

Abstract
Adaptive forms of synaptic plasticity that reduce excitatory synaptic transmission in response to prolonged increases in neuronal activity may prevent runaway positive feedback in neuronal circuits. In hippocampal neurons, for example, glutamatergic presynaptic terminals are selectively silenced, creating “mute” synapses, after periods of increased neuronal activity or sustained depolarization. Previous work suggests that cAMP-dependent and proteasome-dependent mechanisms participate in silencing induction by depolarization, but upstream activators are unknown. We, therefore, tested the role of calcium and G-protein signaling in silencing induction in cultured hippocampal neurons. We found that silencing induction by depolarization was not dependent on rises in intracellular calcium, from either extracellular or intracellular sources. Silencing was, however, pertussis toxin sensitive, which suggests that inhibitory G-proteins are recruited. Surprisingly, blocking four common inhibitory G-protein-coupled receptors (GPCRs) (adenosine A1 receptors, GABAB receptors, metabotropic glutamate receptors, and CB1 cannabinoid receptors) and one ionotropic receptor with metabotropic properties (kainate receptors) failed to prevent depolarization-induced silencing. Activating a subset of these GPCRs (A1 and GABAB) with agonist application induced silencing, however, which supports the hypothesis that G-protein activation is a critical step in silencing. Overall, our results suggest that depolarization activates silencing through an atypical GPCR or through receptor-independent G-protein activation. GPCR agonist-induced silencing exhibited dependence on the ubiquitin-proteasome system, as was shown previously for depolarization-induced silencing, implicating the degradation of vital synaptic proteins in silencing by GPCR activation. These data suggest that presynaptic muting in hippocampal neurons uses a G-protein-dependent but calcium-independent mechanism to depress presynaptic vesicle release.
Introduction
Adaptive forms of plasticity, including homeostatic synaptic plasticity, help maintain neuronal firing rates and prevent overexcitation, excitotoxicity, and information degradation caused by positive feedback inherent in glutamate signaling (Turrigiano, 1999; Turrigiano and Nelson, 2004; Maffei and Fontanini, 2009; Pozo and Goda, 2010). We have described adaptive presynaptic silencing after increased neuronal activity (Moulder et al., 2004, 2006). Indeed, presynaptically silent (mute) synapses have been observed in several preparations, but induction and expression mechanisms remain unclear (Malenka and Nicoll, 1997; Voronin and Cherubini, 2004; Atasoy and Kavalali, 2006).
In hippocampal neurons, depolarization-induced presynaptic silencing is induced rapidly by strong depolarization akin to that generated by stroke, seizure, and spreading depression (Gido et al., 1997; Walz, 2000; Somjen, 2001; Moulder et al., 2004). Physiological action potential firing induces silencing over more protracted periods (Moulder et al., 2006), so presynaptic muting operates over a range of physiological, and perhaps pathophysiological, conditions. Adaptive presynaptic silencing is selective for glutamatergic terminals and is slowly reversible (Moulder et al., 2004). Expression involves altered vesicle priming and requires the ubiquitin proteasome system (UPS) (Moulder et al., 2006; Jiang et al., 2010), but upstream induction mechanisms remain uncertain.
Circumstantial evidence implicates calcium and cAMP alterations in silencing induction. Calcium is involved in many forms of synaptic plasticity (Malenka, 1994; Fisher et al., 1997; Turrigiano, 2008). Prolonged decreases in cAMP signaling produce presynaptic silencing, and cAMP increases occlude depolarization-induced silencing (Moulder et al., 2008). Furthermore, calcium-sensitive adenylyl cyclase activity, which increases cAMP levels, is important for normal recovery from silencing (Moulder et al., 2008). If decreased cAMP signaling is needed for depolarization-induced presynaptic silencing, we might expect involvement of inhibitory G-proteins in its induction. Despite this evidence, double knock-out mice of the two major calcium-sensitive isoforms of adenylyl cyclase exhibit intact depolarization-induced silencing (Moulder et al., 2008), thereby questioning a direct role for either calcium or G-protein signaling. Clarification of how adaptive presynaptic silencing is induced will be crucial in identifying potential targets for therapies aimed at excitotoxicity-related dysfunction.
Here we explored whether calcium and inhibitory G-protein signaling are necessary for hippocampal adaptive presynaptic silencing. Surprisingly, intracellular calcium rises were unnecessary. However, we found that pertussis toxin, which inhibits Gi/o signaling, reduced depolarization-induced silencing. Direct agonist activation of two classes of G-protein-coupled receptors (GPCRs) induced silencing, supporting the hypothesis that silencing is G-protein dependent. In contrast, pharmacological blockade of five candidate receptors, including the two that induced silencing, failed to block depolarization-induced silencing. This suggests that depolarization activates an uncommon GPCR or a pertussis toxin-sensitive G-protein through a receptor-independent mechanism. Nevertheless, depolarization- and GPCR-induced muting share downstream mechanisms because both are sensitive to proteasome inhibition. Thus, adaptive presynaptic silencing is a unique form of calcium-independent, G-protein-dependent synaptic plasticity. Furthermore, we show that prolonged G-protein activation leads to persistent presynaptic silencing, an effect distinct from classical acute presynaptic GPCR effects on vesicle release probability.
Materials and Methods
Cell culture.
Cultures of primary hippocampal neurons were prepared as described previously (Mennerick et al., 1995). Briefly, hippocampi were removed from 0–3 postnatal d male and female Sprague Dawley rat pups and enzymatically treated with papain (1 mg/ml) before mechanical dissociation. Cells were then plated in 35 mm culture dishes precoated with 0.15% agarose as either mass cultures, seeded at ∼650 cells/mm2 on a confluent layer of type I collagen (0.5 mg/ml), or as microisland cultures, seeded at ∼100 cells/mm2 on microdots of collagen. Plating medium contained Eagle's medium (Invitrogen) supplemented with 5% heat-inactivated horse serum, 5% fetal bovine serum, 17 mm d-glucose, 400 μm glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin. Cultures were maintained at 37°C in a humidified incubator under controlled atmospheric conditions (5% CO2/95% air). Cytosine arabinoside (6.7 μm) was added 3–4 d after plating to inhibit cell division. One-half of the culture media was exchanged with Neurobasal medium (Invitrogen) plus B27 supplement 4–5 d after plating. All experiments were performed 10–14 d in vitro, and controls consisted of sibling cultures from the same litter that were treated and/or recorded on the same day to control for synapse development and culture conditions that vary by age and plating conditions. All 4 h treatments were performed in the presence of ionotropic glutamate receptor blockers d-(−)-2-amino-5-phosphonopentanoic acid (d-APV) (50 μm; Tocris Bioscience) and 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX) (1 μm; Tocris Bioscience), and nondepolarized controls always consisted of equimolar NaCl addition to the media to control for osmotic changes. Controls for calcium-free Neurobasal medium (Washington University Medical School Tissue Culture Support Center) supplemented with B27 and calcium chelators were given the identical medium except with 2 mm calcium and without chelators.
Electrophysiology.
All whole-cell voltage-clamp recordings were performed on autaptic neurons in microisland culture. To control for variability in maturity and development, all experiments used similar numbers of neurons from sibling cultures on any given day. Data were collected with an Axopatch 200B or Multiclamp 700B amplifier interfaced to a Digidata 1322A or 1440A data acquisition board (Molecular Devices).
Before recording, the culture medium was exchanged for saline solution containing 138 mm NaCl, 4 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm glucose, 10 mm HEPES, and 25 μm d-APV with a pH of 7.25. For most experiments measuring EPSCs, internal pipette solution consisted of 140 mm K-gluconate, 4 mm NaCl, 0.5 mm CaCl2, 5 mm EGTA, and 10 mm HEPES at a pH of 7.25. For experiments measuring both EPSCs and IPSCs, 140 mm KCl was substituted for K-gluconate to provide a similar PSC driving force at the holding potential of −70 mV.
Electrode pipettes pulled from borosilicate glass (World Precision Instruments) had resistances of 2.5–6 MΩ, and access resistance was compensated 85–100%. Signals were sampled at 5–10 kHz and low-pass filtered at 2–5 kHz. Recordings were performed at room temperature, and membrane potential was typically clamped at −70 mV. Neurons were recorded within 1 h of exchanging the culture medium with the saline solution.
PSCs were evoked by a brief (1.5 ms) depolarization to 0 mV. Paired-pulse responses were evoked by two such depolarizations with an interstimulus interval of 50 ms. Sucrose-evoked EPSCs were elicited by 3 s local application of 0.5 m sucrose. All local solution applications used a multibarrel perfusion system with the common perfusion port placed within 0.5 mm of the neuron under study and yielding solution exchange times of ∼100 ms.
Calcium imaging.
All calcium imaging experiments used mass cultures. For fura-2 experiments, neurons were plated on glass-bottom dishes (MatTek Corporation). Cells were loaded with fura-2 by 60 min incubation with 5 μm fura-2 AM (Invitrogen) and 0.1% Pluronic F-127 (Invitrogen) in Neurobasal medium, pH 7.2, at room temperature, washed with Neurobasal medium, and incubated for another 60 min to allow for ester hydrolysis. After loading, the cells were imaged on an Eclipse TE300 inverted microscope using a 40× (1.3 numerical aperture) oil-immersion objective (Nikon). The microscope was equipped with a 75 W xenon arc lamp and a cooled CCD camera (Cooke Corp.). The fluorescence excitation was provided by a band-specific filter (340 and 380 nm; Semrock) in combination with DM400 dichroic beam splitter (Nikon). Pairs of images were collected at alternate excitation wavelengths. The images were divided by one another to yield ratio values for individual cell bodies after subtracting the matching background. Imaging was performed at room temperature, but cultures were returned to 37°C for the period between images early in the treatment and images at the end of the 4 h treatment. For this reason, the same neurons were represented at baseline and 5 min time points, but separate fields of neurons were used for 4 h time points in Figure 1. MetaFluor software (Molecular Devices) was used for image acquisition and analysis.
Figure 1.

Depolarization-induced presynaptic silencing is not dependent on calcium influx. A, Pseudocolored images of the fura-2 ratio (indicating calcium concentration) in neuronal cell bodies before (baseline) and <1 min after (depolarized) application of 30 mm KCl. Neurons were switched from calcium-containing media to either calcium-containing (control) media or to calcium-free media supplemented with 500 μm EGTA when KCl depolarization began. Scale bar, 25 μm. B, Summary of fura-2 ratio normalized to baseline values in neurons similar to A at two time points after addition of either 30 mm NaCl (control) or 30 mm KCl (depolarized) to calcium-containing media or calcium-free media supplemented with 500 μm EGTA. All comparisons within a single time point were significantly different (p < 0.05 with Bonferroni's correction for multiple comparisons) except those indicated with “N.S.”, meaning not significant (n = 17–62 neurons). Thus, the calcium-free media with EGTA effectively prevented cytosolic free calcium level rises in both control and depolarized neurons. C, vGluT-1 immunoreactivity to identify glutamatergic presynaptic terminals (red) with superimposed FM1-43FX labeling of active presynaptic terminals (green). Neurons were treated 4 h with either 30 mm NaCl (baseline) or 30 mm KCl (depolarized) in either calcium-containing media (control) or calcium-free media supplemented with 500 μm EGTA (calcium-free). Scale bar, 5 μm. D, Summary of the percentage of active glutamatergic presynaptic terminals based on vGluT-1/FM1-43FX correspondence as shown in C. All comparisons were significantly different (p < 0.05 with Bonferroni's correction for multiple comparisons) except those indicated with N.S. (n = 20 fields of 10 vGluT-1 puncta each from 4 independent experiments).
Fluo-4 was loaded into cells by addition of Fluo-4 AM (2 μm; Invitrogen) to the culture media 30 min before the end of the 4 h KCl treatment. Medium was then exchanged for extracellular recording saline containing elevated KCl (34 mm total) and 0 mm CaCl2. Cells were imaged at room temperature using an Eclipse TE2000-S inverted microscope with 40× (0.6 numerical aperture) objective (Nikon) and a cooled 12-bit CCD camera (Photometrics). Epifluorescence was provided by a metal halide lamp, and images were acquired at 1 Hz using MetaMorph software (Molecular Devices). Calcium was briefly elevated to 0.5 mm for 5–10 s after 5 s of baseline in 0 mm calcium.
FM1-43 loading and immunocytochemistry.
All FM1-43 [N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide] imaging experiments used mass cultures plated on glass coverslips. FM1-43 loading and vesicular glutamate transporter 1 (vGluT-1) immunolabeling experiments were performed as described previously (Moulder et al., 2006, 2010). Briefly, within 60 s of replacing media with fresh saline, active presynaptic terminals were labeled for 2 min with 10 μm fixable FM1-43 (FM1-43FX) (Invitrogen) in recording saline supplemented with 45 mm KCl and 1 μm NBQX. Cultures were immediately washed for 5 s with extracellular recording saline containing 500 μm Advasep-7 (CyDex) and 1 μm NBQX, followed by saline plus 1 μm NBQX for 10 min. Afterward, cells were fixed for 10 min in 4% paraformaldehyde/0.2% glutaraldehyde in PBS, washed with PBS, and incubated in a blocking solution (2% normal goat serum/0.08% Triton X-100) for 15 min before 3 h incubation in guinea pig anti-vGluT-1 antibody (Millipore Corporation) diluted 1:2000 in blocking solution. After washing with PBS, cells were incubated for 40 min in Alexa Fluor 647-conjugated anti-guinea pig antibody (Invitrogen) diluted 1:500 in PBS. After washing again with PBS, coverslips were removed from culture dishes and mounted onto glass microscope slides with Fluoromount-G (Southern Biotechnology Associates).
Confocal images were acquired at random locations throughout the dish by an observer naive to the experimental conditions using a C1 scanning confocal laser attached to an inverted Eclipse TE300 microscope with a 60× objective (1.4 numerical aperture) and EZ-C1 software (Nikon). Alternating 543 and 633 nm laser lines were used to obtain z-stack images while pixel dwell time, image resolution, gain settings, field of view size, and z-stack parameters were held constant throughout all conditions from a single experiment. Monochrome images were converted into two-dimensional projected images and analyzed using MetaMorph software.
Trypan blue staining.
For experiments assessing toxicity via trypan blue staining, mass cultures were treated for 5 min with 0.4% trypan blue at 37°C in a humidified incubator under controlled atmospheric conditions (5% CO2/95% air). After one PBS wash at room temperature, neurons were fixed for 10 min in 4% paraformaldehyde/0.2% glutaraldehyde in PBS. After three additional PBS washes, 10 random fields of cells were counted in each dish using a 20× objective. Trypan-positive nuclei were counted under bright field and identified as unhealthy neurons, and phase-bright cell bodies devoid of trypan staining were counted under phase contrast and identified as healthy neurons.
Data analysis.
Electrophysiology data were collected and analyzed with pClamp 9 software (Molecular Devices). For electrically evoked PSCs, leak was subtracted offline before measuring peak amplitude, and the amplitudes of at least three responses were averaged for each cell. For sucrose-evoked EPSCs, currents were integrated over the rise and decay of the response to 10% of the steady-state current. This provided an estimate of the postsynaptic charge transfer corresponding to the readily releasable vesicle pool.
In calcium-imaging experiments using fura-2, the ratio values were normalized to the baseline values (in which no treatment had been applied) to control for differences in baseline calcium concentration. Normalization was achieved by dividing by the first value for each cell under baseline conditions, and 4 h time points were normalized to the average of the ratios at baseline. Values from at least five images were averaged for each neuron at each time point. To calculate the change in fluorescence in fluo-4 experiments, somatic fluorescence intensities from three images were averaged at baseline and subtracted from the average intensity of three images near the peak of fluorescence. The change in fluorescence was normalized to the average baseline fluorescence value to account for variability in fluo-4 loading.
To determine the percentage of active glutamatergic synapses, regions were manually drawn around 10 vGluT-1 puncta each from five images (fields) for each coverslip in a single experiment by an observer naive to experimental conditions. The strategy of using five fields per condition equalized the contribution of a single condition to the final results. Regions were drawn to include the pixels above threshold in the vGluT-1-positive punctum but not larger than the punctum to minimize the contribution of background pixels and/or neighboring puncta to the analysis. Thresholds were applied to all FM1-43FX images before vGluT-1 regions were loaded into the FM1-43FX image. “Active” synapses were defined as those regions that reached at least 10 pixels above threshold in the FM1-43FX image (Moulder et al., 2006). Cluster correlation analyses (Galbraith et al., 2010) and ANOVA statistics applied to the first FM1-43FX/vGluT-1 correspondence dataset (Fig. 1D) found no significant correlations between fields within an experiment (data not shown), so we treated the percentage of active synapses from each field as a statistically independent sample throughout the manuscript.
For trypan blue toxicity experiments, the total numbers of healthy and unhealthy neurons were added together from all 10 fields in each dish. The percentage of healthy neurons was then calculated from the number of healthy neurons relative to the total neurons counted (healthy and unhealthy) in each dish.
Graphs were created using SigmaPlot software (Systat Software). All data were presented as mean ± SEM unless otherwise indicated. To reduce type 1 error that may result from nonsignificant cluster correlations (Galbraith et al., 2010), all experiments used yoked controls (e.g., sibling cultures; see methods above), and we analyzed data conservatively using unpaired statistics with additional corrections (i.e., for multiple comparisons) as appropriate. Unpaired Student's t test was used to determine significance for comparisons of two groups, and Bonferroni's correction for multiple comparisons was applied to analyses of experiments involving more than two conditions. p < 0.05 was required for significance.
Materials.
SCH50911 [(2S)-(+)-5,5-dimethyl-2-morpholineacetic acid], bicuculline, LY341495 [(2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid], (1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid (ACPD), and 2-chloro-N6-cyclopentyladenosine (CCPA) were purchased from Tocris Bioscience. BAPTA-AM [AM derivative of bis(2-aminophenoxy)ethane tetraacetic acid] was purchased from Invitrogen, and 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) was purchased from Research Biochemicals. MG-132 (carbobenzoxy-l-leucyl-l-leucyl-l-leucinal) was purchased from Enzo Life Sciences. All other chemicals and reagents were purchased from Sigma unless otherwise stated.
Results
Depolarization silences presynaptic terminals in the absence of extracellular calcium
Calcium is necessary for multiple forms of hippocampal synaptic plasticity, so we determined the role of calcium influx in adaptive presynaptic silencing by removing calcium from the extracellular media during a depolarizing induction challenge known to produce strong presynaptic muting. We used an induction paradigm of 30 mm KCl for 4 h in the presence of ionotropic glutamate receptor blockers d-APV (50 μm) and NBQX (1 μm). The short, strong induction circumvents inevitable developmental confounds of longer interventions with calcium and G-protein signaling and mimics pathophysiological extracellular potassium concentrations during stroke or seizure (Gido et al., 1997; Walz, 2000). KCl at 30 mm clamps the membrane potential at −20 to −30 mV (Moulder et al., 2003; Crawford et al., 2009) and induces adaptive presynaptic silencing without detectable changes in synaptic structure, vesicle release probability (pr), or postsynaptic function (Moulder et al., 2004, 2006). We incubated mass cultures in either calcium-rich culture media or calcium-free culture media supplemented with 500 μm EGTA to buffer residual free calcium. Using the ratiometric calcium indicator fura-2, we verified that calcium-free media prevented rises in cytosolic free calcium concentration normally produced by KCl depolarization (Fig. 1A,B). Control neurons also exhibited a late rise in cytosolic free calcium concentration (Fig. 1B). We speculate that temperature and pH variations between imaging sessions (at room temperature) and intervening periods (at 37°C) may have contributed to cellular stress and calcium influx in nondepolarized neurons. Regardless, calcium-free media prevented the rise in cytosolic free calcium concentration in both control and depolarized neurons over 4 h (Fig. 1B). We conclude, therefore, that our calcium-free media supplemented with EGTA effectively prevented calcium influx.
To assess presynaptic silencing after depolarization in the absence of calcium influx, we performed FM1-43FX labeling in the presence of calcium followed by retrospective immunostaining for vesicular glutamate transporter 1 (vGluT-1) to identify active glutamatergic terminals. In agreement with previous experiments under normal calcium conditions (Moulder et al., 2004), neurons depolarized in calcium-free media plus EGTA exhibited a decrease in the percentage of vGluT-1 terminals that colocalize with FM1-43FX (Fig. 1C,D). This suggests that calcium influx is not necessary to induce adaptive presynaptic silencing.
Depolarization silences presynaptic terminals in the absence of intracellular free calcium
Although influx of extracellular calcium is not necessary for induction of adaptive presynaptic silencing, calcium rises from intracellular sources may participate. To prevent intracellular calcium rises, we loaded cells with the fast intracellular calcium chelator BAPTA using the cell-permeant precursor BAPTA-AM. Because normal action-potential-driven neurotransmitter release is dependent on fast rises in local presynaptic calcium concentration and would be depressed by BAPTA-AM incubation, we used calcium-independent hypertonic sucrose-evoked EPSCs to determine the size of the readily releasable pool of vesicles (Rosenmund and Stevens, 1996), which is proportional to the total number of active synapses (Moulder et al., 2004). For these experiments, we used autaptic neurons in microisland cultures and applied hypertonic sucrose to evoke EPSCs during whole-cell recordings. Autaptic neurons were used because their isolation eliminates confounding polysynaptic effects during electrophysiological measurements. Similar to previous results (Rosenmund and Stevens, 1996), sucrose-evoked EPSCs were unaffected by 4 h incubation with 20 μm BAPTA-AM in calcium-free media (Fig. 2A,B). In contrast electrically evoked EPSCs, which are dependent on presynaptic calcium influx, were strongly depressed after 4 h BAPTA chelation (Fig. 2A,B). These results suggest that 4 h BAPTA incubation strongly chelates intracellular free calcium.
Figure 2.

Depolarization-induced presynaptic silencing is not dependent on increases in intracellular free calcium from any source. A, Representative examples of sucrose-evoked EPSCs and action potential-evoked EPSCs elicited in autaptic neurons after 4 h treatment in normal calcium-containing media (control) or calcium-free media supplemented with 20 μm of the cell-permeant fast calcium chelator BAPTA-AM. B, Summary of results for sucrose-evoked EPSC charge and action-potential-evoked EPSC amplitude after treatment with control or with calcium-free, BAPTA-AM-supplemented extracellular media in sister cultures (n = 9–11 neurons). *p < 0.05. C, Example fields of neurons filled with the calcium indicator fluo-4 before (0 mm) and after (0.5 mm) perfusion of CaCl2. Neurons were pretreated for 4 h with 30 mm KCl in either calcium-containing media (depolarized) or calcium-free media supplemented with 20 μm BAPTA-AM (depolarized BAPTA-AM). Scale bar, 50 μm. D, Representative examples of sucrose-evoked EPSCs in autaptic neurons after 4 h treatment in calcium-free media supplemented with 20 μm BAPTA-AM and either 30 mm NaCl (baseline) or 30 mm KCl (depolarized).
To test whether intracellular rises in calcium concentration are necessary for presynaptic silencing, we depolarized neurons for 4 h in 30 mm KCl in the presence of 2 mm calcium or in calcium-free media containing 20 μm BAPTA-AM. To ensure that intracellular calcium rises during induction did not saturate the ability of BAPTA to chelate calcium, we performed calcium imaging of treated neurons using the calcium indicator fluo-4. Briefly raising extracellular calcium from 0 to 0.5 mm increased cytosolic free calcium concentration in control neurons after 4 h depolarization in calcium-rich media, as assessed by changes in somatic fluo-4 fluorescence (45.5 ± 4.5% increase; n = 40) (Fig. 2C). This rise in cytosolic free calcium was not observed in neurons after incubation in calcium-free media supplemented with BAPTA-AM (1.0 ± 1.2% increase; n = 40; p = 1.0 × 10−15) (Fig. 2C). BAPTA, therefore, effectively chelates intracellular free calcium, even after a sustained depolarization.
To test whether BAPTA-AM incubation blocks presynaptic silencing, we incubated in BAPTA-AM and compared neurons after the KCl challenge to neurons challenged with an equimolar NaCl control. As shown previously, the 4 h depolarizing challenge in calcium-rich media depressed sucrose-evoked EPSCs by ∼45% (control, 1224.9 ± 147.7 pC; depolarized, 670.1 ± 135.7 pC; n = 29–30 neurons; p < 0.01) (Moulder et al., 2004). Despite BAPTA-AM incubation in calcium-free media, the charge of sucrose-evoked EPSCs was still significantly depressed in KCl-challenged neurons compared with sibling controls (control, 581 ± 100 pC; depolarized, 168 ± 27 pC; n = 26–29; p < 0.0001) (Fig. 2D). These results argue strongly that adaptive presynaptic silencing is calcium independent.
Activation of inhibitory G-proteins is necessary for depolarization-induced presynaptic silencing
Indirect evidence has previously implicated decreased cAMP levels in depolarization-induced presynaptic silencing (Moulder et al., 2008). Thus, prolonged activation of inhibitory G-proteins, which inhibit adenylyl cyclase activity and cAMP production, could participate in adaptive muting. We, therefore, tested the hypothesis that inhibitory G-proteins are necessary for depolarization-induced silencing.
We preincubated neurons for 24 h in 500 ng/ml pertussis toxin, which, by ADP ribosylation of the αi/o subunit, prevents inhibitory G-protein activation. This interferes with α subunit signaling and blocks the association of Gαi/o subunits with their upstream GPCRs (Brown and Sihra, 2008). To confirm that pertussis toxin treatment itself did not induce toxicity, we used trypan blue staining to identify unhealthy neurons. The percentage of healthy neurons did not differ between control neurons and those treated 24 h with 500 ng/ml pertussis toxin (control, 92.2 ± 1.6%; pertussis toxin, 93.5 ± 1.0%; n = 4 dishes; p = 0.54). After 24 h in pertussis toxin, we coincubated with 30 mm KCl or an equimolar control during an additional 4 h in pertussis toxin. After treatment, we assessed synaptic function by measuring EPSC amplitudes in autaptic neurons and vGluT-1/FM1-43FX correspondence in mass cultures. Pertussis toxin had no effect on EPSC amplitude in nondepolarized neurons, confirming that the toxin did not compromise cell health, but it prevented depolarization-induced depression of EPSC amplitude (Fig. 3A,B). Pertussis toxin did not, however, alter paired-pulse depression in any of the experimental groups (control, 29.9 ± 5.6%; pertussis toxin, 18.0 ± 4.3%; depolarized, 8.3 ± 6.2%; depolarized + pertussis toxin, 20.7 ± 5.9%; n = 13–14 neurons per condition; p > 0.05) (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material), as is expected from acute effects of GPCR activation on vesicle release probability (Mennerick and Zorumski, 1995; Dobrunz and Stevens, 1997; Zucker and Regehr, 2002; Brown and Sihra, 2008). Similarly, pertussis toxin prevented the depolarization-induced decrease in active synapses measured via FM1-43FX/vGluT-1 correspondence (Fig. 3C,D). Together, these results suggest that activation of inhibitory G-proteins is necessary for depolarization-induced presynaptic silencing.
Figure 3.
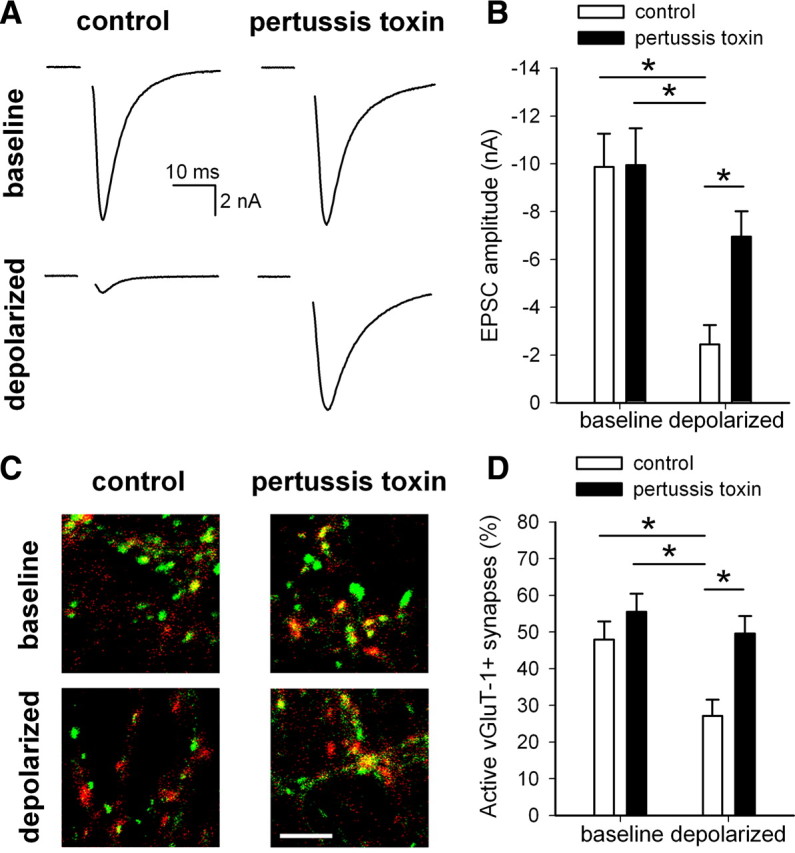
Activation of inhibitory G-proteins is necessary for depolarization-induced presynaptic silencing. A, Example action-potential-evoked EPSCs from autaptic neurons treated 24 h with or without 500 ng/ml pertussis toxin before 4 h coapplication with 30 mm NaCl (baseline) or 30 mm KCl (depolarized). B, Summary of EPSC amplitudes from autaptic neurons treated as in A (n = 13–14 neurons). *p < 0.05 with Bonferroni's correction for multiple comparisons. C, Example images of vGluT-1 (red) and FM1-43FX (green) correspondence in neurons treated 24 h with or without 500 ng/ml pertussis toxin before 4 h NaCl (baseline) or KCI (depolarized). Scale bar, 5 μm. D, Summary of conditions represented in C (n = 25 fields from 5 independent experiments). *p < 0.05 with Bonferroni's correction for multiple comparisons.
A1 receptor activation is not required for depolarization-induced presynaptic silencing
To explore the role of GPCRs upstream of inhibitory G-proteins in presynaptic silencing, we examined whether the G-protein-coupled A1 adenosine receptor is involved. The A1 receptor is an excellent candidate because increased adenosine levels in the extracellular space occur via calcium-independent mechanisms: conversion from ATP (Latini and Pedata, 2001), which is released from astrocytes (Wang et al., 2000), and release by transporters (Dunwiddie and Masino, 2001). Adenosine release has also been implicated in neuroprotection, in part through acute A1 receptor-mediated inhibition of presynaptic glutamate release (Stone et al., 2009). Additionally, A1 receptors selectively inhibit synaptic transmission at excitatory, not inhibitory, terminals in the hippocampus (Yoon and Rothman, 1991), possibly explaining the selectivity of adaptive presynaptic silencing for excitatory over inhibitory terminals (Moulder et al., 2004).
To test whether A1 receptor activation is necessary for depolarization-induced presynaptic silencing, we incubated neurons with depolarizing KCl or equimolar NaCl control in the presence of the selective A1 antagonist DPCPX (200 nm). The decrease in EPSC amplitude in depolarized neurons treated with DPCPX was comparable with ESPC depression from neurons incubated in the absence of DPCPX (Fig. 4A,B). DPCPX incubation also did not alter paired-pulse depression (control, 11.8 ± 4.8%; DPCPX, 16.4 ± 5.1%; depolarized, 11.0 ± 8.7%; depolarized + DPCPX, 7.3 ± 11.7%; n = 9 neurons per condition; p > 0.05) (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Additionally, we assessed presynaptic silencing by measuring the percentage of active glutamatergic presynaptic terminals via FM1-43FX/vGluT-1 correspondence. In neurons treated 4 h with 30 mm KCl, the percentage of vGluT-1-positive presynaptic terminals that colocalize with FM1-43FX fluorescence was decreased from control, regardless of whether the neurons had been coincubated with DPCPX (Fig. 4C,D). We noticed a trend-level difference in the percentage of active synapses in depolarized neurons treated with DPCPX compared with depolarized neurons without DPCPX (depolarized, 10.6 ± 1.5%; depolarized + DPCPX, 17.4 ± 2.1%; p = 0.052 after correction for multiple comparisons), potentially suggesting a minor role of adenosine A1 receptors in depolarization-induced silencing. Despite this trend, however, depolarization still decreased the percentage of active synapses in the presence of DPCPX (DPCPX, 42.6 ± 3.4%; depolarized + DPCPX, 17.4 ± 2.1%; p = 1.7 × 10−8 after correction for multiple comparisons). Together, these data suggest that blocking A1 receptor activation during depolarization does not prevent the induction of adaptive presynaptic silencing.
Figure 4.
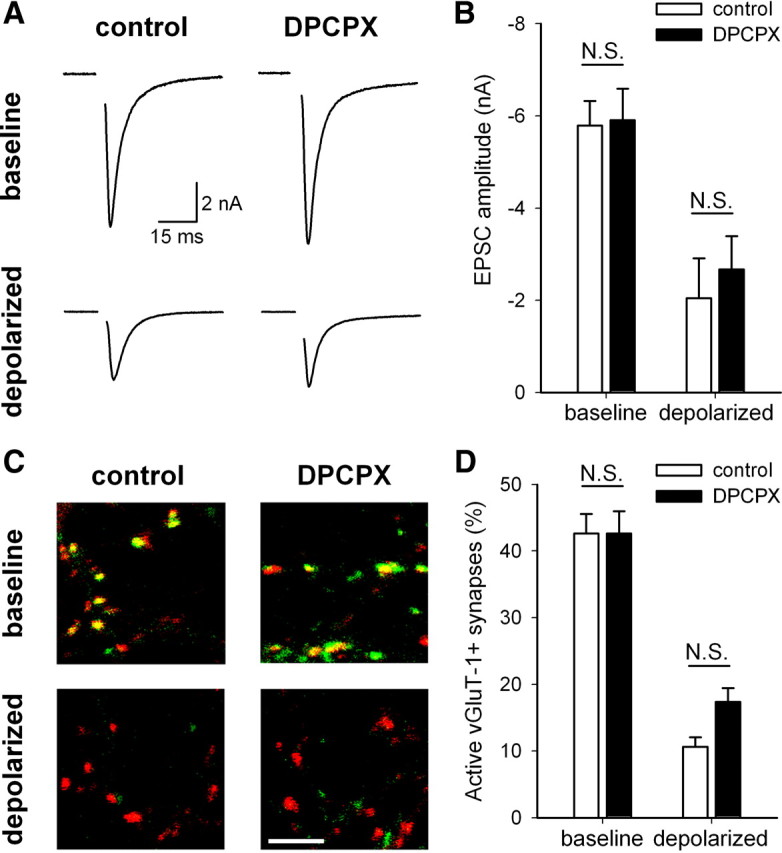
A1 adenosine receptor activation is not necessary for depolarization-induced presynaptic silencing. A, Example action-potential-evoked EPSCs from autaptic neurons treated 4 h with 30 mm NaCl (baseline) or 30 mm KCl (depolarized) in the presence or absence of 200 nm DPCPX, an A1 receptor antagonist. B, Summary of EPSC amplitudes from autaptic neurons treated as in A (n = 9 neurons). All comparisons were significantly different (p < 0.05 with Bonferroni's correction for multiple comparisons) except those indicated (N.S.). C, Example images of vGluT-1 (red) and FM1-43FX (green) correspondence in neurons treated 4 h under baseline or depolarized conditions in the presence or absence of 200 nm DPCPX. Scale bar, 5 μm. D, Summary of conditions represented in C (n = 65 fields from 13 independent experiments). All comparisons were significantly different (p < 0.05 with Bonferroni's correction for multiple comparisons) except those indicated (N.S.).
GABAB, metabotropic glutamate, CB1, and kainate receptors are not required for depolarization-induced presynaptic silencing
Although A1 adenosine receptor activation is not necessary to induce adaptive presynaptic silencing (Fig. 4), activation of inhibitory G-proteins is critical (Fig. 3). Multiple GPCRs linked to inhibitory G-proteins are expressed in rodent hippocampus. Our culture preparation, however, does not contain modulatory projection neurons from outside the hippocampus, so the list of GPCRs with ligands available in vitro is restricted. The calcium independence of induction also excludes transmitters/modulators released by classical calcium-dependent exocytosis. Nevertheless, reverse transport of GABA (Schwartz, 1982; Gaspary et al., 1998), reverse transport of glutamate (Nicholls and Attwell, 1990; Szatkowski et al., 1990), and nonclassical cannabinoid signaling (Di Marzo and Deutsch, 1998; Di Marzo et al., 1998; Wilson and Nicoll, 2001) could elevate ambient modulator levels and induce silencing in response to depolarization.
We first tested whether the G-protein-coupled GABAB receptor is responsible for inducing adaptive presynaptic silencing. To prevent GABAB receptor activation, we incubated neurons in the presence of the GABAB receptor antagonist SCH50911 (50 μm). To test effectiveness of SCH50911 in our system, we coapplied SCH50911 acutely to our autaptic neurons with the GABAB receptor agonist baclofen (10 μm). Baclofen depressed evoked EPSCs and IPSCs by 65.4 ± 8.1%, and addition of SCH50911 recovered the PSCs to 96.0 ± 4.8% of their original amplitude (n = 10 neurons) (Fig. 5A). Incubation of mass cultures in SCH50911 for 4 h significantly increased the percentage of active glutamatergic presynaptic terminals from baseline (Fig. 5B), suggesting a small endogenous GABAB receptor tone in this dataset. Despite this effect on baseline silent synapses, SCH50911 did not prevent the decrease in the percentage of active synapses induced by depolarization (Fig. 5B). These results demonstrate that, despite effectiveness of SCH50911, GABAB receptor activation is not involved in depolarization-induced adaptive presynaptic silencing. Similar results were obtained with the GABAA receptor antagonists bicuculline and picrotoxin (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), confirming that GABA signaling is not involved in depolarization-induced presynaptic silencing.
Figure 5.
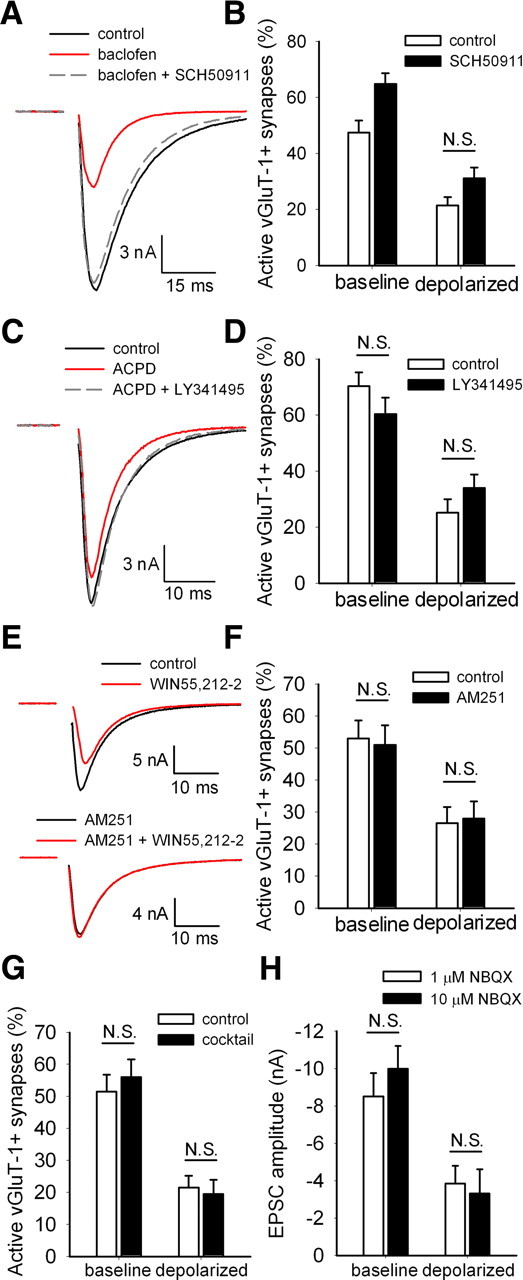
None of likely candidate GPCRs is necessary for depolarization-induced silencing. A, Example action-potential-evoked IPSCs from a single autaptic neuron after acutely applied extracellular saline (control), 10 μm baclofen (a GABAB receptor agonist), or 10 μm baclofen plus 50 μm SCH50911 (a GABAB receptor antagonist). B, Summary of experiments measuring vGluT-1/FM1-43FX correspondence in neurons treated 4 h with 30 mm NaCl (baseline) or 30 mm KCl (depolarized) in the presence or absence of 50 μm SCH50911 (n = 35 fields from 7 independent experiments). All comparisons were significantly different (p < 0.05 with Bonferroni's correction for multiple comparisons) except the one indicated (N.S.). C, Example action-potential-evoked EPSCs from a single autaptic neuron after acutely applied control, 50 μm ACPD (an mGluR agonist), or 50 μm ACPD plus 25 μm LY341495 (a mGluR antagonist). D, Summary of experiments measuring vGluT-1/FM1-43FX correspondence in neurons treated for 4 h under baseline or depolarized conditions in the presence or absence of 25 μm LY341495 (n = 25 fields from 5 independent experiments). All comparisons were significantly different (p < 0.05 with Bonferroni's correction for multiple comparisons) except those indicated (N.S.). E, Example action potential-evoked EPSCs from two different autaptic neurons: one after acutely applied control or 200 nm WIN55,212–2 (a CB1 agonist) and the other after 1–2 h pretreatment with 1 μm AM251 (a CB1 antagonist) and continued AM251 application or AM251 plus WIN55,212-2. The AM251 pretreatment with between-cell comparison was used to mitigate against the very slow reversibility of WIN55,212-2. F, Summary of experiments measuring vGluT-1/FM1-43FX correspondence in neurons treated for 4 h under baseline or depolarized conditions in the presence or absence of 1 μm AM251 (n = 20 fields from 4 independent experiments). All comparisons were significantly different (p < 0.05 with Bonferroni's correction for multiple comparisons) except those indicated (N.S.). G, Summary of experiments measuring vGluT-1/FM1-43FX correspondence in neurons treated for 4 h under baseline or depolarized conditions in the presence or absence of 200 nm DPCPX, 50 μm SCH50911, 25 μm LY341495, and 1 μm AM251 (cocktail; n = 20 fields from 4 independent experiments). All comparisons were significantly different (p < 0.05 with Bonferroni's correction for multiple comparisons) except those indicated (N.S.). H, Summary of experiments measuring EPSC amplitude in neurons treated 4 h under baseline or depolarized conditions in the presence of either 1 μm NBQX or 10 μm NBQX to ensure kainate receptor block (n = 14–16 neurons). Neurons treated for 4 h with 1 μm NBQX were treated acutely (∼1 min) with 10 μm NBQX before recording as a control for any residual acute NBQX effects. All comparisons were significantly different (p < 0.05 with Bonferroni's correction for multiple comparisons) except those indicated (N.S.).
Other plausible candidate GPCRs for the induction of adaptive presynaptic silencing include Gi/o-coupled metabotropic glutamate receptors (mGluRs). LY341495, a nonselective mGluR antagonist at concentrations above 22 μm (Schoepp et al., 1999), reversed acute EPSC depression by a near-saturating concentration of the mGluR agonist ACPD (33.9 ± 7.2% depression in 50 μm ACPD; 92.2 ± 6.8% recovery in ACPD plus 25 μm LY341495; n = 7–8 neurons) (Fig. 5C). However, LY341495 failed to alter baseline active glutamatergic synapses or presynaptic silencing in response to depolarization (Fig. 5D). These results indicate that antagonism of mGluR activation does not prevent depolarization-induced presynaptic silencing.
To test G-protein-coupled CB1 cannabinoid receptor involvement, we incubated mass cultures in 1 μm AM251 [1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide], a CB1 antagonist, during depolarizing or control challenge. To ensure that AM251 inhibited CB1 activation, we applied the CB1 agonist (R)-(+)-WIN55,212-2 [R-(+)-(2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl]pyrol[1,2,3-de]-1,4- benzoxazin-6-yl)(1-naphthalenyl) methanone monomethanesulfonate] (WIN) (200 nm) acutely to autaptic neurons. WIN acutely depressed PSCs by 34.6 ± 8.6% (Fig. 5E), whereas preincubation in AM251 prevented the WIN-induced depression (101.1 ± 2.5% of baseline; n = 8 neurons per condition) (Fig. 5E). AM251 incubation during the 4 h depolarization challenge, however, altered neither the baseline percentage of active glutamatergic synapses nor depolarization-induced silencing (Fig. 5F). We conclude that CB1 activation does not participate in depolarization-induced presynaptic silencing.
It is plausible that multiple GPCRs are simultaneously activated by strong depolarization. If this occurs, then each receptor might contribute individually to a small, statistically insignificant effect on the percentage of active presynaptic terminals. Simultaneous activation, however, may synergistically upregulate the activity of downstream signaling cascades. To test this hypothesis, we incubated mass cultures for 4 h in a mixture of GPCR antagonists (200 nm DPCPX, 50 μm SCH50911, 25 μm LY341495, and 1 μm AM251) to simultaneously block the activation of A1 adenosine receptors, GABAB receptors, mGluRs, and CB1 receptors. This antagonist mixture did not alter the percentage of active glutamatergic presynaptic terminals at baseline (Fig. 5G), indicating that transmitter tone over 4 h was not sufficient to induce additional muting. Additionally, this mixture of GPCR antagonists did not prevent silencing in response to depolarization (Fig. 5G). Interestingly, we failed to observe the trend-level increase in the percentage of active synapses produced in depolarized neurons exposed to DPCPX (Fig. 4D). This suggests that these four receptor classes, although linked to inhibitory G-proteins, are not responsible for the induction of depolarization-induced presynaptic silencing.
Although 1 μm NBQX was present during all depolarization challenges to prevent ionotropic glutamate receptor activation, kainate receptors may not be blocked at this concentration (Bureau et al., 1999; Crowder et al., 2002). Kainate receptors can induce metabotropic effects dependent on G-proteins in addition to their ionotropic effects (Cunha et al., 1999; Lerma, 2003; Rodríguez-Moreno and Sihra, 2007). To ensure that kainate receptors and their downstream metabotropic signaling cascades are not activated during 4 h depolarization, we coincubated with 10 μm NBQX, which should block both kainate receptors and AMPA receptors (Lauri et al., 2001; Delaney and Jahr, 2002). After a 4 h depolarizing or control challenge, we recorded EPSC amplitudes in autaptic neurons. Control cultures were treated acutely for 1 min with 10 μm NBQX just before recording to account for any residual effects of the high NBQX concentration on AMPA receptors. The higher concentration of NBQX did not prevent depolarization-induced silencing and did not alter the EPSC amplitude compared with control neurons (Fig. 5H). This suggests that metabotropic effects downstream of kainate receptor activation are unimportant for depolarization-induced silencing.
A1 receptor activation and GABAB receptor activation induce presynaptic silencing
It is intriguing that A1 receptor, GABAB receptor, mGluR, CB1, and kainate receptor activation are not necessary for induction of presynaptic silencing despite our evidence that depolarization-induced silencing depends on pertussis toxin-sensitive G-proteins. These results suggest that inhibitory G-proteins are activated during strong depolarization but not by activation of these particular receptors. To further test our hypothesis that a G-protein-dependent mechanism, regardless of the source of activation, controls adaptive presynaptic silencing, we tested whether direct activation of several GPCR classes induces silencing.
We first determined whether increases in exogenous extracellular adenosine concentration induce presynaptic silencing. We incubated cultures in the presence of 100 μm adenosine for 4 h. A high concentration was chosen to counteract any degradation that may occur from the natural activity of extracellular enzymes or uptake through transporters (Dunwiddie and Masino, 2001). We used FM1-43FX loading of presynaptic terminals followed by vGluT-1 immunostaining to determine the percentage of active glutamatergic terminals. Incubation for 4 h with 100 μm adenosine decreased the percentage of active synapses (Fig. 6A,B). To determine whether this effect of adenosine resulted from activation of the A1 receptor, we coincubated with DPCPX (200 nm). DPCPX incubation alone did not alter the percentage of active glutamatergic synapses, but it prevented the adenosine-induced silencing (Fig. 6A,B). This result also serves as an effective positive control for experiments testing DPCPX effects on depolarization-induced muting (Fig. 4). Note that FM1-43FX uptake should be immune to the well-known transient, acute effects of adenosine agonists on pr because agonists and antagonists were washed out of the cultures before the FM1-43FX challenge (Wu and Saggau, 1994; Brown and Sihra, 2008). Also, FM1-43FX loading was conducted during 2 min of a strong depolarization designed to empty the entire recycling pool of vesicles regardless of pr. Thus, these results suggest that extracellular adenosine induces presynaptic silencing through an A1 receptor-dependent mechanism.
Figure 6.

A1 adenosine receptor activation and GABAB receptor activation induce presynaptic silencing. A, Example images of vGluT-1 (red) and FM1-43FX (green) correspondence in neurons treated 4 h with control or 100 μm adenosine in the presence or absence of 200 nm DPCPX. Scale bar, 5 μm. B, Summary of conditions represented in A (n = 40 fields from 8 independent experiments). *p < 0.05 with Bonferroni's correction for multiple comparisons. C, Summary of experiments measuring vGluT-1/FM1-43FX correspondence in neurons treated 4 h with control medium, with 10 nm CCPA, or with 10 nm CCPA coapplied with 200 nm DPCPX (n = 35 fields from 7 independent experiments). *p < 0.05 with Bonferroni's correction for multiple comparisons. D, Example action-potential-evoked EPSCs recorded from autaptic neurons after acute (<1 min; control) or 4 h treatment with 10 nm CCPA. E, Summary of EPSC amplitudes from autaptic neurons treated acutely (<1 min; control) or for 4 h with 50 μm baclofen (n = 9–10 neurons) and FM1-43FX/vGluT-1 correspondence after 4 h control or 50 μm baclofen treatment (n = 30 fields from 6 independent experiments). *p < 0.05.
Results with adenosine do not exclude a contribution from A2 or A3 receptors, although this contribution is presumably minimal because the A1-specific antagonist DPCPX blocked the silencing. To confirm that A1 receptor activation alone is sufficient for presynaptic silencing induction, we incubated mass cultures in the presence of the highly selective A1 receptor agonist CCPA. CCPA (10 nm for 4 h) decreased the percentage of active glutamatergic terminals, and the CCPA effect was blocked by coincubation with 200 nm DPCPX (Fig. 6C). We also confirmed that CCPA persistently depressed presynaptic action-potential-driven vesicle release by measuring autaptic EPSC amplitudes after 4 h of CCPA incubation. Electrophysiological recordings were performed in extracellular recording saline in the absence of CCPA, so acute effects are unlikely. However, to ensure that EPSC depression was not caused by residual acute effects of CCPA, a high-affinity ligand that might have failed to dissociate from A1 receptors during the media exchange (Lohse et al., 1988), we used neurons from sibling cultures acutely exposed to 10 nm CCPA (<1 min) as our control. EPSC amplitudes were significantly decreased in neurons recorded after 4 h treatment with CCPA compared with those treated acutely with CCPA (control, −8.92 ± 1.19 nA; 4 h CCPA, −2.59 ± 1.10 nA; n = 10; p = 0.001) (Fig. 6D). Additionally, we measured paired-pulse depression to assess whether lingering CCPA altered vesicle pr. An increased paired-pulse ratio (e.g., a decreased paired-pulse depression) is typically interpreted to reflect decreased pr, which could be caused by presynaptic GPCR activation (Mennerick and Zorumski, 1995; Dobrunz and Stevens, 1997; Zucker and Regehr, 2002; Brown and Sihra, 2008). The paired-pulse depression in neurons treated acutely with CCPA (23.4 ± 5.5%; n = 10) was not significantly different from the depression in neurons treated for 4 h (25.8 ± 7.0%; n = 9; p = 0.79) (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). Together, these results suggest a novel presynaptic muting by prolonged selective activation of A1 adenosine receptors. Presynaptic silencing outlives agonist exposure and does not result from altered pr.
We next incubated neurons in 50 μm baclofen, the GABAB receptor agonist, for 4 h before evaluation in baclofen-free solutions. Baclofen, in the absence of any depolarizing stimulus, persistently depressed autaptic EPSC amplitudes and reduced the percentage of active glutamatergic presynaptic terminals (Fig. 6E). Baclofen did not, however, have a persisting effect on the paired-pulse depression (control, 20.8 ± 7.3%; baclofen, 1.1 ± 7.7%; n = 9; p = 0.08) (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material). Additionally, we have confirmed that the effects of CCPA and baclofen on FM1-43FX/vGluT-1 correspondence do not result from transient actions of these agonists. We coapplied CCPA and baclofen for 4 h and allowed 10 min of recovery in untreated media before FM1-43FX loading. Neurons treated with CCPA and baclofen maintained a decreased percentage of active glutamatergic presynaptic terminals after this period of recovery (control + recovery, 79.0 ± 4.3%; CCPA/baclofen + recovery, 51.5 ± 4.5%; n = 20 fields from 4 coverslips; p = 0.00009). In contrast, saturating concentrations of neither ACPD nor another mGluR agonist, l-AP-4, induced silencing (supplemental Fig. 3A,B, available at www.jneurosci.org as supplemental material). Similarly, the CB1 agonist WIN did not induce silencing at a saturating concentration (supplemental Fig. 3C, available at www.jneurosci.org as supplemental material), and kainate receptor agonist kainic acid did not produce silencing at concentrations of 1 or 10 μm (supplemental Fig. 3D,E, available at www.jneurosci.org as supplemental material). These results suggest that GPCR-induced silencing is induced by only a select group of receptors. The lack of silencing induced by mGluR and CB1 activation may be caused by low receptor expression levels in presynaptic terminals of cultured hippocampal neurons (as suggested by the weak acute effects of near-saturating agonist concentrations in Fig. 5C,E) or by divergent downstream signaling cascades.
GPCR-induced silencing is proteasome dependent
Inhibitory G-protein activation links depolarization-induced silencing and GPCR agonist-induced silencing, but it is unclear whether GPCR-dependent presynaptic silencing invokes similar downstream signaling cascades as depolarization-induced presynaptic silencing. For instance, maximum GPCR agonist-induced silencing tends to be weaker than depolarization-induced silencing (Fig. 6C,E), which could hint at different underlying mechanisms. It was shown recently that depolarization-induced muting is ultimately dependent on activity of the UPS. Depolarization-induced presynaptic silencing is prevented by coincubation with the proteasome inhibitor MG-132 (Jiang et al., 2010). To test whether GPCR agonist-induced silencing also recruits the UPS, we blocked proteasome activity by 30 min preincubation with MG-132 alone before coapplying MG-132 with GPCR agonists for 4 h. To maximize the inhibitory G-protein signal, we coapplied 10 nm CCPA and 50 μm baclofen to simultaneously activate both A1 receptors and GABAB receptors. This treatment did not result in more silencing than either agonist alone (compare Fig. 6C–E with Fig. 7) as expected from convergent downstream signaling in the two receptor systems (Brown and Sihra, 2008). MG-132 and CCPA/baclofen treatment did not alter paired-pulse depression in autaptic neurons (control, 21.1 ± 4.0%; MG-132, 24.3 ± 5.7%; CCPA/baclofen, 22.3 ± 5.8%; CCPA/baclofen + MG-132, 24.7 ± 7.9%; n = 14–15 neurons per condition; p > 0.05) (supplemental Fig. 1E, available at www.jneurosci.org as supplemental material). To confirm that a pr change should have been detected by paired-pulse measurements with our protocols and sample sizes, we tested the effect of acute 10 μm baclofen application on EPSC paired-pulse depression. As expected, paired-pulse modulation was significantly altered by acute baclofen (3.7 ± 5.2% depression in saline control; 32.3 ± 15.6% facilitation in baclofen; n = 12; p = 0.04) (supplemental Fig. 1F, available at www.jneurosci.org as supplemental material). MG-132 alone did not alter the baseline EPSC amplitude, but it prevented silencing normally induced by GPCR agonist application (Fig. 7A,B). This was confirmed with FM1-43FX labeling of presynaptic terminals. The percentage of active glutamatergic presynaptic terminals was not altered by MG-132 alone, but coincubation with GPCR agonists prevented presynaptic silencing (Fig. 7C,D). These results suggest that GPCR agonist-induced silencing also recruits a proteasome-dependent mechanism, supporting our hypothesis that persistent GPCR-dependent depression uses similar signaling cascades as depolarization-induced silencing. This also supports our model that depolarization-induced silencing occurs through a calcium-independent inhibitory G-protein-dependent mechanism that ultimately depends on activation of the UPS.
Figure 7.
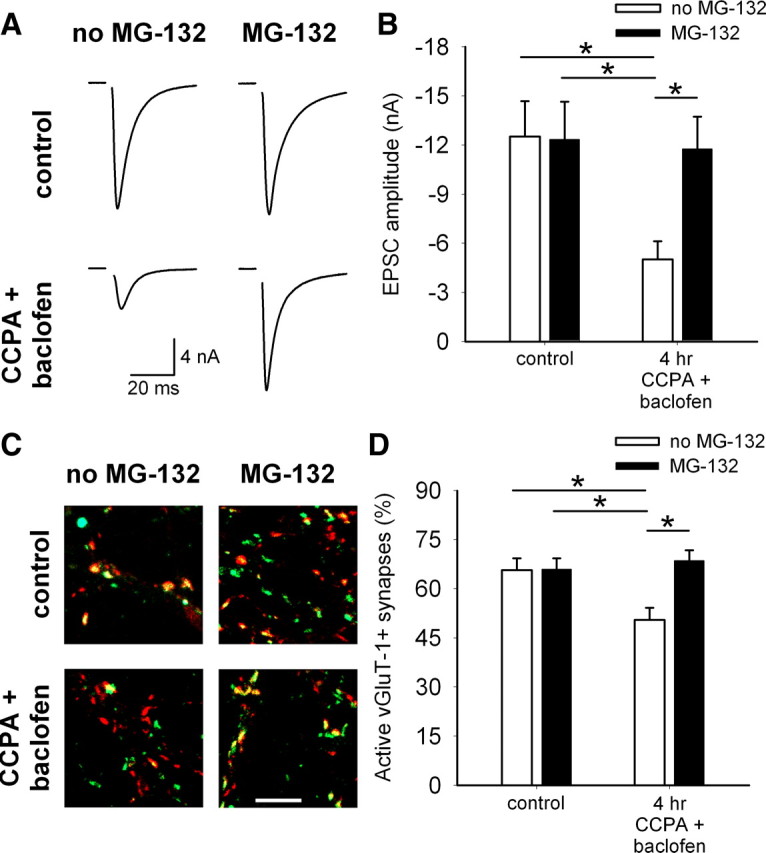
GPCR-dependent silencing requires proteasome activity. A, Example action-potential-evoked EPSCs from autaptic neurons treated acutely (<1 min; control) or for 4 h with 10 nm CCPA and 50 μm baclofen with or without 3 μm MG-132. MG-132 was added 30 min before the start of (and remained during) the 4 h treatment. B, Summary of EPSC amplitudes from autaptic neurons treated as in A. *p < 0.05 with Bonferroni's correction for multiple comparisons (n = 15 neurons). C, Example images of vGluT-1 (red) and FM1-43FX (green) correspondence in neurons treated 4 h with or without 10 nm CCPA and 50 μm baclofen in the presence or absence of 3 μm MG-132. MG-132 was added 30 min before the start of the 4 h treatment. Scale bar, 5 μm. D, Summary of experiments measuring vGluT-1/FM1-43FX correspondence in neurons treated as in C. *p < 0.05 with Bonferroni's correction for multiple comparisons (n = 35 fields from 7 independent experiments).
Discussion
Adaptive modulation at synapses has been studied extensively, but induction mechanisms remain unclear (see Pozo and Goda, 2010). We tested whether calcium and G-protein signaling, two potential upstream regulators of cAMP, are necessary for presynaptic muting. Surprisingly, depolarization-induced presynaptic silencing is not dependent on calcium rises from extracellular or intracellular sources. Depolarization-induced muting is pertussis toxin sensitive, implicating Gi/o signaling, and prolonged, direct activation of two classes of Gi/o-linked GPCRs induces silencing, suggesting novel presynaptic Gi/o-induced modulation. Blocking five receptor classes failed to prevent depolarization-induced silencing, suggesting that G-protein signaling is activated by a pathway that remains untested. Furthermore, GPCR-dependent silencing is proteasome dependent, as documented previously for depolarization-induced silencing. Our results support the model that prolonged depolarization upregulates inhibitory G-protein signaling, possibly independent of receptor activation, to ultimately degrade vital regulators of neurotransmission.
Calcium and persistent forms of synaptic plasticity
Calcium from neither the extracellular space (Fig. 1) nor intracellular stores (Fig. 2) is necessary for adaptive presynaptic silencing. This result is surprising because multiple persistent forms of hippocampal synaptic plasticity depend on calcium (Dunwiddie et al., 1978; Wigström et al., 1979; Lynch et al., 1983; Williams and Johnston, 1989; Wickens and Abraham, 1991; Mulkey and Malenka, 1992; Xie et al., 1992; Tzounopoulos et al., 1998; Patenaude et al., 2003; Izumi et al., 2008; Kellogg et al., 2009). Other adaptive forms of plasticity, including synaptic scaling and plasticity of intrinsic excitability, require changes in intracellular calcium (Thiagarajan et al., 2002; Cudmore and Turrigiano, 2004; Frank et al., 2006; Ibata et al., 2008; Wu et al., 2008). Although it is possible that depolarization-induced presynaptic silencing requires postsynaptic depolarization, our BAPTA-AM experiments exclude an important induction role for any source of calcium, including presynaptic, postsynaptic, and glial sources. These results are consistent with our previous finding that calcium-sensitive adenylyl cyclase isoforms play no role in silencing induction (Moulder et al., 2008).
Depolarization-induced muting is among a few examples of calcium-independent plasticity. These include short-term, frequency-dependent synaptic depression (Betz, 1970; Stevens and Tsujimoto, 1995; Garcia-Perez et al., 2008), transient modulation by nontraditional messengers such as nitric oxide and steroid hormones (Meffert et al., 1996; Zadran et al., 2009), and some forms of hippocampal mGluR-dependent long-term depression (Fitzjohn et al., 2001; Ireland and Abraham, 2009). Because none of these mechanisms is implicated in presynaptic muting, the silencing described herein appears to be unique.
Presynaptic inhibition by G-protein activation
Acute activation of pertussis toxin-sensitive presynaptic and postsynaptic inhibitory G-proteins induces well-known, rapidly reversible effects on calcium currents and inwardly rectifying potassium channels (Brown and Sihra, 2008). Although acute presynaptic depression elicited by Gi/o stimulation persists for at least 4 h in the continued presence of agonist (Wetherington and Lambert, 2002a,b), several major features distinguish the depolarization-induced and GPCR-induced presynaptic silencing observed here from this classical presynaptic GPCR-induced depression.
First, acute Gi/o activation causes synaptic depression with rapid onset and rapid offset during agonist withdrawal (Brown and Sihra, 2008). Presynaptic muting, in contrast, requires prolonged stimulation, and recovery requires hours (Moulder et al., 2004). GPCR agonist-induced silencing failed to recover within 10 min in imaging experiments and persisted for at least 1 h after agonist removal in our electrophysiology experiments (Figs. 6, 7). Furthermore, only two of five receptor classes tested induced presynaptic silencing. It is possible that persistent silencing requires stronger Gi/o stimulation than acute depression, because CB1 and mGluR stimulation yielded weak acute depression and no detectable silencing (Fig. 5C,E and supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Second, acute depression depends on Gβγ-dependent presynaptic inhibition of calcium channels to reduce vesicle pr (Brown and Sihra, 2008). Decreases in pr are reflected as increased paired-pulse ratios (Dobrunz and Stevens, 1997; Zucker and Regehr, 2002). Depolarization-induced and GPCR-induced muting did not alter paired-pulse ratios (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) and may involve degradation of important presynaptic proteins through the UPS (Jiang et al., 2010). This is an unusual profile for presynaptic change and might explain why GPCR-induced silencing has been overlooked. Thus, persistent GPCR-dependent presynaptic silencing invokes different mechanisms than acute GPCR-dependent synaptic depression.
Other forms of slowly induced synaptic depression dependent on GPCRs have been described in the hippocampus. For example, GPCR activation is necessary for certain forms of long-term depression or depotentiation of long-term potentiation (Oliet et al., 1997; Palmer et al., 1997; Chevaleyre and Castillo, 2004; Izumi and Zorumski, 2008; Kellogg et al., 2009). Although a presynaptic change has been implicated in some of these examples, presynaptic muting has not been implicated because these are calcium-dependent and/or require activation of receptors that we have shown are unnecessary for depolarization-induced presynaptic silencing.
The finding that prolonged A1 and GABAB receptor activation induces silencing is reminiscent of evidence for tonic GPCR activation in situ. A1 receptors and GABAB receptors are constitutively active at low levels under physiological conditions (Canning and Leung, 2000; Thümmler and Dunwiddie, 2000; Jensen et al., 2003; Kukley et al., 2005) (but see Ariwodola and Weiner, 2004). Thus, GPCR-induced silencing may contribute to basally silent terminals in situ. Evidence for basal GPCR contributions to presynaptically mute synapses in our cultures was limited (Fig. 5B), possibly because of much larger dilution of transmitters than occurs in situ. Because evoked accumulation of adenosine and GABA likely achieves higher local concentrations over more prolonged periods in intact tissue (Mitchell et al., 1993; Masino and Dunwiddie, 1999; de Groote and Linthorst, 2007; Ghijsen et al., 2007), GPCR-induced silencing could play a dynamic role in sculpting the pattern of silent and active terminals.
Relationship between G-proteins and proteasome activity
Multiple downstream signaling cascades are activated by GPCRs. Our data implicate that, ultimately, GPCR agonist-induced presynaptic silencing is dependent on activity of the UPS because the proteasome inhibitor MG-132 blocked silencing induction (Fig. 7). One potential model is that inhibition of adenylyl cyclase by G-proteins decreases cAMP accumulation and associated protein phosphorylation by protein kinase A. We speculate that phosphorylation may protect key presynaptic proteins from degradation by the UPS. This view is consistent with evidence that downregulation of cAMP signaling leads to silencing (Moulder et al., 2008) and that depolarization-induced silencing is proteasome dependent (Jiang et al., 2010). In addition Munc13-1 and Rim1α, presynaptic proteins essential for vesicle priming (Augustin et al., 1999; Betz et al., 2001; Koushika et al., 2001), are selectively degraded relative to other presynaptic proteins after stimuli that induce presynaptic silencing (Jiang et al., 2010). This mirrors work in Drosophila showing that the Munc13 ortholog Dunc13 is regulated in a G-protein-dependent, cAMP-dependent, and proteasome-dependent manner (Aravamudan and Broadie, 2003) as well as work at the calyx of Held showing that GABAB receptor activation inhibits vesicle recruitment via reduced cAMP levels (Sakaba and Neher, 2003). Because Rim1α is a protein kinase A and UPS substrate (Lonart et al., 2003; Yao et al., 2007), phosphorylation-induced Rim1α stability could link increased cAMP levels to preserved presynaptic function. This speculative scenario does not preclude the possibility that induction stimuli modulate proteasome activity directly in a cAMP-independent manner (Jiang et al., 2010). Future work may clarify whether cAMP-independent G-protein signaling upregulates proteasome activity or whether depolarization increases proteasome activity by a pathway divorced from G-protein signaling.
G-protein signaling in depolarization-induced silencing
Although depolarization-induced presynaptic silencing requires activation of inhibitory G-proteins, how G-proteins are activated during prolonged depolarization remains unclear. We excluded four primary candidate GPCRs as well as metabotropic actions of kainate receptors (Figs. 4, 5). Depolarization may cause release of a ligand for Gi/o-linked GPCRs that we did not test. For example, neuromodulatory GPCRs such as dopamine and serotonin receptors are possible candidates (Catapano and Manji, 2007), but monoaminergic cells are absent from our cultures, making ligand presence extremely unlikely. Furthermore, the calcium independence of presynaptic muting excludes an important role of synaptic release of these potential mediators. Clearly, if a ligand is involved in induction of depolarization-induced muting, it is released through a nonclassical mechanism. Alternatively, cell-autonomous mechanisms such as voltage-sensitive proteins, including inhibitory GPCRs activated directly by voltage, could mediate silencing induction (Reddy et al., 1995; Ben-Chaim et al., 2006; Parnas and Parnas, 2007; Okamura et al., 2009). One intriguing possibility is that of receptor-independent activation of G-proteins, for instance, through recently described activators of G-protein signaling (AGS) (Blumer et al., 2007). AGS proteins interact with G-protein subunits to initiate downstream signaling in the absence of receptor activation. Some AGS proteins are expressed in hippocampal neurons, but it is unclear how they are activated (Fang et al., 2000; Blumer et al., 2002; Takahashi et al., 2003).
Although we have learned much about the induction of presynaptic silencing in excitatory neurons (Tong et al., 1996; Ma et al., 1999; Moulder et al., 2004, 2006, 2008; Voronin and Cherubini, 2004; Atasoy and Kavalali, 2006; Yao et al., 2006; Jiang et al., 2010), many segments of the signaling cascade have remained obscure. We show here that calcium-independent, G-protein-dependent, and proteasome-dependent mechanisms induce presynaptic muting, suggesting a unique form of synaptic plasticity. Because adaptive synaptic plasticity may counteract potentially damaging changes in neuronal activity (Turrigiano and Nelson, 2004), additional clarification of silencing induction pathways may lead to new therapeutic strategies.
Footnotes
This work was supported by National Institutes of Health Grants GM08151 (D.C.C.), NS066611 (D.C.C.), NS032636 (Mark P. Goldberg, supporting K.L.H.), NS036265 (Mark P. Goldberg, supporting K.L.H.), and MH78823 (S.M.) and National Institutes of Health Neuroscience Blueprint Core Grant P30NS057105 (to Washington University in St. Louis, St. Louis, MO). We thank Ann Benz and Amanda Taylor for assistance with cultures, Xiaoping Jiang for contributions to the imaging experiments, Charles Zorumski for helpful comments on this manuscript, and Timothy Holy, Robert Gereau, Narendrakumar Ramanan, and laboratory members for advice and discussion.
References
- Aravamudan B, Broadie K. Synaptic Drosophila UNC-13 is regulated by antagonistic G-protein pathways via a proteasome-dependent degradation mechanism. J Neurobiol. 2003;54:417–438. doi: 10.1002/neu.10142. [DOI] [PubMed] [Google Scholar]
- Ariwodola OJ, Weiner JL. Ethanol potentiation of GABAergic synaptic transmission may be self-limiting: role of presynaptic GABAB receptors. J Neurosci. 2004;24:10679–10686. doi: 10.1523/JNEUROSCI.1768-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Kavalali ET. Presynaptic unsilencing: searching for a mechanism. Neuron. 2006;50:345–346. doi: 10.1016/j.neuron.2006.04.018. [DOI] [PubMed] [Google Scholar]
- Augustin I, Rosenmund C, Südhof TC, Brose N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature. 1999;400:457–461. doi: 10.1038/22768. [DOI] [PubMed] [Google Scholar]
- Ben-Chaim Y, Chanda B, Dascal N, Bezanilla F, Parnas I, Parnas H. Movement of “gating charge” is coupled to ligand binding in a G-protein-coupled receptor. Nature. 2006;444:106–109. doi: 10.1038/nature05259. [DOI] [PubMed] [Google Scholar]
- Betz A, Thakur P, Junge HJ, Ashery U, Rhee JS, Scheuss V, Rosenmund C, Rettig J, Brose N. Functional interaction of the active zone proteins Munc13-1 and RIM1 in synaptic vesicle priming. Neuron. 2001;30:183–196. doi: 10.1016/s0896-6273(01)00272-0. [DOI] [PubMed] [Google Scholar]
- Betz WJ. Depression of transmitter release at the neuromuscular junction of the frog. J Physiol. 1970;206:629–644. doi: 10.1113/jphysiol.1970.sp009034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumer JB, Chandler LJ, Lanier SM. Expression analysis and subcellular distribution of the two G-protein regulators AGS3 and LGN indicate distinct functionality. Localization of LGN to the midbody during cytokinesis. J Biol Chem. 2002;277:15897–15903. doi: 10.1074/jbc.M112185200. [DOI] [PubMed] [Google Scholar]
- Blumer JB, Smrcka AV, Lanier SM. Mechanistic pathways and biological roles for receptor-independent activators of G-protein signaling. Pharmacol Ther. 2007;113:488–506. doi: 10.1016/j.pharmthera.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Sihra TS. Presynaptic signaling by heterotrimeric G-proteins. Handb Exp Pharmacol. 2008;2008:207–260. doi: 10.1007/978-3-540-74805-2_8. [DOI] [PubMed] [Google Scholar]
- Bureau I, Bischoff S, Heinemann SF, Mulle C. Kainate receptor-mediated responses in the CA1 field of wild-type and GluR6-deficient mice. J Neurosci. 1999;19:653–663. doi: 10.1523/JNEUROSCI.19-02-00653.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canning KJ, Leung LS. Excitability of rate dentate gyrus granule cells in vivo is controlled by tonic and evoked GABAB receptor-mediated inhibition. Brain Res. 2000;863:271–275. doi: 10.1016/s0006-8993(00)02124-7. [DOI] [PubMed] [Google Scholar]
- Catapano LA, Manji HK. G protein-coupled receptors in major psychiatric disorders. Biochim Biophys Acta. 2007;1768:976–993. doi: 10.1016/j.bbamem.2006.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Endocannabinoid-mediated metaplasticity in the hippocampus. Neuron. 2004;43:871–881. doi: 10.1016/j.neuron.2004.08.036. [DOI] [PubMed] [Google Scholar]
- Crawford DC, Moulder KL, Gereau RW, 4th, Story GM, Mennerick S. Comparative effects of heterologous TRPV1 and TRPM8 expression in rat hippocampal neurons. PLoS ONE. 2009;4:e8166. doi: 10.1371/journal.pone.0008166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowder TL, Ariwodola OJ, Weiner JL. Ethanol antagonizes kainate receptor-mediated inhibition of evoked GABAA inhibitory postsynaptic currents in the rat hippocampal CA1 region. J Pharmacol Exp Ther. 2002;303:937–944. doi: 10.1124/jpet.102.038471. [DOI] [PubMed] [Google Scholar]
- Cudmore RH, Turrigiano GG. Long-term potentiation of intrinsic excitability in LV visual cortical neurons. J Neurophysiol. 2004;92:341–348. doi: 10.1152/jn.01059.2003. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Malva JO, Ribeiro JA. Kainate receptors coupled to Gi/Go proteins in the rat hippocampus. Mol Pharmacol. 1999;56:429–433. doi: 10.1124/mol.56.2.429. [DOI] [PubMed] [Google Scholar]
- de Groote L, Linthorst AC. Exposure to novelty and forced swimming evoke stressor-dependent changes in extracellular GABA in the rat hippocampus. Neuroscience. 2007;148:794–805. doi: 10.1016/j.neuroscience.2007.06.030. [DOI] [PubMed] [Google Scholar]
- Delaney AJ, Jahr CE. Kainate receptors differentially regulate release at two parallel fiber synapses. Neuron. 2002;36:475–482. doi: 10.1016/s0896-6273(02)01008-5. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Deutsch DG. Biochemistry of the endogenous ligands of cannabinoid receptors. Neurobiol Dis. 1998;5:386–404. doi: 10.1006/nbdi.1998.0214. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Melck D, Bisogno T, De Petrocellis L. Endocannabinoids: endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 1998;21:521–528. doi: 10.1016/s0166-2236(98)01283-1. [DOI] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- Dunwiddie T, Madison D, Lynch G. Synaptic transmission is required for initiation of long-term potentiation. Brain Res. 1978;150:413–417. doi: 10.1016/0006-8993(78)90293-7. [DOI] [PubMed] [Google Scholar]
- Fang M, Jaffrey SR, Sawa A, Ye K, Luo X, Snyder SH. Dexras1: a G protein specifically coupled to neuronal nitric oxide synthase via CAPON. Neuron. 2000;28:183–193. doi: 10.1016/s0896-6273(00)00095-7. [DOI] [PubMed] [Google Scholar]
- Fisher SA, Fischer TM, Carew TJ. Multiple overlapping processes underlying short-term synaptic enhancement. Trends Neurosci. 1997;20:170–177. doi: 10.1016/s0166-2236(96)01001-6. [DOI] [PubMed] [Google Scholar]
- Fitzjohn SM, Palmer MJ, May JE, Neeson A, Morris SA, Collingridge GL. A characterisation of long-term depression induced by metabotropic glutamate receptor activation in the rat hippocampus in vitro. J Physiol. 2001;537:421–430. doi: 10.1111/j.1469-7793.2001.00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron. 2006;52:663–677. doi: 10.1016/j.neuron.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith S, Daniel JA, Vissel B. A study of clustered data and approaches to its analysis. J Neurosci. 2010;30:10601–10608. doi: 10.1523/JNEUROSCI.0362-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Perez E, Lo DC, Wesseling JF. Kinetic isolation of a slowly recovering component of short-term depression during exhaustive use at excitatory hippocampal synapses. J Neurophysiol. 2008;100:781–795. doi: 10.1152/jn.90429.2008. [DOI] [PubMed] [Google Scholar]
- Gaspary HL, Wang W, Richerson GB. Carrier-mediated GABA release activates GABA receptors on hippocampal neurons. J Neurophysiol. 1998;80:270–281. doi: 10.1152/jn.1998.80.1.270. [DOI] [PubMed] [Google Scholar]
- Ghijsen WE, Zuiderwijk M, Lopes da Silva FH. Electrically evoked GABA release in rat hippocampus CA1 region and its changes during kindling epileptogenesis. Brain Res. 2007;1135:69–76. doi: 10.1016/j.brainres.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Gidö G, Kristián T, Siesjö BK. Extracellular potassium in a neocortical core area after transient focal ischemia. Stroke. 1997;28:206–210. doi: 10.1161/01.str.28.1.206. [DOI] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Ireland DR, Abraham WC. Mechanisms of group I mGluR-dependent long-term depression of NMDA receptor-mediated transmission at Schaffer collateral-CA1 synapses. J Neurophysiol. 2009;101:1375–1385. doi: 10.1152/jn.90643.2008. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Zorumski CF. Direct cortical inputs erase long-term potentiation at Schaffer collateral synapses. J Neurosci. 2008;28:9557–9563. doi: 10.1523/JNEUROSCI.3346-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Tokuda K, Zorumski CF. Long-term potentiation inhibition by low-level N-methyl-d-aspartate receptor activation involves calcineurin, nitric oxide, and p38 mitogen-activated protein kinase. Hippocampus. 2008;18:258–265. doi: 10.1002/hipo.20383. [DOI] [PubMed] [Google Scholar]
- Jensen K, Chiu CS, Sokolova I, Lester HA, Mody I. GABA transporter-1 (GAT1)-deficient mice: differential tonic activation of GABAA versus GABAB receptors in the hippocampus. J Neurophysiol. 2003;90:2690–2701. doi: 10.1152/jn.00240.2003. [DOI] [PubMed] [Google Scholar]
- Jiang X, Litkowski PE, Taylor AA, Lin Y, Snider BJ, Moulder KL. A role for the ubiquitin-proteasome system in activity-dependent presynaptic silencing. J Neurosci. 2010;30:1798–1809. doi: 10.1523/JNEUROSCI.4965-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg R, Mackie K, Straiker A. Cannabinoid CB1 receptor-dependent long-term depression in autaptic excitatory neurons. J Neurophysiol. 2009;102:1160–1171. doi: 10.1152/jn.00266.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koushika SP, Richmond JE, Hadwiger G, Weimer RM, Jorgensen EM, Nonet ML. A post-docking role for active zone protein Rim. Nat Neurosci. 2001;4:997–1005. doi: 10.1038/nn732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukley M, Schwan M, Fredholm BB, Dietrich D. The role of extracellular adenosine in regulating mossy fiber synaptic plasticity. J Neurosci. 2005;25:2832–2837. doi: 10.1523/JNEUROSCI.4260-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latini S, Pedata F. Adenosine in the central nervous system: release mechanisms and extracellular concentrations. J Neurochem. 2001;79:463–484. doi: 10.1046/j.1471-4159.2001.00607.x. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Bortolotto ZA, Bleakman D, Ornstein PL, Lodge D, Isaac JT, Collingridge GL. A critical role of a facilitatory presynaptic kainate receptor in mossy fiber LTP. Neuron. 2001;32:697–709. doi: 10.1016/s0896-6273(01)00511-6. [DOI] [PubMed] [Google Scholar]
- Lerma J. Roles and rules of kainate receptors in synaptic transmission. Nat Rev Neurosci. 2003;4:481–495. doi: 10.1038/nrn1118. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Klotz KN, Schwabe U, Cristalli G, Vittori S, Grifantini M. 2-Chloro-N6-cyclopentyladenosine: a highly selective agonist at A1 adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol. 1988;337:687–689. doi: 10.1007/BF00175797. [DOI] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Südhof TC, Linden DJ. Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell. 2003;115:49–60. doi: 10.1016/s0092-8674(03)00727-x. [DOI] [PubMed] [Google Scholar]
- Lynch G, Larson J, Kelso S, Barrionuevo G, Schottler F. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983;305:719–721. doi: 10.1038/305719a0. [DOI] [PubMed] [Google Scholar]
- Ma L, Zablow L, Kandel ER, Siegelbaum SA. Cyclic AMP induces functional presynaptic boutons in hippocampal CA3-CA1 neuronal cultures. Nat Neurosci. 1999;2:24–30. doi: 10.1038/4525. [DOI] [PubMed] [Google Scholar]
- Maffei A, Fontanini A. Network homeostasis: a matter of coordination. Curr Opin Neurobiol. 2009;19:168–173. doi: 10.1016/j.conb.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC. Synaptic plasticity in the hippocampus: LTP and LTD. Cell. 1994;78:535–538. doi: 10.1016/0092-8674(94)90517-7. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Silent synapses speak up. Neuron. 1997;19:473–476. doi: 10.1016/s0896-6273(00)80362-1. [DOI] [PubMed] [Google Scholar]
- Masino SA, Dunwiddie TV. Temperature-dependent modulation of excitatory transmission in hippocampal slices is mediated by extracellular adenosine. J Neurosci. 1999;19:1932–1939. doi: 10.1523/JNEUROSCI.19-06-01932.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meffert MK, Calakos NC, Scheller RH, Schulman H. Nitric oxide modulates synaptic vesicle docking fusion reactions. Neuron. 1996;16:1229–1236. doi: 10.1016/s0896-6273(00)80149-x. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Zorumski CF. Paired-pulse modulation of fast excitatory synaptic currents in microcultures of rat hippocampal neurons. J Physiol. 1995;488:85–101. doi: 10.1113/jphysiol.1995.sp020948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Que J, Benz A, Zorumski CF. Passive and synaptic properties of hippocampal neurons grown in microcultures and in mass cultures. J Neurophysiol. 1995;73:320–332. doi: 10.1152/jn.1995.73.1.320. [DOI] [PubMed] [Google Scholar]
- Mitchell JB, Lupica CR, Dunwiddie TV. Activity-dependent release of endogenous adenosine modulates synaptic responses in the rat hippocampus. J Neurosci. 1993;13:3439–3447. doi: 10.1523/JNEUROSCI.13-08-03439.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder KL, Cormier RJ, Shute AA, Zorumski CF, Mennerick S. Homeostatic effects of depolarization on Ca2+ influx, synaptic signaling, and survival. J Neurosci. 2003;23:1825–1831. doi: 10.1523/JNEUROSCI.23-05-01825.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder KL, Meeks JP, Shute AA, Hamilton CK, de Erausquin G, Mennerick S. Plastic elimination of functional glutamate release sites by depolarization. Neuron. 2004;42:423–435. doi: 10.1016/s0896-6273(04)00184-9. [DOI] [PubMed] [Google Scholar]
- Moulder KL, Jiang X, Taylor AA, Olney JW, Mennerick S. Physiological activity depresses synaptic function through an effect on vesicle priming. J Neurosci. 2006;26:6618–6626. doi: 10.1523/JNEUROSCI.5498-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder KL, Jiang X, Chang C, Taylor AA, Benz AM, Conti AC, Muglia LJ, Mennerick S. A specific role for Ca2+-dependent adenylyl cyclases in recovery from adaptive presynaptic silencing. J Neurosci. 2008;28:5159–5168. doi: 10.1523/JNEUROSCI.5317-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder KL, Jiang X, Taylor AA, Benz AM, Mennerick S. Presynaptically silent synapses studied with light microscopy. J Vis Exp. 2010;pii:1676. doi: 10.3791/1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- Nicholls D, Attwell D. The release and uptake of excitatory amino acids. Trends Pharmacol Sci. 1990;11:462–468. doi: 10.1016/0165-6147(90)90129-v. [DOI] [PubMed] [Google Scholar]
- Okamura Y, Murata Y, Iwasaki H. Voltage-sensing phosphatase: actions and potentials. J Physiol. 2009;587:513–520. doi: 10.1113/jphysiol.2008.163097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Palmer MJ, Irving AJ, Seabrook GR, Jane DE, Collingridge GL. The group I mGlu receptor agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacology. 1997;36:1517–1532. doi: 10.1016/s0028-3908(97)00181-0. [DOI] [PubMed] [Google Scholar]
- Parnas H, Parnas I. The chemical synapse goes electric: Ca2+- and voltage-sensitive GPCRs control neurotransmitter release. Trends Neurosci. 2007;30:54–61. doi: 10.1016/j.tins.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Patenaude C, Chapman CA, Bertrand S, Congar P, Lacaille JC. GABAB receptor- and metabotropic glutamate receptor-dependent cooperative long-term potentiation of rat hippocampal GABAA synaptic transmission. J Physiol. 2003;553:155–167. doi: 10.1113/jphysiol.2003.049015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo K, Goda Y. Unraveling mechanisms of homeostatic synaptic plasticity. Neuron. 2010;66:337–351. doi: 10.1016/j.neuron.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy R, Smith D, Wayman G, Wu Z, Villacres EC, Storm DR. Voltage-sensitive adenylyl cyclase activity in cultured neurons. A calcium-independent phenomenon. J Biol Chem. 1995;270:14340–14346. doi: 10.1074/jbc.270.24.14340. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, Sihra TS. Metabotropic actions of kainate receptors in the CNS. J Neurochem. 2007;103:2121–2135. doi: 10.1111/j.1471-4159.2007.04924.x. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Direct modulation of synaptic vesicle priming by GABAB receptor activation at a glutamatergic synapse. Nature. 2003;424:775–778. doi: 10.1038/nature01859. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- Schwartz EA. Calcium-independent release of GABA from isolated horizontal cells of the toad retina. J Physiol. 1982;323:211–227. doi: 10.1113/jphysiol.1982.sp014069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev. 2001;81:1065–1096. doi: 10.1152/physrev.2001.81.3.1065. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Tsujimoto T. Estimates for the pool size of releasable quanta at a single central synapse and for the time required to refill the pool. Proc Natl Acad Sci U S A. 1995;92:846–849. doi: 10.1073/pnas.92.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone TW, Ceruti S, Abbracchio MP. Adenosine receptors and neurological disease: neuroprotection and neurodegeneration. Handb Exp Pharmacol. 2009;2009:535–587. doi: 10.1007/978-3-540-89615-9_17. [DOI] [PubMed] [Google Scholar]
- Szatkowski M, Barbour B, Attwell D. Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature. 1990;348:443–446. doi: 10.1038/348443a0. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Umeda N, Tsutsumi Y, Fukumura R, Ohkaze H, Sujino M, van der Horst G, Yasui A, Inouye ST, Fujimori A, Ohhata T, Araki R, Abe M. Mouse dexamethasone-induced RAS protein 1 gene is expressed in a circadian rhythmic manner in the suprachiasmatic nucleus. Brain Res Mol Brain Res. 2003;110:1–6. doi: 10.1016/s0169-328x(02)00543-0. [DOI] [PubMed] [Google Scholar]
- Thiagarajan TC, Piedras-Renteria ES, Tsien RW. alpha- and betaCaMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron. 2002;36:1103–1114. doi: 10.1016/s0896-6273(02)01049-8. [DOI] [PubMed] [Google Scholar]
- Thümmler S, Dunwiddie TV. Adenosine receptor antagonists induce persistent bursting in the rat hippocampal CA3 region via an NMDA receptor-dependent mechanism. J Neurophysiol. 2000;83:1787–1795. doi: 10.1152/jn.2000.83.4.1787. [DOI] [PubMed] [Google Scholar]
- Tong G, Malenka RC, Nicoll RA. Long-term potentiation in cultures of single hippocampal granule cells: a presynaptic form of plasticity. Neuron. 1996;16:1147–1157. doi: 10.1016/s0896-6273(00)80141-5. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends Neurosci. 1999;22:221–227. doi: 10.1016/s0166-2236(98)01341-1. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Tzounopoulos T, Janz R, Südhof TC, Nicoll RA, Malenka RC. A role for cAMP in long-term depression at hippocampal mossy fiber synapses. Neuron. 1998;21:837–845. doi: 10.1016/s0896-6273(00)80599-1. [DOI] [PubMed] [Google Scholar]
- Voronin LL, Cherubini E. 'Deaf, mute and whispering' silent synapses: their role in synaptic plasticity. J Physiol. 2004;557:3–12. doi: 10.1113/jphysiol.2003.058966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int. 2000;36:291–300. doi: 10.1016/s0197-0186(99)00137-0. [DOI] [PubMed] [Google Scholar]
- Wang Z, Haydon PG, Yeung ES. Direct observation of calcium-independent intercellular ATP signaling in astrocytes. Anal Chem. 2000;72:2001–2007. doi: 10.1021/ac9912146. [DOI] [PubMed] [Google Scholar]
- Wetherington JP, Lambert NA. GABAB receptor activation desensitizes postsynaptic GABAB and A1 adenosine responses in rat hippocampal neurones. J Physiol. 2002a;544:459–467. doi: 10.1113/jphysiol.2002.023093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetherington JP, Lambert NA. Differential desensitization of responses mediated by presynaptic and postsynaptic A1 adenosine receptors. J Neurosci. 2002b;22:1248–1255. doi: 10.1523/JNEUROSCI.22-04-01248.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickens JR, Abraham WC. The involvement of L-type calcium channels in heterosynaptic long-term depression in the hippocampus. Neurosci Lett. 1991;130:128–132. doi: 10.1016/0304-3940(91)90244-n. [DOI] [PubMed] [Google Scholar]
- Wigström H, Swann JW, Andersen P. Calcium dependency of synaptic long-lasting potentiation in the hippocampal slice. Acta Physiol Scand. 1979;105:126–128. doi: 10.1111/j.1748-1716.1979.tb06323.x. [DOI] [PubMed] [Google Scholar]
- Williams S, Johnston D. Long-term potentiation of hippocampal mossy fiber synapses is blocked by postsynaptic injection of calcium chelators. Neuron. 1989;3:583–588. doi: 10.1016/0896-6273(89)90268-7. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of hippocampus. Neuron. 1994;12:1139–1148. doi: 10.1016/0896-6273(94)90321-2. [DOI] [PubMed] [Google Scholar]
- Wu WW, Chan CS, Surmeier DJ, Disterhoft JF. Coupling of L-type Ca2+ channels to KV7/KCNQ channels creates a novel, activity-dependent, homeostatic intrinsic plasticity. J Neurophysiol. 2008;100:1897–1908. doi: 10.1152/jn.90346.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Berger TW, Barrionuevo G. Isolated NMDA receptor-mediated synaptic responses express both LTP and LTD. J Neurophysiol. 1992;67:1009–1013. doi: 10.1152/jn.1992.67.4.1009. [DOI] [PubMed] [Google Scholar]
- Yao I, Takagi H, Ageta H, Kahyo T, Sato S, Hatanaka K, Fukuda Y, Chiba T, Morone N, Yuasa S, Inokuchi K, Ohtsuka T, Macgregor GR, Tanaka K, Setou M. SCRAPPER-dependent ubiquitination of active zone protein RIM1 regulates synaptic vesicle release. Cell. 2007;130:943–957. doi: 10.1016/j.cell.2007.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Qi J, Chen G. Actin-dependent activation of presynaptic silent synapses contributes to long-term synaptic plasticity in developing hippocampal neurons. J Neurosci. 2006;26:8137–8147. doi: 10.1523/JNEUROSCI.1183-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KW, Rothman SM. Adenosine inhibits excitatory but not inhibitory synaptic transmission in the hippocampus. J Neurosci. 1991;11:1375–1380. doi: 10.1523/JNEUROSCI.11-05-01375.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadran S, Qin Q, Bi X, Zadran H, Kim Y, Foy MR, Thompson R, Baudry M. 17-Beta-estradiol increases neuronal excitability through MAP kinase-induced calpain activation. Proc Natl Acad Sci U S A. 2009;106:21936–21941. doi: 10.1073/pnas.0912558106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]