ineffective erythropoiesis, or induced enterohepatic cycling (EHC) of unconjugated bilirubin (UCB), which we focus on in this review. Other causes of ‘black’ stones are gallbladder hypomotility secondary to diabetus mellitus [2], total parenteral nutrition [2] and truncal vagotomy [4]. Neither intensive blue-light phototherapy for hyperbilirubinemia in neonates and Crigler-Najjar syndrome Type 1 patients [5] nor excessive sun exposure have been proven to cause black pigment gallstone formation [6]. Blue light leads to biliary and urinary excretion of water-soluble configurational as well as structural UCB photoisomers. These rapidly revert to the more stable but insoluble natural Z-Z photoisomer in bile [7,8]. It is not surprising, therefore, that cholestasis can result from phototherapy due to massive precipitation of bile pigment in the small intrahepatic bile ducts [9,10]. The ‘bronze baby syndrome’ is due to retention of the geometric isomer, photobilirubin II, in the skin of infants with phototherapy-induced cholestasis [11].
‘Brown’ gallstones are invariably laminated, containing unpolymerized Ca(HUCB)2 coprecipitated with amorphous calcium salts of palmitate and stearate (calcium ‘soaps’) derived from bacterial phospholipase A1 hydrolysis of biliary phosphatidylcholines [12]. They also contain unconjugated (free) bile acids [2] and Ca bile salts and variable amounts of cholesterol. This contrasts with ‘black’ stones, in which the bile sterol is often absent [2].
‘Brown’ stones may occur anywhere in the biliary tree but rarely in the gallbladder. They are associated with anaerobic bacterial infection secondary to obstruction and/or stasis of any etiology [13], including parasitic infestation from nematodes and flukes [14]. Thick section electron microscopy reveals bacterial cytoskeletons within the stones’ structures [15]. A migratory cholesterol/black pigment gallstone that escapes the gallbladder and becomes impacted in the bile duct is frequently the cause of stasis and secondary bacterial infection [16]. An often overlooked cause of biliary stasis-associated brown stones is a juxtapapillary duodenal diverticulum harboring anaerobes [17]. In developing and tropical countries the bileseeking worms that are initiating factors are Ascaris lumbricoides, Clonorchis sinensis [18] and Opisthorchis viverrini [19]. In contrast to black pigment stones that are usually radiopaque, brown pigment gallstones are radiolucent.
For brown gallstones to form, the biliary tree must become infected with colonic anaerobic microbiota producing β-glucuronidase, an enzyme that hydrolyzes bisglucuronosyl bilirubin to UCB. Bacterial production of detergent-resistant phospholipase A1 produces free palmitic and stearic acids, the products of hydrolysis of the sn-1 ester linkage of biliary phosphatidylcholines [12]. These precipitate with calcium counter-ions as long-chain insoluble soaps [2]. The amide linkage of conjugated bile salts is hydrolysed by cholylglycylamidase, which is secreted by many anaerobic strains of bacteria (e.g., Streptococcus faecalis, Clostridium spp., Bacteroides spp.) [20], forming free (i.e., unconjugated) bile acids. These are capable of precipitating as insoluble bile acids per se or less likely as calcium bile salts [21]. Provided marked stasis is present, ‘brown’ pigment gallstones may develop exceptionally under apparently ‘sterile’ conditions, such as throughout the biliary tree in patients with Caroli´s syndrome or in isolated choledochal cysts [22].
Because hyperbilirubinbilia is the critical risk factor, black stones are associated frequently with all major hemolytic anemias, e.g., spherocytosis [23], sickle cell disease [24], thalassemia [25], and also with subclinical hemolysis from prosthetic valve replacements [26], malaria [27], hypersplenism from hepatic cirrhosis [28], and foot trauma in long-distance runners [29]. Ineffective erythropoiesis, typically secondary to folate and vitamin B12 deficiencies [30], is now rare but can complicate thalassemia as well as sickle cell disease. Other conditions associated with increased prevalence of black pigment gallstones are Gilbert’s syndrome [31], associated with enhanced biliary secretion of monoglucuronosyl bilirubin, and hyperparathyroidism [32], associated with higher levels of ionized calcium in bile.
Acquired EHC of UCB as a cause of hyperbilirubinbilia and black pigment stone formation is a newly described pathophysiological and clinical entity [33]. Its principal causes are ileal dysfunction, disease, resection or bypass, and various dietary influences on ileal function, such as high carbohydrate or cholesterol intake, or alcohol abuse [33]. Although absence of biliary infection has always been the basic prerequisite for black pigment gallstone formation, recent data suggest that they might also be primed by biles infected with ‘nanobacteria’ that produce hydroxyapatite [34], as was also suggested for nephrolithiasis [35]. These claims have not been further elucidated clinically or experimentally. Figure 1 summarizes the proven concepts underlying the pathobiology of black (A) and brown (B) pigment stone formation in humans.
Fig 1.
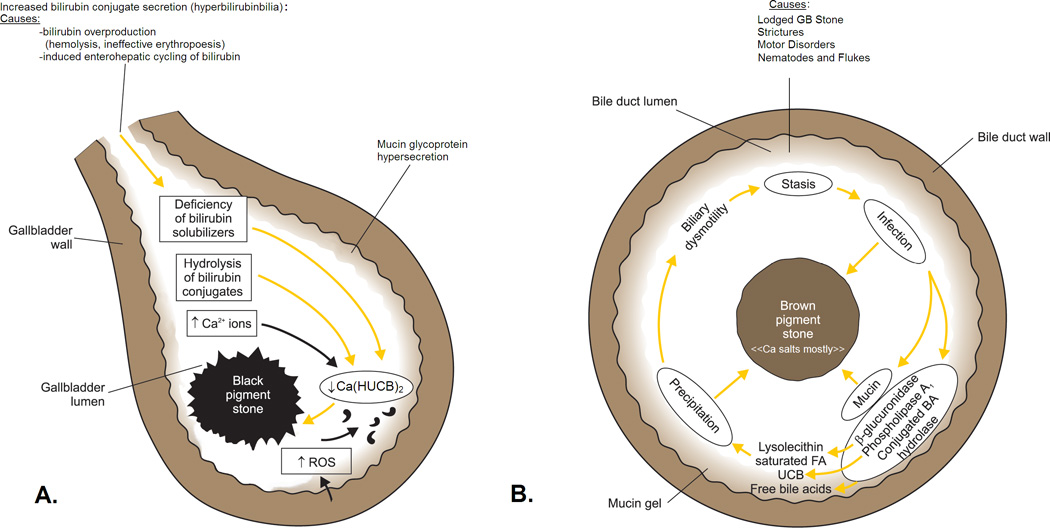
Schematic outline of the pathogenesis of: A, ‘black’ pigment stones in sterile gallbladder bile and B, ‘brown’ pigment stones in an obstructed biliary tree (gallbladder infrequently) infected with a mixed anaerobic microflora derived from the colon. In A, increased HUCB− levels in bile arise from endogenous acidic β-glucuronidase, principally of hepatic origin, that hydrolyzes excess bilirubin glucuronides in bile (hyperbilirubinbilia) to UCB. Deficiency of solubilizers for Ca2+, monoacidic unconjugated bilirubin (HUCB−), and Ca bilirubinates (bile salt-cholesterol micelles, bile salt-lecithin-cholesterol micelles, and unilamellar lecithin-cholesterol vesicles) leads to increases in free Ca2+ ions and free bilirubinate anions. Likewise, increases in free Ca2+ ions may be due to increased secretion into the biliary tree. In slightly alkaline bile, the concentrations of other Ca2+-sensitive anions, such as carbonate and phosphate, may also increase. When the ion products of the Ca-anion salts exceed their equilibrium solubility products in bile, the biliary system becomes supersaturated and precipitation is thermodynamically possible. Mucin glycoproteins are hypersecreted by the gallbladder mucosa like in other lithogenic settings and form a biliary gel providing a nucleation matrix for the precipitated pigment salts. Cholesterol may or may not phase-separate depending on its level in bile and is absent in stones when the cholesterol saturation index (CSI) of bile is less than 1, as occurs in most black pigment stone gallbladder biles. Solid-state ‘attack’ by free radicals or singlet oxygen (reactive oxygen species, ROS), from an inflamed gallbladder mucosa leads to tetrapyrrole polymerization and oxidation, which imparts the black spiculated appearance to most of these stones.
In B, stasis, due to motor disorders of the sphincter of Oddi, strictures from biliary surgery or foreign bodies such as gallstones from the gallbladder, sutures, parasites and their eggs and carcasses (principally from nematodes and flukes), facilitate anaerobic bacterial invasion and overgrowth. Lithogenesis, stasis and infection lead to hypersecretion of biliary tree mucin glycoproteins that form a gel in bile. Bacterial enzymes catalyze the hydrolysis of ester and amide linkage in all biliary lipids: Phospholipase A1 catalyzes hydrolysis of biliary phosphatidylcholines at the sn-1 position to produce palmitic and stearic acids plus lysophosphatidylcholine; β-glucuronidase catalyzes hydrolysis of bilirubin glucuronosides to produce UCB and glucuronic acid which acidifies bile further; conjugated bile salt hydrolase (cholylglycylamidase) catalyzes hydrolysis of the amide linkage of conjugated bile salts to produce free (unconjugated) bile acids, taurine, or glycine. Saturated fatty acids can precipitate per se but typically form insoluble calcium soaps with Ca2+ ions; UCB forms insoluble acidic calcium bilirubinate salts Ca(HUCB)2, and free secondary bile acids may precipitate as such, or less commonly as calcium salts. Lysophosphatidylcholine may act as a fusogen for cholesterol-containing unilamellar phospholipid vesicles, leading to nucleation of cholesterol monohydrate crystals that are invariably co-precipitated with brown stones. The calcium salts, cholesterol, and biliary tree mucins, the major components of ‘brown’ pigment stones, also act as a ‘trap’ for the anaerobic bacteria, making their elimination difficult; bacterial ‘skeletons’ are usually visualized by electron microscopy of thick-sections of these stones. Ductal obstruction by brown stones themselves perpetuates the vicious cycle of stasis and chronic anaerobic bacterial infection. (Figure and legends A/B reproduced in modified form from reference 2 with written permission of the copyright owner: Thieme Medical Publishers, Inc. New York and Stuttgart, and redrawn by Mr. Martin Vyhnanovský, Prague, Czech Republic, and Ms. Jessica Y. Xia, Boston, MA, USA).
Physical-chemistry of ‘black’ and ‘brown’ pigment gallstone formation
UCB, the end product of heme catabolism, is an open-chain tetrapyrrole molecule whose outer monopyrroles are interconnected by rigid –CH= linkages, and the dipyrrole units by a flexible central –CH2–bridge. The central pyrrole rings subtend two carboxyethyl side-chains, which in aqueous systems ionize only at alkaline pH values [36]. Because of six intra-molecular hydrogen bonds [36], UCB, which is in the H2UCB form at physiological pH values, is highly insoluble in aqueous media (<70 nM) [37]. However, at pH values of human hepatic (7.3–8.7) and gallbladder (6.2–7.8) biles [2], UCB is a monoacidic species (HUCB−) principally because of the presence of high concentrations of bile salts. Nevertheless, it is exquisitely sensitive to precipitation as the monoacid salt with typical biliary concentrations of ionized Ca [2,38]. In contrast, mono- and bisglucuronosyl bilirubins, the predominant conjugated bile pigment species in human and most laboratory animal biles, bind ionized calcium as soluble salts and are not precipitated with physiological calcium concentrations [39]. Increased plasma ionized calcium concentrations, a hallmark of hyperparathyroidism, also seems to be a risk factor for black pigment gallstone disease [32] due to the rapid equilibration of ionized calcium, which has a small hydrated radius, between intravascular and biliary compartments.
Several physiological and biochemical mechanisms solubilize UCB in aqueous environments: a) binding to albumin or HDL in plasma; b) binding to bile salt monomers, dimers and micelles [40], mucin glycoproteins in bile and intestinal lumen [41]; c) covalent linkage of one or both –COOH groups with glucuronic acid catalyzed by hepatic bilirubin UDP-glucuronosyl transferase (UGT1A1) producing monoglucuronosyl bilirubins and bisglucuronosyl bilirubins; d) disruption of internal hydrogen bonding by phototherapy-induced isomerization of UCB, forming water-soluble structural and configurational isomers [42]; or e) binding to unilamellar vesicles in cholesterol supersaturated bile [2].
Increased proportions of biliary monoglucuronosyl bilirubin to bisglucuronosyl bilirubin are the rule in biles of patients with black pigment gallstones [43,44]. Monoglucuronosyl bilirubin can more easily be deconjugated in bile by endogenous acidic β-glucuronidase that is secreted by hepatic parenchymal cells [45], or less frequently by biliary epithelial β-glucuronidase and non-enzymatic hydrolysis [46]. An increase in the absolute concentration of biliary UCB has been reported in humans and animals with black pigment gallstones as well as early calcium bilirubinate precipitates [47,48]. Calcium bilirubinate precipitates may also serve as seed nuclei during the initiation of cholesterol cholelithiasis [48,49]. In addition, human ‘biliary sludge’ consists of calcium, monoconjugated and unconjugated bilirubins plus mucin glycoproteins, and may serve as a matrix in which pigment or cholesterol stones develop [49]. Because human pigment gallstones invariably contain a mucin glycoprotein matrix, or ‘scaffolding’ [50], it is likely that hypersecretion of mucin by the gallbladder, as observed in animal models of pigment gallstones [51], leads to mucin gel-Ca-HUCB− interactions. Similar association of pigment gallstones has also been reported for osteopontin, a lithogenic Ca-binding glycoprotein secreted by human gallbladder mucosa [52].
Intestinal pathobiology of bile salts and bilirubin in relation to ‘black’ pigment gallstone formation
Important pathophysiological perturbations in the gut-liver axis controlling the homeostasis of both bilirubin and bile salts may lead to hyperbilirubinbilia, the critical risk factor in formation of ‘black’ pigment gallstones [33]. In all of these scenarios the common denominator appears to be ileal dysfunction from any cause with spillage of malabsorbed bile salts into the cecum and large intestine.
Under physiological conditions, the bile salt pool enjoys a very efficient EHC. Bile salt molecules are retrieved from the gut via the sodium-coupled apical membrane cotransporters (SLC10A2) in the distal ileum, which is responsible for >96% of bile salt conservation within the enterohepatic axis. In health, an EHC of UCB does not exist or is negligible [2]. The conjugates are too bulky and polar, intestinal deconjugation of bilirubin conjugates to UCB is rapid [2], and efficient reduction of bilirubin to urobilinoids prevent passive resorption of UCB [33]. Deconjugation in the gut can occur via a β-glucuronidase of bacterial origin but also by the mammalian enzyme of sloughed enterocytes which contain a neutral β-glucuronidase. Luminal solubilization is crucial for induced resorption of UCB. This can occur with: a) higher rates of bilirubin deconjugation due to prolonged intestinal transit time such as during calorie deprivation with overgrowth of β-glucuronidase-producing bacteria, or a supply of β-glucuronidase from extraintestinal sources, such as breast milk, resulting in ‘breast milk jaundice’ in premature infants; b) slow or absent intestinal reduction of UCB by bacteria, such as present in neonates or in adults during oral antibiotic therapy; c) more alkaline luminal pH, higher bile salt concentrations, or agents preventing complexing of UCB with Ca2+ in the intestinal lumen. Malabsorbed bile salts are the most obvious cause of an induced EHC of UCB. They elevate colonic detergent levels that solubilize UCB, promoting its passive resorption from the intestine [53]. Even in the absence of ileal disease, ileal resorption of conjugated bile salts via SLC10A2 is dependent on intraluminal pH. A 0.5 pH unit decrease in ileal lumenal pH diminishes sodium taurocholate transport by 9% per enterohepatic cycle [54]. Finally, complexing of UCB with Ca2+ in the intestinal lumen blocks its resorption, and a diminution in Ca2+ concentration may lead to higher UCB levels. Bile salts also bind intraluminal calcium, forming soluble calcium complexes with similar results on elevating soluble UCB levels [55]. Another example of insoluble calcium salt formation depleting intraluminal Ca2+ ions occurs in the setting of dietary fat malabsorption. In this regard, intraluminal calcium precipitates as calcium long-chain fatty acid soaps in the proximal small intestine and becomes unavailable to bind cation-sensitive anions, such as UCB, in the distal intestine, thereby increasing its solution levels. It has been proposed [53] that hyperbilirubinbilia from an induced EHC of UCB represents a parallel pathophysiology to that of enteric hyperoxaluria in the setting of fat malabsorption [56].
Apart from its primary lithogenic effect, hyperbilirubinbilia may lead to ¨uncoupling¨ of biliary lipids from bile salts at the canalicular level with diminution in their biliary secretion rates [57]. Diminished biliary secretion of biliary phosphatidylcholine and cholesterol molecules leads, in turn, to a lessening of their protective role on bile salt detergency. As a result, there is a higher likelihood of bile salt-induced injury to the mucosa of the bile ducts and gallbladder, further facilitating pigment gallstone formation via production of mucosal β-glucuronidase from biliary epithelial cells, and possibly also mucin glycoproteins and reactive oxygen species (ROS) [54].
The intestinal microflora, dietary factors and pigment gallstone formation
Intestinal bacterial overgrowth occurs in several pathologic conditions and may result in increased bile salt and bilirubin deconjugation in the small intestine. This could contribute further to bile salt malabsorption and enhanced solubilization and EHC of UCB from the colon [33]. This is exemplified by intestinal hypomotility from any cause including chronic constipation [33], total parenteral nutrition [33], cystic fibrosis [33], and spinal cord injury [58]. Furthermore, bile salt malabsorption may also be caused by undigested starch and β-cyclodextrin, a starch degradation product in subjects ingesting high carbohydrate diets [59]. Likewise, high intake of refined sugars affects metabolism of intestinal microflora and prolongs intestinal transit time [60], again potentially affecting an EHC of UCB. Moreover, hypertonic solutions of sucrose increase intestinal mucosa permeability [61]. Indeed, high sucrose diets induce pigment gallstones in prairie dogs [62], a species known otherwise as a facile model of cholesterol gallstone formation [63]. Contrariwise, high dietary fibre may prevent pigment gallstone formation because of the production of abundant short-chain fatty acids from bacterial fermentation [62]. These diminish distal intestinal pH, precipitate UCB and sequester bile acids as insoluble species in the large intestine. Such a scenario is common in subjects where an abundance of dietary fibre is a food staple, such as with vegetarians and sub-Saharan Africans known to have very low prevalences of gallstone disease [64,65]. Additionally, resorption of UCB may be impaired because of bulk-induced increases in colonic transit times [66,67]. Besides direct effects of bile salts on UCB solubilization, secondary bile salt production from bacterial overgrowth may also promote release of intracellular microbiotal β-glucuronidase by enhancing plasma membrane permeability of bacteria [68].
Decreased concentrations of biliary β-glucuronolactone, an inhibitor of β-glucuronidase [69], was reported to be associated with black pigment gallstone disease in post-WWII Japan because of low-protein diets. Impaired intestinal barrier function has been proposed as playing a role in passive UCB resorption during experimental pigment lithogenesis in hamsters [70], as is also evidenced by high serum endotoxin levels and indices of immune cell activation in the intestinal mucosa [70,71]. In germ-free Swiss Webster mice, intestinal barrier dysfunction may also be a critical factor facilitating an EHC of UCB, which, in the germ-free state, cannot be degraded to urobilinoids [72].
In black pigment gallstone formation, another potential lithogenic factor is slow or absent bacterial reduction of intestinal UCB, as evidenced by the dearth of intestinal microflora in neonates and in patients requiring prolonged oral antibiotic therapy. Absence of UCB reduction in the intestinal lumen is a contributing factor to neonatal jaundice [73], but also for pigment gallstone disease as well as the hyperbilirubinemia associated with oral antibiotic therapy [74]. As alluded to above, germ-free Swiss Webster mice exhibit extremely high prevalences of spontaneous black pigment gallstone formation [72,75], apparently secondary to what appear to be at least four factors: inability to degrade UCB to urobilinoids, decreased intestinal motility, gallbladder hypomotility from a pre-diabetic state, and diminution in intestine mucosal barrier function [72,75].
Since gallbladder inflammation is implicated in black pigment gallstone formation, it is tempting to speculate that UCB per se might be specifically involved in the immune response ultimately leading to pigment cholelithiasis. In fact, UCB has been shown to be involved in regulatory T-cell differentiation [76].
Putative role of ROS in ‘black’ stone polymerization and oxidation
It is likely that polymerization of the HUCB− tetrapyrrole, a key event in the transition of soft yellow to hard black pigment gallstones, is initiated by free radicals or singlet oxygen species and occurs in the solid or semi-solid state. A hepatic source of ROS is unlikely since Ca(HUCB)2 in ‘brown’ stones remains unpolymerized [2]. ROS are more likely products of mucosal macrophages or neutrophils from an inflamed gallbladder that accompanies pigment stone disease [2,72,75]. This is consistent with observations of bacterially-derived oxysterols in human bile as well as in black pigment gallstones [77]. In addition, oxidative stress induces formation of bilirubin free radicals, which are prone to polymerize easily, as evidenced by ESR and NMR studies of human pigment stones [78]. Indeed, administration of the free radical scavenger melatonin (N-acetyl-5-methoxytryptamine) is reported to prevent pigment gallstone formation in bile duct-ligated guinea pigs [79]. This also appears to be the case for methionine, which acts as a free radical scavenger. Methionine deficiency in the diet of domestic dogs is associated with increased formation of black pigment gallstones, whereas methionine supplementation counteracts lithogenesis [80]. Moreover, as inferred from ex vivo studies [81], suppression of oxidative stress in the gallbladder may also play a role in ameliorating cholesterol gallstone formation.
Conclusions
‘Black‘ pigment gallstones form only in the gallbladder and are due to ‘hyperbilirubinbilia’ usually from three conditions: (i) hemolysis from any cause including unsuspected conditions such as malaria, hypersplenism and prosthetic neovasculature; (ii) ineffective erythropoiesis from vitamin B12 and folate deficiencies, which are now rare in humans; and (iii) induced EHC of UCB, a new pathophysiologic concept, which has multiple predisposing factors highlighted in this review. With respect to the latter, any or all of the factors that make more UCB soluble and available in the large intestine facilitate its resorption to return to the liver for reconjugation and resecretion into bile. UCB resorption may be aided by an increased permeability and residence time of UCB in the gut as well as low levels of ionized Ca2+.
The primary source of free UCB in gallbladder bile is from endogenous biliary β-glucuronidase hydrolysis of bilirubin conjugates. This enzyme is of hepatic origin but, with gallbladder inflammation, can also be secreted from a biliary mucosal source. In the concentrated bile salt milieu of hepatic and gallbladder bile, UCB at biliary pH values is a monoacid, HUCB−. Two molecules of HUCB− bind with ionized biliary Ca2+ to form a divalent salt that has miniscule solubility in aqueous systems and precipitates from solution in bile. The ROS secreted by an inflamed gallbladder mucosa apparently causes Ca(HUCB)2 polymerization and oxidation [82]. The germ-free Swiss Webster mouse, a newly described animal model of ‘black‘ pigment gallstone formation [72,75], promises to answer many of the vexing questions regarding the pathophysiology and physical chemistry of this disease.
‘Brown’ stones are a phase-separated artifact of bile that can form in any part of the biliary tree secondary to chronic stasis from any cause and anaerobic bacterial infection. The anaerobes secrete multiple enzymes that hydrolyse ester and amide linkages of all biliary lipids and lipopigments into calcium-sensitive anions that phase-separate as insoluble protonated anions or calcium salts. These precipitates deposit on obstructing ‘nuclei’ such as small migratory cholesterol or ‘black’ stones formed in the gallbladder, parasite eggs and dead worms or flukes. The Oriental hepatolithiasis syndrome is the most serious manifestation of ‘brown‘ pigment stone disease. Beginning in early life, it is initiated by nematode or fluke infestation within the extrahepatic and intrahepatic bile ducts usually favoring the left lobe of the liver; worldwide, Ascaris lumbricoides, Clonorchis sinensis and Opisthorchis viverrini appear to be the principal parasites involved.
Acknowledgments
Grant support: This work was supported by grants CZ:GA CR:P206/11/0836, from the Research Granting Agency of the Czech Republic and NIH Grant R37DK36588 from the US Public Health Service.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement:
The authors have no relevant financial disclosures.
References
- 1.Schafmayer C, Hartleb J, Tepel J, Albers S, Freitag S, Völzke H, et al. Predictors of gallstone composition in 1025 symptomatic gallstones from Northern Germany. BMC Gastroenterol. 2006;6:36–44. doi: 10.1186/1471-230X-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cahalane MJ, Neubrand MW, Carey MC. Physical-chemical pathogenesis of pigment gallstones. Semin Liver Dis. 1988;8:317–328. doi: 10.1055/s-2008-1040553. [DOI] [PubMed] [Google Scholar]
- 3.Suzuki N, Nakamura Y, Kobayashi N, Sato T. On metal elements in pure pigment gallstones. Tohoku J Exp Med. 1975;116:233–240. doi: 10.1620/tjem.116.233. [DOI] [PubMed] [Google Scholar]
- 4.Tsunoda K, Shirai Y, Wakai T, Yokoyama N, Akazawa K, Hatakeyama K. Increased risk of cholelithiasis after esophagectomy. J Hepatobiliary Pancreat Surg. 2004;11:319–323. doi: 10.1007/s00534-004-0914-7. [DOI] [PubMed] [Google Scholar]
- 5.Strauss KA, Robinson DL, Vreman HJ, Puffenberger EG, Hart G, Morton DH. Management of hyperbilirubinemia and prevention of kernicterus in 20 patients with Crigler-Najjar disease. Eur J Pediatr. 2006;165:306–319. doi: 10.1007/s00431-005-0055-2. [DOI] [PubMed] [Google Scholar]
- 6.Pavel S, Thijs CT, Potocky V, Knipschild PG. Fair, and still a sun lover: risk of gallstone formation. J Epidemiol Community Health. 1992;46:425–427. doi: 10.1136/jech.46.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lund HT, Jacobsen J. Influence of phototherapy on unconjugated bilirubin in duodenal bile of newborn infants with hyperbilirubinemia. Acta Paediatr Scand. 1972;61:693–696. doi: 10.1111/j.1651-2227.1972.tb15968.x. [DOI] [PubMed] [Google Scholar]
- 8.Agati G, Fusi F, Pratesi S, Galvan P, Donzelli GP. Bilirubin photoisomerization products in serum and urine from a Crigler—Najjar type I patient treated by phototherapy. J Photochem Photobiol B. 1998;47:181–189. doi: 10.1016/s1011-1344(98)00221-8. [DOI] [PubMed] [Google Scholar]
- 9.Wolkoff AW, Chowdhury JR, Gartner LA, Rose AL, Biempica L, Giblin DR, et al. Crigler-Najjar syndrome (Type 1) in an adult male. Gastroenterology. 1979;76:840–848. [PubMed] [Google Scholar]
- 10.Ostrow JD. Photocatabolism of labeled bilirubin in the congenitally jaundiced (Gunn) rat. J Clin Invest. 1971;50:707–718. doi: 10.1172/JCI106541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Onishi S, Itoh S, Isobe K, Togari H, Kitoh H, Nishimura Y. Mechanism of development of bronze baby syndrome in neonates treated with phototherapy. Pediatrics. 1982;69:273–276. [PubMed] [Google Scholar]
- 12.Robins SJ, Fasulo JM, Patton GM. Lipids of pigment gallstones. Biochim Biophys Acta. 1982;712:21–25. doi: 10.1016/0005-2760(82)90079-0. [DOI] [PubMed] [Google Scholar]
- 13.Kaufman HS, Magnuson TH, Lillemoe KD, Frasca P, Pitt HA. The role of bacteria in gallbladder and common duct stone formation. Ann Surg. 1989;209:584–591. doi: 10.1097/00000658-198905000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rana SS, Bhasin DK, Nanda M, Singh K. Parasitic infestations of the biliary tract. Curr Gastroenterol Rep. 2007;9:156–164. doi: 10.1007/s11894-007-0011-6. [DOI] [PubMed] [Google Scholar]
- 15.Stewart L, Smith AL, Pellegrini CA, Motson RW, Way LW. Pigment gallstones form as a composite of bacterial microcolonies and pigment solids. Ann Surg. 1987;206:242–250. doi: 10.1097/00000658-198709000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sauerbruch J, Stellaard F, Soehendra N, Paumgartner G. Cholesterol content of bile-duct stones. Dtsch Med Wochenschr. 1983;108:99–102. doi: 10.1055/s-2008-1069700. [DOI] [PubMed] [Google Scholar]
- 17.Sandstad O, Osnes T, Skar V, Urdal P, Osnes M. Common bile duct stones are mainly brown and associated with duodenal diverticula. Gut. 1994;35:1464–1467. doi: 10.1136/gut.35.10.1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jang JS, Kim KH, Yu JR, Lee SU. Identification of parasite DNA in common bile duct stones by PCR and DNA sequencing. Korean J Parasitol. 2007;45:301–306. doi: 10.3347/kjp.2007.45.4.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sripa B, Kanla P, Sinawat P, Haswell-Elkins MR. Opisthorchiasis-associated biliary stones: light and scanning electron microscopic study. World J Gastroenterol. 2004;10:3318–3321. doi: 10.3748/wjg.v10.i22.3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akiyoshi T, Nakayama F. Bile acid composition in brown pigment stones. Dig Dis Sci. 1990;35:27–32. doi: 10.1007/BF01537218. [DOI] [PubMed] [Google Scholar]
- 21.Gu JJ, Hofmann AF, Ton-Nu HT, Schteingart CD, Mysels KJ. Solubility of calcium salts of unconjugated and conjugated natural bile acids. J Lipid Res. 1992;33:635–646. [PubMed] [Google Scholar]
- 22.Nakanuma Y, Terada T, Nagakawa T, Kakita A, Yoshikawa T, Ohta G, et al. Pathology of hepatolithiasis associated with biliary malformation in Japan. Liver. 1988;8:287–292. doi: 10.1111/j.1600-0676.1988.tb01006.x. [DOI] [PubMed] [Google Scholar]
- 23.del Giudice EM, Perrotta S, Nobili B, Specchia C, d'Urzo G, Iolascon A. Coinheritance of Gilbert syndrome increases the risk for developing gallstones in patients with hereditary spherocytosis. Blood. 1999;94:2259–2262. [PubMed] [Google Scholar]
- 24.Chaar V, Kéclard L, Diara JP, Leturdu C, Elion J, Krishnamoorthy R, et al. Association of UGT1A1 polymorphism with prevalence and age at onset of cholelithiasis in sickle cell anemia. Haematologica. 2005;90:188–199. [PubMed] [Google Scholar]
- 25.Premawardhena A, Fisher CA, Fathiu F, de Silva S, Perera W, Peto TEA, et al. Genetic determinants of jaundice and gallstones in haemoglobin E beta thalassaemia. Lancet. 2001;357:1945–1946. doi: 10.1016/s0140-6736(00)05082-0. [DOI] [PubMed] [Google Scholar]
- 26.Azemoto R, Tsuchiya Y, Ai T, Murayama H, Nakagawa Y, Saisho H, et al. Does gallstone formation after open cardiac surgery result only from latent hemolysis by replaced valves? Am J Gastroenterol. 1996;91:2185–2189. [PubMed] [Google Scholar]
- 27.Chawla LS, Sidhu G, Sabharwal BD, Bhatia KL, Sood A. Jaundice in Plasmodium falciparum malaria. J Assoc Physicians India. 1989;37:390–391. [PubMed] [Google Scholar]
- 28.Diehl AK, Schwesinger WH, Holleman DR, Jr, Chapman JB, Kurtin WE. Clinical correlates of gallstone composition: distinguishing pigment from cholesterol stones. Am J Gastroenterol. 1995;90:967–972. [PubMed] [Google Scholar]
- 29.Weight LM, Byrne MJ, Jacobs P. Haemolytic effects of exercise. Clin Sci (Lond) 1991;81:147–152. doi: 10.1042/cs0810147. [DOI] [PubMed] [Google Scholar]
- 30.Aydogdu I, Sari R, Ulu R, Sevinc A. The frequency of gallbladder stones in patients with pernicious anemia. J Surg Res. 2001;101:120–123. doi: 10.1006/jsre.2001.6269. [DOI] [PubMed] [Google Scholar]
- 31.Buch S, Schafmayer C, Völzke H, Seeger M, Miquel JF, Sookoian SC, et al. Loci from a genome-wide analysis of bilirubin levels are associated with gallstone risk and composition. Gastroenterology. 2010;139:1942–1951. doi: 10.1053/j.gastro.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 32.Broulik PD, Haas T, Adámek S. Analysis of 645 patients with primary hyperparathyroidism with special references to cholelithiasis. Intern Med. 2005;44:917–921. doi: 10.2169/internalmedicine.44.917. [DOI] [PubMed] [Google Scholar]
- 33.Vítek L, Carey MC. Enterohepatic cycling of bilirubin as a cause of 'black' pigment gallstones in adult life. Eur J Clin Invest. 2003;33:799–810. doi: 10.1046/j.1365-2362.2003.01214.x. [DOI] [PubMed] [Google Scholar]
- 34.Wang L, Shen W, Wen J, An X, Cao L, Wang B. An animal model of black pigment gallstones caused by nanobacteria. Dig Dis Sci. 2006;51:1126–1132. doi: 10.1007/s10620-006-8019-6. [DOI] [PubMed] [Google Scholar]
- 35.Shiekh FA, Khullar M, Singh SK. Lithogenesis: induction of renal calcifications by nanobacteria. Urol Res. 2006;34:53–57. doi: 10.1007/s00240-005-0034-0. [DOI] [PubMed] [Google Scholar]
- 36.Ostrow JD, Mukerjee P. Revalidation and rationale for high pK'a values of unconjugated bilirubin. BMC Biochem. 2007;8:7. doi: 10.1186/1471-2091-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ostrow JD, Pascolo L, Brites D, Tiribelli C. Molecular basis of bilirubin-induced neurotoxicity. Trends Mol Med. 2004;10:65–70. doi: 10.1016/j.molmed.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 38.Ostrow JD, Celic L. Bilirubin chemistry, ionization and solubilization by bile salts. Hepatology. 1984;4:38S–45S. doi: 10.1002/hep.1840040807. [DOI] [PubMed] [Google Scholar]
- 39.Ostrow JD, Murphy NH. Isolation and properties of conjugated bilirubin from bile. Biochem J. 1970;120:311–327. doi: 10.1042/bj1200311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ostrow JD, Celic L, Mukerjee P. Molecular and micellar associations in the pH-dependent stable and metastable dissolution of unconjugated bilirubin by bile salts. J Lipid Res. 1988;29:335–348. [PubMed] [Google Scholar]
- 41.Smith BF, Lamont JT. Bovine gallbladder mucin binds bilirubin in vitro. Gastroenterology. 1983;85:707–712. [PubMed] [Google Scholar]
- 42.McDonagh AF, Lightner DA. Phototherapy and the photobiology of bilirubin. Semin Liver Dis. 1988;8:272–283. doi: 10.1055/s-2008-1040549. [DOI] [PubMed] [Google Scholar]
- 43.Goresky CA, Gordon ER, Hinchey EJ, Fried GM. Bilirubin conjugate changes in the bile of gallbladders containing gallstones. Hepatology. 1995;21:373–382. [PubMed] [Google Scholar]
- 44.Boonyapisit ST, Trotman BW, Ostrow JD. Unconjugated bilirubin, and the hydrolysis of conjugated bilirubin, in gallbladder bile of patients with cholelithiasis. Gastroenterology. 1978;74:70–74. [PubMed] [Google Scholar]
- 45.Whiting JF, Narcisco JP, Chapman V, Ransil BJ, Swank RT, Gollan JL. Deconjugation of bilirubin-IX alpha glucuronides: a physiologic role of hepatic microsomal beta-glucuronidase. J Biol Chem. 1993;268:23197–23201. [PubMed] [Google Scholar]
- 46.Spivak W, DiVenuto D, Yuey W. Non-enzymic hydrolysis of bilirubin mono- and diglucuronide to unconjugated bilirubin in model and native bile systems. Potential role in the formation of gallstones. Biochem J. 1987;242:323–329. doi: 10.1042/bj2420323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Donovan JM, Carey MC. Physical-chemical basis of gallstone formation. Gastroenterol Clin North Am. 1991;20:47–66. [PubMed] [Google Scholar]
- 48.Ostrow JD. Unconjugated bilirubin and cholesterol gallstone formation. Hepatology. 1990;12:219S–226S. [PubMed] [Google Scholar]
- 49.Malet PF, Williamson CE, Trotman BW, Soloway RD. Composition of pigmented centers of cholesterol gallstones. Hepatology. 1986;6:477–481. doi: 10.1002/hep.1840060326. [DOI] [PubMed] [Google Scholar]
- 50.Lamont JT, Ventola AS, Trotman BW, Soloway RD. Mucin glycoprotein content of human pigment gallstones. Hepatology. 1983;3:377–382. doi: 10.1002/hep.1840030316. [DOI] [PubMed] [Google Scholar]
- 51.Lamont JT, Turner BS, Bernstein SE, Trotman B. Gallbladder glycoprotein secretion in mice with hemolytic anemia and pigment gallstones. Hepatology. 1983;3:198–200. doi: 10.1002/hep.1840030211. [DOI] [PubMed] [Google Scholar]
- 52.Imano M, Satou T, Itoh T, Takeyama Y, Yasuda A, Peng YF, et al. An immunohistochemical study of osteopontin in pigment gallstone formation. Am Surg. 2010;76:91–95. [PubMed] [Google Scholar]
- 53.Brink MA, Méndez Sánchez N, Carey MC. Bilirubin cycles enterohepatically after ileal resection in the rat. Gastroenterology. 1996;110:1945–1957. doi: 10.1053/gast.1996.v110.pm8964422. [DOI] [PubMed] [Google Scholar]
- 54.Freudenberg F, Broderick AL, Yu BB, Leonard MR, Glickman JN, Carey MC. Pathophysiological basis of liver disease in cystic fibrosis employing a {Delta}F508 mouse model. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1411–G1420. doi: 10.1152/ajpgi.00181.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Donovan JM, Leonard MR, Batta AK, Carey MC. Calcium affinity for biliary lipid aggregates in model biles: complementary importance of bile salts and lecithin. Gastroenterology. 1994;107:831–846. doi: 10.1016/0016-5085(94)90134-1. [DOI] [PubMed] [Google Scholar]
- 56.Chadwick VS, Modha K, Dowling RH. Mechanism for hyperoxaluria in patients with ileal dysfunction. N Engl J Med. 1973;289:172–176. doi: 10.1056/NEJM197307262890402. [DOI] [PubMed] [Google Scholar]
- 57.Verkade HJ, Havinga R, Kuipers F, Vonk RJ. Mechanism of biliary lipid secretion. In: Hofmann AF, Paumgartner G, Stiehl A, editors. Bile Acids in Gastroenterology: Basic and Clinical Advances. Dordrecht: Kluwer Academic Publishers; 1995. pp. 230–246. [Google Scholar]
- 58.Apstein MD, Dalecki-Chipperfield K. Spinal cord injury is a risk factor for gallstone disease. Gastroenterology. 1987;92:966–968. doi: 10.1016/0016-5085(87)90971-1. [DOI] [PubMed] [Google Scholar]
- 59.Abadie C, Hug M, Kübli C, Gains N. Effect of cyclodextrins and undigested starch on the loss of chenodeoxycholate in the faeces. Biochem J. 1994;299:725–730. doi: 10.1042/bj2990725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kruis W, Forstmaier G, Scheurlen C, Stellaard F. Effect of diets low and high in refined sugars on gut transit, bile acid metabolism, and bacterial fermentation. Gut. 1991;32:367–371. doi: 10.1136/gut.32.4.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laker MF, Menzies IS. Increase in human intestinal permeability following ingestion of hypertonic solutions. J Physiol. 1977;265:881–894. doi: 10.1113/jphysiol.1977.sp011750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Conter RL, Roslyn JJ, Pitt HA, DenBesten L. Carbohydrate diet-induced calcium bilirubinate sludge and pigment gallstones in the prairie dog. J Surg Res. 1986;40:580–587. doi: 10.1016/0022-4804(86)90101-0. [DOI] [PubMed] [Google Scholar]
- 63.Holzbach RT. Animal models of cholesterol gallstone disease. Hepatology. 1984;4:191S–198S. doi: 10.1002/hep.1840040836. [DOI] [PubMed] [Google Scholar]
- 64.Parekh D, Lawson HH, Kuyl JM. Gallstone disease among black South Africans. S Afr Med J. 1987;72:23–26. [PubMed] [Google Scholar]
- 65.Rahman GA. Cholelithiasis and cholecystitis: changing prevalence in an African Community. J Natl Med Assoc. 2005;97:1534–1538. [PMC free article] [PubMed] [Google Scholar]
- 66.Cummings JH, Southgate DA, Branch WJ, Wiggins HS, Houston H, Jenkins DJ, et al. The digestion of pectin in the human gut and its effect on calcium absorption and large bowel function. Br J Nutr. 1979;41:477–485. doi: 10.1079/bjn19790062. [DOI] [PubMed] [Google Scholar]
- 67.Walters RL, Baird IM, Davies PS, Hill MJ, Drasar BS, Southgate DA, et al. Effects of two types of dietary fibre on faecal steroid and lipid excretion. Br Med J. 1975;2:536–538. doi: 10.1136/bmj.2.5970.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fujisawa T, Mori M. Influence of bile salts on beta-glucuronidase activity of intestinal bacteria. Lett Appl Microbiol. 1996;22:271–274. doi: 10.1111/j.1472-765x.1996.tb01159.x. [DOI] [PubMed] [Google Scholar]
- 69.Nagase M, Hikasa Y, Soloway RD, Tanimura H, Setoyama M, Kato H. Gallstones in Western Japan. Factors affecting the prevalence of intrahepatic gallstones. Gastroenterology. 1980;78:684–690. [PubMed] [Google Scholar]
- 70.Fan Y, Wu SD, Sun L, Fu BB, Su Y. Possible relationship between intestinal barrier function and formation of pigment gallstones in hamsters. Hepatobiliary Pancreat Dis Int. 2008;7:529–532. [PubMed] [Google Scholar]
- 71.Su Y, Wu S, Fan Y, Jin J, Zhang Z. The preliminary experimental and clinical study of the relationship between the pigment gallstone and intestinal mucosal barrier. J Gastroenterol Hepatol. 2009;24:1451–1456. doi: 10.1111/j.1440-1746.2009.05842.x. [DOI] [PubMed] [Google Scholar]
- 72.Whary MT, Woods SE, Muthupalani S, Fox JG. Pigment gallstones in germfree Swiss Webster mice. Gastroenterology. 2010;138 Suppl 1:S211. [Google Scholar]
- 73.Vítek L, Kotal P, Jirsa M, Malina J, Cerná M, Chmelar D, et al. Intestinal colonization leading to fecal urobilinoid excretion may play a role in the pathogenesis of neonatal jaundice. J Pediatr Gastroenterol Nutr. 2000;30:294–298. doi: 10.1097/00005176-200003000-00015. [DOI] [PubMed] [Google Scholar]
- 74.Vítek L, Zelenka J, Zadinová M, Malina J. The impact of intestinal microflora on serum bilirubin levels. J Hepatol. 2005;42:238–243. doi: 10.1016/j.jhep.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 75.Leonard MR, Andrade JD, Whary MT, Fox JG, Carey MC. Striking alterations in bilirubin and bile salt solution chemistry of gallbladder bile in a germ-free mouse model of "black" pigment gallstones. Gastroenterology. 2011;140 Suppl 1:S68. [Google Scholar]
- 76.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 77.Haigh WG, Lee SP. Identification of oxysterols in human bile and pigment gallstones. Gastroenterology. 2001;121:118–123. doi: 10.1053/gast.2001.25513. [DOI] [PubMed] [Google Scholar]
- 78.Liu XT, Hu J. Relationship between bilirubin free radical and formation of pigment gallstone. World J Gastroenterol. 2002;8:413–417. doi: 10.3748/wjg.v8.i3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shiesh SC, Chen CY, Lin XZ, Liu ZA, Tsao HC. Melatonin prevents pigment gallstone formation induced by bile duct ligation in guinea pigs. Hepatology. 2000;32:455–460. doi: 10.1053/jhep.2000.16332. [DOI] [PubMed] [Google Scholar]
- 80.Christian JS, Rege RV. Methionine, but not taurine, protects against formation of canine pigment gallstones. J Surg Res. 1996;61:275–281. doi: 10.1006/jsre.1996.0116. [DOI] [PubMed] [Google Scholar]
- 81.Braganza JM, Worthington H. A radical view of gallstone aetiogenesis. Med Hypotheses. 1995;45:510–516. doi: 10.1016/0306-9877(95)90232-5. [DOI] [PubMed] [Google Scholar]
- 82.Baig SJ, Biswas S, Das S, Basu K, Chattopadhyay G. Histopathological changes in gallbladder mucosa in cholelithiasis: correlation with chemical composition of gallstones. Trop Gastroenterol. 2002;23:25–27. [PubMed] [Google Scholar]