

Abstract
Purpose
Patients with anaplastic lymphoma kinase (ALK) gene rearrangements often manifest dramatic responses to crizotinib, a small molecule ALK inhibitor. Unfortunately, not every patient responds and acquired drug resistance inevitably develops in those that do respond. This study aimed to define molecular mechanisms of resistance to crizotinib in ALK+ non-small cell lung cancer (NSCLC) patients.
Experimental Design
We analyzed tissue obtained from 14 ALK+ NSCLC patients demonstrating evidence of radiologic progression while on crizotinib in order to define mechanisms of intrinsic and acquired resistance to crizotinib.
Results
Eleven patients had material evaluable for molecular analysis. Four patients (36%) developed secondary mutations in the tyrosine kinase domain of ALK. A novel mutation in the ALK kinase domain, encoding a G1269A amino acid substitution that confers resistance to crizotinib in vitro, was identified in two of these cases. Two patients, one with a resistance mutation, exhibited new onset ALK copy number gain (CNG). One patient demonstrated outgrowth of EGFR mutant NSCLC without evidence of a persistent ALK gene rearrangement. Two patients exhibited a KRAS mutation, one of which occurred without evidence of a persisting ALK gene rearrangement. One patient demonstrated the emergence of an ALK gene fusion negative tumor compared to the baseline sample, but with no identifiable alternate driver. Two patients retained ALK positivity with no identifiable resistance mechanism.
Conclusions
Crizotinib resistance in ALK+ NSCLC occurs through somatic kinase domain mutations, ALK gene fusion CNG, and emergence of separate oncogenic drivers.
Keywords: oncogene fusion, anaplastic lymphoma kinase, protein kinase inhibitors, drug resistance, non-small cell lung cancer
Introduction
Non-small cell lung cancer (NSCLC) is increasingly recognized as a heterogeneous set of diseases at the molecular level and these differences can drive therapeutic decision-making (1–3). Transforming rearrangements of the anaplastic lymphoma kinase (ALK) gene were initially identified in anaplastic large cell lymphoma (4). In 2007, an ALK gene rearrangement (ALK+) in which the 5’ end of the echinoderm microtubule-associated protein-like 4 (EML4) gene was fused to the 3’ portion of ALK was identified in NSCLC (5).
Recently, crizotinib has gained FDA approval for the treatment of ALK+ NSCLC. Crizotinib approval was based in part on data from the phase I clinical trial which demonstrated an overall response rate of 57% and a probability of progression free survival at 6 months of 72% (6). Unfortunately, some ALK+ patients will not derive any benefit from crizotinib (intrinsic resistance), while patients who initially derive benefit later develop resistance (acquired resistance).
Several groups have explored molecular mechanisms of acquired resistance in a comparable clinical scenario involving resistance to EGFR TKIs in EGFR mutant NSCLC (7–9). The most common mechanism noted in these patients involves a change in the target of the drug, specifically a second EGFR mutation, T790M, that alters the binding kinetics of the reversible TKIs to the target molecule (10). In addition, clinical examples of second oncogenic drivers within the same cell harboring the EGFR mutation, notably MET gene amplification, bypassing the block caused by the TKI have also been reported (8, 11).
Isolated case reports have recently identified mutations in ALK+ NSCLC that occur following crizotinib therapy (12, 13). However, the frequencies with which these and any other mechanisms of resistance occur remain unknown. Here we report on the clinical and molecular details of 14 ALK+ NSCLC patients with intrinsic or acquired resistance to crizotinib.
Materials and Methods
Patients
Patients with ALK+ NSCLC, who were treated on a phase I study of crizotinib, were considered for re-biopsy upon progression on crizotinib therapy (6). Patients were consented for tissue banking under the University of Colorado Lung Cancer SPORE tissue banking protocol and all research tests were conducted under IRB approval. Formalin-fixed paraffin-embedded (FFPE), fresh frozen and/or tissue for the initiation of cell lines were collected from each patient as deemed safe and feasible by the interventional radiologist/pulmonologist. Diagnostic sampling was prioritized over research sampling in each case. No sample size was pre-specified and the analysis performed was descriptive.
ALK molecular testing
ALK positivity was ascertained by FISH using ALK break-apart probes (Vysis LSI ALK (2p23) Dual Color, Break Apart Rearrangement Probe; Abbott Molecular). The FISH assays and analyses were performed as described previously with minor modifications (14). Using the ALK beak-apart probe, 3’ (red) and 5’ (green) signals physically separated by ≥2 signal diameters were considered split. Specimens were considered positive for ALK rearrangement if <15% of tumor cells showed split signals or single red signals. Copy number of ALK rearrangement was based upon determination of the mean of split and isolated red signals per tumor cell (15). At least 100 tumor cells were analyzed per specimen.
RNA from either FFPE or Frozen tissue was processed using the RecoverAll™ Total Nucleic Acid Isolation Kit from Ambion (Austin, TX). RT-PCR was carried out using the SuperScript III One-Step RT-PCR System with Platinum Taq DNA Polymerase from Invitrogen (Carlsbad, CA) with primers to EML4-ALK. PCR products were either resolved on an agarose gel or analyzed with a Bioanalyzer from Agilent Technology (Santa Clara, CA), excised, and purified using Wizard SV Gel and PCR Clean Up Kit from Promega (Madison, WI) then sequenced as described below. Primers used for RT-PCR or multiplexed RT-PCR of the EML4-ALK gene fusion transcript are listed in Supplementary Table 1 or have been previously described (16, 17).
Table 1.
ALK FISH comparison before and after crizotinib on evaluable patients
| Pre-crizotinib | Pre-crizotinib | |||||||
|---|---|---|---|---|---|---|---|---|
| Patient # |
ALK FISH % cells positive |
ALK FISH pattern§ |
AbnormalALK copy number/cell* |
ALK FISH |
ALK FISH % cells positive |
ALK FISH pattern§ |
Abnormal ALK copy number/cell* |
ALK Change |
| 4 | 78% | sR | 1.2 sR | Positive | 90% | sR | 1.5 sR | Same |
| 5 | 37% | split | 0.4sR,sG | Positive | 51% | split | 0.5 sR,sG | Same |
| 6 | 86% | split | 0.8 sR,sG | Positive | 70% | split | 0.8 sR,sG | Same |
| 7 | 28% | split | 0.3 sR,sG | Positive | 82% | split | 1.5 sR,sG | CNG |
| 8 | 48% | split | 0.5 sR,sG | Positive | 66% | split | 2.2 sR,sG | CNG |
| 9a | 80% | sR | 1.2sR | Negative | 2% | NA | Loss | |
| 9b | Positive | 56% | sR | 0.9 sR | Same | |||
| 10¶ | 28% | mix | 0.3 sR, 0.2 sG | Positive | 30% | mix | 0.3 sR, 0.2 sG | Same |
| 11 | 48% | split | 0.5 sR,sG | Positive | 56% | split | 0.7 sR,sG | Same |
| 12 | 26% | split | 0.3 sR,sG | Negative | 8% | NA | Loss | |
| 13¶ | 60% | split | 0.6 sR, sG | Positive | 48% | split | 0.6 sR, sG | Same |
| 14 | 68% | sR | 1.2sR | Positive | 92% | sR | 1.5 sR | Same |
Patients with intrinsic resistance
sR = single red, sG = single green, split = split red/green, mix = split red/green and single red
Rounded to the nearest tenth decimal
EGFR, KRAS, ALK and Short Tandem Repeat Genotyping
Genomic DNA was isolated from manually microdissected FFPE tumor samples using the QiaAmp FFPE DNA isolation kit from Qiagen (Valencia, CA). Samples were PCR amplified using custom primer sets for exon 2 of KRAS, exons 19 through 21 of EGFR, or exons 21–25 of ALK (see Supplementary Table 1 for primers) and directly sequenced using the ABI Big Dye Thermocycle Sequencing kit and analyzed on an ABI 3730 DNA Sequencer (16). Mutation analysis was assisted by the use of Mutation Surveyor software v3.97–4.0.0 from SoftGenetics (State College, PA). The reference sequence for ALK sequence comparison used was NM_004304.4, for EML4 NM_019063.3, for EGFR NM_005228.3, and for KRAS NM_004985.3. We have used the colloquial single letter amino acid substitution for mutations described here rather than the official nomenclature for simplicity (e.g., L1196M rather than p.Leu1196Met).
The SNaPshot assay for evaluation of multiple oncogenic mutations in APC, AKT1, BRAF, CTNNB1, EGFR, FLT3, JAK2, KIT, KRAS, MAP2K1 (MEK1), NOTCH1, NRAS, PIK3CA, PTEN, and TP53 was performed by amplification using 13 multiplexed PCR reactions followed by single nucleotide base extension reactions. The products were separated by capillary electrophoresis and analyzed using GeneMapper 4.0 as has been previously described (18). To confirm patient identity across multiple samples in select cases, short tandem repeat analysis was performed on extracted genomic DNA using the AmpFlSTR® Identifiler™ PCR Amplification Kit (Applied Biosystems, Carlsbad, CA).
Cell lines and Reagents
Primary cell lines were derived from patients by placing fresh tumor tissue into sterile tissue culture dishes and culturing in RPMI supplemented with 10% FBS. A new cell line, CUTO-1 (Colorado University Thoracic Oncology), derived from a tumor biopsy sample of patient #10 is described here. Ba/F3 cells (kindly provided by James DeGregori) were cultured in RPMI supplemented with 10% FBS and 1ng/ml of IL-3 from R&D Systems (Minneapolis, MN). 293T human embryonic kidney cells and NIH3T3 mouse fibroblast cells were obtained from ATCC (Manassas, VA) and grown in DMEM supplemented with 5% FBS. NCI-H3122 and NCI-H2228 (kindly provided by John D. Minna and Adi F. Gazdar, UTSW) were grown in RPMI with 10% FBS.
Mouse monoclonal against total ALK (#3791), rabbit polyclonal against phosphorylated ALK Tyr 1278/1282/1283 (#3983), mouse monoclonal against AKT (#2920), mouse monoclonal against total ERK p42/44 (#9107), rabbit polyclonal against phosphorylated ERK Thr202/Tyr204 (#9101), mouse monoclonal against total STAT3 (#9139), rabbit polyclonal against phosphorylated STAT3 Tyr705 (#9145) were purchased from Cell Signaling Technology (Beverly, MA). Rabbit polyclonal antibodies against phosphorylated AKT Ser474 (sc-135651) and mouse monoclonal antibodies against α-tubulin (#sc-8035) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). IRDye® 680LT- and 800CW-conjugated goat anti-mouse secondary antibodies were purchased from LI-COR Biotechnology (Lincoln, NE). PF-02341066 (crizotinib) was kindly provided by Pfizer and dissolved in DMSO for experiments.
Lentiviral constructs and transduction
The EML4-ALK gene fusion containing EML4 exons 1–6 variant and ALK exons 20–29 (E6a;A20) was cloned by RT-PCR from mRNA isolated from the cell line, H2228, and inserted into the lentiviral expression plasmid, pCDH-MCS1-EF1-puromycin from Systems Biosciences (Mountain View, CA). Point mutations were introduced into the EML4-ALK fusion gene using the QuikChange II XL Site-Directed Mutagenesis Kit from Agilent Technology (see Supplementary Table 1 for primers). Lentiviral transduction of Ba/F3, NIH3T3 and H3122 were carried out as previously described (19).
Immunoblotting
Immunoblotting was performed as previously described with minor modifications (20). Briefly, cells were lysed in modified RIPA buffer supplemented with Halt Protease and Phosphatase Inhibitor Cocktail purchased from Thermo Scientific (Waltham, MA). Total protein was separated by SDS-PAGE, transferred to nitrocellulose, and stained with the indicated primary antibodies. Protein detection was achieved by imaging with an Odyssey Imager and Odyssey Version 3.0 image analysis software from LI-COR (Lincoln, NE).
Proliferation and Soft Agar Colony Assays
Proliferation of Ba/F3 cells was measured using the Cell-titer 96 Aqueous Proliferation Assay from Promega (Madison, WI) according to the manufacturer’s instructions. Briefly, cells were seeded into 96-well plates 24 hours prior to drug treatment and proliferation was measured 72 hours after treatment. The absorbance at 490nm was measured in 96-well plates using a Microplate Reader from Molecular Devices (Sunnyvale, CA). The IC50 values were calculated using Prism v5.02 from GraphPad Software (La Jolla, CA).
In order to measure anchorage-independent growth and its inhibition by crizotinib, Ba/F3 cells expressing non-mutated or mutated cDNAs encoding EML4-ALK (E6a;A20) were suspended in media containing 0.4% agar and plated in 6-well plates containing media + 0.5% agar per well. Wells were fed every 3 days with media with the indicated concentrations of crizotinib and incubated for 14 days. Colonies were stained for 24 hours with NBT and photographs were taken for colony quantification with Metamorph Software from Molecular Devices (Sunnyvale, CA).
Results
Re-biopsy of patients following progression on crizotinib
Fourteen ALK+ patients underwent 15 re-biopsy procedures following radiologic evidence of disease progression/lack of response on crizotinib (Fig. 1B and S1A, Table S1). Typically, diagnostic biopsies were used as the pre-crizotinib (baseline) sample for comparison purposes within this study. Two patients were biopsied following disease progression at the first evaluation on crizotinib (intrinsic resistance). Twelve patients underwent biopsy following initial benefit then progression (acquired resistance) after median time on crizotinib of 8.9 months (range, 3.8–21.1 months). One patient underwent two separate biopsy procedures post-initiation of crizotinib, one after a lack of response (stable disease), followed by biopsy of a separate lesion after disease progression per RECIST (version 1.0).
Figure 1. Identification and characterization of a novel ALK Kinase domain mutation in patients.

Surface (A) and ribbon (B) models of the ALK kinase domain in complex with crizotinib (PDB 2XP2). Insets to the right represent a magnified view of the ATP- binding pocket where crizotinib is located. (A) and (B) were generated using the PyMol molecular graphics system. (C) Ba/F3 cells expressing wild-type EML4-ALK (E6;A20), or the same EML4-ALK construct with the specified ALK kinase domain mutations or empty vector were treated with the indicated concentration of crizotinib and viable cells were measured after 72 hours and then plotted relative to untreated controls. Ba/F3 cells expressing empty vector were grown in the presence of IL-3. (D) SDS-PAGE and immunoblotting analysis to detect the indicated proteins in cell lysates from NIH3T3 cells expressing wild-type EML4-ALK (E6;A20) and EML4-ALK (E6;A20) G1269A treated with the indicated doses of crizotinib for 5 hours. NIH3T3 with empty vector are also shown as a control. (E) Quantitation of western blot in (D) is graphically represented using LiCor image analysis software. (F) Ba/F3 cells expressing wild-type EML4-ALK (E6;A20), or the same EML4-ALK construct with the specified ALK kinase domain mutations or empty vector (with and without IL-3) were plated and viable cells were measured at the indicated time points and plotted.
Three patients (#1–3) failed to yield evaluable material from their biopsy. Frozen tumor tissue was collected from 4 patients and FFPE was collected on 11 patients. All eleven patients with evaluable tissue underwent repeat ALK FISH testing (Table 1). Attempts to propagate a cell line occurred for 8 patients. Currently only one cell line, from patient #10, is propagating at a rate permitting evaluation.
ALK kinase domain mutations as a resistance mechanism to crizotinib
Acquired mutations clustering around the kinase domain ATP binding site are a well-recognized mechanism of acquired resistance to tyrosine kinase inhibitors (TKIs) (7, 21, 22). We therefore performed direct sequencing of ALK exons 21–25, encoding the kinase domain. Four patients (#4–7) were found to have point mutations in the kinase domain of ALK (Table 2). None of these patients had sufficient tissue in their pre-crizotinib biopsy to determine if these mutations were detectable prior to crizotinib therapy. Patients #4 and #5 were found to have the previously described mutation encoding the L1196M substitution (data not shown). Two patients (#6 and 7) demonstrated the presence of a novel mutation that encodes a G1269A substitution (Table 2 Supplemental Fig. S2A, and data not shown).
Table 2.
Molecular analysis of re-biopsy samples of evaluable patients
| Mechanisms of Resistance |
|||||
|---|---|---|---|---|---|
| Patient # | ALK FISH | ALK FISH CNG | ALK kinase domain sequencea |
EGFR | KRAS |
| 4 | Positive | Negative | L1196M | WTa | WTa |
| 5 | Positive | Negative | L1196M | WTa | WTa |
| 6 | Positive | Negative | G1269A* | WTa,b | WTa,b |
| 7 | Positive | Positive | G1269A | WTb | WTb |
| 8 | Positive | Positive | WT | WTa | WTa |
| 9a | Negative | Negative | NA | L858R a,b | WTa,b |
| 9b | Positive | Negative | WT* | WTb | WTb |
| 10¶ | Positive/Negative§ | Negative | WT | ND | G12C§b |
| 11 | Positive | Negative | WT | WTa | G12Va |
| 12 | Negative | Negative | WT | WTb | WTb |
| 13¶ | Positive | Negative | WT* | ND | ND |
| 14 | Positive | Negative | WT | WTa | WTa |
Patients with intrinsic resistance
Direct sequencing
SNaPshot® includes APC, AKT1, BRAF, CTNNB1, EGFR, FLT3, JAK2, KIT, KRAS, MAP2K1 (MEK1), NOTCH1, NRAS, PIK3CA, PTEN, TP53
Heterozygous for rs3795850 ALK SNP
From patient-derived cell line with loss of ALK gene rearrangement by FISH or RT-PCR .The FFPE tissue was ALK FISH +.
A multiplexed RT-PCR assay for EML4-ALK was performed when possible to determine the variant of the ALK gene fusion and we successfully confirmed the presence of EML4-ALK in 4 patients. Interestingly, patient #7 demonstrated a new variant fusing exon 6 of EML4 to exon 19 of ALK (E6;A19) (Supplemental Fig. S3, Table 2). An EML4-ALK specific RT-PCR was performed on mRNA in order to selectively amplify and sequence the expressed transcripts of EML4-ALK (Supplemental Fig. S3A).
The amino acid substitutions encoded by the observed mutations were mapped onto the crystal structure of the ALK kinase domain (Fig. 1A and B) (23). All of the mutations detected in this series were found in, or near, the long narrow groove that comprises the binding pocket for both ATP and crizotinib. The L1196M substitution has been previously described in a patient with crizotinib resistance and is homologous to the gatekeeper mutations identified in BCR-ABL (T315I) and EGFR (T790M) (7, 12, 21).
The Gly 1269 residue is positioned at the end of the narrow ATP-binding pocket of ALK (Fig. 1A and B). Substitution of Gly1269 with the larger Ala residue would be expected to reduce binding of crizotinib due to steric hindrance. To determine whether the G1269A substitution produced resistance to crizotinib, the mutation encoding this substitution was generated in a cDNA encoding the E6a;A20 variant of EML4-ALK. Lentiviral vectors encoding wild-type EML4-ALK (E6a;E20) or the same cDNA encoding G1269A, C1156Y, or L1196M substitutions or empty vector were introduced into Ba/F3 cells. Proliferation assays in the presence of increasing doses of crizotinib were performed (Fig. 1C). The G1269A mutation induced crizotinib resistance that was intermediate between the previously identified mutations, C1156Y and L1196M. Similar results were obtained when these constructs were introduced into the NIH3T3 cell line and colony formation was measured in soft agar (Supplemental Fig. S4). Consistent with the observed cell line resistance, ALK phosphorylation and phosphorylation of downstream effectors, ERK, STAT3, and AKT were preserved at higher doses of crizotinib in the G1269A mutant compared to wild-type EML4-ALK (Fig. 1D and E).
Preclinical and clinical evidence suggests that the gatekeeper mutation T790M in EGFR mutant NSCLC may provide a growth disadvantage compared to NSCLC with an EGFR mutation lacking T790M (24, 25). We therefore examined the relative fitness of the G1269A, C1156Y, and L1196M mutations compared to wild-type EML4-ALK (variant E6a;A20) in the Ba/F3 cell system (Fig. 1F). We did not detect a growth disadvantage for any of the resistant mutations compared to wild-type. Indeed, the EML4-ALK constructs harboring resistance mutations induced increased proliferation compared to non-mutated EML4-ALK in the absence of crizotinib.
Copy number gain of the ALK gene rearrangement as a mechanism of crizotinib resistance
A gain in ALK gene fusion copy number has recently been implicated as a mechanism of resistance to crizotinib in vitro (26). In addition to standard FISH analysis for ALK, we measured the number of copies per cell of the ALK gene rearrangement before and after crizotinib treatment (15). Copy number gain (CNG) was defined as more than two-fold increase in the mean of the rearranged gene per cell in the post-treatment specimen compared with the pre-treatment specimen. Two patients demonstrated a marked increase in abnormal signal copy number (#7 at 5-fold and #8 at >4 fold), consistent with CNG of the ALK gene fusion (Table 1Fig. 2A and B). The CNG in these two patients was due both to more copies of the ALK rearrangement per cell and more cells displaying the rearrangement pattern.
Figure 2. ALK FISH pattern changes from pre- to post-crizotinib tumor samples.
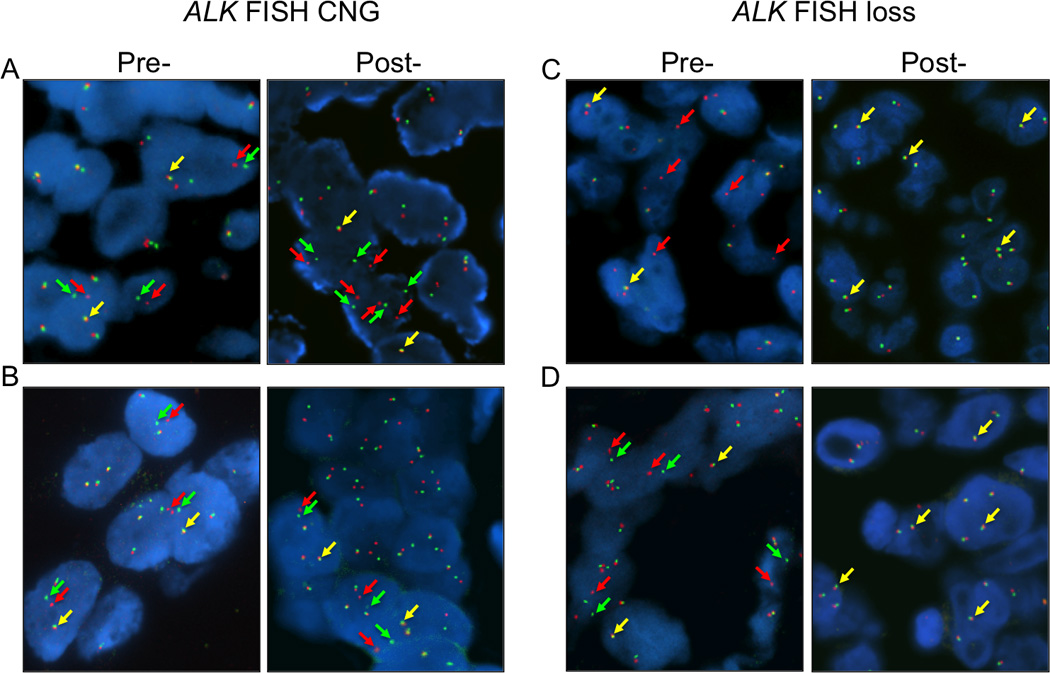
FISH analysis of patients #6 (A) and #7 (B) before crizotinib treatment (left) and following progression on crizotinib (right) demonstrating a gain of split green (5’) and red (3’) ALK signals per each tumor cell. FISH analysis of patients #8a (C) and 11 (D) before crizotinib treatment (left) and following progression on crizotinib (right) demonstrating loss of split green (5’) and red (3’) ALK signals.
EGFR mutations as a mechanism of crizotinib resistance
Patient #9 underwent two separate biopsy procedures each of different lesions. The first was performed after 61 days on crizotinib after the initial re-staging scans demonstrated stable disease, which was considered unusual given the responses seen in the majority of ALK positive patients treated with crizotinib (6). This biopsy demonstrated a lack of an ALK gene rearrangement by FISH (Fig. 2C). Further evaluation by direct sequencing demonstrated the presence of an EGFR exon 21 mutation encoding the L858R substitution (Table 2 Supplemental Fig. S2B) that was not present in the initial transbronchial biopsy used to establish the ALK+ diagnosis (data not shown). The EGFR mutation on this specimen was confirmed by SNaPshot analysis (data not shown). Interestingly, a second biopsy performed on a progressing liver lesion after 113 days on crizotinib did show an ALK gene rearrangement, but no evidence of an ALK kinase domain mutation, an EGFR mutation or of any other abnormal oncogenes as assessed by SNapSHOT (Tables 1 and 2, data not shown). To confirm that all samples were indeed from the same patient, the original diagnostic biopsy and both re-biopsy samples were fingerprinted using STR analysis, confirming the common genetic origin of the samples (data not shown).
Of note, Patient #1, who underwent a post-crizotinib biopsy but had no evaluable material (Supplemental Table S1), demonstrated the presence of an EGFR exon 20 mutation (S768I) in the pre-crizotinib biopsy sample in addition to the presence of an ALK gene rearrangement by FISH analysis. This patient received erlotinib therapy for more than 9 months before discontinuing it and beginning crizotinib.
KRAS mutations as a mechanism for crizotinib resistance
Patient #10 received only 27 days of crizotinib before disease progression was evident (Supplemental Table S1). Testing of the re-biopsy sample showed a persistent ALK+ FISH test but no further molecular testing was performed as the remainder of the tumor sample was used to initiate a cell line (Supplementary Fig. S5). Initial analysis of this cell line demonstrated marked resistance to crizotinib in comparison to two known EML4-ALK positive NSCLC cell lines (Fig. 3A). FISH analysis of this cell line later demonstrated no evidence of an ALK gene rearrangement and repeated RT-PCR analysis failed to show evidence of an EML4-ALK gene transcript (data not shown). SNaPshot analysis of the cell line demonstrated the presence of a KRAS G12C mutation and this was confirmed by direct sequencing (Supplemental Fig. S3C). Given the short duration until progression we asked whether this mutation was detectable in the pre-crizotinib sample. Direct sequencing of the microdissected pre-crizotinib biopsy, which had not been previously analyzed for KRAS, demonstrated the presence of the KRAS G12C mutation (Supplemental Fig. S3C).
Figure 3. Alternate activating oncogenes in patients with ALK+ NSCLC.
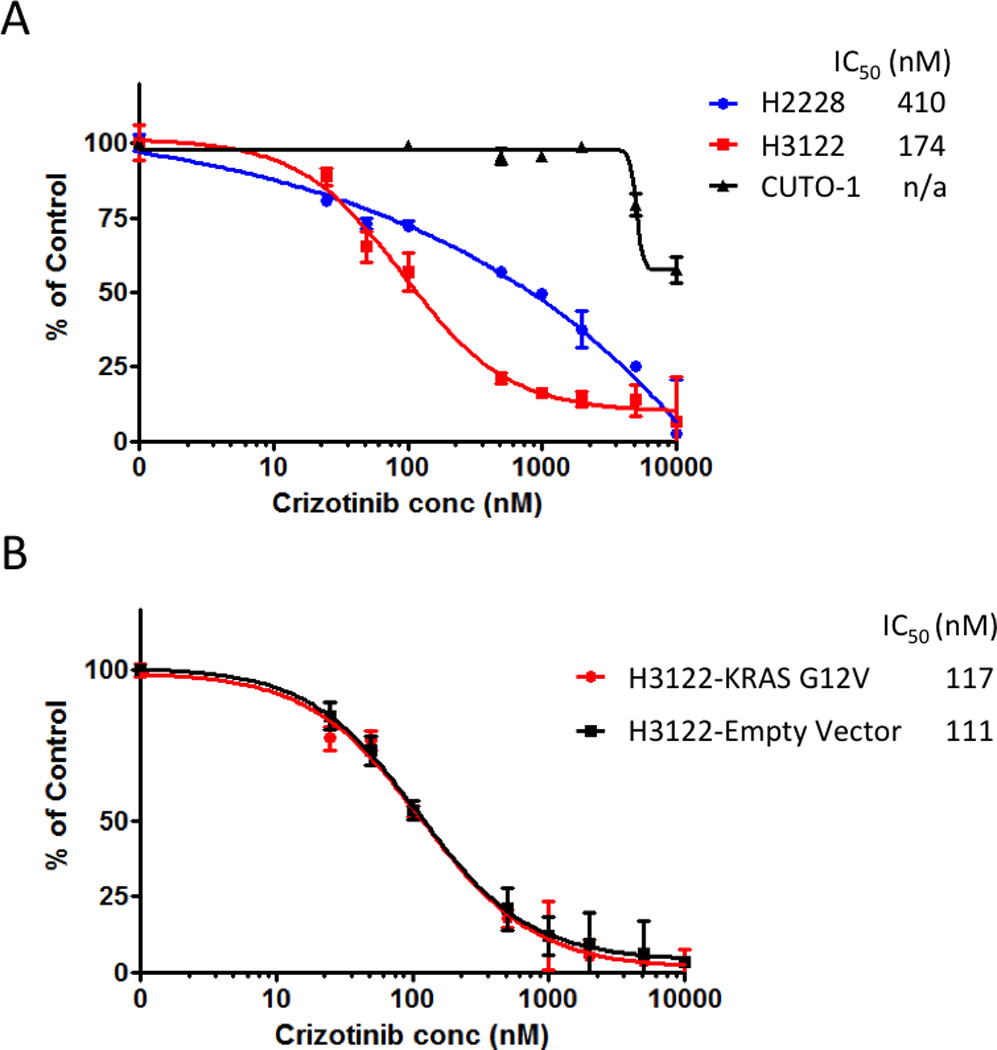
(A) H3122, H2228, and CUTO-1 cells (from patient #10) were treated with the indicated concentration of crizotinib and viable cells were measured after 72 hours and then plotted relative to untreated controls. (B) H3122 cells with expression of KRAS G12V or empty vector were treated with the indicated concentration of crizotinib and viable cells were measured after 72 hours and then plotted relative to untreated controls.
Patient #11 experienced a partial response to treatment, before progression in the liver after 7 months, when a biopsy was performed and demonstrated persistence of an ALK gene rearrangement as well as a KRAS mutation encoding the G12V substitution (Tables 1 and 2, Supplemental Fig. S3D). Analysis of the diagnostic biopsy showed no evidence of a pre-existing mutation in EGFR or KRAS (Supplemental Table S1).
In patient #11, we were unable to determine whether the ALK gene rearrangement and KRAS mutation occurred in the same or different tumor cells. Whereas in the case of patient #10, given that an ALK negative, KRAS positive cell line could be generated from a lesion in whom both abnormalities were present in the biopsy this means that ALK and KRAS positive cells must have existed as separate subclones within the same tumor. To further explore whether acquisition of a KRAS mutation could serve as a direct mechanism of acquired resistance to crizotinib in ALK positive cells, we asked whether expression of a mutant KRAS G12V could elicit crizotinib resistance in an EML4-ALK positive cell line (H3122), which is normally sensitive to crizotinib. KRAS G12V was selected because it was the form observed in patient #11, where the co-existence in the same cells could not be formally evaluated. Mutant KRAS G12V or empty vector was introduced into H3122 and cell proliferation was measured after exposure to increasing doses of crizotinib (Fig. 3B). The IC50 of H3122 expressing KRAS G12V was not significantly different from H3122 harboring the empty vector.
Emergence of an ALK gene fusion negative tumor
All post-crizotinib biopsy samples with evaluable tumor tissue underwent repeat ALK FISH testing. In patient #10, an ALK gene fusion was not observed in the cells which were propagated from the rebiopsied supraclavicular fossa lesion, which were later shown to be KRAS mutant. In patient #9, an ALK gene fusion was not observed in the rebiopsied supraclavicular fossa lesion, which was later shown to harbor and EGFR mutation. Patient #12 also lacked an ALK gene fusion as detected by FISH in the rebiopsy of the infraclavicular lymph node (Table 1Fig. 2D). RT-PCR of the post-crizotinib sample from patient #12 using a multiplexed assay to detect different EML4-ALK variants also failed to demonstrate the presence of an ALK gene fusion (data not shown). Unlike with patients #10 and #11, no other abnormality was detected in the evaluated genes in this patient using the SNaPshot assay.
Unknown mechanisms of crizotinib resistance
Patients #13 and #14 demonstrated the presence of an ALK gene rearrangement by FISH analysis following progression on crizotinib (Table 1). Patient #13 showed no evidence of ALK gene CNG or loss and no evidence of an ALK kinase domain mutation. Patient #14 showed no evidence of ALK gene CNG or loss, no evidence of an ALK kinase domain mutation, and no evidence of an EGFR or KRAS mutation (Table 1 and 2).
Discussion
Here we describe the molecular mechanisms of resistance in a large series of ALK+ NSCLC patients with progression o n crizotinib therapy. Previous studies have described the development of in vitro resistance mechanism using cell lines grown in the presence of crizotinib (26–29). Selected clinical cases of crizotinib resistance have been described, but without a denominator to estimate the frequency of the given abnormalities (12, 13). In this study, we successfully obtained molecular data on 11 of 14 rebiopsied ALK+ NSCLC patients with progression on crizotinib. A specific potential resistance mechanism was identified in 9 of these cases (Fig. 4A).
Figure 4. Relative frequencies of crizotinib resistance mechanisms in ALK+ NSCLC patients and models for potential mechanisms of alternate oncogene acquisition.

(A) The wedges represent different molecular mechanisms of resistance identified in ALK+ NSCLC patients in this study. The blue arc represents presumed or confirmed presence of an alternate oncogene. The yellow arc represents copy number gain (CNG). The red arc represents the presence of an ALK kinase domain mutation. The grey wedge represents those patients where an ALK gene rearrangement was observed, but no mechanism of resistance was identified. *Denotes inclusion of one patient with intrinsic resistance within this category. (B) Model #1 depicts the low level presence of a second oncogenic driver in the same cell as an ALK gene rearrangement, which following treatment with crizotinib becomes the dominant clone. Model #2 depicts the presence of separate clonal populations, some with an ALK gene rearrangement as the driver and others with an alternate oncogene driver (e.g., KRAS or EGFR). Following treatment with crizotinib, the non-ALK clones become the dominant clone.
Precedent exists for the emergence of kinase domain mutations as mechanisms of resistance, most notably T315I in BCR-ABL and T790M in EGFR (7, 21, 22). We identified four patients (36%) with acquired resistance mutations in ALK. Two patients had the previously described L1196M mutation, in the classical gatekeeper position homologous to T315I and T790M (12). L1196M levels were low in one patient but were confirmed by RFLP analysis (data not shown). Two patients demonstrated a novel G1269A mutation. One of these patients also harbored a novel ALK gene fusion involving exon 6 of EML4 and exon 19 of ALK (Supplementary Fig. 3). All previously published ALK gene fusions involve exon 20 of ALK. Mutations at position G1269 have previously been identified using an in vitro mutagenesis screen (29). In vitro studies with G1269A demonstrate persistent ALK phosphorylation and downstream effector phosphorylation at higher doses of crizotinib than in the wild-type. Decreased growth inhibition from crizotinib was also observed with G1269A relative to the wild-type. Interrogation of the ALK crystal structure bound to crizotinib reveals G1269 to be critically situated in the ATP-binding pocket (Fig. 1A and B). Crizotinib binding in this pocket is unlikely to be able to tolerate larger amino acid substitutions in either G1269 or L1196. Replacement of the Leu side chain with a longer thioether side chain (L1196M) has been shown to significantly compromise the interaction with crizotinib (12). Similarly, a bulkier Ala in place of Gly1269 would preclude proper binding of the halogenated aromatic ring of crizotinib (Fig. 1A and B). One of the patients with a G1269A mutation also demonstrated a novel EML4-ALK fusion variant (E6; A19). All previously described gene fusions involving ALK in NSCLC fuse different exons of EML4 to exon 20 of ALK.
The observation that 4 of 10 resistant samples had ALK kinase domain mutation suggests that resistance mutations in ALK+ NSCLC will likely be a common mechanism of resistance to crizotinib, similar to that of EGFR TKI resistance (9). However, unlike EGFR TKI resistance in NSCLC, there appears to be a greater diversity of resistance mutations as 4 different mutations have now been identified in ALK+ NSCLC patients (12, 13). This is reminiscent of CML patients treated with imatinib, in which multiple different mutations in the ABL kinase domain have been identified (30). One can speculate that the kinase domain of EGFR may be more constrained given the presence of an existing activating mutation and may only be able to tolerate a very limited set of mutations that inhibit drug binding but still allow constitutive activation. In contrast EML4-ALK, like BCR-ABL, has a native kinase domain structure that may be able to tolerate a wider spectrum of resistance mutations that inhibit crizotinib binding but still allow constitutive activation. Consistent with this hypothesis is the in vitro data demonstrating that the ALK resistance mutations studied here exhibited enhanced growth compared to the non-mutated EML4-ALK in an isogenic cell line. In contrast, EGFR T790M mutations confer impaired growth compared to cells carrying EGFR activating mutations alone (25).
The existence of multiple different resistance mutations in ALK may have important clinical implications. First, testing for resistance mutations will require direct sequencing of multiple exons or a multiplexed assay looking for different specific mutations as data emerge. Second, the possibility of different mutations existing in the same patient has to be considered, increasing the difficulty of detecting each mutation. This is in addition to multiple other mechanisms that could affect detection including allelic dilution or a mixture of mutant and non-mutant cells in the biopsy sample (31, 32). Consequently, some resistance mutations may be missed and could conceivably be present in either of the patients in our study who retained ALK but appeared to have an unknown mechanism of resistance, or in the patient with CNG alone as their apparent mechanism of resistance. CNG alone has been described as a potential mechanism of resistance in vitro, however this appeared to be a precursor state to the development of a resistance mutation (26). Finally, based on our own and others preclinical work different mutations appear to have different sensitivity rank orderings to crizotinib and this ordering may differ between ALK inhibitors (Fig. 1C and Supplemental Fig. S4) (29, 33). Therefore, the optimal exposure of any ALK inhibitor in the TKI naïve setting may differ from that in the acquired resistance setting. In addition, beyond dosing and toxicity issues, different ALK inhibitors may have to be prioritized depending on the specific mutation involved.
Seven out of 11 patients in this study did not have ALK kinase domain mutations, which led us to investigate alternate oncogenes as contributors to resistance. Prior studies have demonstrated the coexistence of both an EGFR activating mutation or a KRAS mutation and an ALK gene rearrangement in the same tumor sample (13, 34–36). Here we describe 2 cases with a KRAS mutation (one of which was retrospectively detected in the pre-crizotinib specimen) and one patient with an EGFR activating mutation at the time of resistance. An additional patient had a coexistent EGFR S768I mutation and an ALK gene rearrangement in the pre-crizotinib tumor sample, but no evaluable tissue following progression on crizotinib. The presence of EGFR and ALK, or KRAS and ALK in two of the pre-crizotinib tumor samples is consistent with results from a large cohort of ALK+ patients showing that 3 of 38 (8%) ALK+ NSCLC patients also demonstrated the presence of a KRAS or EGFR mutation (37).
The presence of multiple oncogenes in a tumor sample raises the question of whether these occur in the same tumor cells or different tumor cells and how these different oncogenic drivers arise (Fig. 4B). In EGFR mutant NSCLC with acquired resistance to EGFR TKIs via MET gene amplification, in vitro data suggest that MET gene amplification occurs as a secondary event in the same tumor cell as depicted in model #1 (Acquisition of Second Oncogenic Driver) (11). This can be detected at low levels in the biopsies taken prior to EGFR TKI treatment (38). Treatment with an EGFR TKI selects for those clones with an EGFR activating mutation and MET amplification. We suggest a different model by which an alternate oncogene provides resistance to crizotinib in ALK+ NSCLC. In model #2 (Emergence of a Separate Oncogenic Driver), EGFR or KRAS mutations exist in separate subclonal populations that lack an ALK gene rearrangement. The presence of EGFR/ALK or KRAS/ALK double positive results in biopsies prior to crizotinib treatment cannot distinguish model #1 from model #2. However, the outgrowth of a KRAS mutant, ALK negative cell line from patient #10 confirms that model #2 can occur clinically. Similarly, although we did not have cell line data from patient #11 who manifested both ALK and KRAS positivity following progression, the fact that the introduction of the KRAS mutation seen in patient #11 into an ALK positive cell line did not appear to alter their sensitivity to crizotinib in vitro argues against model #1, at least with regard to KRAS and ALK. In contrast to KRAS activating mutations, introduction of an EGFR activating mutation into H3122 cells in vitro is sufficient to induce crizotinib resistance, suggesting that in some situations the acquisition of a second driver within the same ALK positive cells could act as a mechanism of resistance clinically (model #1) (13). However, as an EGFR activating mutation was noted in progressing lesions without evidence of a persistent ALK gene rearrangement in patient #9, clinically, even for EGFR, model #2 can also exist. We also cannot exclude the presence of two separate primary cancers in this patient. Formally, whether separate oncogenic driver subclones arise completely independently, whether they share a common progenitor that lacks either driver and these drivers are developed independently as later events, or whether both drivers co-exist within the same cell, and then one or other is lost in subclonal evolution is unclear.
Emergence of an ALK gene fusion negative tumor was observed in one patient where another oncogenic driver was not identified. Given the limited tumor sample and lack of a cell line in this patient, we were unable to query the presence of other oncogenic drivers beyond selected alleles of the genes evaluated in the SNaPshot panel. However, given that the emergence of an ALK negative tumor was associated with definite evidence of a separate oncogenic driver in both patients #9 and #10, the assumption is that some other as yet unidentified oncogenic driver is present in these cells for them to persist. It should also be noted that in cases of the emergence of an ALK FISH negative tumor that the percent positive cells was not zero, consistent with background noise in the break apart FISH assay as described previously (39).
ALK CNG was observed in two patients. One patient had ALK CNG in conjunction with an ALK mutation and one had CNG without another detectable oncogene or mutation being present. The percentage cells positive for a rearrangement was greater following CNG consistent with previous findings (15). A cell line that was partially crizotinib resistant in vitro apparently due to ALK gene fusion amplification alone was recently described (26). Increase of BCR-ABL copy number in CML serves as a precedent for this mechanism of resistance (40). In contrast, EGFR CNG has been more associated with EGFR TKI sensitivity rather than resistance (41, 42).
We have described a series of ALK positive NSCLC patients with intrinsic or acquired resistance to crizotinib. Multiple different mechanisms appear to occur (Fig. 4). We would predict that in patients in whom ALK remains the dominant driver of their cancers (those with kinase domain mutations and ALK CNG), different therapies primarily directed towards the ALK protein, either TKIs or HSP90 inhibitors, may be beneficial (43, 44). However, when separate or second drivers occur – drug combinations or broader based treatments such as cytotoxic chemotherapies may be required (13, 45). If understanding the heterogeneity present in NSCLC is important enough to direct patients to the correct initial therapy then it is becoming clear that re-biopsying and re-analyzing cancers as they stop responding may be equally important as resistance is not occurring through a single mechanism (37). Fully understanding the basis and frequency of the different mechanisms of resistance to crizotinib that are emerging will help us to continue to exploit personalized medicine approaches when considering how to overcome crizotinib resistance in ALK+ NSCLC patients in the future.
Translational Relevance.
Crizotinib is an orally bioavailable, small molecule tyrosine kinase inhibitor (TKI) that was approved by the FDA in 2011 for use in patients with ALK FISH+ NSCLC. Crizotinib provides significant clinical benefit for ALK positive NSCLC. Unfortunately, it is expected that not all ALK+ patients will benefit and those patients who do respond will eventually experience resistance of their NSCLC to crizotinib. In the same way that using targeted therapies in patients with molecularly defined cancer has been successful, it is anticipated that understanding the molecular mechanisms of resistance to targeted therapies in cancer will lead to therapeutic strategies to overcome resistance in the same population of patients. Here we describe the molecular mechanisms of resistance to crizotinib in a series of ALK+ NSCLC patients.
Supplementary Material
Supplemental Figure S1. (A) Schema for ALK+ NSCLC patients included in this study of crizotinibresistance. (B) PET/CT Imaging of patient #7 demonstrating a left adrenal gland metastasis prior to crizotinib (left), complete response by PET after 41 days of crizotinib (center), followed by regrowth after 197 days of crizotinib therapy at which time the adrenal gland was resected.
Supplemental Figure S2. DNA sequence analysis of ALK, EGFRand KRAS mutations. (A) Forward (top) and reverse (middle) chromatograms from direct sequencing of ALK exon 25 amplified from genomic DNA (gDNA) demonstrating at GGA→GCA mutation (G1269A) in patient #7. Direct sequencing of the EML4-ALK (E6;A19) RT-PCR product is also shown (bottom). (B) Chromatogram from direct sequencing of EGFR exon 21 amplified from gDNA demonstrating a CTG→CGG mutation (L858R) in patient #9a. (C) Chromatogram from direct sequencing of KRAS exon 2 demonstrating at GGT→TGT mutation (G12C) in the cell line CUTO-1 derived from patient #10 (left). Chromatogram from direct sequencing of KRAS exon 2 demonstrating at GGT→TGT mutation (G12C) in pre-crizotinib biopsy from patient #10 (right). (D) Chromatogram from direct sequencing of KRAS exon 2 demonstrating at GGT→GTT mutation (G12V) in patient #11.
Supplemental Figure S3. A novel ALK gene rearrangement. (A) Chromatogram from direct sequencing of an RT-PCR reaction demonstrating exon 6 of EML4 fused to exon 19 of ALK from patient #7. (B) Chromatogram from direct sequencing of an RT-PCR reaction demonstrating exon 6 of EML4 fused to exon 20 of ALK from patient #6 for comparison.
Supplemental Figure S4. Soft agar colony formation by NIH3T3 cells expressing EML4-ALK (E6;A20) with non-mutated ALK, G1269A, L1196M, or C1156Y in the presence of increasing doses of crizotinib. Photographs of soft agar plates are displayed in (A). Colony counts are plotted against crizotinib dose in (B).
Supplemental Figure S5. FISH analysis of patient #10 before crizotinib treatment (A) and after progression on crizotinib treatment (B) demonstrating a similar pattern of split green (5’) and red (3’) ALK signals in tumor cells.
Acknowledgements
We would like to thank Barbara A. Helfrich and Christopher Korch for technical assistance and Delee A. Maxson for administrative assistance on this manuscript.
Grant Support
Financial Support: This research was supported by the University of Colorado Lung Cancer SPORE grant (P50CA058187) to RCD, MVG, and LEH, by a research grant from Eli Lilly & Co. to RCD and DRC, and by funds from the Boettcher Foundation’s Webb-Waring Biomedical Research Program to RCD.
Footnotes
Conflict of Interest Statement: RCD has research grants from Pfizer, Eli Lilly, and ImClone. RCD and MVG have received speaker’s fees from Abbott Molecular. RCD, MVG, DRC, KLK, AJW, and DLA have served as consultants for Pfizer. MVG has served as a consultant for Abbott Molecular. DRC has served as a consultant/advisory board member for Chugai, Ariad, and Eli Lilly. DLA has served as a consultant for GSK.
References
- 1.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 3.Scagliotti GV, Parikh P, von Pawel J, Biesma B, Vansteenkiste J, Manegold C, et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26:3543–3551. doi: 10.1200/JCO.2007.15.0375. [DOI] [PubMed] [Google Scholar]
- 4.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994;263:1281–1284. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 5.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 6.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104:20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 12.Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363:1734–1739. doi: 10.1056/NEJMoa1007478. [DOI] [PubMed] [Google Scholar]
- 13.Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, et al. A Novel ALK Secondary Mutation and EGFR Signaling Cause Resistance to ALK Kinase Inhibitors. Cancer Res. 2011;71:6051–6060. doi: 10.1158/0008-5472.CAN-11-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, Felicioni L, et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol. 2009;27:1667–1674. doi: 10.1200/JCO.2008.19.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Camidge DR, Theodoro M, Maxson DA, Skokan M, O'Brien T, Lu X, et al. Correlations between the percentage of tumor cells showing an ALK gene rearrangement, ALK signal copy number and response to crizotinib therapy in ALK FISH positive non-small cell lung cancer. Cancer. 2011 doi: 10.1002/cncr.27411. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455:971–974. doi: 10.1038/nature07399. [DOI] [PubMed] [Google Scholar]
- 17.Takeuchi K, Choi YL, Soda M, Inamura K, Togashi Y, Hatano S, et al. Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clin Cancer Res. 2008;14:6618–6624. doi: 10.1158/1078-0432.CCR-08-1018. [DOI] [PubMed] [Google Scholar]
- 18.Dias-Santagata D, Akhavanfard S, David SS, Vernovsky K, Kuhlmann G, Boisvert SL, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010;2:146–158. doi: 10.1002/emmm.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doebele RC, Schulze-Hoepfner FT, Hong J, Chlenski A, Zeitlin BD, Goel K, et al. A novel interplay between Epac/Rap1 and mitogen-activated protein kinase kinase 5/extracellular signal-regulated kinase 5 (MEK5/ERK5) regulates thrombospondin to control angiogenesis. Blood. 2009;114:4592–4600. doi: 10.1182/blood-2009-04-217042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong J, Doebele RC, Lingen MW, Quilliam LA, Tang WJ, Rosner MR. Anthrax edema toxin inhibits endothelial cell chemotaxis via Epac and Rap1. J Biol Chem. 2007;282:19781–19787. doi: 10.1074/jbc.M700128200. [DOI] [PubMed] [Google Scholar]
- 21.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 22.Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A. 2005;102:7665–7670. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee CC, Jia Y, Li N, Sun X, Ng K, Ambing E, et al. Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem J. 2010;430:425–437. doi: 10.1042/BJ20100609. [DOI] [PubMed] [Google Scholar]
- 24.Oxnard GR, Arcila ME, Sima CS, Riely GJ, Chmielecki J, Kris MG, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res. 2011;17:1616–1622. doi: 10.1158/1078-0432.CCR-10-2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chmielecki J, Foo J, Oxnard GR, Hutchinson K, Ohashi K, Somwar R, et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci Transl Med. 2011;3:90ra59. doi: 10.1126/scitranslmed.3002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011;108:7535–7540. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heuckmann JM, Holzel M, Sos ML, Heynck S, Balke-Want H, Koker M, et al. ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin Cancer Res. 2011 doi: 10.1158/1078-0432.CCR-11-1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sasaki T, Okuda K, Zheng W, Butrynski J, Capelletti M, Wang L, et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010;70:10038–10043. doi: 10.1158/0008-5472.CAN-10-2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang S, Wang F, Keats J, Zhu X, Ning Y, Wardwell SD, et al. Crizotinib-Resistant Mutants of EML4-ALK Identified Through an Accelerated Mutagenesis Screen. Chem Biol Drug Des. 2011;78:999–1005. doi: 10.1111/j.1747-0285.2011.01239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von Bubnoff N, Schneller F, Peschel C, Duyster J. BCR-ABL gene mutations in relation to clinical resistance of Philadelphia-chromosome-positive leukaemia to STI571: a prospective study. Lancet. 2002;359:487–491. doi: 10.1016/S0140-6736(02)07679-1. [DOI] [PubMed] [Google Scholar]
- 31.Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borras AM, Gale CM, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006;116:2695–2706. doi: 10.1172/JCI28656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yatabe Y, Matsuo K, Mitsudomi T. Heterogeneous distribution of EGFR mutations is extremely rare in lung adenocarcinoma. J Clin Oncol. 2011;29:2972–2977. doi: 10.1200/JCO.2010.33.3906. [DOI] [PubMed] [Google Scholar]
- 33.Heuckmann JM, Holzel M, Sos ML, Heynck S, Balke-Want H, Koker M, et al. ALK Mutations Conferring Differential Resistance to Structurally Diverse ALK Inhibitors. Clin Cancer Res. 2011;17:7394–7401. doi: 10.1158/1078-0432.CCR-11-1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tiseo M, Gelsomino F, Boggiani D, Bortesi B, Bartolotti M, Bozzetti C, et al. EGFR and EML4-ALK gene mutations in NSCLC: a case report of erlotinib-resistant patient with both concomitant mutations. Lung Cancer. 2011;71:241–243. doi: 10.1016/j.lungcan.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 35.Martelli MP, Sozzi G, Hernandez L, Pettirossi V, Navarro A, Conte D, et al. EML4-ALK rearrangement in non-small cell lung cancer and non-tumor lung tissues. Am J Pathol. 2009;174:661–670. doi: 10.2353/ajpath.2009.080755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuo YW, Wu SG, Ho CC, Shih JY. Good response to gefitinib in lung adenocarcinoma harboring coexisting EML4-ALK fusion gene and EGFR mutation. J Thorac Oncol. 2010;5:2039–2040. doi: 10.1097/JTO.0b013e3181f43274. [DOI] [PubMed] [Google Scholar]
- 37.Kris MG, Johnson BE, Kwiatkowski DJ, Iafrate AJ, Wistuba II, Aronson SL, et al. Identification of driver mutations in tumor specimens from 1,000 patients with lung adenocarcinoma: The NCI’s Lung Cancer Mutation Consortium (LCMC) J Clin Oncol. 2011;29 abstr CRA7506. [Google Scholar]
- 38.Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Camidge DR, Kono SA, Flacco A, Tan AC, Doebele RC, Zhou Q, et al. Optimizing the detection of lung cancer patients harboring anaplastic lymphoma kinase (ALK) gene rearrangements potentially suitable for ALK inhibitor treatment. Clin Cancer Res. 2010;16:5581–5590. doi: 10.1158/1078-0432.CCR-10-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.le Coutre P, Tassi E, Varella-Garcia M, Barni R, Mologni L, Cabrita G, et al. Induction of resistance to the Abelson inhibitor STI571 in human leukemic cells through gene amplification. Blood. 2000;95:1758–1766. [PubMed] [Google Scholar]
- 41.Hirsch FR, Varella-Garcia M, Cappuzzo F, McCoy J, Bemis L, Xavier AC, et al. Combination of EGFR gene copy number and protein expression predicts outcome for advanced non-small-cell lung cancer patients treated with gefitinib. Ann Oncol. 2007;18:752–760. doi: 10.1093/annonc/mdm003. [DOI] [PubMed] [Google Scholar]
- 42.Gandhi J, Zhang J, Xie Y, Soh J, Shigematsu H, Zhang W, et al. Alterations in genes of the EGFR signaling pathway and their relationship to EGFR tyrosine kinase inhibitor sensitivity in lung cancer cell lines. PLoS One. 2009;4:e4576. doi: 10.1371/journal.pone.0004576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Normant E, Paez G, West KA, Lim AR, Slocum KL, Tunkey C, et al. The Hsp90 inhibitor IPI-504 rapidly lowers EML4-ALK levels and induces tumor regression in ALK-driven NSCLC models. Oncogene. 2011;30:2581–2586. doi: 10.1038/onc.2010.625. [DOI] [PubMed] [Google Scholar]
- 44.Zhang S, Wang F, Keats J, Zhu X, Ning Y, Wardwell SD, et al. Crizotinib-Resistant Mutants of EML4-ALK Identified Through an Accelerated Mutagenesis Screen. Chem Biol Drug Des. 2011 doi: 10.1111/j.1747-0285.2011.01239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Camidge DR, Kono SA, Lu X, Okuyama S, Baron AE, Oton AB, et al. Anaplastic lymphoma kinase gene rearrangements in non-small cell lung cancer are associated with prolonged progression-free survival on pemetrexed. J Thorac Oncol. 2011;6:774–780. doi: 10.1097/JTO.0b013e31820cf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. (A) Schema for ALK+ NSCLC patients included in this study of crizotinibresistance. (B) PET/CT Imaging of patient #7 demonstrating a left adrenal gland metastasis prior to crizotinib (left), complete response by PET after 41 days of crizotinib (center), followed by regrowth after 197 days of crizotinib therapy at which time the adrenal gland was resected.
Supplemental Figure S2. DNA sequence analysis of ALK, EGFRand KRAS mutations. (A) Forward (top) and reverse (middle) chromatograms from direct sequencing of ALK exon 25 amplified from genomic DNA (gDNA) demonstrating at GGA→GCA mutation (G1269A) in patient #7. Direct sequencing of the EML4-ALK (E6;A19) RT-PCR product is also shown (bottom). (B) Chromatogram from direct sequencing of EGFR exon 21 amplified from gDNA demonstrating a CTG→CGG mutation (L858R) in patient #9a. (C) Chromatogram from direct sequencing of KRAS exon 2 demonstrating at GGT→TGT mutation (G12C) in the cell line CUTO-1 derived from patient #10 (left). Chromatogram from direct sequencing of KRAS exon 2 demonstrating at GGT→TGT mutation (G12C) in pre-crizotinib biopsy from patient #10 (right). (D) Chromatogram from direct sequencing of KRAS exon 2 demonstrating at GGT→GTT mutation (G12V) in patient #11.
Supplemental Figure S3. A novel ALK gene rearrangement. (A) Chromatogram from direct sequencing of an RT-PCR reaction demonstrating exon 6 of EML4 fused to exon 19 of ALK from patient #7. (B) Chromatogram from direct sequencing of an RT-PCR reaction demonstrating exon 6 of EML4 fused to exon 20 of ALK from patient #6 for comparison.
Supplemental Figure S4. Soft agar colony formation by NIH3T3 cells expressing EML4-ALK (E6;A20) with non-mutated ALK, G1269A, L1196M, or C1156Y in the presence of increasing doses of crizotinib. Photographs of soft agar plates are displayed in (A). Colony counts are plotted against crizotinib dose in (B).
Supplemental Figure S5. FISH analysis of patient #10 before crizotinib treatment (A) and after progression on crizotinib treatment (B) demonstrating a similar pattern of split green (5’) and red (3’) ALK signals in tumor cells.