

Abstract
Rheumatoid arthritis (RA) is an autoimmune synovitis characterized by the formation of pannus and the destruction of cartilage and bone in the synovial joints. Although immune cells, which infiltrate the pannus and promote inflammation, play a prominent role in the pathogenesis of RA, other cell types also contribute. Proliferation of synovial fibroblasts, for example, underlies the formation of the pannus, while proliferation of endothelial cells results in neovascularization, which supports the growth of the pannus by supplying it with nutrients and oxygen. The synovial fibroblasts also promote inflammation in the synovium by producing cytokines and chemokines. Finally, osteoclasts cause the destruction of bone. Here we show that erlotinib, an inhibitor of the tyrosine kinase EGFR, reduces the severity of established collagen-induced arthritis (CIA), a mouse model of RA—and that it does so by targeting synovial fibroblasts, endothelial cells, and osteoclasts. Erlotinib-induced attenuation of autoimmune arthritis was associated with a reduction in number of osteoclasts and blood vessels, and erlotinib inhibited the formation of murine osteoclasts and the proliferation of human endothelial cells in vitro. Erlotinib also inhibited the proliferation and cytokine production of human synovial fibroblasts in vitro. Moreover, EGFR was highly expressed and activated in the synovium of mice with CIA and patients with RA. Together, these findings suggest that EGFR plays a central role in the pathogenesis of RA and that EGFR inhibition may provide benefit in the treatment of RA.
Keywords: Rheumatoid Arthritis, Erlotinib, Epidermal Growth Factor Receptor, Synovial Fibroblast
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease characterized by synovial inflammation and joint destruction. T cells, B cells, macrophages, and dendritic cells infiltrate the inflamed synovium and drive disease by producing cytokines and autoantibodies (1). For this reason, current biological therapies for RA (such as TNF inhibitors, abatacept, rituximab and tocilizumab) directly target these immune cells and proinflammatory cytokines, dampening the immune response (2). However, these therapies also increase the risk of infections such as tuberculosis, indicating that other therapies—especially ones targeting non-immune cells—may be beneficial (2). In addition to the typical immune cells, synovial fibroblasts, endothelial cells, and osteoclasts have been highlighted for their roles in promoting RA. Synovial fibroblasts form pannus and produce inflammatory mediators and chemokines that promote immune cell infiltration of the synovium. Proliferation of endothelial cells results in neovascularization, which supports the growth of the pannus by supplying it with nutrients and oxygen (3); and osteoclasts mediate adjacent bone destruction (4).
A recent case study described the prolonged remission of RA symptoms in an RA patient who was treated for head and neck cancer with focal radiation and cetuximab, a mAb against EGFR (5). Moreover, adenoviral expression of soluble herstatin, an alternative splice variant of ErbB2 that naturally inhibits dimerization of ErbB receptors, effectively reduced the severity of collagen-induced arthritis (CIA), a mouse model of RA (6). Similarly, RB200, a ligand trap that inhibits responses from EGFR, ErbB2 and ErbB3, reduced CIA severity (7). These findings suggest that inhibiting EGFR may be beneficial in RA.
EGFR is a widely expressed member of the ErbB family of tyrosine kinase receptors (8) and is overexpressed in many tumors (8). Activation of EGFR promotes survival, proliferation, cytokine production, cell adhesion, blood vessel recruitment and invasion depending on the cell type (8). Like a tumor, RA synovium is hyperplastic, invasive, and expresses EGFR. Specifically, EGFR is expressed on RA fibroblast cell lines and on the vascular endothelial cells and subsynovial fibroblasts of certain RA patients (9, 10). Moreover, levels of EGFR ligands, such as EGF, amphiregulin or TGFα, are significantly higher in synovial fluid, serum, or synovium of RA patients than in that of osteoarthritis patients or non-inflammatory controls (9, 11, 12)
Here we elucidate the role of EGFR in RA. We show that levels of EGF ligands are abnormally high in the serum of RA patients and that EGFR is expressed and activated in RA synovial tissue. Moreover, we demonstrate that erlotinib hydrochloride (Tarceva), a small-molecule EGFR inhibitor that is FDA approved for the treatment of metastatic non-small cell lung cancer (13), can attenuate CIA. Findings from our in situ and in vitro studies suggest that erlotinib ameliorates autoimmune arthritis by inhibiting the EGFR-dependent proliferation and cytokine production of synovial fibroblasts, proliferation of endothelial cells, and formation of osteoclasts.
Materials and Methods
Serum and join tissue samples
Serum and joint tissue samples were collected from RA patients who met the 1987 American College of Rheumatology criteria and from healthy individuals under protocols approved by the Stanford University Institutional Review Board. Joint tissue was collected from RA patients undergoing knee arthroplasty and snap-frozen in OCT freezing media.
Reagents
For animal studies, we used erlotinib tablets (OSI Pharmaceuticals, Melville, NY) purchased from the Stanford Inpatient Pharmacy Service. Erlotinib tablets were formulated as a fine suspension with 0.5% (w/v) hydroxypropylmethyocellulose (Dow) and 0.1% (v/v) Tween-80 in distilled water. For in vitro studies, we used 99% pure, chemically synthesized erlotinib (LC Laboratories) stored as a 100 mM stock solution at -20 °C in DMSO. PD153035 (TOCRIS) was stored as a 10 mM stock solution at -20 °C in DMSO. LA-1 (Milipore) was reconstituted in water. For all studies, we used recombinant murine EGF, murine platelet-derived growth factor bb (PDGFbb), human EGF, human vascular endothelial growth factor (VEGF), murine M-CSF, and murine RANKL (Peprotech) reconstituted in water.
Immunohistochemistry
Seven micrometer sections of synovial tissue from patients with RA were fixed in 4% (v/v) paraformeldehyde, permeabilized with triton X-100, and probed with antibodies against total EGFR (Clone LA-1, Millipore) or EGFR phosphorylated at tyrosine 1068 (p-EGFR(Y1068); Abcam). Hind limbs of mice with CIA were fixed in formalin and embedded as described in the histology section. Seven micrometer sections of mouse paws were rehydrated, subjected to antigen retrieval using ficin solution (Invitrogen), and probed with antibodies against total EGFR (Rockland), p-EGFR(Y1068) (Abcam), von-Willebrand Factor (Millipore), or tartrate-resistant acid phosphatase (TRAP) (Abcam). Staining was developed using Vectastain Elite ABC kit for mouse and rabbit IgG (Vector Labs). To quantify the number of vessels and osteoclasts in the mouse paw, we took pictures of the ankle and the calcaneous-cuboid joint, respectively, and counted the vessels or cells per square millimeter. Pictures and quantification was performed in a blinded manner.
CIA and K/BxN studies
CIA was generated in six- to eight-week-old male DBA/1 mice (The Jackson Laboratory) weighing approximately 20 g. Mice were used under protocols approved by the Administrative Panel on Laboratory Animal Care and in accordance with NIH guidelines. For the induction of CIA, mice were first immunized with a s.c. tail injection of 100 μg of bovine CII (Chondrex) emulsified in 100 μL of CFA containing 250 μg/mouse of heat-killed Mycobacterium tuberculosis H37Ra (BD). Twenty-one days after the primary immunization, mice were boosted by s.c. injection at the base of the tail with 100 μg bovine CII emulsified in 100 μL IFA. Mice were assessed daily for signs of inflammatory arthritis. As each mouse reached a total paw score between 2 and 4, it was randomly assigned to a treatment group. Arthritis severity was assessed using both a visual scoring system and paw thickness measurements. For the visual scoring system, each mouse could have a maximal score of 16 because each limb was given a score of 0-4. Grade 0 indicated no swelling or erythema; grade 1, mild swelling and erythema or digit inflammation; grade 2, moderate swelling and erythema confined distal to the mid-paw; grade 3, more pronounced swelling and erythema with extension to the ankle; and grade 4, severe swelling, erythema, and joint rigidity of the ankle, foot, and digits. Paw thickness was determined by measuring the thickness of the hind paws above the metatarsal region using 0- to 10-mm calipers. Upon randomization into a treatment group, mice were given 0.2 ml of vehicle, 10 mg/kg of erlotinib, or 50 mg/kg of erlotinib twice daily by oral gavage using a 1-ml syringe and a 20-gauge gavage needle (Fisher Scientific). All mouse groups were treated until inflammation in the vehicle-treated mice peaked and began to decrease, as assessed by visual score and paw thickness. At that time, feet and ankles were collected in formalin for histology.
K/BxN serum transfer arthritis was induced in Balb/c mice by dual injections of 50 μL KRN serum diluted 1:1 in 1X PBS on day 0 and day 2. Mice were treated with either vehicle or 50 mg/kg of erlotinib twice daily by oral gavage. Mice were scored as described above.
Histopathology
Mouse hind limbs were fixed in formalin, decalcified in Cal-Ex II (Fischer Scientific), and embedded in paraffin. Sections were stained with hematoxylin and eosin, and scored by an investigator blinded to the treatment group. Sections were evaluated for synovitis, pannus formation, and bone and/or cartilage destruction using a previously described scoring system: grade 0, normal; grade 1, mild inflammation, mild hyperplasia of the synovial lining layer, mild cartilage destruction without bone erosion; grades 2–4, increasing degrees of inflammatory cell infiltrates, synovial lining hyperplasia, and pannus formation and cartilage and bone destruction (14).
Isolation and stimulation of synovial fibroblasts
Synovial fibroblasts were isolated from remnant pannus obtained during knee arthroplasty, as previously described (14), and cultured in RPMI 1640 containing 10% FCS, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin sulfate at 37°C, 5% CO2. Cells were used for experiments between passages 5 and 8 and exhibited characteristic fibroblast appearance. Moreover, cells were checked at passage 6 for purify, and did not exhibit staining above background for CD3, CD14, CD20 and CD11c (Fig. S1). For experiments, 1 × 103 cells/cm2 were plated in media containing 5% FBS, RPMI-1640, P/S and allowed to adhere for 24 hours. Media were replenished, cells were pretreated with erlotinib, or PD153035 for 30 minutes, and EGF or PDGFbb was added to cells for another 48 hours. Supernatants were collected after 48 hours for ELISA analysis.
Endothelial cell stimulation
HUVEC (Lonza) were cultured in endothelial growth media-2 (Lonza) on plates coated with 10 μg/cm2 of rat tail collagen I (Sigma-Aldrich). To evaluate HUVEC proliferation, we plated cells at a density of 1 × 103 cells/cm2 in complete media and allowed them to adhere overnight. Cells were then washed with 1× PBS and incubated with reduced serum media containing endothelial basal media-2 (Lonza), 1% FBS, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate at 37°C, 5% CO2 for 24 hours. Cells were pre-treated with erlotinib for 30 minutes, and EGF or VEGF were added for 72 hours.
Proliferation assays
BrdU was added during the last 24 hours of incubation to evaluate synovial fibroblast and HUVEC proliferation. BrdU incorporation was assessed as a marker of proliferation by using the BrdU Cell Proliferation Assay (Calbiochem) according to the manufacturer’s instructions.
ELISAs
Levels of EGF, betacellulin (Peprotech), amphiregulin, HB-EGF, and TGFα (R&D Systems) were determined by ELISA in serum samples diluted 1:2 to 1:200 in assay diluent. Levels of VEGF, IL-8, MCP-1 (Peprotech) and MMP-3 (R&D Systems) were determined by ELISA in supernatants collected from synovial fibroblasts cultures 48 hours after stimulation. For IgG ELISAs, plates were coated with 100 ug/mL of bovine collagen II. Mouse serum diluted 25-fold was then added followed by anti-IgG1-HRP or anti-IgG2a-HRP (Southern Biotech) diluted 10,000 fold. Absorption was read at 450 nm.
Osteoclast assays
Bone marrow cells were isolated from naïve, 7-week-old DBA/1J mice. Twenty-four hours after being isolated, undifferentiated (non-adherent) cells were collected and replated at 15 × 103 cells/cm2. Cells were stimulated with 40 ng/ml of M-CSF (Peprotech) in complete α-MEM for three days. Differentiation media containing 40 ng/ml of M-CSF and 100 ng/ml of RANKL (Peprotech) was then added for another 3-4 days, and osteoclast formation was monitored visually. For these studies, culture media was refreshed every two days. Erlotinib or PD153035 was added to the cultures at the specified concentrations at the beginning of the differentiation period. For identification of TRAP+ cells, cells were washed with 1X PBS and stained using the acid phosphatase leukocyte kit (Sigma-Aldrich). Eight photographs of each well at 40X and 100X were analyzed in a blinded manner. The number of osteoclasts per picture and the number of nuclei per osteoclast were counted. Image J was used to calculate the surface area of the osteoclasts formed. For measurement of TRAP enzyme activity, cells were lysed at 4°C in a buffer containing 90 mM of citrate, 0.1% Triton X-100, and 80 mM of sodium tartrate for 10 minutes. Substrate solution consisting of 20 mM of p-nitrophenyl phosphate was then added to the wells for 1 hour at room temperature. The reaction was stopped by the addition of 0.5 M NaOH, and plates were read at 405 nm. For bone resorption assays, osteoclasts were grown on dentin discs (Boldon) for three days with 40 ng/mL M-CSF and six days with 40 ng/mL M-CSF, 100 ng/mL RANKL and erlotinib. Media was changed every two days. After nine days, discs were washed with distilled water, gently rubbed with a cotton swab to remove attached cells and stained with toluidine blue. Resorbed areas were photographed and analyzed using Image J software.
Quantification of mRNA expression
RNA isolation was performed using the RNeasy kit (Qiagen), and cDNA was made using the Qscript CDNA Synthesis Kit (VWR). Real-time PCR was performed using Perfecta Sybr 2X mix (VWR). Primers, purchased from IDT, were as follows: Human COX-2, forward 5’-CCGGGTACAATCGCACTTAT-3’, reverse 5’-GGCGCTCAGCCATACAG-3’; human GAPDH, forward 5’-GAAGGCTGGGGCTCATTT-3’, reverse 5’-ATGGTTCACACCCATGACG-3’; murine Acp5, forward 5’-GCTGGAAACCATGATCACCT-3’, reverse 5’-GCAAACGGTAGTAAGGGCTG-3’; murine Ctsk, forward 5’-GCCGTGGCGTTATACATACA-3’, reverse 5’CTTCCAATACGTGCAGCAGA-3’; murine Fos, forward 5’-TGGCACTAGAGACGGACAGA-3’, reverse 5’-TCCTACTACCATTCCCCAGC-3’; murine Hprt1, forward 5’-TGTTGTTGGATATGCCCTTG-3’, reverse 5’-TGGCAACATCAACAGGACTC-3’; murine Itgb3, forward 5’-CGCCTC GTGTGGTACAGAT-3’, reverse 5’-AGTGGCCGGGACAACTCT-3’; murine Mmp3, forward 5’-AGCCTT GGCTGAGTGGTAGA-3’, reverse 5’CGATGATGAACGATGGACAG-3’; murine Mmp9, forward 5’-CTGTCGGCTGTGGTTCAG T-3’, reverse 5’-AGACGACATAGACGGCATCC-3’; murine Mmp14, forward 5’-GTGAGCGTTGTGTGTGGGTA-3’, reverse 5’-CCCAAGGCAGCAACTTCAG-3’; murine Nfatc1, forward 5’-AATTAGGAGTGGGGGATCGT-3’, reverse 5’-ATCCAACCCAACTCGCCT-3’. COX-2 expression was normalized to GAPDH levels, and murine gene expression was normalized to Hprt1 levels.
Western blotting
Cells were serum starved in serum free media for three hours prior to stimulation. Whole-cell lysates were generated using a buffer containing 1% NP-40 (Sigma), 0.1% SDS (Fluka), 0.5% Sodium deoxycholate (Sigma), 10 mM EDTA (Promega), and 1/100 Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific). Lysates were analyzed using standard immunoblotting procedures. Briefly, lysates were separated on 4-12% Bis-Tris gels (Bio-Rad), transferred to PVDF membranes, blocked with 5% (w/v) BSA or milk, and probed with primary and secondary antibodies in BSA or milk. Signal was detected with SuperSignal West Fempto Chemiluminescent Substrate (Pierce Biotechnology). Antibodies used were against EGFR (Rockland), phospho-EGFR(Y1068) (Abcam), Akt1 (Cell Signaling), phospho-Akt(S473) (Cell Signaling), Erk1/2 (Cell Signaling), and phospho-Erk1/2 (T202, Y204) (Cell Signaling).
Flow cytometry
Human peripheral blood mononuclear cells (PBMCs) and RA synovial fibroblasts were stained with antibodies including CD3-PE, CD20-PerCP-Cy5.5 and CD11c-PE (BD) and CD14-PeCy7 (ebioscience). Samples were analyzed on a BD LSR Fortessa.
Cell death assays
Synovial fibroblasts, HUVEC, and osteoclasts were treated with erlotinib or PD153035 for twenty-four hours in the same media and at the same treatment concentrations as used in the cytokine, proliferation, and osteoclast formation assays. Tween 20 was added for the last hour to induce a loss of membrane integrity and release of lactate dehydrogenase (LDH). Cell supernatants were analyzed using a LDH Cytotoxicity Assay Kit (Cayman Chemical). Briefly, supernatants were incubated for 30 minutes at room temperature with a reaction buffer containing LDH Diaphorase, NAD+, Lactic acid, and tetrazolium salt. Absorbance was then read at 490 nm.
Statistics
Visual arthritis scores, paw thicknesses, and histology scores were analyzed by the Mann-Whitney U test using GraphPad InStat version 3.0 (GraphPad Software). All other data were analyzed, using an unpaired two-tailed Student’s t-tests (GraphPad Software).
Results
Levels of EGF are increased in RA serum
Which EGFR ligand(s) play a role in RA is unclear, with differing reports each suggesting that EGF, TGFα, or amphiregulin is the important EGFR ligand (9, 11, 12). We measured the concentration of the five best characterized EGFR ligands in serum from RA patients and normal controls. Levels of EGF and betacellulin were significantly higher in sera from RA patients than in that from healthy individuals (Fig. 1A and S2A). In contrast, serum levels of TGFα, amphiregulin, or HB-EGF did not differ significantly between the two groups (Fig. S2B- S2D). Thus, there is increased capacity for activation of EGFR signaling in patients with RA.
FIGURE 1.
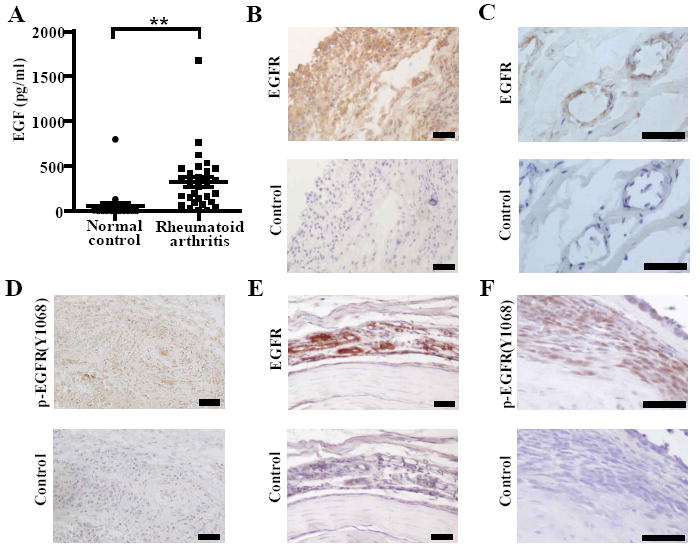
EGF and EGFR are expressed in RA and CIA. A, EGF concentration was measured in serum from normal controls (n = 20) and RA patients (n = 30) by ELISA (**P <0.01). RA synovial tissue was sectioned and examined for EGFR staining (B) in the synovial lining and (C) in the subsynovial tissue. RA synovial tissue was examined for (D) phosphorylated (p)-EGFR (Y1068) expression. E, Paws from mice with CIA (score 4) were stained for EGFR and (F) p-EGFR (Y1068). Pictures represent areas of pannus surrounding the bone next to inflammatory foci. B-F, Control sections indicate adjacent sections stained with appropriate isotype control antibodies. A-C, Data represent two experiments. E and F, Data represent at least four experiments. Bars represent 50 μm.
EGFR is expressed in RA and CIA joint tissue
We next examined whether the synovial tissue of patients with RA and of mice with CIA express EGFR, and whether the EGFR in these tissues is activated. We found that RA synovial lining expresses high levels of both total EGFR and tyrosine-1068-phosphorylated (Y1068) EGFR, the activated form of EGFR (Fig. 1B-D). Deeper within the RA synovial tissue, EGFR was expressed around high-endothelial venules and blood vessels (Fig. 1C). Likewise, EGFR and P-EGFR(Y1068) were highly expressed in areas of synovitis and around vessels in joint tissues of mice with CIA (Fig. 1F, 1G). Thus, EGFR is expressed and activated in synovial tissue in RA, as well as in the CIA model of RA.
EGFR inhibition reduces the severity of CIA
To investigate the role of EGFR in CIA, we administered 10 or 50 mg/kg of erlotinib twice daily to mice with established CIA. These doses of erlotinib produce plasma concentrations in the same range as that achieved with human dosing (15, 16) Erlotinib treatment dose-dependently reduced the visual arthritis score and paw thickness of mice with CIA (Fig. 2A, 2B). Histologically, it dose-dependently reduced the degree of pannus, synovitis, and erosion in the paws of mice with CIA (Fig. 2C, 2D). This was not accompanied by a significant change in the IgG1 or IgG2a titers (Fig. S3A, S3B). Similarly, erlotinib treatment also reduced the severity of disease in the K/BxN serum transfer model (Fig. S3C). Thus, erlotinib, at pharmacologically relevant doses, attenuates autoimmune arthritis in mice.
FIGURE 2.
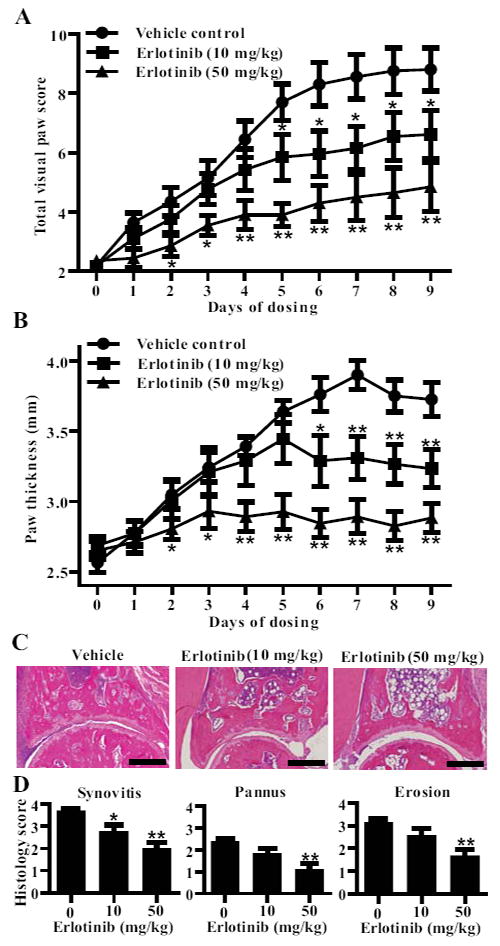
Erlotinib treatment ameliorates CIA. Following the development of arthritis, DBA1/J mice with CIA were randomly enrolled in a treatment group when they reached a mean arthritic score between 2 and 4. Mice were administered vehicle (n = 10), 10 mg/kg (n = 12), 50 mg/kg (n = 12) erlotinib suspended in vehicle twice daily by oral gavage. A, Arthritis severity was assessed using a visual arthritis scoring system and (B) caliper measurements of paw thickness (*P < 0.05; **P < 0.01). Data represent one experiment of at least three independent experiments. To analyze disease pathology, (C) representative pictures of paw sections were taken, and (D) blinded histological scores of synovitis, pannus formation and bone and cartilage erosion were made (*P < 0.05; **P < 0.01). Bars represent 300 μm. Groups were compared at each time point using a Mann Whitney analysis comparing erlotinib treated mice to vehicle-treated mice. Error bars indicate ± SEM.
Erlotinib reduces EGF-induced synovial fibroblast proliferation and cytokine production
To elucidate erlotinib’s mechanism of action in treating autoimmune arthritis, we first examined its effects on synovial fibroblasts derived from RA patients. In RA, synovial fibroblasts become hyperplastic and invade the surrounding bone and cartilage, forming the pannus (17). EGFR ligands may increase the mitogenic capacity of synovial fibroblasts (9, 18-21), and erlotinib-induced attenuation of CIA was associated with reduction in pannus formation (Fig. 2D). We therefore tested erlotinib’s effect on EGF-induced proliferation of synovial fibroblasts. We concurrently tested the effect of PD153035, another small-molecule inhibitor of EGFR. To assess the specificity of erlotinib, we also tested erlotinib’s effect on synovial fibroblast proliferation induced by PDGFbb, which is known to promote synovial fibroblast proliferation and cytokine production through PDGFR, rather than EGFR (14). Erlotinib and PD153035 reduced the proliferation induced by EGF, but not that induced by PDGFbb (Fig. 3A).
FIGURE 3.
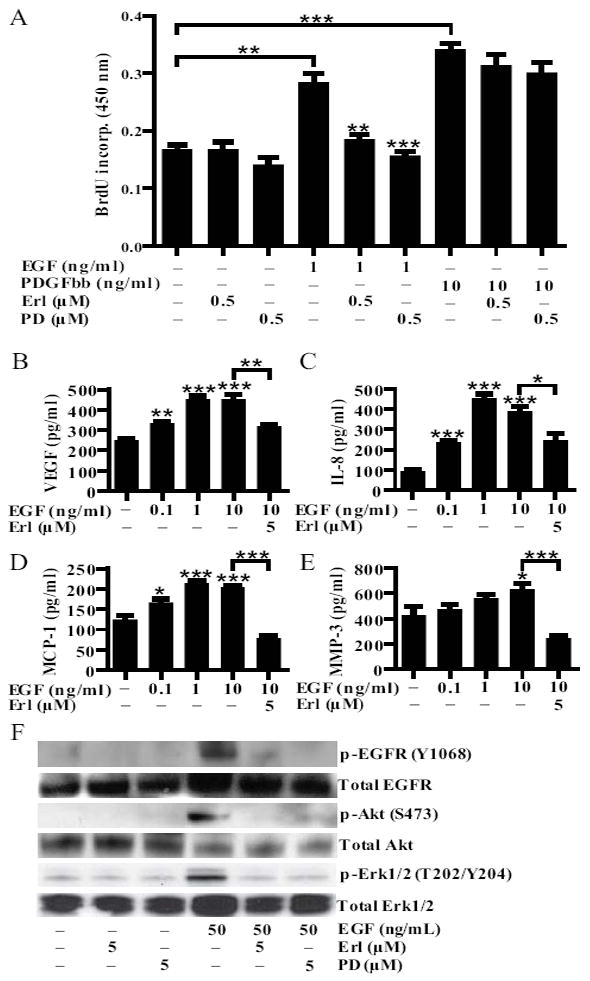
Erlotinib reduces RA synovial fibroblasts proliferation and cytokine production. A, RA synovial fibroblasts were stimulated with EGF (1 ng/ml) and PDGFbb (10 ng/ml) and concurrently treated with erlotinib (0.5 μM) or PD153035 (0.5 μM). After 48 hours of incubation, proliferation was analyzed by BrdU incorporation. B-E, RA synovial fibroblasts were stimulated with increasing concentrations of EGF and concurrently treated with erlotinib (5 μM). After 48 hours of incubation, the concentrations of (B) VEGF, (C) IL-8, (D) MCP-1 and (E) MMP-3 were determined by ELISA. Data represent one experiment of at least three independent experiments (*P < 0.05; **P < 0.01; *** P < 0.001). Error bars indicate ± SEM. E, To examine EGFR signaling in the cells, cells were pretreated with erlotinib (5 μM) or PD153035 (5 μM) for 30 minutes, stimulated with EGF (50 ng/ml) for 5 minutes, and cell lysates were examined via western blotting. Data represent one experiment of at least three independent experiments. PD = PD153035.
EGFR ligands may also stimulate the production of proinflammatory cytokine production of synovial fibroblast (9, 20). We found that EGF induces RA synovial fibroblasts to produce VEGF, IL-8, MCP-1 and MMP-3, and concurrent erlotinib treatment reduced the production of these factors (Fig. 3B-E). All of these EGF-induced cytokines are implicated in RA: VEGF drives angiogenesis (22); IL-8 promotes neutrophil infiltration into the joint space (23); MCP-1 promotes the infiltration of monocytes into the synovium (24); and MMP-3 aids the destruction of cartilage (25).
Treating cells with erlotinib or PD153035 did not induce the release of LDH, indicating that erlotinib’s effect on proliferation and cytokine production was not due to the induction of cell death (Fig. S4A). Thus, erlotinib can reduce not only proliferation but also cytokine production of synovial fibroblasts (Fig. 3A-E), indicating that erlotinib may directly reduce pannus formation and indirectly reduce angiogenesis and synovitis by acting on these cells.
We found that erlotinib inhibited the EGF-induced phosphorylation of EGFR, as well as the EGF-induced phosphorylation of Akt and Erk1/2 (Fig. 3F), kinases that mediate two different signaling pathways downstream of EGFR (8). These findings confirm that erlotinib suppresses proliferation and cytokine production by inhibiting EGFR signaling.
Erlotinib reduces EGF-induced endothelial cell proliferation and COX-2 expression
As we show in Fig. 1, endothelial cells in RA and CIA synovium express EGFR. Blood vessels, which are lined with endothelial cells, increase in number in RA synovium as compared to osteoarthritis (OA) or healthy synovium (26). Targeting the formation of these blood vessels in RA is hypothesized to have three beneficial consequences: it may reduce the growth of the synovium by decreasing the blood supply that provides nutrients, decrease the inner blood vessel wall surface area for lymphocyte ingress from the blood into the synovium, and diminish the production of chemoattractants by the endothelial cells (3). Interestingly, EGF and HB-EGF have been shown to promote angiogenesis by promoting endothelial cell proliferation or migration (27-30). To determine whether erlotinib affects neoangiogenesis in CIA, we quantified the number of blood vessels per square millimeter in the ankle of mice treated with vehicle or erlotinib, by staining paw sections for Von-Willebrand factor. We found that the attenuation of CIA was associated with a reduction in the number of blood vessels formed in the ankles of erlotinib-treated mice, indicating the erlotinib reduces neo-angiogenesis in vivo (Fig. 4A, 4B). Furthermore, we show that erlotinib markedly suppresses EGF-induced, but not VEGF-induced, proliferation of HUVEC in vitro, as does PD153035 (Fig. 4D).
FIGURE 4.
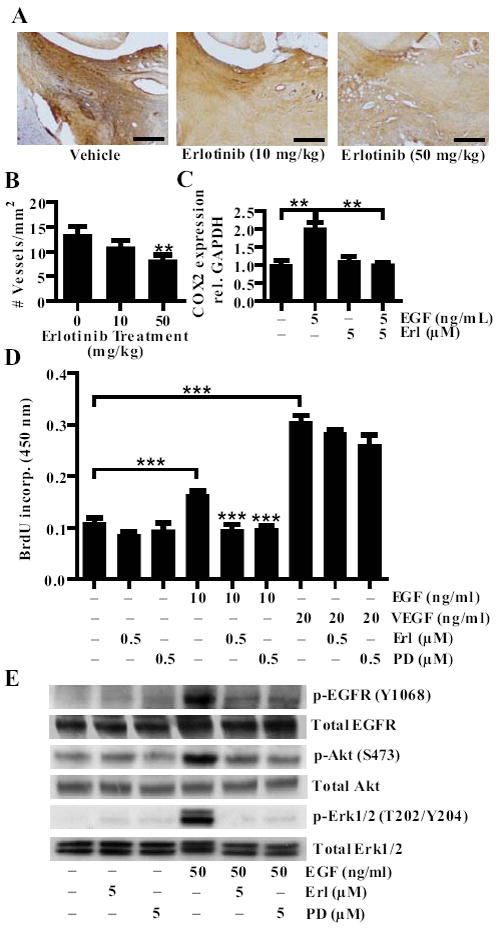
Erlotinib inhibits HUVEC proliferation and COX-2 expression. A, Mice with CIA were treated with vehicle, 10 or 50 mg/kg of erlotinib twice daily by oral gavage. Paws were collected, sectioned and stained for von willebrand factor (VWF). Bars represent 150 μm. B, The number of VWF positive vessels in the ankle was quantified per square area for each treatment group (**P < 0.01). C, HUVEC were stimulated with 5 ng/ml of EGF for 45 minutes with or without concurrent treatment with 5 μM of erlotinib. Cells were collected in Trizol and RTPCR was performed to determine COX-2 mRNA expression. D, HUVEC were stimulated with EGF (10 ng/ml) or VEGF (20 ng/ml) and treated with either erlotinib (0.5 μM) or PD153035 (0.5 μM). Proliferation was measured by BrdU incorporation. E, To examine signaling in the cells, cells were pretreated with erlotinib (5 μM) or PD153035 (5 μM) for 30 minutes, stimulated with EGF (50 ng/ml) for 5 minutes, and cell lysates were examined via western blotting. C-D, Data represent one experiment of at least three independent experiments (**P < 0.01; *** P < 0.001). Error bars indicate ± SEM.
COX-2 inhibitors can reduced swelling and pain in the joints of RA patients (31), and COX2 expression is significantly greater in endothelial cells in RA synovium than in OA synovium (32). Moreover, angiogenesis is dependent on COX-2 activity, as COX-2 specific inhibitors strongly reduce tube formation (33). Previous studies have indicated that EGF induces COX-2 expression in RA synovial fibroblasts (21). We found that erlotinib inhibits the EGF-induced expression of COX-2 in HUVEC, suggesting that erlotinib may be reducing angiogenesis in vivo by modulating COX-2 expression in endothelial cells (Fig. 4C).
Erlotinib inhibited the EGF-induced phosphorylation of EGFR, as well as the EGF-induced phosphorylation of Akt and Erk1/2 in HUVEC (Fig. 4E), signifying that erlotinib suppresses HUVEC proliferation and COX-2 expression by inhibiting EGFR signaling. Erlotinib and PD153035 did not induce LDH release after 24 hours of treatment (Fig. S4B), indicating that their suppression of HUVEC proliferation is not due to cell death.
Erlotinib reduces osteoclastogenesis
RA and CIA are characterized by the progressive destruction of bone and cartilage in the synovial joints (34, 35). This destruction is mediated by osteoclasts, which resorb mineralized bone and cartilage (4). Numerous osteoclasts are found in and adjacent to the inflamed synovium and are thought to derive from monocyte/macrophage lineage cells in the synovium (4). Because erlotinib treatment reduced the erosion of bone and cartilage in the joints of mice with CIA (Fig. 2), we compared the number of osteoclasts in the paws of CIA mice treated with vehicle and those treated with erlotinib. To this end, we performed immunohistochemical analysis for the expression of tartrate-resistant acid phosphatase (TRAP), a characteristic of osteoclasts. As with cartilage and bone erosion, there were more TRAP-positive cells in vehicle-treated than in erlotinib-treated mice. In fact, erlotinib reduced the number of TRAP-positive cells in a dose-dependent fashion (Fig. 5A). We next asked whether erlotinib could directly reduce the in vitro formation of osteoclasts, which we defined as TRAP+ cells with three or more nuclei. Erlotinib significantly reduced the number of osteoclasts formed from bone marrow-derived monocytes; it also reduced the size of the osteoclasts (Fig. 5B-D). Erlotinib treatment significantly decreased the expression of four genes characteristic of osteoclasts: Acp5 which encodes TRAP, Ctsk which encodes Cathepsin K, Itgb3 which encodes integrin beta-3, and Nfatc1 which encodes nuclear factor of activated T –cells, cytoplasmic 1 (Fig. 5E) (36). Erlotinib treatment also reduced the expression of several MMPs, including Mmp3, 9 and 14 which mediate breakdown of the non-mineralized parts of the bone (Fig. 5E). Moreover, treatment with erlotinib significantly decreased osteoclast mediated bone resorption (Fig. 5F) and did not alter LDH release (Fig. S4C). This indicates that erlotinib inhibits the formation of the osteoclast from mononuclear cells. Together, this showed that EGFR inhibition reduces osteoclastogenesis.
FIGURE 5.
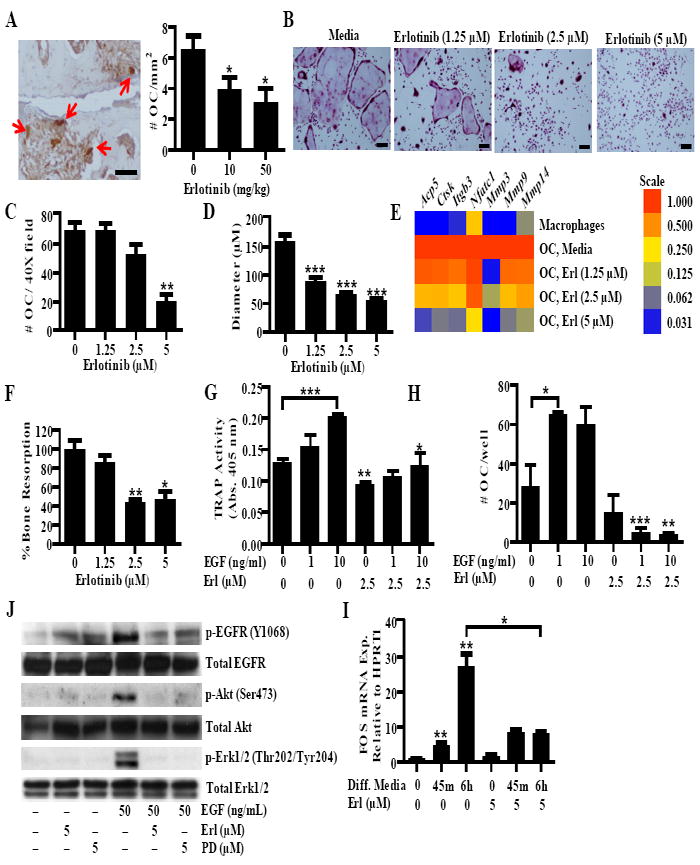
Erlotinib inhibits osteoclastogenesis. A, Paw sections from CIA mice treated with vehicle or erlotinib were stained for TRAP. Arrows indicate osteoclasts defined as large, TRAP+ cells adjacent to bone. The number of osteoclasts was quantified for each treatment group. B-E, Osteoclasts were treated with erlotinib during the differentiation period and stained for TRAP. B, Representative pictures were taken, and the (C) number of osteoclasts and (D) relative diameter was determined using Image J analysis. For quantification, osteoclasts were defined as TRAP+ cells with three or more nuclei. E, Expression of osteoclast specific genes and mmps was quantified in macropahges and osteoclasts treated with erlotinib. Gene expression was normalized to Hprt1 and the level was arbitrarily set to 1 in untreated osteoclasts. F, Osteoclasts were grown on dentin discs and bone resorption was analyzed by toluidine blue staining and image analysis. Data is normalized to the level of untreated osteoclasts. G and H, Osteoclasts were differentiated in the presence of EGF. At day seven, TRAP activity was measured by an (G) enzymatic reaction and the (H) number of osteoclasts per well was counted. I, Osteoclasts were pretreated with either erlotinib (5 μM) or PD153035 (5 μM) and stimulated with EGF (50 ng/ml) for 5 minutes and western blots were performed. J, Monocytes were treated with serum free differentiation media containing M-CSF and RANKL for 45 minutes (min) and six hours (h). Fos expression was determined by QPCR. B-D, G-J, Data represent one experiment of at least three independent experiments E-F, Data represent one experiment of at least two independent experiments. For all experiments (*P < 0.05; ** P < 0.01; *** P < 0.001). A and B, bars represents 150 μm. OC, osteoclast. Error bars indicate ± SEM.
TNF promotes osteoclastogenesis both directly and indirectly, by promoting RANKL expression and binding TNFα-receptor type 1 on osteoclasts (4). To determine whether EGF promotes osteoclastogenesis directly, we treated the bone marrow-derived monocytes with EGF in the presence of osteoclast differentiation media containing a reduced concentration of RANKL, and then measured TRAP enzymatic activity and quantified the number of osteoclasts formed. A reduced concentration of RANKL was used for these experiments because sub-optimal osteoclast formation was required in order to discriminate the effects of EGF. EGF significantly increased the cells’ TRAP activity and the number of osteoclasts formed, suggesting that it promotes osteoclastogenesis. Moreover, concurrent treatment with erlotinib blocked the increase in TRAP activity and formation of osteoclasts induced by EGF, and partially blocked the TRAP activity induced by RANKL alone (Fig. 5G-H).
EGF induced the phosphorylation of EGFR, Akt and Erk1/2 in osteoclasts, and this was reduced by treatment with erlotinib and PD153035 (Fig. 5I), suggesting that erlotinib suppresses osteoclastogenesis by inhibiting EGFR-induced gene expression. FOS is a member of the AP-1 family of transcription factors and is required for osteoclast formation (37). RANKL activates c-Fos transcription (37), possibly by transactivating EGFR (38). Moreover, EGFR signaling independently upregulates c-Fos expression in HeLa cells (39). We therefore tested the effect of erlotinib on monocytes incubated with differentiation media containing M-CSF and RANKL. We found that the osteoclast differentiation media caused a strong upregulation of Fos mRNA expression at six hours, and that this was reduced by erlotinib (Fig. 5J). Given that FOS is necessary for osteoclast development (37), inhibiting FOS may be one way in which erlotinib reduces osteoclastogenesis.
Discussion
In this study, we show that EGF levels are abnormally high in the serum of RA patients, and that EGFR is both highly expressed and activated in synovial tissue from RA patients and from mice with autoimmune arthritis. Moreover, we find that the EGFR inhibitor erlotinib attenuates autoimmune arthritis in mice, reducing synovitis, pannus formation, and cartilage and bone erosion in their synovial joints. Erlotinib-induced attenuation of autoimmune arthritis was associated with a reduction in number of osteoclasts and blood vessels formed in the mouse paws, and erlotinib directly inhibited the formation of osteoclasts and the proliferation of endothelial cells in vitro, as did two other EGFR inhibitors. Erlotinib also inhibited the proliferation and cytokine production of synovial fibroblasts in vitro, an effect that may explain its ability to reduce pannus formation and synovitis in vivo.
Previous findings are conflicting as to which EGFR ligand is upregulated in RA. Studies of bone marrow-derived mononuclear cells and synovial fluid have implicated EGF, TGFα, and amphiregulin in RA (9, 12, 40, 41). However, we find that only levels of EGF and betacellulin, and not TGFα, HB-EGF, or amphiregulin, are higher in serum from RA patients than in serum from healthy individuals. This suggests that the exact EGF ligand involved in RA may differ between serum and synovial fluid, or may depend on RA subtype, disease duration, or the treatment regimen. Although we focused on EGF in this study, it will be interesting to further define the activity of betacellulin in RA. Betacellulin is known to promote angiogenesis indicating that it may promote the growth of the synovium (42).
Previous studies found that EGFR is expressed on RA fibroblast cell lines, and on the vascular endothelial cells and subsynovial fibroblasts of certain RA patients (9, 10). Here, we show that EGFR is not only expressed but also activated in RA and CIA synovium. Furthermore, we confirm that EGFR is expressed and can be transactivated by EGF in cultured synovial fibroblasts.
Our finding that erlotinib, an EGFR inhibitor that is FDA-approved for the treatment of cancer, attenuated CIA complements a recent report indicating that soluble herstatin, which inhibits the dimerization of ErbB receptors, attenuates CIA (6). Together, these findings suggest that inhibition of EGFR, and possibly ErbB2 (which is a frequent binding partner of EGFR), may provide benefit in RA.
Amphiregulin has been shown to increase the proliferation of RA synovial fibroblasts and to increase their expression of VEGF, IL-8, GM-CSF, and IL-6 (9, 40). EGF on its own was found to have little effect on synovial fibroblasts, but to synergize with TNF and IL-1β to increase mRNA expression of MMPs and proinflammatory molecules (19). We find that EGF alone can in fact induce the proliferation of synovial fibroblasts in a dose-dependent manner, an effect that could promote the growth of the pannus in RA. Moreover, we demonstrate that EGF alone can induce the production of IL-8, VEGF, MMP-3 and MCP-1 protein by RA synovial fibroblasts, and that concurrent EGFR inhibition with erlotinib or PD153035 reduces the production of these factors. Thus, erlotinib may indirectly have widespread effects on RA though its actions on RA synovial fibroblasts. These include decreasing IL-8 and MCP-1 induced neutrophil and monocyte migration into the joint space (23, 24), decreasing angiogenesis driven by VEGF (22), and reducing cartilage destruction promoted by MMP-3 (25).
RA synovium contains significantly greater number of blood vessels than OA synovium (26). Here, we demonstrate that erlotinib reduces the number of blood vessels in CIA and that this may occur through both direct and indirect mechanisms. Erlotinib may directly reduce angiogenesis by impeding endothelial cell proliferation and COX-2 expression, and indirectly reduce angiogenesis by decreasing VEGF production by RA syovial fibroblasts.
RA and CIA are characterized by the progressive destruction of bone and cartilage in the synovial joints, a process mediated by osteoclasts (34, 35). Osteoclast development is driven by excessive production of RANKL and TNF in the RA synovium, which interact with their receptors on monocytes and macrophages driving osteoclast formation through the expression of transcription factors, such as FOS of the AP-1 complex and NFATc1 (4). EGF and EGFR may also promote bone destruction in RA, as bone formation is abnormal in EGFR-deficient mice, suggesting that EGFR is important for osteoclast formation (43). The EGFR inhibitors AG1478 and PD153035 have been shown to reduce osteoclast formation (38, 43); here we show that EGFR inhibition with erlotinib also reduces osteoclast formation. EGFR inhibition reduced the number of osteoclasts formed, as well as their size, osteoclast-specific gene expression and bone resorption capacity. Moreover, we demonstrate that EGF increases the TRAP enzymatic activity and number of osteoclasts formed during low-RANKL conditions. This indicates that EGF, like TNF, promotes osteoclast activity in RA and CIA. Erlotinib reduced the osteoclast activity induced by EGF as well as that induced in the absence of added EGF. Furthermore, we demonstrate that erlotinib acts on osteoclasts by reducing EGF-induced phosphorylation of EGFR, Erk and Akt, as well as RANKL-induced FOS expression. EGF may therefore supplement the transactivation of EGFR by RANKL, and thereby promote osteoclast formation. This is important because it indicates that EGFR in addition to RANK might be a beneficial target to reduce bone loss in RA.
Together, our findings suggest that targeting EGFR has potential as a novel therapeutic strategy for RA. Rational targeting of EGFR inhibitors to specific phases of RA, limiting the duration of dosing, and combining EGFR inhibitors with other disease-modifying agents may be important in maximizing the benefit of EGFR inhibitors in RA. Patients with early, active RA may benefit most from EGFR inhibitor therapy. Patients with undifferentiated arthritis who go on to develop RA exhibit a distinct and transient increase in EGF expression in synovial fluid (44). This suggests that, early in the course of arthritis, there may be a window of opportunity where EGFR inhibition could be beneficial in those patients with significantly heightened EGF levels. EGF levels may also correlate with disease activity and severity. EGF levels in synovial fluid correlated with C-reactive protein and IgM levels in RA patients (12). High pretreatment levels of EGF and MCP-1 distinguished patients who responded to etanercept therapy (45). Thus, as disease progresses, EGFR inhibition may continue to be of benefit to RA patients with more destructive disease courses.
EGFR inhibitors such as erlotinib are known to cause adverse side effects, e.g. rash and diarrhea (13), which make them less amenable to treating chronic, non-terminal diseases such as RA. However, several treatment strategies may increase the tolerability and efficacy of EGFR inhibitor therapy. First, pulsed, rather than chronic, therapy may suffice during early disease or during a disease flare when EGF levels appear to peak. Second, patients with high levels of EGFR ligands in their serum may be specifically targeted for EGFR inhibitor therapy. Third, next-generation EGFR inhibitors being developed for the treatment of cancer may better therapeutic index. These include agents that bind EGFR irreversibly, target multiple ErbB family members, and inhibit additional kinases receptors such as those for VEGF and FGF which are known to drive disease (46).
In conclusion, we show that EGFR inhibition using erlotinib reduces the severity of autoimmune arthritis in mice. The mechanism of action appears to be through inhibition of synovial fibroblast, endothelial cell, and osteoclast inflammatory and degradative responses. Our findings demonstrate the importance of EGFR in the pathogenesis of autoimmune arthritis, and provide evidence that EGFR, a molecule long targeted in cancer, should be considered a “new” molecular target in RA.
Supplementary Material
Acknowledgments
We thank members of the WHR laboratory for many rewarding discussions. Lauren Lahey, Orr Sharpe and Yann Chong Tan particularly aided in figure development, western blots and PCR troubleshooting, respectively. Eun-Ju Chang and Qian Wang provided valuable insight on osteoclast development and analysis, and the Butcher lab members generously supplied endothelial cell resources and knowledge.
Abbreviations
- b.i.d
twice per day
- CIA
collagen-induced arthritis
- COX-2
cyclooxygenase-2
- EGF
epidermal growth factor
- EGFR
EGF-receptor
- ERBB2
erythroblastic leukemia viral oncogene homolog 2
- FLS
fibroblast-like synoviocytes
- HB-EGF
heparin-binding EGF-like growth factor
- HPRT1
hypoxanthine phosphoribosyltransferase 1
- LDH
lactate dehydrogenase
- M-CSF
- MMP
matrix metalloproteinase
- P-EGFR
phosphorylated-EGFR
- PDGFbb
platelet-derived growth factor bb
- RANKL
receptor activator of nuclear factor kappa-B ligand
- RA
rheumatoid arthritis
- TRAP
tartrate-resistant acid phosphatase
- VEGF
vascular endothelial growth factor
- Y
tyrosine
References
- 1.Cope AP. T cells in rheumatoid arthritis. Arthritis Res Ther. 2008;10(Suppl 1):S1. doi: 10.1186/ar2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. 2010;376:1094–1108. doi: 10.1016/S0140-6736(10)60826-4. [DOI] [PubMed] [Google Scholar]
- 3.Firestein GS. Starving the synovium: angiogenesis and inflammation in rheumatoid arthritis. J Clin Invest. 1999;103:3–4. doi: 10.1172/JCI5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schett G. Cells of the synovium in rheumatoid arthritis. Osteoclasts. Arthritis Res Ther. 2007;9:203. doi: 10.1186/ar2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sullivan T, Benjamin CG, Kempf PW, Deeken JF. Cetuximab in the treatment of rheumatoid arthritis. J Clin Rheumatol. 2010;16:32–33. doi: 10.1097/RHU.0b013e3181c8e176. [DOI] [PubMed] [Google Scholar]
- 6.Sumariwalla PF, Jin P, Zhang J, Ni I, Crawford D, Shepard HM, Paleolog EM, Feldmann M. Antagonism of the human epidermal growth factor receptor family controls disease severity in murine collagen-induced arthritis. Arthritis Rheum. 2008;58:3071–3080. doi: 10.1002/art.23885. [DOI] [PubMed] [Google Scholar]
- 7.Luke L, Gompels NM, Madden Leigh, Jin Pei, Feldmann Marc, Shepard Michael, Paleolog Ewa M. Human epidermal growth factor receptor bispecific ligand trap RB200: abrogation of collagen-induced arthritis in combination with tumour necrosis factor blockade. Arthritis Research & Therapy. 2011;13 doi: 10.1186/ar3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004;59:21–26. doi: 10.1016/j.ijrobp.2003.11.041. [DOI] [PubMed] [Google Scholar]
- 9.Yamane S, Ishida S, Hanamoto Y, Kumagai K, Masuda R, Tanaka K, Shiobara N, Yamane N, Mori T, Juji T, Fukui N, Itoh T, Ochi T, Suzuki R. Proinflammatory role of amphiregulin, an epidermal growth factor family member whose expression is augmented in rheumatoid arthritis patients. J Inflamm (Lond) 2008;5:5. doi: 10.1186/1476-9255-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Young CL, Adamson TC, 3rd, Vaughan JH, Fox RI. Immunohistologic characterization of synovial membrane lymphocytes in rheumatoid arthritis. Arthritis Rheum. 1984;27:32–39. doi: 10.1002/art.1780270106. [DOI] [PubMed] [Google Scholar]
- 11.Farahat MN, Yanni G, Poston R, Panayi GS. Cytokine expression in synovial membranes of patients with rheumatoid arthritis and osteoarthritis. Ann Rheum Dis. 1993;52:870–875. doi: 10.1136/ard.52.12.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kusada J, Otsuka T, Matsui N, Hirano T, Asai K, Kato T. Immunoreactive human epidermal growth factor (h-EGF) in rheumatoid synovial fluids. Nippon Seikeigeka Gakkai Zasshi. 1993;67:859–865. [PubMed] [Google Scholar]
- 13.Lu JF, Eppler SM, Wolf J, Hamilton M, Rakhit A, Bruno R, Lum BL. Clinical pharmacokinetics of erlotinib in patients with solid tumors and exposure-safety relationship in patients with non-small cell lung cancer. Clin Pharmacol Ther. 2006;80:136–145. doi: 10.1016/j.clpt.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 14.Paniagua RT, Sharpe O, Ho PP, Chan SM, Chang A, Higgins JP, Tomooka BH, Thomas FM, Song JJ, Goodman SB, Lee DM, Genovese MC, Utz PJ, Steinman L, Robinson WH. Selective tyrosine kinase inhibition by imatinib mesylate for the treatment of autoimmune arthritis. J Clin Invest. 2006;116:2633–2642. doi: 10.1172/JCI28546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Higgins B, Kolinsky K, Smith M, Beck G, Rashed M, Adames V, Linn M, Wheeldon E, Gand L, Birnboeck H, Hoffmann G. Antitumor activity of erlotinib (OSI-774, Tarceva) alone or in combination in human non-small cell lung cancer tumor xenograft models. Anticancer Drugs. 2004;15:503–512. doi: 10.1097/01.cad.0000127664.66472.60. [DOI] [PubMed] [Google Scholar]
- 16.Hidalgo M, Bloedow D. Pharmacokinetics and pharmacodynamics: maximizing the clinical potential of Erlotinib (Tarceva) Semin Oncol. 2003;30:25–33. [PubMed] [Google Scholar]
- 17.Abeles AM, Pillinger MH. The role of the synovial fibroblast in rheumatoid arthritis: cartilage destruction and the regulation of matrix metalloproteinases. Bull NYU Hosp Jt Dis. 2006;64:20–24. [PubMed] [Google Scholar]
- 18.Goddard DH, Grossman SL, Moore ME. Autocrine regulation of rheumatoid arthritis synovial cell growth in vitro. Cytokine. 1990;2:149–155. doi: 10.1016/1043-4666(90)90009-i. [DOI] [PubMed] [Google Scholar]
- 19.Huber LC, Kunzler P, Boyce SH, Michel BA, Gay RE, Ink BS, Gay S. Effects of a novel tyrosine kinase inhibitor in rheumatoid arthritis synovial fibroblasts. Ann Rheum Dis. 2008;67:389–394. doi: 10.1136/ard.2007.072330. [DOI] [PubMed] [Google Scholar]
- 20.Taylor DJ, Feldmann M, Evanson JM, Woolley DE. Comparative and combined effects of transforming growth factors alpha and beta, interleukin-1 and interferon-gamma on rheumatoid synovial cell proliferation, glycolysis and prostaglandin E production. Rheumatol Int. 1989;9:65–70. doi: 10.1007/BF00270247. [DOI] [PubMed] [Google Scholar]
- 21.Nah SS, Won HJ, Ha E, Kang I, Cho HY, Hur SJ, Lee SH, Baik HH. Epidermal growth factor increases prostaglandin E2 production via ERK1/2 MAPK and NF-kappaB pathway in fibroblast like synoviocytes from patients with rheumatoid arthritis. Rheumatol Int. 2010;30:443–449. doi: 10.1007/s00296-009-0976-6. [DOI] [PubMed] [Google Scholar]
- 22.Maruotti N, Cantatore FP, Crivellato E, Vacca A, Ribatti D. Angiogenesis in rheumatoid arthritis. Histol Histopathol. 2006;21:557–566. doi: 10.14670/HH-21.557. [DOI] [PubMed] [Google Scholar]
- 23.Endo H, Akahoshi T, Takagishi K, Kashiwazaki S, Matsushima K. Elevation of interleukin-8 (IL-8) levels in joint fluids of patients with rheumatoid arthritis and the induction by IL-8 of leukocyte infiltration and synovitis in rabbit joints. Lymphokine Cytokine Res. 1991;10:245–252. [PubMed] [Google Scholar]
- 24.Ogata H, Takeya M, Yoshimura T, Takagi K, Takahashi K. The role of monocyte chemoattractant protein-1 (MCP-1) in the pathogenesis of collagen-induced arthritis in rats. J Pathol. 1997;182:106–114. doi: 10.1002/(SICI)1096-9896(199705)182:1<106::AID-PATH816>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 25.Ainola MM, Mandelin JA, Liljestrom MP, Li TF, Hukkanen MV, Konttinen YT. Pannus invasion and cartilage degradation in rheumatoid arthritis: involvement of MMP-3 and interleukin-1beta. Clin Exp Rheumatol. 2005;23:644–650. [PubMed] [Google Scholar]
- 26.FitzGerald O, Soden M, Yanni G, Robinson R, Bresnihan B. Morphometric analysis of blood vessels in synovial membranes obtained from clinically affected and unaffected knee joints of patients with rheumatoid arthritis. Ann Rheum Dis. 1991;50:792–796. doi: 10.1136/ard.50.11.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu LW, Mayo LD, Dunbar JD, Kessler KM, Baerwald MR, Jaffe EA, Wang D, Warren RS, Donner DB. Utilization of distinct signaling pathways by receptors for vascular endothelial cell growth factor and other mitogens in the induction of endothelial cell proliferation. J Biol Chem. 2000;275:5096–5103. doi: 10.1074/jbc.275.7.5096. [DOI] [PubMed] [Google Scholar]
- 28.Mehta VB, Besner GE. HB-EGF promotes angiogenesis in endothelial cells via PI3-kinase and MAPK signaling pathways. Growth Factors. 2007;25:253–263. doi: 10.1080/08977190701773070. [DOI] [PubMed] [Google Scholar]
- 29.Mehta VB, Zhou Y, Radulescu A, Besner GE. HB-EGF stimulates eNOS expression and nitric oxide production and promotes eNOS dependent angiogenesis. Growth Factors. 2008;26:301–315. doi: 10.1080/08977190802393596. [DOI] [PubMed] [Google Scholar]
- 30.Semino CE, Kamm RD, Lauffenburger DA. Autocrine EGF receptor activation mediates endothelial cell migration and vascular morphogenesis induced by VEGF under interstitial flow. Exp Cell Res. 2006;312:289–298. doi: 10.1016/j.yexcr.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 31.Baraf HS. Efficacy of the newest COX-2 selective inhibitors in rheumatic disease. Curr Pharm Des. 2007;13:2228–2236. doi: 10.2174/138161207781368819. [DOI] [PubMed] [Google Scholar]
- 32.Siegle I, Klein T, Backman JT, Saal JG, Nusing RM, Fritz P. Expression of cyclooxygenase 1 and cyclooxygenase 2 in human synovial tissue: differential elevation of cyclooxygenase 2 in inflammatory joint diseases. Arthritis Rheum. 1998;41:122–129. doi: 10.1002/1529-0131(199801)41:1<122::AID-ART15>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 33.Woods JM, Mogollon A, Amin MA, Martinez RJ, Koch AE. The role of COX-2 in angiogenesis and rheumatoid arthritis. Exp Mol Pathol. 2003;74:282–290. doi: 10.1016/s0014-4800(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 34.Choi Y, Arron JR, Townsend MJ. Promising bone-related therapeutic targets for rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:543–548. doi: 10.1038/nrrheum.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi S, Lee YA, Hong SJ, Lee GJ, Kang SW, Park JH, Park HK. Evaluation of inflammatory change and bone erosion using a murine type II collagen-induced arthritis model. Rheumatol Int. 2010 doi: 10.1007/s00296-009-1333-5. [DOI] [PubMed] [Google Scholar]
- 36.Rho J, Altmann CR, Socci ND, Merkov L, Kim N, So H, Lee O, Takami M, Brivanlou Ali H. Gene expression profiling of osteoclast differentiation by combined suppression subtractive hybridizatioin (SSH) and cDNA microarray analysis. DNA and Cell Biology. 2002;21:541–549. doi: 10.1089/104454902320308915. [DOI] [PubMed] [Google Scholar]
- 37.Boyce BF, Yamashita T, Yao Z, Zhang Q, Li F, Xing L. Roles for NF-kappaB and c-Fos in osteoclasts. J Bone Miner Metab. 2005;23(Suppl):11–15. doi: 10.1007/BF03026317. [DOI] [PubMed] [Google Scholar]
- 38.Yi T, Lee HL, Cha JH, Ko SI, Kim HJ, Shin HI, Woo KM, Ryoo HM, Kim GS, Baek JH. Epidermal growth factor receptor regulates osteoclast differentiation and survival through cross-talking with RANK signaling. J Cell Physiol. 2008;217:409–422. doi: 10.1002/jcp.21511. [DOI] [PubMed] [Google Scholar]
- 39.Leaman DW, Pisharody S, Flickinger TW, Commane MA, Schlessinger J, Kerr IM, Levy DE, Stark GR. Roles of JAKs in activation of STATs and stimulation of c-fos gene expression by epidermal growth factor. Mol Cell Biol. 1996;16:369–375. doi: 10.1128/mcb.16.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakamura N, Shimaoka Y, Tougan T, Onda H, Okuzaki D, Zhao H, Fujimori A, Yabuta N, Nagamori I, Tanigawa A, Sato J, Oda T, Hayashida K, Suzuki R, Yukioka M, Nojima H, Ochi T. Isolation and expression profiling of genes upregulated in bone marrow-derived mononuclear cells of rheumatoid arthritis patients. DNA Res. 2006;13:169–183. doi: 10.1093/dnares/dsl006. [DOI] [PubMed] [Google Scholar]
- 41.Hallbeck AL, Walz TM, Briheim K, Wasteson A. TGF-alpha and ErbB2 production in synovial joint tissue: increased expression in arthritic joints. Scand J Rheumatol. 2005;34:204–211. doi: 10.1080/03009740510017715. [DOI] [PubMed] [Google Scholar]
- 42.Kim HS, Shin HS, Kwak HJ, Cho CH, Lee CO, Koh GY. Betacellulin induces angiogenesis through activation of mitogen-activated protein kinase and phosphatidylinositol 3’-kinase in endothelial cell. FASEB J. 2003;17:318–320. doi: 10.1096/fj.02-0570fje. [DOI] [PubMed] [Google Scholar]
- 43.Wang K, Yamamoto H, Chin JR, Werb Z, Vu TH. Epidermal growth factor receptor-deficient mice have delayed primary endochondral ossification because of defective osteoclast recruitment. J Biol Chem. 2004;279:53848–53856. doi: 10.1074/jbc.M403114200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raza K, Falciani F, Curnow SJ, Ross EJ, Lee CY, Akbar AN, Lord JM, Gordon C, Buckley CD, Salmon M. Early rheumatoid arthritis is characterized by a distinct and transient synovial fluid cytokine profile of T cell and stromal cell origin. Arthritis Res Ther. 2005;7:R784–795. doi: 10.1186/ar1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fabre S, Dupuy AM, Dossat N, Guisset C, Cohen JD, Cristol JP, Daures JP, Jorgensen C. Protein biochip array technology for cytokine profiling predicts etanercept responsiveness in rheumatoid arthritis. Clin Exp Immunol. 2008;153:188–195. doi: 10.1111/j.1365-2249.2008.03691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gazdar AF. Epidermal growth factor receptor inhibition in lung cancer: the evolving role of individualized therapy. Cancer Metastasis Rev. 2010;29:37–48. doi: 10.1007/s10555-010-9201-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.