

Summary
The nature and site of tumor-antigen presentation to immune T cells by bone-marrow-derived cells within the tumor microenvironment remains unresolved. We generated a fluorescent mouse model of spontaneous immuno-evasive breast cancer and identified a subset of myeloid cells with significant similarity to dendritic cells and macrophages that constitutively ingest tumor-derived proteins and present processed tumor antigens to reactive T cells. Using intravital live-imaging, we determined that infiltrating tumor-specific T cells engage in long-lived interactions with these cells, proximal to the tumor. In vitro, these cells capture cytotoxic T cells in signaling-competent conjugates, but do not support full-activation or sustain cytolysis. The spatiotemporal dynamics revealed here implicate non-productive interactions between T cells and antigen presenting cells on the tumor margin.
Introduction
Despite the recruitment of tumor-specific CD8+ tumor-infilitrating lymphocytes (TILs) to the tumor microenvironment, the immune response is limited in its ability to clear tumors (Drake et al. 2006). Numerous lines of evidence suggest that tolerance to tumors relies on presentation of peptide antigens on major histocompatibility complex (MHC) molecules on the surface of bone-marrow-derived antigen presenting cells (APC) as is the case for other peripheral tissues (Adler et al. 1998; Heath and Carbone 2001; Kusmartsev et al. 2005; Sotomayor et al. 2001). In recent years, broad classes of cell types derived from the mononuclear phagocytic lineage (MPS), such as myeloid-derived suppressor cells (MDSC) and tumor associated macrophages (TAM), have been implicated in promoting tumor growth and metastasis while inhibiting a productive immune response by T cells (Gabrilovich and Nagaraj 2009; Pollard 2009). TAM increase metastasis in the PyMT breast cancer model (Lin et al. 2006), partly as a result of T cell skewing toward a Th2 phenotype (DeNardo et al. 2009). Isolated MDSC bearing the marker Gr-1, in contrast, inhibit T cell activation and suppress the activation of T cells by secondary APCs or other stimuli (Gabrilovich et al. 2009; Terabe et al. 2003). There has, however, been no direct identification of the cell type that physically mediates antigen uptake and presentation in the tumor microenvironment.
Dendritic cells (DC), another cell type derived from the MPS that are very similar to macrophages, are the pre-eminent APC for T cells in lymphoid organs and in tissues. DCs in this setting are clearly integral in activating T cells but may also serve to tolerize them (Hawiger et al. 2001). The role of DCs in tumors (TuDC) is less well understood, but their presence is extensively documented. While the elicitation of a potent T cell expansion by DCs is clearly an integral part of a successful immunotherapy and can be augmented by transferring antigen-pulsed DC to hosts (Mayordomo et al. 1995), DCs in the tumor microenvironment may also be an important aspect of immune dysregulation. For example, the presence of specific subsets of DC, especially CD123+ plasmacytoid DC (pDC), are associated with negative prognosis in human patients (Ambe et al. 1989; Treilleux et al. 2004).
Delineating cell types of the MPS purely on surface phenotype has proven difficult because TAM, MDSC and DC share many common lineage markers. While direct staining of these cells from sections provides information about the populations in aggregate, it is likely that the MPS contributes a variety of cell types to the microenvironment, all of which might be the primary APC for T cells. Real-time intravital imaging has shed light on key processes during priming in the lymph node including the priming of T cells on DC (Miller et al. 2002; Mempel et al. 2004), and holds promise for delineating subsets of APC that are responsible for interacting with T cells. It has the benefit of complementing phenotypic surface marker data with morphological and behavioral phenotypes. Imaging of ectopic thymomas (EL4) using labeled TCR transgenic cells has revealed long-lived antigen-dependent contacts as well as effective killing between T cells and the tumor cells during tumor rejection (Mrass et al. 2006). However, in those models, there was no possibility to visualize or define the APCs of the microenvironment, or to study T cell interaction with such bone-marrow-derived APCs. Furthermore, introduction of tumor-specific T cells in these models leads to tumor regression, unlike the case in typical refractory tumors. Thus, the T-APC interactions that accompany tumor tolerance have, as yet, remained largely unresolved.
To gain insights into the nature of tumor antigen-presentation and the nature of T cell-APC interactions in refractory tumors, we have generated a spontaneous tumor model of human breast cancer based on the well-described MMTV-PyMT model (Guy et al. 1992) that incorporates tags for both imaging and for T cell characterization. This model allows us to track uptake of a co-expressed protein-fluorophore from tumor into the APC compartment. This study effectively exposes a key cell type and its antigen uptake, presentation and activation capacities, as well as placing these activities spatially within the tumor microenvironment.
Results
Recruitment and Inactivity of Tumor-specific T cells in a Spontaneous Model of Breast Cancer
To provide a method for tracking the flow of antigens from tumor to T cell, we generated a spontaneous mouse model for breast cancer, in which the initiating oncogene and a neo tumor antigen are co-expressed with the stable fluorescent protein mCherry, under a common breast epithelium-specific promoter. For this, we adapted the extensively characterized mouse mammary tumor virus-polyoma middle T (MMTV-PyMT) transgenic cassette (Guy et al. 1992), whose expression gives rise to disease in mice that closely models many aspects of spontaneous luminal breast cancer, including characteristic stages from hyperplasia to adenoma to carcinoma and ultimately to metastatic disease (Lin et al., 2003). We introduced two self-cleaving P2A sequences (de Felipe et al. 2003) downstream of the PyMT cDNA to produce the mCherry and ovalbumin proteins (OVA) (Fig. 1A). We included mCherry expression to aid in visualization of transformed cells and to track antigen uptake in the microenvironment. We included OVA to generate germline-encoded self-antigens that were specific to cells expressing the oncogene (“tumor self”) and could be recognized by well-characterized CD8 T cells expressing the OT-I T cell receptor. By co-expressing these genes in a common MMTV-driven transcript we ensured the co-production of each protein, in concert with onset of transformation, and yet allowed both mCherry and OVA to be processed independently, thus preventing premature degradation. The expression of these proteins as distinct polypeptides was demonstrated by western blotting (Fig. 1B) where each individual protein is detected as a single band at the expected molecular weight. The onset of tumors in this ‘PyMT ChOVA’ line was approximately 20 weeks of age on the C57BL/6 background (Fig. S1). The >95% penetrance of tumors and onset at times similar to the parental PyMT strain on C57/BL6 background (data not shown) suggested that the inclusion of the mCherry and OVA proteins provided little benefit to the immune system's ability to recognize or control the tumor.
Figure 1. Recruitment and Ineffectiveness of Tumor-specific T cells in a Spontaneous Model of Breast Cancer.
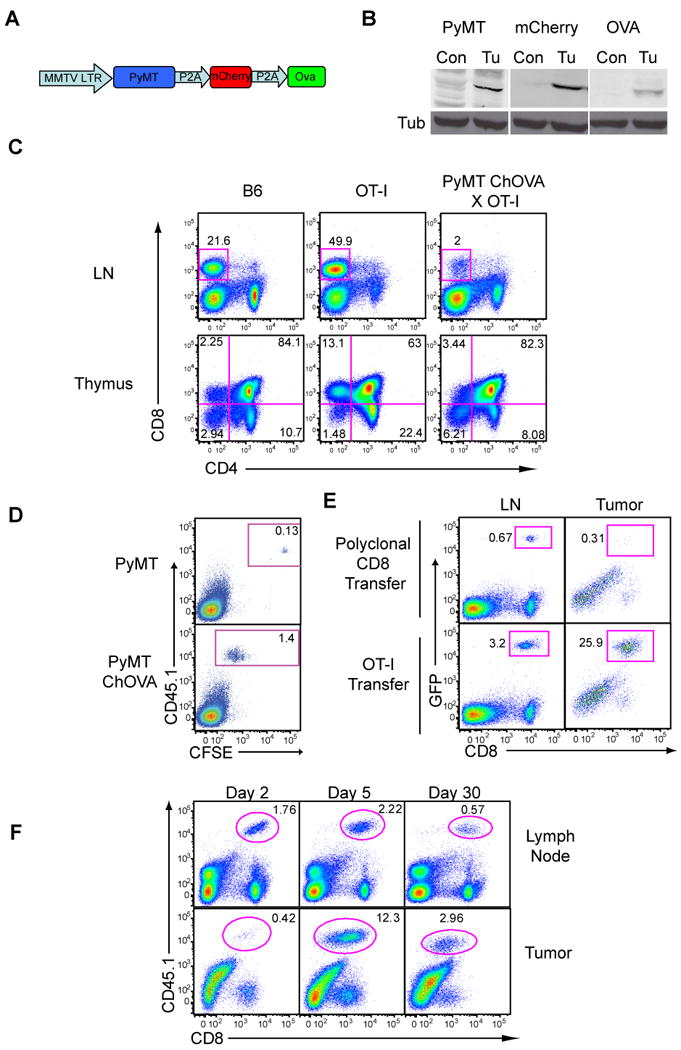
A. Schematic of PyMT ChOVA transgenic construct.
B. Western blots displaying the expression of PyMT, mCherry and Ova from tumor cells and control T cells in PyMT ChOVA mice.
C. Thymic negative selection of high-affinity TCRs in the PyMTChOVA spontaneous breast cancer model. Lymph nodes and thymii from either wild type B6, OT-I or PyMT ChOVA × OT-I mice were analyzed by flow cytometry for CD4 and CD8 expression.
D. Flow cytometry of tumor-draining lymph nodes 48 hours after 1×106 CFSE labeled CD45.1 OT-I T cells were transferred to either a PyMT tumor-bearing mouse or a PyMT ChOVA tumor-bearing mouse.
E. 5×106 OT-I-UbGFP T cells or polyclonal Ub-GFP CD8 T cells were transferred to a PyMT ChOVA tumor bearing mice. 5 days post-transfer tumor draining lymph nodes and tumors were removed and analyzed by flow cytometry. Plots from tumor were previously gated for CD45+ leukocytes.
F. Tumor-bearing PyMT ChOVA mice received 1×106 CD45.1 OT-I cells i.v., and were sacrificed at the specified day post-transfer. Tumor draining lymph nodes and tumor were removed and analyzed by flow cytometry. Plots from tumor were previously gated for CD45+ leukocytes.
See also Figure S1.
A sizeable fraction of the tumor antigens that have been isolated from human tumors to date are non-mutated germline-encoded tissue-specific proteins that are more dominantly expressed by transformed cells (Finn et al. 1995; Kawakami et al. 1994; van der Bruggen et al. 1991). In these cases, their presumed expression in the thymus apparently leads to negative selection of T cells bearing high-affinity T cell receptors (TCRs), and data shows a prevalence of cytotoxic T cells (CTL) with low avidity to these tumor antigens (Gervois et al. 1996) with some notable exceptions identified using tetramers (Lee et al. 1999; Yee et al. 1999). The MMTV promoter used in the PyMT ChOVA model also drove expression in the thymus, as indicated by the negative selection of developing anti-OVA/anti-tumor OT-I TCR transgenic T cells (Fig. 1C). Thus, OVA in this mouse replicates partial central-tolerance to tumor antigens.
We next investigated the ability of high affinity anti-OVA OT-I T cells, introduced into mice already bearing tumors, to respond in situ. Tumor-antigen presentation in PyMT ChOVA lymph nodes was quite robust, as evidenced by proliferation-dependent dilution of the vital dye CFSE specifically in naïve tumor-specific T cells (OT-I) in PyMT ChOVA mice, but not in the PyMT mouse strain (Fig. 1D). Similarly, introduced GFP-labeled tumor-specific OT-I T cells were present in the tumor, and specifically proliferated and accumulated in the lymph node, whereas non-specific T cells did not expand or significantly populate the tumor (Fig. 1E). That the tumor antigen-specific T cells expanded to over 10% of the CD45+ cells and then persisted at high percentages at lymph nodes and tumor for over 30 days (Fig. 1F) indicates that the primed cells were not subject to strong deletional tolerance.
Tumor-specific T cells are Incapable of Eliminating PyMT ChOVA Tumors
Tumor reactive naïve T cells are efficiently activated in the secondary lymphoid organs of PyMT ChOVA mice. However, naïve high-avidity T cells had only a slight effect in slowing tumor growth, and were unable to eliminate tumors when transferred to tumor-bearing hosts (Fig. 2A-B). These results are consistent with adoptive transfer of tumor-specific T cells in another spontaneous mouse model, TRAMP, in which lymph node CTL activation fails to be sufficient to induce rejection (Anderson et al. 2007). Notably, in our model, whereas tumor-specific T cells primed for 5 days in the lymph node of tumor-bearing mice demonstrated excellent cytolytic function against antigen-bearing targets, CTL found in the tumor at that time were effectively non-lytic (Fig. 2C). That the latter were derived from the former is suggested by the earlier kinetics of accumulation and CFSE dilution in the lymph node relative to the tumor (Fig. 1F and data not shown). Overall, this result is similar to human cancers such as melanoma in which tumor-antigen specific Melan-A/MART-1 tetramer staining demonstrates expanded levels of tumor-specific cells in TILs relative to peripheral sites, coupled with the same profound failure to reject the tumor (Lee et al. 1999; Romero et al. 1998).
Figure 2. Tumor-specific T cells are Defective in their Ability to Eliminate PyMT ChOVA tumors.
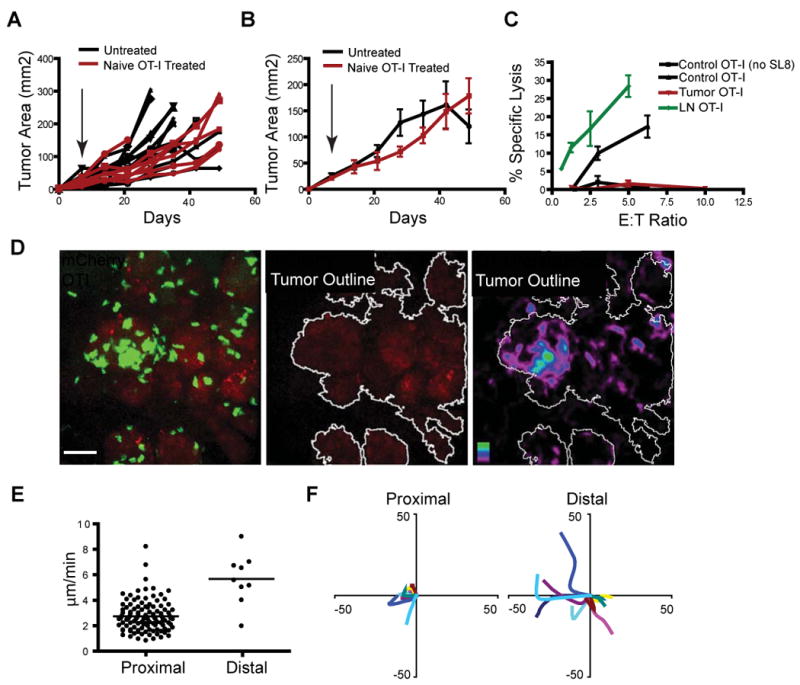
A. Tumor burden in individual PyMT ChOVA mice following adoptive transfer of 5×106 naïve OT-I cells (red lines, N=7) or untreated controls (black line, N=12). Arrow indicates date of adoptive transfer.
B. Average combined tumor burden in PyMT ChOVA mice following adoptive transfer of 5×106 naïve OT-I cells (red line mean +/- SEM, N=7) or untreated controls (black line mean +/- SEM, N=12).
C. Cytotoxic activity of OT-I T cells isolated from the LNs (green), or tumor (red) of PyMTChOva mice 5 days after adoptive transfer, compared to in vitro activated control OT-I T cells (black line with triangles) or control OT-I lysis of unpulsed EL4s (black line with squares). 104 EL-4 cells (+/- 100 ng/ml SL8 peptide-pulse) were used as targets. All lines mean +/- SEM, N=3.
D. Spinning disc confocal live-imaging of OT-I GFP T cells five days after adoptive transfer into PyMT ChOVA mice. Representative image of T cell localization at the site of a mCherry fluorescent tumor (left), image displaying tumor area outlined using a threshold mask (middle), and time average image of T cell persistence at the tumor site with tumors outlined (right). Scale bar represents 50 μm.
E. Average velocity of individual T cell tracks of cells located proximal (within 5 μm) or distal to the tumor.
F. Representative displacement tracks from T cells located either proximal (within 5 μm) or distal to the tumor border.
See also Movie S1
We thus sought to determine the in vivo behavior of T cells in our model. To this end, we turned to intravital imaging (Egeblad et al., 2008), adoptively transferring naïve GFP-labeled OT-I T cells, allowing them to activate and traffic to tumors and subsequently imaging their interactions in the tumor. We observed significant accumulations of T cells, often in multicellular clusters and at distinct foci along the tumor borders (Fig. 2D and Movie S1). As motility appeared much slower near the tumor, we defined a cutoff at 5 μm and considered cells within this radius ‘proximal’ whereas those further away are considered ‘distal’. In tumor-distal regions, T cells were largely motile, moving at 6 μm/min (Fig. 2E), a speed only slightly slower than those reported for unrestricted movements in lymph nodes (Miller et al. 2002). However, in the proximal regions, where clusters formed on the tumor margins, speeds were largely reduced to an average of 3 μm/min, with many cells much slower. Radial tracking plots of randomly selected cells supported that most of these cells were in fact swarming or jittering in the proximal clustered regions (Fig. 2F), whereas distally located cells quickly diverged from the origin. We did not observe any evidence for destruction of tumor cells by OVA-specific (OT-I) T cells in these tumors, such as single-step loss of mCherry fluorescence in cell-sized voxel regions, consistent with tumor measurements that had previously shown that T cells had little effect on tumor growth.
Phenotypic Characterization of Tumor Antigen Cross-presenting Dendritic Cells
In engineering the PyMT ChOVA model to co-express mCherry, we exploited this protein's apparent resistance to degradation to allow us to track antigens that are taken up from tumor cells for presentation by neighboring cells. By flow cytometry, we detected a population of mCherry+/CD45+ cells of hematopoietic origin in single-cell suspensions of PyMT ChOVA but not original PyMT tumors (Fig. 3A). A majority of CD45+ cells positive for mCherry above background also expressed high levels of CD11c, an integrin enriched in the dendritic cell lineage. To demonstrate that the mCherry in these cells derived from ingestion of the protein, as opposed to production, we generated bone marrow chimeras in which non-transgenic CD45.1+ bone-marrow introduced into PyMT ChOVA mice resulted in mCherry fluorescence in CD11c+ cells of the adopted (transgene negative) lineage (Fig. 3B). We observed mCherry fluorescence as puncta in isolated cells consistent with phagosomes (data not shown), but were unable to detect significant levels of mCherry transcripts in these cells (data not shown), consistent with uptake.
Figure 3. Phenotypic Characterization of Tumor-antigen Cross-presenting Dendritic Cells.
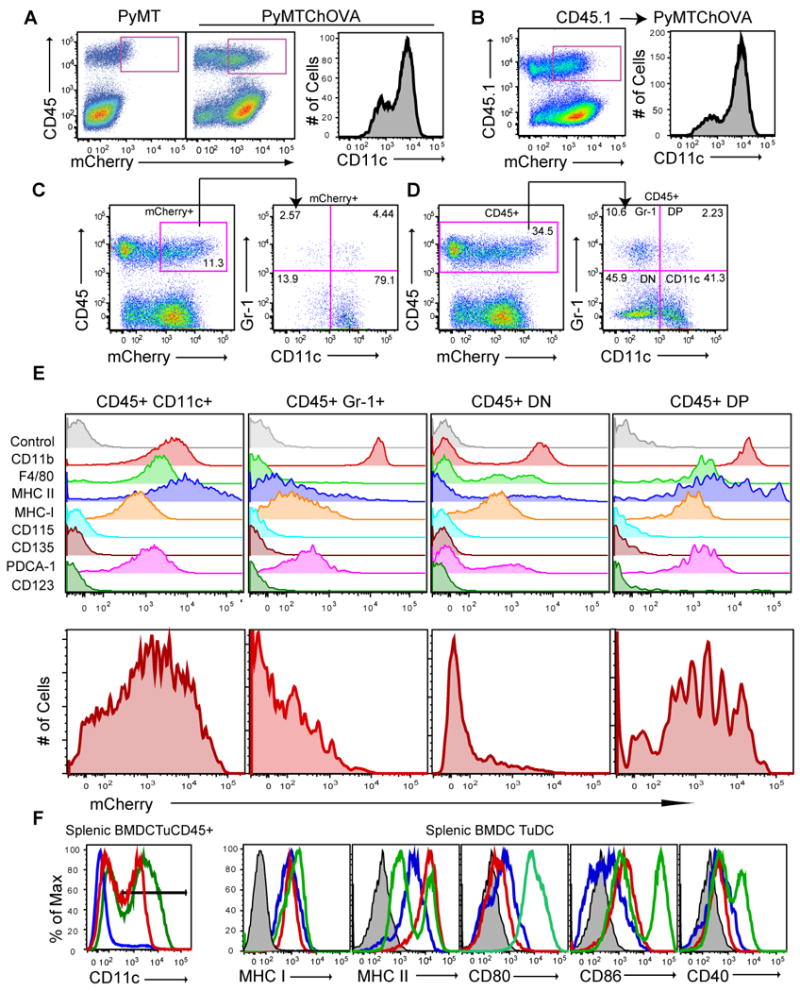
A. Flow cytometry of CD45 expression versus mCherry levels from digested tumors from PyMT (left) or PyMT ChOVA (middle) mice. CD11c levels of gated CD45+ mCherry+ cells (right) from previous dot plot.
B. As A, but from bone marrow chimera made by adoption of CD45.1 bone marrow cells into an irradiated PyMT ChOVA mouse.
C. CD45 expression versus mCherry levels from digested tumor from PyMT ChOVA mouse, gated CD45+ mCherryhi cells were propagated to subsequent dot plot, and analyzed for their expression of CD11c and Gr-1.
D. CD45 expression versus mCherry levels from digested tumor from PyMT ChOVA mouse, gated CD45+ cells were propagated to subsequent dot plot and analyzed for their expression of CD11c and Gr-1.
E. Gated and labeled populations from D. were analyzed for their expression of the listed cell surface markers or for their mCherry fluorescence level.
F. CD11c+ cells from either the spleen of B6 mice, BMDC cultures or the tumor of PyMT ChOVA mice. Gate in left histogram propagated to subsequent histograms.
Analysis of CD11c versus Gr-1 staining in the CD45+ mCherryhi cells, demonstrated that approximately 3/4 of the mCherryhi expressed CD11c and less than 5% of them expressed Gr-1, a marker closely associated with MDSC (Fig. 3C). We performed a similar analysis of CD11c and Gr-1 from the entire CD45+ gate (Fig.3D) to characterize the infiltrate in aggregate and thereby defined four populations: CD11c+Gr-1- (“CD11c+”), CD11c- Gr-1+ (“Gr-1+”), double-positives (“CD11c+, Gr-1+”) or double-negative (“DN”). Staining for additional surface proteins showed that none of these subsets expressed CD123, a marker for pDCs, CD135, a marker for bone marrow progenitors, or CD115, the CSF-1R. CD11c+ subsets co-expressed moderate CD11b, whereas Gr-1+ cells expressed distinctly high levels (Fig. 3E). The CD11c+ populations also expressed high levels of F4/80 and MHC II. The F4/80 expression together with the CD11b positivity would also qualify this subset as ‘TAM’ (Ojalvo et al. 2009). The CD11c+ population also had taken up significantly more of the tumor derived mCherry than the Gr-1+ or DN populations, although we note that this is bimodal, suggesting the population is not homogeneous in this respect (Fig. 3E). The higher levels of both MHC II and mCherry suggests that the CD11c+ cells are most likely to be effective antigen presenting cells for incoming effector T cells. We also note that they are considerably more abundant within the tumor relative to the other three populations. On an absolute scale, the DP populations were most homogeneously mCherry positive but represented less than 1% of the total isolate. Less than 1% of CD11c+ cells expressed the pDC markers, CD123 (Fig. 3E) or B220 (data not shown). The best correlation with mCherry positivity was the marker F4/80 (data not shown).
As we were interested in these cells as putative APC, we next compared the entire lineage of CD11c-positive cells from tumors to splenic DC, and to in vitro matured, bone marrow-derived DC (BMDC). CD11c levels were just slightly lower on tumor APC as compared to BMDC or splenic DC (left panel). When CD11c+ “TuDC” cells were analyzed, we observed roughly similar levels of MHC and costimulatory molecule expression in the tumor-associated cells and splenic DC, suggesting they would be similarly capable of presenting antigen to naïve T cells (Fig. 3F). Although many CD11c+ cells at the tumor site were mCherry+ there was still significant heterogeneity within the CD11c+ population for the amount of mCherry that had been ingested. This could reflect that not all cells are in a position to effectively take up the antigen or that the cells degrade the protein over time.
Live Imaging of Tumor Dendritic Cell Behavior
Given the presence of the CD11c protein on the majority of the mCherryhi cells of interest, we interbred PyMT ChOVA mice with mice that express YFP under the control of the CD11c promoter(Lindquist et al. 2004), permitting us to visualize the location and behavior of these APCs directly through real-time intravital live-imaging of exposed tumor masses. Using the same 5 μm proximity cutoffs as in Figure 2, we characterized tumor-“proximal” CD11c+ cells that were closely juxtaposed with the tumor and had very low motility compared to “distal” cells (Fig. 4A, B, C and H and Movie S2 and S3). The proximal CD11c-YFP+ cells of the tumors also had the highest level of mCherry fluorescence (Fig. 4D, E, G and Movie S3). The observation of the behavior of these cells in vivo supported our previous observation that CD11c+ cells ingested tumor antigens and eliminated the possibility that the mCherry fluorescence we observe by flow cytometry is simply an artifact of in vitro digestion of the tumor. Flow cytometry of tumor from PyMT ChOVA x CD11c YFP mice also demonstrated that CD11c-YFP+ cells from the tumor had taken up mCherry to a similar extent as in cells stained with antibodies against CD11c supporting that the transgene is generally faithful to the native protein (Fig. 4F). Proximal CD11c-YFP+ contained significant amounts of mCherry signal in their cytoplasm, often in puncta suggestive of endocytic vesicles, while distal CD11c-YFP+ did not contain these structures(Fig. 4G). Despite not moving their cell bodies along the tumor margin, these proximal cells dynamically extended and retracted dendrites, suggestive of ongoing sampling of the environment (Fig. 4H and Movie S3). Morphological data complement phenotypic markers in supporting an assignment of CD11c+ cells from the tumor as DCs, although it is important to remember that the lineages are highly plastic as previously discussed and these cells may equally be called TAMs on the basis of flow cytometry.
Figure 4. Live Imaging of Tumor-Dendritic Cell Behavior.
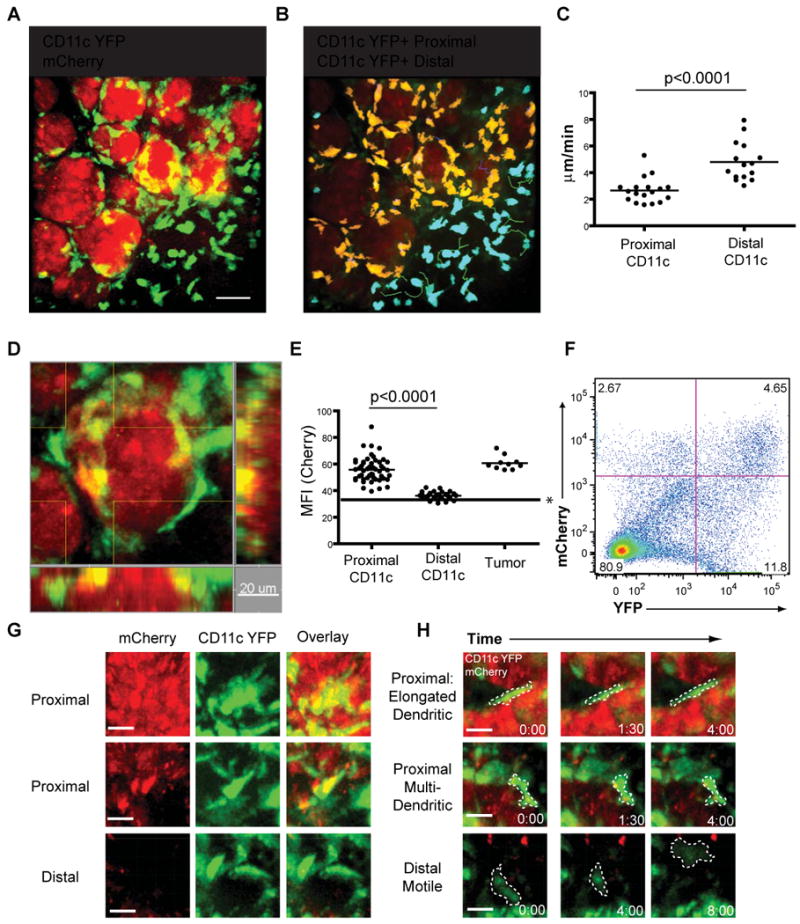
A. Representative still image acquired by intravital spinning disc confocal microscopy of mCherry+ CD11c YFP+ dendritic cells in PyMT ChOVA × CD11c YFP mice. Scale bar represents 10 μm.
B. DCs are color coded based on their proximity to the tumor border. Proximal DCs (within 5μm) are coded yellow, distal DCs (>5μm from tumor border) are coded blue. Color coded DCs with representative tracks of their movement during imaging.
C. Average velocity of individual CD11c YFP+ cells located proximal or distal to the tumor border.
D. Representative still image acquired by intravital spinning disc confocal microscopy of mCherry+ CD11c YFP+ dendritic cells in PyMT ChOVA × CD11c YFP mice.
E. Mean fluorescent intensity of CD11c YFP+ cells located proximal or distal to the tumor border, and tumor masses. * line represents background fluorescence of image.
F. Flow cytometry analysis of CD11c-YFP × PyMT ChOVA mouse displaying CD11c YFP+ mCherry+ DCs.
G. Representative YFP, mCherry and overlay still images of DCs either proximal or distal to the tumor. Scale bar represents 10μm.
H. Representative still images of YFP+ cells displaying either dendritic like or motile cell behaviors. Cell outlines are marked in dashed line. Scale bar represents 10 μm.
Changes in Myeloid Cell Populations During Tumor Development
Having identified CD11c+ cells as important in ingesting tumor antigens, we sought to characterize whether they were a consistent feature of the tumor across multiple stages. An advantage of the PyMT model is that tumors of distinct developmental stages exist in the same mouse. We identified mice with large carcinomas (area greater than 100 mm2) and adenomas (barely palpable tumors < 9 mm2) at different mammary glands (Fig. 5A) and dissociated these tumors for analysis by flow cytometry. After gating on the CD45+ leukocytic infiltrate, we again analyzed the levels of the common myeloid markers, Gr-1, CD11c, CD11b and F4/80 (Fig. 5B). We found that the numbers of cells positive for Gr-1 were significantly higher in carcinomas than adenomas, while CD11c and F4/80 were only modestly increased (Fig. 5C). Together these data suggest that the CD11c+ population is a consistent component of tumors across multiple stages and not restricted to early or late tumors.
Figure 5. Alteration of Leukocytic Infiltrate with Tumor Development.
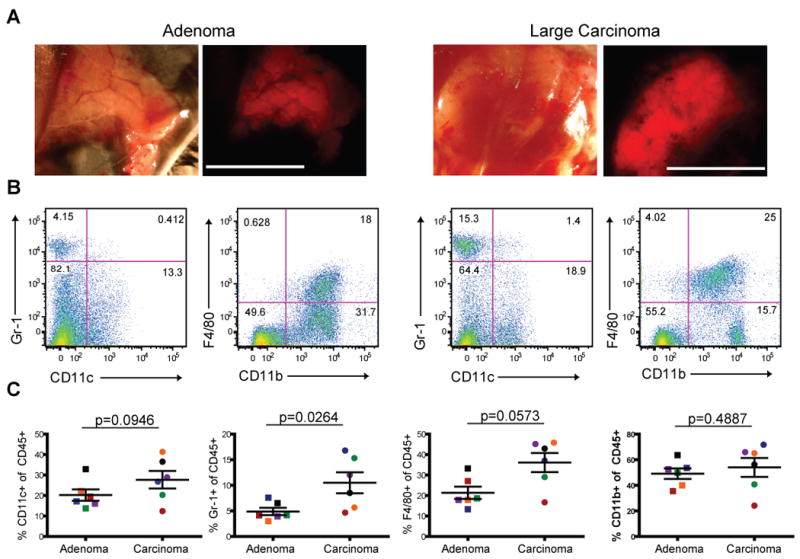
A. Transmitted and mCherry-fluorescent images of tumors from PyMT ChOVA mouse taken on a dissecting scope. Scale bar represents 1cm.
B. Cells from either an adenoma or a late carcinoma tumor were dissociated and analyzed by flow cytometry. CD45+ cells were analysed for the cell surface markers CD11c, Gr-1, CD11b and F4/80.
C. Plots depict percent+ of specified cell surface markers out of total CD45+ cells from different stage tumors. Each data point represents one tumor, and matched colors indicate tumors were from the same mouse.
Tumor DCs Interact with Tumor-Specific T cells in vitro and in vivo
Armed with the knowledge that this population of CD11c+ cells ingest tumor antigens specifically along the tumor margin, we noted that this location would also ideally place them in a position to interact with the swarming/arrested antigen-specific T cells that we had visualized earlier. To test whether this was the case, we modified our cell-labeling approach to allow direct assessment of T cell- CD11c+ interactions. For this, we transferred OT-I cells derived from mice interbred with the CD2RFP strain, which expresses the RFP protein at a very high level in T cells. We thereby could co-image reactive T cells and the proximal CD11c+ DC in a PyMT ChOVA × CD11c-YFP mouse. In this setting, the mCherry expression of the tumor was considerably less than the RFP-expressing T cells, allowing the latter to be viewed distinctly. We observed both clusters and individual T cells interacting for prolonged periods with tumor-marginal DC (Fig. 6A and Movie S4). We also observed that, whereas large T cell-DC cluster interactions were stable (white arrows), single T-DC conjugates also formed (blue arrow) or dissipated (yellow arrow) over time.
Figure 6. Tumor DCs Interact with Tumor-specific T cells in vitro and in vivo.
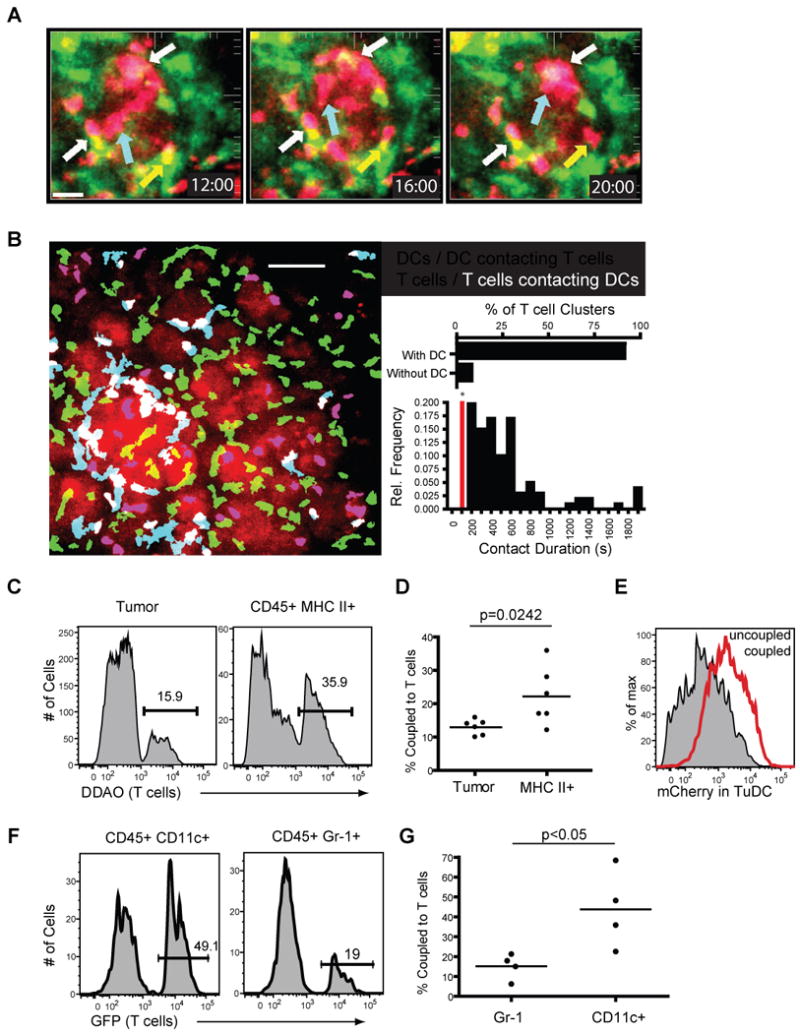
A. Spinning disc confocal imaging of CD2RFP OT-I T cells 5 days after adoptive transfer to PyMT ChOVA × CD11c-YFP mice. A representative image sequence of CD2RFP T cells (red) interacting with CD11c -YFP (green) dendritic cells at the tumor site, white arrows indicate T cells interacting with DCs throughout the time lapse, yellow arrows indicate T cell leaving a DC interaction, blue arrows indicate a T cell moving to a DC. Scale bar represents 30 μm.
B. Representative image displaying T cells (red) and DCs (green) at the tumor site. Blue cells indicate DCs contacting T cells (defined by overlapping red and green fluorescence), white cells indicate T cells contacting DCs. Scale bar represents 50 μm. Inset/top. Graph of the number of T cell clusters (defined as stable groups of greater than 2 T cells) that occur with DCs present or without DCs being present Inset/bottom. Relative frequency of contact duration between OT-I T cells and TuDCs. Pairs with contact durations lower than 200 s were not considered.
C. Histogram displaying the percentage of tumor cells (left) or MHC II+ cells (right) from PyMT ChOVA tumors that form couples with DDAO labeled T cells.
D. Results from 6 separate coupling assays plotted as percent of tumor or MHC II+ cells coupled to T cells. Bar represents mean.
E. mCherry fluorescence of CD45+ MHC class II+ cells that were not coupled to T cells (shaded histogram) or were coupled to T cells (red histogram).
F. Histogram displaying the percentage of mCherry CD11c+ or CD45+ Gr-1+ cells that form couples with OT-I GFP T cells.
G. Results from 4 separate coupling assays plotted as percent of CD11c or GR-1+ cells coupled to T cells. (Bar represents mean.)
Analysis of larger fields, using color-coding that differentiates lone T cells and DCs from those that are interacting, visually demonstrated that T-DC interactions were less frequent and more transient in the distal region of the tumor region as compared to the proximal (Fig. 6B and Movie S5). Quantification of T cell behaviors over multiple sites demonstrated that the majority of those in clusters, as observed in Fig. 6A, were attached to a tumor-proximal DC. Additionally, when individual T-DC contacts were scored, a large fraction persisted in contact with one another for more than 5 minutes, with some persisting beyond the 30 minutes of our standard imaging session. Many of the T cells in the dense clusters persisted there for at least the 30-minute periods of our observation. Additionally, when we analyzed whether individual T cells had interacted with a DC during the course of our 30 minute imaging session, we found that by 30 minutes 96% (147/153) of T cells had interacted with DCs (defined as contacting DCs) while at the start of acquisition 56% (85/153) were in contact with DCs. This evidence supports that tumor-proximal T cells are preferentially localized together with the mCherryhi CD11c+ cells in this local environment and that they are effectively engaged in both prolonged and ongoing interactions.
We thus sought to test specifically whether these APCs were more likely to form stable interactions with tumor-specific T cells compared to the OVA+ tumor cells themselves. As it was not possible to distinguish all cell types simultaneously during intravital imaging we turned to an in vitro system to study T-cell coupling frequencies with specific cells of the tumor microenvironment. We prepared single cell suspensions of all cells from a PyMT ChOVA tumor and allowed the collections of tumor and stromal cells to form couples with a significant excess of in vitro activated OT-I T cells, and then stained for surface markers to delineate the tumor-derived populations that had interacted. Using CD45+ and MHC II+ as a strategy to highlight all potential APCs with tumor, we demonstrated that T cells preferentially coupled with bone marrow-derived APC by this definition compared to tumor cells, even though the latter vastly outnumber them(Fig. 6C-D). Strikingly, T cell-engaged APC had higher levels of mCherry fluorescence than those that failed to couple, suggesting the T cells differentially couple to the mCherryhi cells (Fig. 6E); corresponding to the high frequency of T cell arrest on those “proximal” mCherryhi cells in vivo (Fig.6B). We applied the same technique to determine if CD11c+ or GR-1+ cells were more likely to interact with OT-I T cells, and found that CD11c+ cells were significantly better at forming couples with T cells (Fig. 6F and G). These results corroborate the observations made during intravital imaging and confirm that T cells preferentially interact with CD11c+ cells when given the opportunity to choose their partner.
Finally, we investigated the status of TCR levels and the ability to bind pMHC; since overall downregulation of the complex is associated with recent signaling (Valitutti et al. 1995), but absence of pMHC binding with presence of the α/β complex has been previously associated with tumor tolerance (Nagaraj et al. 2007). We thus tested the ability of both lymph node and tumor infiltrating T cells to bind to OVA labeled Kb pentamers five days after adoptive transfer into tumor bearing hosts. We found that both lymph node and tumor OT-I cells both showed decreased levels of pentamer binding in comparison to OT-I cells that were stimulated and then rested for 6 days in vitro, with tumor OT-I showing the lowest level of staining (Fig. S2). We similarly determined by staining for the Vβ5 of the OTI TCR that this was decreased in a similar hierarchy in these cells compared to blasted/rested OT-I (Fig. S2). We interpret this data as being consistent with ongoing TCR engagement at both lymph node and tumor, leading to downregulation of the entire TCR complex, but not a specific alteration of TCR binding capabilities (e.g. Nagaraj et al.).
Tumor DCs Activate Naïve but not Previously Activated OT-I T cells
Since activated T cells preferentially interact with the CD11c+ cell populations both in vitro and in vivo, we sought to define the ability of these CD11c+ cells to interact with and stimulate T cells in vitro. Given their cell surface similarity to splenic DC and their mCherry positive phenotype, we were not surprised to find that a total CD11c+/MHC II+ population, freshly isolated from tumors and without added antigen (hereafter TuDC), stimulated the proliferation of naïve OVA-specific (OT-I) T cells in vitro (Fig. 7A) at lower, but still significantly above background levels compared to BMDC that had been pulsed with exogenous peptides. This finding established that the TuDCs process and display antigens in peptide-MHC complexes, and are capable of stimulating naïve T cells, a feature sometimes associated with mature myeloid populations such as mature DC. However, when assayed for the ability to support proliferation of established CTL, the tumor-derived antigen-bearing DCs proved unexpectedly incapable of driving cell division. This deficit was true across a wide range of concentrations of DC and even when exogenous peptides were added to the TuDC (Fig. 7B), suggesting that the block was profound.
Figure 7. Tumor DCs Activate Naïve but not Previously Activated OT-I T cells.
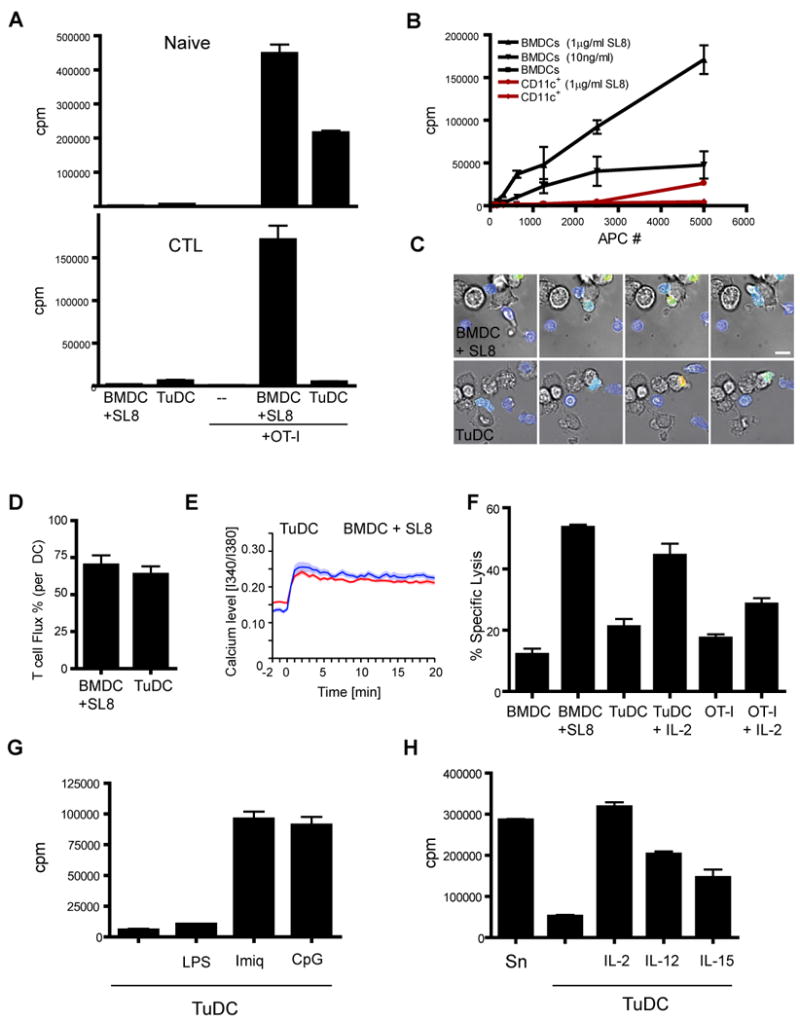
A. Proliferation of either naïve (upper) or previously activated (lower) OT-I T cells activated with either sorted TuDCs (50,000) or BMDCs (5,000) pulsed with 100 ng/ml SL8 peptide. N=3 mean +/- SEM.
B. Proliferation of previously activated OT-I T cells cultured with varying numbers of TuDCs or BMDCs pulsed with the specified amount of SL8 peptide. N=3 mean +/- SEM.
C. Live imaging of previously activated and Fura-2 labeled OT-I T cells interacting with BMDCs pulsed with 100 ng/ml SL8 peptide or with TuDCs. Brightfield images are overlaid with a pseudocolor image of the ratiometric Fura-2 fluorescence values, representing low intracellular calcium levels in blue and high intracellular calcium in red. Scale bar represents 10 μm.
D. Frequency of cell contacts with SL8 peptide-pulsed BMDCs or TuDCs that lead to calcium transients in previously activated OT-I T cells. N=3 mean +/- SEM.
E. Intracellular calcium levels of previously activated OT-I T cells contacting BMDCs pulsed with 100 ng/ml SL8 peptide or TuDC. Mean values with standard error are shown.
F. Cytolytic activity of in vitro activated T cells after overnight culture with BMDCs, TuDCs or BMDCs pulsed with 100 ng/ml SL8 peptide. N=3 mean +/- SEM.
G. Proliferation of OT-I T cells cultured with TuDCs alone or TuDCs with the TLR ligands, LPS (1 μg/ml), Imiquimod (2.5 μg/ml) or CpG (10 μg/ml). N=3 mean +/- SEM.
H. Proliferation of OT-I T cells cultured with either control splenocytes pulsed with 100 ng/ml SL8 peptide or TuDCs alone or in the presence of IL-2 (3U/ml), IL-12(10ng/ml) or IL-15(10ng/ml). N=3 mean +/- SEM
However, live-imaging of CTL, labeled with the calcium dye FURA-2, interacting with TuDC, demonstrated that proximal activation occurred in CTL and that peptide-MHC triggering of TCRs at the site of these interactions was not defective (Fig. 7C and Movie S6 and S7). Both the percentage of cells that generated calcium transients during interaction (Fig. 7D) and the magnitude of calcium curves generated by a panel of cells (Fig. 7E) were similar between TuDC and the stimulatory BMDC population pulsed with exogenous peptides. These results are consistent with peptide-MHC expression on these cells but suggest that TuDC apparently might bear inhibitory ligands that function distal to synapse-formation and TCR-signaling and inhibit productive responses. We also tested the ability of TuDCs to induce upregulation of CD69 on previously activated T cells and found that CD69 is upregulated by T cells stimulated by both BMDCs pulsed with peptide and TuDCs (Fig. S3). This data supports the conclusion that TuDCs are indeed antigen positive and are also able to initiate early events of TCR signaling. We screened a large number of candidate molecules and pathways and eliminated the canonical T cell inhibitory pathways that are suggested to function in MDSC, M2 macrophages and other APC, including both surface receptors and soluble factors (Fig. S3). That none of these restored proliferation suggests an unknown mechanism of T cell inhibition.
Given the ability of these TuDC to support antigen-dependent coupling with CTL in vitro and in vivo, we sought to determine the downstream effects of these signaling interactions. We therefore tested the ability of CTL encountering the TuDC to maintain CTL function, a key deficit in T cells that had homed to the tumor (Fig. 2). Comparisons of cytolysis by T cells cultured with TuDC showed that this interaction does not sustain cytolytic function, similar to T cells cultured alone or T cells stimulated with BMDCs without peptide. In contrast T cells cultured with BMDC pulsed with peptides, or T cells cultured alone or with TuDCs in the presence of IL-2 did maintain cytolytic acitivity (Fig. 7F).
As we were unable to restore proliferation effectively by blocking established inhibitory pathways in these assays, we also sought to reverse the phenotype by modulating the DC. Maturing the TuDC by adding either Imiquimod or CpG to the stimulation reaction strikingly relicensed them to stimulate CTLs to further divide (Fig. 7G). These two agents are agonists for APC expressed toll-like receptor (TLR) 7and 9 respectively and neither agent had significant effects on the T cells (data not shown). As a control, cells were treated without effect by applying lipopolysaccharide (LPS), an agonist to TLR4, which is poorly expressed by TuDC (data not shown). Supplementing the stimulation reaction with exogenous cytokines, either IL-2, IL-12 or IL-15 also was able to rescue the proliferative defect (Fig. 7H). Together our data suggest a specific stimulatory defect of TuDCs in their interactions with CTL, separate from antigen processing and presentation.
Given the ability of these TuDC to support antigen-dependent interactions with CTL, we finally sought to determine whether TuDC suppress CTL from responding to other antigen presenting cells. While in some experiments inhibition was profound, it was inconsistent over 13 experiments with an average of just 30% inhibition (Fig. S3). Since we do not yet understand the mechanisms utilized, any dominant suppression, if present, may also be sensitive to culture conditions or may be reversible (e.g see Fig. 7G). Alternatively, given the paucity of other effective APCs in the tumor microenvironment, tolerance may result purely from ineffective or defective re-priming.
Discussion
Delineating APCs in Tumors
Using a spontaneous mouse model of breast cancer, we have identified a population of myeloid cells that is optimized for ingesting and presenting tumor antigens to CD8 T cells, and thus is a clear and relevant APC of the tumor microenvironment. This formal identification is an important finding since many cells are likely capable of antigen presentation in vitro when pulsed appropriately with peptides, yet no previous study has been able to unequivocally define the cells that interact with tumor-specific T cells within tumors. The direct demonstration of their presence at the site of T cell arrest along the margin suggest that they fill this roll in vivo as well as in vitro.
Ojalvo et al., recently described TAMs as c-fmsGFP+ F4/80+ and dextran+ (Ojalvo et al. 2009); by this definition the mCherry+ cells we describe here could also be classified as TAMs, as they are CD11b+ and F4/80+ and their mCherry positivity largely substitutes for dextran+ as a marker of phagocytic ability (JE and MFK, unpublished). Although we do not find significant expression of CD115, the protein product of the c-fms GFP reporter, the TuDC population we describe otherwise can be considered a substantial subset of TAMs as well and the nomenclature discrepancy in the literature may ultimately resolve these on the basis of data such as ours. The plasticity and overlap of cell surface markers makes the study and classification of these cells difficult, but we suggest to classify these cells as dendritic cells for immunological purposes, based on their cell surface marker expression and in vivo morphology and behavior. This apparent equivalency in the literature is then intriguing as some of the same cells that are implicated in tolerizing T cells are also those implicated in remodeling and angiogenesis (Schoppmann et al. 2002; Qian and Pollard 2010).
We have excluded tumor APCs from the MDSC population because of their lack of expression of the defining marker for this population, Gr-1, and further because the CD11c+ cells do not function in analogous manner, namely defective proliferation is not restored with iNOS and/or Arginase inhibition (Fig S2) (Gabrilovich et al. 2009). Although we identify CD11c+ cells to be a distinct subset of cells from the entire lineage of the mononuclear phagocyte system (MPS), we acknowledge that some of these cells have previously been described by others as TAMs (DeNardo et al. 2009) or MDSCs, or that some of our cells may share phenotypic or functional characteristics of these populations. However, the method that we have used clearly highlights this population of cells on the basis of location, morphology, phagocytosis and their unique interactions with infiltrating T cells. It is also important to note that transient TCR-triggering interactions might occur with other APCs at sites besides the proximal region, and we can not rule out interactions between T cells and the few CD11c- mCherry+ APCs. We have demonstrated in the lymph nodes that even transient interactions can trigger TCR clustering and/or internalization (Friedman et al. 2010). Although, with our in vitro coupling experiments and in vivo behavioral studies, we established that CD11c+ TuDC are the predominant partners for T cells, this by no means excludes other members of the MPS from ever acting on T cells via antigen receptors.
Despite the robust mCherry signal in tumor CD45+ cells, we were unable to detect mCherry+ DCs in the tumor draining lymph nodes (data not shown). It thus remains unknown how and by which cells antigen is being presented to lymph node T cells. DCs from the tumor site may be trafficking to lymph nodes but degrading the mCherry protein, or soluble tumor antigens may travel through the lymph directly.
The Behavior of Tumor-Specific T cells in Refractory Tumors
In addition to identifying the tumor antigen cross-presenting DCs at the tumor site, we were also able to study the behavior of tumor antigen-specific T cells in a spontaneous and progressive model that is refractory to large numbers of infiltrating tumor-specific T cells. While activation and proliferation of naïve tumor-specific T cells was robust in the lymph node, these T cells were shown to be defective at lysis of targets after exposure to the tumor microenvironment This lack of cytolytic ability correlated with a lack of therapeutic benefit to adoptive transfer of T cells. This inactivity is similar to what is seen in human vaccination trials where large numbers of tumor-specific T cells can be elicited without eliminating tumors (Gattinoni et al. 2006; Rosenberg et al. 2005), suggesting a later defect, as we are observing in this model. The ability of exogenous γc cytokines to restore proliferation of T cells is also consistent with higher levels of efficacy of adoptive cell therapy when combined with IL-2 or IL-15 administration (Gattinoni et al. 2005; Overwijk et al. 2003; Epardaud et al. 2008).
Intravital imaging in our model revealed strong long-lasting interactions between tumor specific T cells and DCs at the tumor site. The importance of T cell-DC interactions at the effector site of an immune response is an emerging field of study, in the case of viral infections, the stimulation of memory or effector cells by DCs at peripheral sites is thought to be important for effective viral immunity (Wakim et al. 2008). The behavior of these ineffective tumor specific T cells is different from the behavior of tumor-rejecting T cells described in previous studies of ectopic tumor models (Mrass et al. 2006). While that study highlighted the activity of a strongly productive T cell response to a tumor, our study represents the activity occurring in a tumor setting that is more representative of naturally occurring human disease, where TILs are not effective in controlling tumor growth. It remains to be determined why T cells preferentially interact with TuDC as compared to other cells of the tumor and it is intriguing to conjecture that a specific chemokine-driven interaction may also contribute both to cell positioning within the tumor and, perhaps, to the limited stimulation capacity.
Tolerance Induction through T-APC Interactions within the Tumor
The concept that a bone marrow derived cell serves to tolerize T cells that interact with them as cognate APCs is not unprecedented. Tolerance in the lymph node occurs when antigens are directed to DEC205+ DC in the lymph nodes in the absence of additional stimuli (Hawiger et al. 2001). Pardoll and co-workers broadly described the requirement for MHC-matched bone-marrow-derived cells in the tolerance to a tissue-restricted antigen (Adler et al. 1998) and induction of tolerance to an A20 lymphoma required matched bone marrow cells and not the tumors themselves (Sotomayor et al. 2001).
Based on our in vitro studies, we propose that the interaction between T cells and DCs may in fact inhibit the ongoing T cell response, or alternatively may simply fail to effectively restimulate T cells to control the tumor while diverting them into such ‘sterile’ interactions. After isolation of tumor DCs we found that they were capable of activating naïve T cells to proliferate, but not in vitro generated activated T cells. A recent report describing TuDCs isolated from the NeuT model of mammary carcinoma demonstrated an inhibitory phenotype for these cells when exogenously pulsed with peptides and used to stimulate naïve CD8 T cells (Norian et al. 2009). While similar to our findings, the differences, that our cells are able to stimulate naïve CD8s while theirs are not and that their inhibition was mediated by arginine metabolism suggest that the mechanism of inhibition is distinct.
The defect in T cell restimulation exhibited by TuDCs could be rescued by treatment of the cultures with the TLR agonists CpG DNA and Imiquimod. These data suggest that TuDCs are either specifically lacking a stimulatory signal or are actively giving an inhibitory signal that is distinct from other stimulatory DCs. These data are consistent with a previous study in another spontaneous tumor model, RIP-Tag, where adoptive cell therapy was only effective in conjunction with CpG administration (Garbi et al. 2004) Similarly, topical administration of Imiquimod was effective against some established tumors in an ectopic model (Lu et al. 2010), and it is tempting to speculate that this TuDC/CTL axis is being modulated in that case. Topical application of imiquimod to breast tissue was not effective in our hands (data not shown). TuDCs from imiquimod treated mice or following treatment in vitro had similar levels of MHC II, CD80 and CD86 when compared to untreated TuDC. Our highest doses of imiquimod sometimes led to tumor hemorrhaging and so we suspect a more directed approach toward modifying TuDC function will be necessary.
Additional in vitro data correlates the functionally inactive T cells at the tumor site to DC interactions. T cells cultured with tumor DC, but not stimulatory BMDCs are unable to maintain lytic activity against targets, again suggesting that this interaction is deficient in its ability to promote a strong tumor response. Both the intravital imaging detailing interactions between T cells and DCs at the tumor site and in vitro coupling assays indicate that T cells are preferentially drawn to DCs as they enter the tumor microenvironment. Since this interaction fails to promote T cell effector function, it may be a significant impediment to the ongoing tumor response.
Does this represent a new mechanism of T cell tolerance? We have examined a large variety of surface receptor and soluble mediators and find that these do not revert inhibition and allow proliferation. Interestingly, we can demonstrate that proximal signaling, both as assessed by calcium signaling and, indirectly, through the formation of stable T-TuDC couples, is intact. Germain and co-workers had previously described a state of ‘split anergy’ in CTL clones; T cells retained cytotoxicity but lost proliferation and IL-2 secretion, when these lines engaged partially-fixed APCs (Otten and Germain 1991). The situation in response to TuDC has interesting parallels, though inverted; the key defect for tumor rejection may in fact be the loss of CTL function.
Implications for Immunotherapy
Current immunotherapies focus on promoting a strong anti-tumor T cell response by altering the T cells themselves, either by increasing tumor specific T cell frequency by adoptive cell transfer (Dudley et al. 2002), increasing the reactivity of responding T cells by engineering of high-avidity TCRs specific for tumors (Park et al. 2011), or by eliciting more potent T cells by blockade of inhibitory co-stimulatory molecules such as CTLA-4 or PD-1. While CTLA-4 blockade in particular has shown promise in treating human melanoma, many patients still show no clinical response after treatment (Hodi et al. 2010). The reasons behind this are unknown, but our data suggest that tumor-based APC may function by a unique mechanism and that combination therapy to boost T cell responses via CTLA-4 blockade in conjunction with treatments that alter TuDC stimulatory capacity may be particularly effective. Our model in many ways represents a best-case scenario for immunotherapy, in that we have available a large number of high-avidity, tumor specific T cells for adoptive therapy. That treatment with this therapy still fails highlights the need to search for other ways to enhance the T cell response within tumors.
Experimental Procedures
Mice and Genotyping
PyMT ChOVA transgenic C57BL/6 founder mice were generated as described in the associated supplement. All other mice are as described in the supplement. All mice were maintained in microisolator cages and treated in accordance with NIH and American Association of Laboratory Animal Care standards, and consistent with the animal care and use regulations of the Institutional Animal Care and Use Committee of the University of California, San Francisco.
Cell tracking and Imaging Analysis
Data was visualized and analyzed using Imaris 5.7.2 and 7.0 Software (Bitplane). Individual cells were identified and tracked by Imaris, and cell speed and displacement were calculated from tracks. Mean fluorescence intensity of mCherry in CD11c YFP DCs was calculated using iso- surfaces of masked DCs from MATLAB segmentation. Contact duration was determined by calculated track duration of masked T cell-DC couples that were tracked using Imaris. T cell clusters for analysis of presence of DCs in couples were defined as clusters of T cells containing more than 2 T cells. The presence of DCs contacting these clusters was determined by visual inspection. Statistical analysis of speeds, stopping times, mean fluorescence intensity, and interaction times was done using Prism 4.0 (GraphPad Software) employing an unpaired t-test and a two-tailed 95% confidence interval.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism. Unless specifically noted all data are representative of >3 separate experiments. Error bars represent SEM calculated using Prism, and are derived from triplicate experimental conditions. Specific statistical tests used were paired and unpaired T tests and all p values less than 0.05 were considered statistically significant.
Supplementary Material
Significance.
The cells involved in presenting antigens to T cells at the tumor site are not defined. To better study the T cell response to tumors, we developed a fluorescent- and antigen-linked spontaneous model of breast cancer. The primary cells responsible for ingesting tumor antigens and presenting them to T cells are low-motility myeloid cells localized along tumor margins. These cells fail to stimulate T cells, and may serve as a barrier to an effective T cell response. This model demonstrates the behavior of tolerized T cells within tumors and provides a target for immunotherapies. Marker analysis demonstrates that the cells presenting antigens to T cells are a large subset of cells implicated in tumor remodeling.
Highlights.
In a spontaneous mouse model, T cells prime and traffic to the tumor effectively.
Tumor antigen presenting cells ingest and present tumor-derived antigens.
Tumor specific T cells form long-lived interactions with tumor dendritic cells.
Despite engaging CD8 T cells, tumor APCs fail to effectively restimulate them.
Acknowledgments
We thank A. Ma, L. Lanier and N. Matsumoto for advice and the generous sharing of materials. We also thank Nigel Killeen and the UCSF Transgenic Core for assistance in the production of transgenic mice, and Cliff MacArthur and Shu-Wei Jiang for assistance with cell sorting. This work was supported by NIH grants R01CA134622, R01CA129523, U01CA141451, and the Cancer Research Institute. B. Boldajipour was supported by an EMBO fellowship and J Engelhardt was supported by NIH training grant CA108462. We thank Mark Anderson, Alexis Madrid, and Emily Thornton for critical reading of the manuscript. We thank Lisa Coussens, David Denardo, Brian Ruffell and Vicki Plaks for helpful discussions. The authors do not have competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adler AJ, Marsh DW, Yochum GS, Guzzo JL, Nigam A, Nelson WG, Pardoll DM. CD4+ T cell tolerance to parenchymal self-antigens requires presentation by bone marrow-derived antigen-presenting cells. J Exp Med. 1998;187:1555–1564. doi: 10.1084/jem.187.10.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambe K, Mori M, Enjoji M. S-100 protein-positive dendritic cells in colorectal adenocarcinomas. Distribution and relation to the clinical prognosis. Cancer. 1989;63:496–503. doi: 10.1002/1097-0142(19890201)63:3<496::aid-cncr2820630318>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Anderson MJ, Shafer-Weaver K, Greenberg NM, Hurwitz AA. Tolerization of tumor-specific T cells despite efficient initial priming in a primary murine model of prostate cancer. Journal of Immunology. 2007;178:1268–76. doi: 10.4049/jimmunol.178.3.1268. [DOI] [PubMed] [Google Scholar]
- Bruggen P van der, Traversari C, Chomez P, Lurquin C, Plaen E De, Eynde B Van den, Knuth A, Boon T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643–1647. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, Coussens LM. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer cell. 2009;16:91–102. doi: 10.1016/j.ccr.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Advances in immunology. 2006;90:51–81. doi: 10.1016/S0065-2776(06)90002-9. [DOI] [PubMed] [Google Scholar]
- Dudley ME, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M, Ewald AJ, Askautrud HA, Truitt ML, Welm BE, Bainbridge E, Peeters G, Krummel MF, Werb Z. Visualizing stromal cell dynamics in different tumor microenvironments by spinning disk confocal microscopy. Disease models & mechanisms. 2008;1:155–67. doi: 10.1242/dmm.000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epardaud M, Elpek KG, Rubinstein MP, Yonekura A ris, Bellemare-Pelletier A, Bronson R, Hamerman JA, Goldrath AW, Turley SJ. Interleukin-15/interleukin-15R alpha complexes promote destruction of established tumors by reviving tumor-resident CD8+ T cells. Cancer research. 2008;68:2972–83. doi: 10.1158/0008-5472.CAN-08-0045. [DOI] [PubMed] [Google Scholar]
- Felipe P de, Hughes LE, Ryan MD, Brown JD. Co-translational, intraribosomal cleavage of polypeptides by the foot-and-mouth disease virus 2A peptide. The Journal of biological chemistry. 2003;278:11441–8. doi: 10.1074/jbc.M211644200. [DOI] [PubMed] [Google Scholar]
- Finn OJ, Jerome KR, Henderson RA, Pecher G, Domenech N, Magarian-Blander J, Barratt-Boyes SM. MUC-1 epithelial tumor mucin-based immunity and cancer vaccines. Immunological reviews. 1995;145:61–89. doi: 10.1111/j.1600-065x.1995.tb00077.x. [DOI] [PubMed] [Google Scholar]
- Friedman RS, Beemiller P, Sorensen CM, Jacobelli J, Krummel MF. Real-time analysis of T cell receptors in naive cells in vitro and in vivo reveals flexibility in synapse and signaling dynamics. J Exp Med. 2010;207:2733–49. doi: 10.1084/jem.20091201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nature reviews. Immunology. 2009;9:162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbi N, Arnold B, Gordon S, Hämmerling GJ, Ganss R. CpG motifs as proinflammatory factors render autochthonous tumors permissive for infiltration and destruction. Journal of Immunology. 2004;172:5861–9. doi: 10.4049/jimmunol.172.10.5861. [DOI] [PubMed] [Google Scholar]
- Gattinoni L, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. The Journal of clinical investigation. 2005;115:1616–26. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Powell DJ, Jr, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6:383–393. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervois N, Guilloux Y, Diez E, Jotereau F. Suboptimal activation of melanoma infiltrating lymphocytes (TIL) due to low avidity of TCR/MHC-tumor peptide interactions. J Exp Med. 1996;183:2403–7. doi: 10.1084/jem.183.5.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, Ravetch JV, Steinman RM, Nussenzweig MC. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med. 2001;194:769–79. doi: 10.1084/jem.194.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annual review of immunology. 2001;19:47–64. doi: 10.1146/annurev.immunol.19.1.47. [DOI] [PubMed] [Google Scholar]
- Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. The New England journal of medicine. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Rivoltini L, Topalian SL, Miki T, Rosenberg SA. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci U S A. 1994;91:3515–3519. doi: 10.1073/pnas.91.9.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusmartsev S, Nagaraj S, Gabrilovich DI. Tumor-associated CD8+ T cell tolerance induced by bone marrow-derived immature myeloid cells. Journal of immunology. 2005;175:4583–92. doi: 10.4049/jimmunol.175.7.4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PP, et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med. 1999;5:677–685. doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- Lin EY, Jones JG, Li P, Zhu L, Whitney KD, Muller WJ, Pollard JW. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163:2113–2126. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue X nan, Pollard JW. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer research. 2006;66:11238–46. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- Lindquist RL, Shakhar G, Dudziak D, Wardemann H, Eisenreich T, Dustin ML, Nussenzweig MC. Visualizing dendritic cell networks in vivo. Nat Immunol. 2004;5:1243–1250. doi: 10.1038/ni1139. [DOI] [PubMed] [Google Scholar]
- Lu H, et al. Treatment failure of a TLR-7 agonist occurs due to self-regulation of acute inflammation and can be overcome by IL-10 blockade. Journal of Immunology. 2010;184:5360–7. doi: 10.4049/jimmunol.0902997. [DOI] [PubMed] [Google Scholar]
- Mayordomo JI, et al. Bone marrow-derived dendritic cells pulsed with synthetic tumour peptides elicit protective and therapeutic antitumour immunity. Nature medicine. 1995;1:1297–302. doi: 10.1038/nm1295-1297. [DOI] [PubMed] [Google Scholar]
- Mempel TR, Henrickson SE, Andrian UH Von. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–159. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- Miller MJ, Wei SH, Parker I, Cahalan MD. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science. 2002;296:1869–1873. doi: 10.1126/science.1070051. [DOI] [PubMed] [Google Scholar]
- Mrass P, et al. Random migration precedes stable target cell interactions of tumor-infiltrating T cells. J Exp Med. 2006;203:2749–2761. doi: 10.1084/jem.20060710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nature Medicine. 2007;13:828–35. doi: 10.1038/nm1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norian LA, Rodriguez PC, O'Mara LA, Zabaleta J, Ochoa AC, Cella M, Allen PM. Tumor-infiltrating regulatory dendritic cells inhibit CD8+ T cell function via L-arginine metabolism. Cancer research. 2009;69:3086–94. doi: 10.1158/0008-5472.CAN-08-2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojalvo LS, King W, Cox D, Pollard JW. High-density gene expression analysis of tumor-associated macrophages from mouse mammary tumors. The American journal of pathology. 2009;174:1048–64. doi: 10.2353/ajpath.2009.080676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otten GR, Germain RN. Split anergy in a CD8+ T cell: Receptor-dependent cytolysis in the absence of Interleukin-2 production. Science. 1991;251:1228–1231. doi: 10.1126/science.1900952. [DOI] [PubMed] [Google Scholar]
- Overwijk WW, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–80. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park TS, Rosenberg SA, Morgan RA. Treating cancer with genetically engineered T cells. Trends in biotechnology. 2011;29:1–8. doi: 10.1016/j.tibtech.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard JW. Trophic macrophages in development and disease. Nature reviews. Immunology. 2009;9:259–70. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero P, et al. Ex vivo staining of metastatic lymph nodes by class I major histocompatibility complex tetramers reveals high numbers of antigen-experienced tumor-specific cytolytic T lymphocytes. J Exp Med. 1998;188:1641–50. doi: 10.1084/jem.188.9.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. Journal of immunology. 2005;175:6169–76. doi: 10.4049/jimmunol.175.9.6169. [DOI] [PubMed] [Google Scholar]
- Schoppmann SF, et al. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. The American journal of pathology. 2002;161:947–56. doi: 10.1016/S0002-9440(10)64255-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotomayor EM, Borrello I, Rattis FM, Cuenca AG, Abrams J, Staveley-O'Carroll K, Levitsky HI. Cross-presentation of tumor antigens by bone marrow-derived antigen-presenting cells is the dominant mechanism in the induction of T-cell tolerance during B-cell lymphoma progression. Blood. 2001;98:1070–7. doi: 10.1182/blood.v98.4.1070. [DOI] [PubMed] [Google Scholar]
- Terabe M, et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med. 2003;198:1741–52. doi: 10.1084/jem.20022227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treilleux I, et al. Dendritic cell infiltration and prognosis of early stage breast cancer. Clin Cancer Res. 2004;10:7466–7474. doi: 10.1158/1078-0432.CCR-04-0684. [DOI] [PubMed] [Google Scholar]
- Valitutti S, Muller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature. 1995;375:148–151. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- Wakim LM, Waithman J, Rooijen N van, Heath WR, Carbone FR. Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science. 2008;319:198–202. doi: 10.1126/science.1151869. [DOI] [PubMed] [Google Scholar]
- Yee C, Savage PA, Lee PP, Davis MM, Greenberg PD. Isolation of high avidity melanoma-reactive CTL from heterogeneous populations using peptide-MHC tetramers. J Immunol. 1999;162:2227–2234. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.