

Abstract
Long-term memory formation requires selective changes in gene expression. Here, we determined the contribution of chromatin remodeling to learning-induced changes in brain-derived neurotrophic factor (bdnf) gene expression in the adult hippocampus. Contextual fear learning induced differential regulation of exon-specific bdnf mRNAs (I, IV, VI, IX) that was associated with changes in bdnf DNA methylation and altered local chromatin structure. Infusions of zebularine (a DNA methyltransferase inhibitor) significantly altered bdnf DNA methylation and triggered changes in exon-specific bdnf mRNA levels, indicating that altered DNA methylation is sufficient to drive differential bdnf transcript regulation in the hippocampus. In addition, NMDA receptor blockade prevented memory-associated alterations in bdnf DNA methylation, resulting in a block of altered bdnf gene expression in hippocampus and a deficit in memory formation. These results suggest epigenetic modification of the bdnf gene as a mechanism for isoform-specific gene readout during memory consolidation.
Keywords: DNA methylation, histone acetylation, memory, chromatin remodeling, hippocampus, DNA methyltransferase
Introduction
Brain-derived neurotrophic factor (BDNF) has been shown to contribute to neuronal activity-dependent processes such as long-term potentiation (Bramham and Messaoudi, 2005; Soulé et al., 2006), a leading candidate cellular mechanism for associative memory formation (Bliss and Collingridge, 1993; Whitlock et al., 2006). Similarly, there is increasing behavioral evidence indicating that bdnf gene expression is induced in the hippocampus after contextual and spatial learning and that this mechanism is essential for normal learning and memory (Linnarsson et al., 1997; Hall et al., 2000). However, investigations into the transcription-regulating mechanisms mediating changes in bdnf gene expression in memory formation are lacking, partly because of the complex structure of the bdnf gene. The bdnf gene consists of nine 5′ noncoding exons each linked to individual promoter regions, and a 3′ coding exon (IX), which codes for the BDNF preprotein amino acid sequence (Liu et al., 2006; Aid et al., 2007). The neuronal activity-dependent regulation of specific promoter regions within the bdnf gene dictates the spatial and temporal expression of specific bdnf transcript isoforms (Lauterborn et al., 1996; Nanda and Mack, 1998), which can regulate subsequent trafficking and targeting of the transcript (Blichenberg et al., 1999; Pal et al., 2003; Aranda-Abreu et al., 2005).
Chromatin remodeling can control gene transcription in the nervous system and modulates long-term memory formation (Guan et al., 2002; Alarcón et al., 2004; Korzus et al., 2004; Levenson et al., 2004, 2006; Kumar et al., 2005; Chwang et al., 2006; Bredy et al., 2007; Fischer et al., 2007; Lubin and Sweatt, 2007). DNA methyltransferases (DNMTs) regulate de novo methylation or maintenance of methylation at CpG island sites within DNA which can direct the transcription of genes by altering local chromatin structure. CpG islands are regions of DNA near and in ∼40% of promoters of mammalian genes (Goldberg et al., 2007) and are stretches of DNA in which there are a large number of cytosine-guanine dinucleotide sequences linked by phosphodiester bonds in DNA. Recent studies suggest that DNA methylation is a crucial mechanism for controlling chromatin remodeling in the adult mammalian nervous system (Levenson et al., 2006; Nelson et al., 2008). Furthermore, dysregulation of DNA methylation has been implicated in mental illnesses, such as schizophrenia, depression, bipolar disorder, Rett syndrome, and fragile X mental retardation (Das et al., 1997; Abdolmaleky et al., 2005; Grayson et al., 2005). Importantly, aberrant bdnf gene expression has also been specifically implicated in the etiology of several of these mental illnesses, including schizophrenia, depression, and bipolar disorder (Weickert et al., 2003; Angelucci et al., 2005; Tsankova et al., 2007). Thus, DNA methylation represents a provocative epigenetic mechanism for potentially contributing to ongoing regulation of bdnf transcription in the CNS. Here, we investigated whether DNA methylation and regulation of chromatin structure regulates bdnf transcription in the hippocampus in a contextual fear memory model.
Materials and Methods
Animals.
Adult male Sprague Dawley rats (250–300 g) were used for all experiments. Animals were housed under a 12 h light/dark cycle and allowed access to rodent chow and water ad libitum. Animals were allowed to acclimate to laboratory conditions and handled at least 5 d before use in behavioral experiments. All procedures were performed with the approval of the University of Alabama at Birmingham Institutional Animal Care and Use Committee and according to national guidelines and policies.
Behavioral procedures.
Animals were handled for 5 d, and on the day of experiments they were transported to the laboratory at least 2 h before fear conditioning. For contextual fear conditioning, animals were placed into the training chamber and allowed to explore for 2 min, after which they received an electric shock (1 s, 0.5 mA). The 2 min/1 s shock paradigm was repeated for a total of three shocks. After the last shock, animals were allowed to explore the context for an additional 1 min before removal from the training chamber. The context-alone-treated animals were exposed to the fear conditioning chamber for the same duration as the fear-conditioned animals but received no footshock. The immediate shock-alone group received three consecutive 1 s, 0.5 mA footshocks and were immediately removed from the fear conditioning chamber. For experiments investigating the effect of inhibition of DNMTs, intra-CA1 infusions of zebularine were performed 1 h before training. Animals received an infusion of either 10% DMSO (vehicle) or zebularine (600 ng/μl) (Calbiochem). Zebularine was freshly prepared in DMSO diluted to the appropriate concentration in sterile saline. For experiments investigating the effect of direct inhibition of DNMTs, intra-CA1 infusions of RG108 (N-phthalyl-l-tryptophan) were performed 1 h before training. Animals received an infusion of either 100% DMSO (vehicle) or RG108 (200 ng/μl) (Calbiochem). For (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK801) injection experiments, animals received saline (0.9% NaCl, 1.25 ml/kg) or MK801 (300 μg/kg) (Sigma) injections intraperitoneally 1 h before being placed in the chamber. For behavioral experiments, freezing behavior was measured at 24 h after fear conditioning. Freezing behavior was measured by observing the animals for 5 min. For biochemistry studies, age-matched animals that were handled but did not receive any experimental manipulations (naive) were used as controls in all fear-conditioning experiments.
Intra-CA1 cannula implantation.
During surgery, each rat was implanted with a 26 gauge bilateral stainless steel guide cannula (Plastics One) from which the injector extended 1 mm to end in area CA1. Stereotaxic coordinates used were as follows: anteroposterior, −3.6 mm from bregma, ±1.7 mm lateral from the midline, and −2.6 mm from dura (Paxinos and Watson, 1998) measured from the tip of the cannula guide. Cannula placements were verified in brains collected from animals and 20 μm sections were prepared through area CA1 and stained with cresyl violet to confirm the location of the infusion needle tips. Animals were habituated to dummy cannula removal and given 5 d of recovery and handling before the start of the experimental procedure.
Isolation of area CA1.
Whole brains were immersed in oxygenated (95%/5% O2/CO2) ice-cold cutting saline [containing the following (in mm): 110 sucrose, 60 NaCl, 3 KCl, 1.25 NaH2PO4, 28 NaHCO3, 0.5 CaCl2, 7 MgCl2, 5 glucose, 0.6 ascorbate] before isolation of CA1 subfields. Area CA1 of hippocampus was microdissected and immediately frozen in dry ice and stored at −80°C until RNA or DNA extracts were prepared.
Measuring mRNA levels by real-time, reverse transcriptase PCR.
The CA1 hippocampal subfield from vehicle- or zebularine-treated animals was collected for RNA quantification. Total RNA was extracted using TRIzol (Invitrogen) following the manufacturer's instructions. mRNA was reverse transcribed using the iScript RT-PCR iQ SYBR Green Supermix (Bio-Rad). Specific primers were used to amplify specific cDNA regions of the transcripts of interest. RT-PCR amplifications were performed in an iQ5 real-time PCR system (Bio-Rad) at 50°C for 30 min, 95°C for 15 min, followed by 40 cycles of 94°C for 60 s, 57°C for 60 s, 72°C for 60 s, and then incubation at 70°C for 10 min, using primers specific to the rat bdnf exons (I, II, IV, VI, and IX). β-Tubulin-4 quantification was used as an internal control for normalization. See supplemental Table 2, available at www.jneurosci.org as supplemental material, for primer sets used for the assessment of bdnf exon-specific and β-tubulin-4 mRNA levels. PCR products were electrophoresed on 2% agarose gels and stained with ethidium bromide.
Methylation-specific real-time PCR analysis.
DNA was isolated from hippocampal tissue, purified, and processed for bisulfite modification. For all our methylation-specific real-time PCR (MSP) data, we performed bisulfite treatment of genomic DNA an average of 5–6 times. Bisulfite treatment of genomic DNA converts cytosine to uracil, but leaves methylated 5′-cytosines unchanged. Quantitative real-time PCR was used to determine the DNA methylation status of the bdnf gene. Methylation-specific PCR primers were designed using Methprimer software (http://www.urogene.org/methprimer/) (Li and Dahiya, 2002). CpG islands were found within promoters 1 and 2 and the exonic regions of the bdnf gene immediately after the transcriptional start site of exons II, IV, VI, and IX (see Fig. 2A). This was expected, because region-specific DNA methylation is primarily found in 5′-CpG-3′-dinucleotides within promoters or in the first exon of genes (Goll and Bestor, 2005). Methylation-specific and unmethylated PCR primers were designed to target putative CpG islands detected in silico in promoter or non-promoter regions of the bdnf gene. Detection of methylated DNA associated with bdnf exons was performed with the primers listed in supplemental Table 1, available at www.jneurosci.org as supplemental material. It is important to note that in a circumstance in which a specific promoter is in a highly methylated or unmethylated basal state, the two different primer sets will be preferentially sensitive to alterations in methylation (Li and Dahiya, 2002). For example, a specific CpG island that is highly methylated in the basal state is much more likely to reveal demethylation changes when using the “unmethylated” primers and vice versa. We also evaluated β-tubulin-4 DNA after context exposure or contextual fear conditioning and found no significant alterations in unmethylated β-tubulin-4 DNA. Thus, we used unmethylated β-tubulin-4 levels for normalization of bdnf DNA methylation in the studies presented here. PCRs were in an iQ5 real-time PCR system (Bio-Rad) using the following cycling conditions: 95°C for 3 min, 40 cycles of 95°C for 15 s, and 58–60°C for 1 min. Detection of the fluorescent products occurred at the end of the 15 s temperature step. For a melting curve analysis, PCR products were melted by increasing the temperature in 1°C increments beginning at 60°C. To further verify specificity of the final product, 10 μl of the amplified products were analyzed by electrophoresis on a 2% agarose gel stained with ethidium bromide and visualized under UV light. Samples were normalized to β-tubulin-4 and the comparative cycle threshold method was used to calculate differences in gene expression between samples (Livak and Schmittgen, 2001).
Figure 2.

Altered bdnf DNA methylation with contextual fear conditioning. A, The positions of the bdnf CpG islands are indicated relative to the transcription start site of exon I (−658 to −447 bp), exon II (+33 to +304 bp), exon IV (−123 to + 161 bp), and exon VI (−141 to +348 bp). The locations of the methylated PCR primer pairs (M1, M2, M4, and M6) are indicated by the arrows, and primer sequences can be found in supplemental Table 1, available at www.jneurosci.org as supplemental material. To minimize confusion the newest structure of the bdnf gene is depicted in the figure (I, II, IV, VI, IX) with the older nomenclature displayed above (I, II, III, IV, V). In the immediate shock-alone group, we observed no significant changes in methylation at the bdnf CpG islands. With context exposure alone, there were decreases in methylation at exons I and VI (I, t(7) = 7.53, p = 0.001; IV, t(4) = 9.04, p = 0.0008, n = 5–8). No significant changes in DNA methylation were observed at exons II and IV after context exposure. With fear conditioning, there were increases in methylation at exon VI (t(6) = 2.34, p = 0.0362, n = 7), but a decrease in methylation at exons I and IV (II, t(4) = 3.09, p = 0.0367; IV, t(4) = 11.57, p = 0.0003, n = 5–6). Context + shock had no effect on promoter 2 methylation. B, Bisulfite sequencing analysis performed on 12 CpG sites near the transcription initiation site of exon IV show percentage of cytosine residues that were methylated with shock alone, context exposure alone, or fear conditioning. A two-way ANOVA revealed a highly significant effect of the behavior group (F = 128.8, p < 0.0001) and region (F = 3.07, p < 0.0009). The solid line across the bars represents normalized naive control levels [one-sample t test, *p < 0.05, **p < 0.01, ***p < 0.001, compared with naive controls; Student's t test; not significant (ns), #p < 0.05, ##p < 0.01, compared with context alone]. Error bars indicate SEM.
Bisulfite sequencing PCR.
Bisulfite-treated samples (as described above) were amplified by primers that amplify the same region of exon IV DNA, but independent of methylation status. The thermocycler protocol involved an initial denaturation cycle (5 min, 95°C), 50 cycles of denaturation (1 min, 95°C), annealing (1 min, 60°C), and extension (1 min, 72°C), followed by a final extension cycle (5 min, 72°C) terminating at 4°C, resulting in a 258 bp PCR product for exon IV (see supplemental Table 1, available at www.jneurosci.org as supplemental material, for primer sets used for the assessment of bdnf cytosine residues). The PCR product for this region was then purified using a gel extraction kit (Qiagen) and sequenced using the reverse primer at the University of Alabama at Birmingham Genomics Core Facility of the Heflin Center for Human Genetics. The percentage methylation of each CpG site within the region amplified was determined by the ratio between peaks values of guanine (G) and adenine (A) (G/[G + A]), and these levels on the electropherogram were determined using Chromas software.
Chromatin immunoprecipitation assay.
The CA1 regions of hippocampus were microdissected on ice and held in ice-cold PBS solution containing protease inhibitors [1 mm phenylmethysulfonyl fluoride, 1 μg/ml protease inhibitor mixture, and phosphatase inhibitors (1 mm Na3VO4 and 20 mm NaF)]. CA1 was immediately incubated in 1% formaldehyde in PBS at 37°C for 10 min. The tissue was washed with an ice-cold PBS solution containing protease inhibitors (1 mm phenylmethylsulfonyl fluoride, 1 μg/ml protease inhibitor mixture). Tissue was homogenized in SDS lysis buffer (50 mm Tris, pH 8.1, 10 mm EDTA, 1% SDS). Chromatin was sheared using a Branson sonifier 250 at 1.5 power and constant duty cycle. Lysates were centrifuged to pellet debris and then diluted 1:10 in chromatin immunoprecipitation (ChIP) dilution buffer (16.7 mm Tris, pH 8.1, 0.01% SDS, 1.1% Triton X-100, 167 mm NaCl, 1.2 mm EDTA). Extracts were precleared for 45 min with a 50% suspension of salmon sperm-saturated protein A. Immunoprecipitations were performed at 4°C overnight with primary antibodies (anti-acetyl H3, anti-acetyl H4, anti-phosphoacetyl H3) or no antibody (control). Immune complexes were collected with protein A and sequentially washed with low salt buffer (20 mm Tris, pH 8.0, 0.1% SDS, 1% Triton X-100, 2 mm EDTA, 150 mm NaCl), high salt buffer (20 mm Tris, pH 8.1, 0.1% SDS, 1% Triton X-100, 500 mm NaCl, 1 mm EDTA), LiCl immune complex buffer (0.25 M LiCl, 10 mm Tris, pH 8.1, 1% deoxycholic acid, 1% IGEPAL-CA 630, 500 mm NaCl, 2 mm EDTA), and Tris-EDTA (TE) buffer. Immune complexes were extracted in 1× TE containing 1% SDS, and protein–DNA cross-links were reverted by heating at 65°C overnight. After proteinase K digestion (100 μg; 2 h at 37°C), DNA was extracted by phenol/chloroform/isoamyl alcohol and then precipitated in ethanol. Immunoprecipitated DNA was subjected to quantitative real-time PCR using primers specific to the rat bdnf promoters (supplemental Table 2, available at www.jneurosci.org as supplemental material). The cumulative fluorescence for each amplicon was normalized to input amplification.
Methylated DNA immunoprecipitation.
Rat tissue (∼400 mg) was homogenized in buffer [containing the following (in mm): 10 KCl, 20 HEPES, 1 MgCl2, 1 dithiothreitol, supplemented with protease inhibitors] with a Dounce homogenizer using 10 strokes. Samples were then incubated on ice for 5 min and centrifuged at 1500 rpm for 10 min at 4°C. Centrifuged pellet was resuspended in 0.5 ml of 1× TE with protease inhibitor. Genomic DNA was extracted by phenol and chloroform. DNA was then precipitated with ethanol containing 75 mm sodium acetate and resuspended in TE buffer (20 μg/ml). Genomic DNA was sheared by sonication using a Branson sonifier 250 at 1.5 power and constant duty cycle. Immunoprecipitations using 4 μg of DNA were performed at 4°C overnight with 10 μg of monoclonal antibody against 5-methylcytosine (Epigentek), or with no antibody, or an equivalent amount of normal IgG (anti-mouse; Santa Cruz Biotechnology). A portion of the sonicated DNA was left untreated to serve as input control. Immune complexes were collected with protein A beads and sequentially washed two times with low salt buffer (20 mm Tris, pH 8.1, 0.1% SDS, 1% Triton X-100, 2 mm EDTA, 150 mm NaCl), high salt buffer (20 mm Tris, pH 8.1, 0.1% SDS, 1% Triton X-100, 500 mm NaCl, 2 mm EDTA), LiCl immune complex buffer (0.25 M LiCl, 10 mm Tris, pH 8.1, 1% deoxycholic acid, 1% Nonidet P-40, 1 mm EDTA), and TE buffer. After proteinase K digestion (100 μg; 2 h at 37°C), DNA was extracted by phenol/chloroform/isoamyl alcohol and then ethanol-precipitated. Immunoprecipitated DNA was subjected to quantitative real-time PCR using primers specific for 200 bp segments corresponding to CpG island sites within rat BDNF-exon IX (supplemental Table 1, available at www.jneurosci.org as supplemental material). The cumulative fluorescence for each amplicon was normalized to input amplification.
Statistics.
Methylation and mRNA data were analyzed with a one-sample t test, one-way ANOVA, with Tukey post hoc test as appropriate. Differences in bisulfite sequencing data were analyzed by two-way ANOVA with Bonferroni's post hoc tests. Specific comparisons between treatment groups were made using Student's t test. GraphPad Prism software was used for statistical analysis of the data. Data are shown as the mean ± SEM.
Results
Regulation of bdnf gene expression in hippocampus during fear memory consolidation
We trained animals using a rodent contextual fear-conditioning paradigm wherein a novel context (training chamber) is paired with a footshock (Fig. 1A).We chose to investigate contextual fear conditioning because long-term memory for this association requires de novo gene transcription and protein synthesis (McGaugh, 2000). Twenty-four hours after training, exposing the animal to the training chamber again without the footshock triggered retrieval of the associative memory as quantitated by measuring the animals' freezing behavior (Fig. 1B) (McGaugh, 2000). As controls we also tested freezing in animals that received an immediate footshock or were exposed to the novel context alone. Neither control group exhibited significant freezing when re-placed into the training context (Fig. 1B).
Figure 1.

Increased bdnf gene expression after context exposure and fear conditioning. A, Schematic representation of the contextual fear-conditioning test protocol. Animals were exposed to the training chamber and either received a series of footshocks after being exposed to the context (Context + Shock), being exposed to the context alone (Context + No Shock), or receiving only the footshock without being exposed to the context (No Context + Shock). B, Animals were reexposed to the training chamber 24 h later and tested for freezing behavior. n = 8–9/group; *p < 0.001 compared with shock-alone controls. C, bdnf mRNA in area CA1 of hippocampus is increased within 0.5 h of context exposure (t(8) = 2.48, p = 0.0381, n = 5) and context + shock (t(8) = 2.41, p = 0.0425, n = 5) compared with naive controls. At 2 h, bdnf mRNA expression peaks in area CA1 of hippocampus in fear-conditioned animals (t(9) = 3.15, p = 0.0117, n = 5–6). At 24 h, bdnf mRNA levels returned to baseline in area CA1 of hippocampus from both context (t(6) = 0.42, p = 0.6887, n = 4) and context + shock (t(7) = 1.97, p = 0.0894, n = 4–5) animals relative to naive controls. D, After context exposure alone, exon I and VI bdnf mRNA increased in area CA1 (exon I, t(3) = 3.42, p = 0.0418, n = 4; exon VI, t(5) = 2.66, p = 0.0449, n = 6) with a corresponding increase in total bdnf mRNA levels (exon IX, t(4) = 3.38, p = 0.0279, n = 5) relative to naive control. After fear conditioning, exon IV bdnf mRNA increased (t(3) = 5.88, p = 0.0098, n = 4) in area CA1 of hippocampus with an increase in total bdnf gene expression as assessed by exon IX mRNA (t(5) = 3.49, p = 0.0175, n = 6). No significant changes in exon II bdnf mRNA were observed with context alone or context + shock relative to naive control. The solid line across the bars represents normalized naive control levels [one-sample t test, *p < 0.05, **p < 0.01, compared with naive controls; Student's t test, not significant (ns), #p < 0.05, ##p < 0.01, compared with context alone]. Error bars indicate SEM.
To evaluate bdnf gene regulation after fear learning, we performed a time course analysis (0.5, 2, and 24 h) for total bdnf gene expression in area CA1 of rat hippocampus, using quantitative real-time PCR. We specifically assessed bdnf exon IX mRNA levels because this allows for the evaluation of total bdnf transcription (i.e., the sum of all isoforms transcribed). Within 0.5 h of fear conditioning, total bdnf mRNA levels had increased in area CA1 (Fig. 1C), and mRNA levels further increased at 2 h after training relative to naive controls. At 24 h after fear conditioning, total bdnf mRNA returned to baseline levels. These results indicate that bdnf gene expression is induced in hippocampus after fear learning. This is in agreement with previous reports that indicate that altered bdnf transcription is involved in contextual learning (Hall et al., 2000; Bredy et al., 2007; Ou and Gean, 2007).
An interesting finding from these studies is that introducing an animal to a novel context alone elicits a modest increase in total bdnf mRNA levels in area CA1, both at the 0.5 and 2 h time points (Fig. 1C). In contrast, immediate shock-alone controls elicited no changes in bdnf gene expression in hippocampus. Thus, our results indicate that altered bdnf gene transcription occurs both after novel context exposure and associative fear learning, with a much more pronounced upregulation being associated with associative pairing of stimuli. Furthermore, these results are consistent with a role for altered bdnf transcription as a mechanism for consolidation of contextual memories, be they associative (fear conditioning) or episodic (context learning).
The bdnf gene structure is complex, with multiple initiation start sites allowing isoform-specific transcription of multiple mRNA transcripts. We therefore investigated whether memory-associated alterations in bdnf gene expression might involve isoform-specific transcription initiation at the bdnf gene locus. We measured exon-specific bdnf mRNA levels in hippocampus at 2 h after fear conditioning using quantitative real-time PCR. There were no significant changes in exon I, II, and VI mRNA levels in hippocampus after fear conditioning. However, exon IV mRNA levels were increased in area CA1 of hippocampus after fear conditioning relative to naive or shock-alone controls (Fig. 1D). We also observed differential regulation of bdnf exons in hippocampus with context exposure alone. At 2 h after context exposure, transcription from exons I and VI was increased in area CA1 relative to naive or shock-alone controls (Fig. 1D). There were no changes in transcription from exons II and IV after context exposure. Overall, these data indicate that the increase in total bdnf mRNA levels seen after context exposure is attributable to increases in exon I- and VI-containing bdnf transcripts and, after associative fear conditioning, attributable to exon IV-containing transcripts. This highlights an interesting, differential, site-specific initiation of bdnf transcription in novel context learning versus associative contextual conditioning.
DNA methylation changes at the bdnf gene after context exposure and fear conditioning
Our observations of memory-associated regulation of exon-specific bdnf transcripts led us to speculate that methylation of CpG island sites in the promoter or non-promoter regions of the bdnf gene might be a mechanism mediating bdnf gene expression in the adult hippocampus during memory formation. Thus, we next examined the effect of context exposure and contextual fear conditioning on bdnf DNA methylation in hippocampus. In silico analysis of the bdnf gene revealed CpG islands located within promoter and exonic regions of the bdnf gene (Fig. 2A) (see Materials and Methods).
Data generated using MSP indicated that context exposure alone elicited a decrease in methylated DNA levels associated with exons I and VI relative to naive or shock-alone controls (Fig. 2A). No changes in methylated DNA levels associated with exons II and IV were observed with context exposure alone compared with naive or shock-alone controls. With contextual fear conditioning, we found significant decreases in methylated DNA levels associated with exons I and IV in area CA1 of hippocampus at 2 h compared with naive controls with no change at exon II (Fig. 2A). We also observed prominent increases in methylated exon VI DNA at 2 h compared with naive controls (Fig. 2A). As a control, we measured levels of unmethylated DNA within the bdnf gene after context exposure or fear conditioning using primer sequences designed to amplify unmethylated CpG island sites (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Overall, these data support the idea that DNA methylation controls exon-specific readout of the bdnf gene, because there is a correlation of decreased methylation at specific exon-associated CpG islands (Fig. 2A) with increased exon transcription (Fig. 1D). In addition, they indicate a surprising complexity to the control of DNA methylation at the bdnf gene locus with specific decreases and increases in methylation at individual initiation start sites being associated with specific bdnf transcripts regulated in response to a novel context versus associative fear conditioning.
As another independent assessment of altered methylation, we confirmed our MSP data using direct bisulfite sequencing PCR (BSP) to examine site-specific methylation within the exon IV region. We examined exon IV specifically because it was the only bdnf transcript isoform we found significantly increased in area CA1 with fear conditioning (Fig. 1D), and our MSP data suggested that this increase in transcription was associated with decreased methylation at this locus (Fig. 2A). A schematic of the 12 CpG dinucleotides within the exon IV region screened by MSP is shown in Figure 2B. This exon IV region contains a cAMP response element (5′-TCACGTCA-3′, located between base pairs −38 and −31) site for the transcription factor cAMP response element-binding protein (Shieh et al., 1998; Tao et al., 1998), which encompasses CpG site 1. Sequencing data confirm active demethylation of bdnf exon IV after fear conditioning (Fig. 2B). Interestingly, although MSP failed to detect bdnf exon IV methylation changes with context exposure alone, BSP analysis revealed active demethylation of exon IV after context exposure. However, post hoc analyses revealed that demethylation of exon IV is greater at specific CpG sites with fear conditioning versus context exposure alone, which is consistent with the hypothesis of differential regulation of this site in response to different behavioral experiences. Thus, the bisulfite DNA sequencing data reiterate active regulation of DNA methylation within the bdnf gene, and furthermore demonstrate that bdnf methylation is regulated in response to fear conditioning or context exposure.
Effect of DNMT inhibition on bdnf DNA methylation in hippocampus
To determine whether decreased DNA methylation is sufficient to trigger altered bdnf gene transcription, we examined the effect of DNMT inhibition on bdnf mRNA levels in vivo. For these experiments, animals received intra-CA1 infusions of the DNMT inhibitor, zebularine, or vehicle. At 40 min after zebularine infusions, the CA1 tissue immediately surrounding the infusion site was microdissected and bdnf mRNA levels were quantified using real-time PCR. As a control, we confirmed the location of the infusion needle tips in a separate set of cannulated animals; histology showed that the infusion needles effectively targeted area CA1 of hippocampus (Fig. 3A). Analysis of exon-specific bdnf mRNA revealed that DNMT inhibition significantly increased exon I, IV, and VI mRNA levels with a corresponding increase in total bdnf gene expression (exon IX) in area CA1 of hippocampus, relative to vehicle controls (Fig. 3B). Levels of exon II mRNA did not change after DNMT inhibition (Fig. 3B). These results indicate that DNMT inhibition regulates basal bdnf gene expression in hippocampus in vivo.
Figure 3.
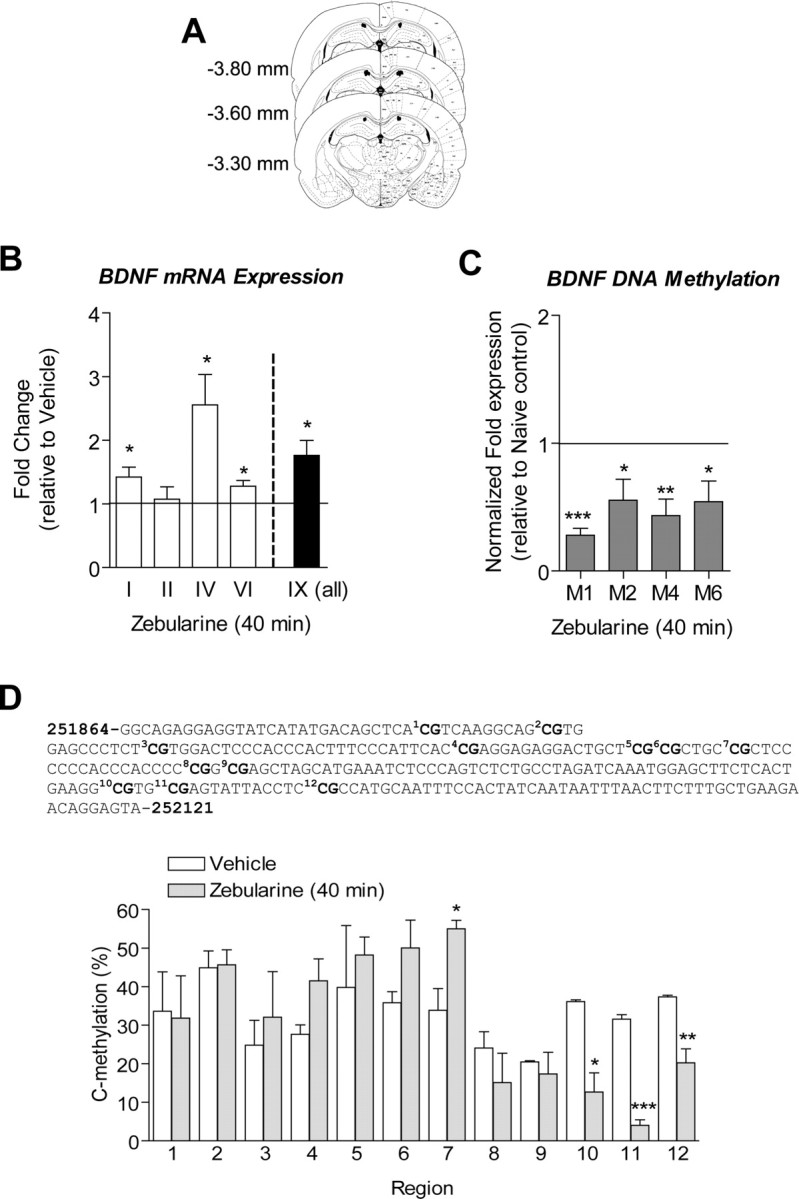
Inhibition of DNMT alters bdnf gene expression and DNA methylation in hippocampus in vivo. A, The diagram represents the histology from animals for which needle tips for intra-CA1 infusions were confirmed before biochemistry studies in B–D. B, Reverse transcriptase quantitative real-time PCR was used to determine the effect of intra-CA1 infusions of zebularine (DNMT inhibitor) on basal bdnf mRNA levels relative to saline control. cDNA products for bdnf exons I, IV, VI, and IX indicate that bdnf mRNA is artificially increased in area CA1 of hippocampus with zebularine treatment in vivo (I, t(6) = 2.71, p = 0.0352; IV, t(6) = 3.27 p = 0.0171; VI, t(5) = 3.07, p = 0.0278; IX, t(6) = 3.26, p = 0.0173, n = 6–7). Products for exon II mRNA did not change with zebularine. C, Correlative studies from the same animals used in B. Zebularine significantly decreased levels of methylated bdnf exons I, II, IV, and VI DNA in area CA1 of hippocampus (I, t(6) = 13.33, p = 0.0001; II, t(5) = 2.73, p = 0.0411; IV, t(6) = 4.371, p = 0.0047; VI, t(6) = 2.82, p = 0.0304, n = 6–7). The solid line across the bars represents normalized naive control levels. D, Bisulfite sequencing analysis performed on 12 CpG sites near the transcription initiation site of exon IV show percentage of cytosine residues at specific CpG sites that were demethylated after zebularine treatment (Student's t test, *p < 0.05, **p < 0.01, ***p < 0.001, compared with naive controls).
In correlation with the observed increases in exon I, IV, and VI mRNA levels after DNMT inhibition, MSP analysis suggested that there was a decrease in DNA methylation associated with exons I, IV, and VI in area CA1 of hippocampus relative to vehicle controls (Fig. 3C). Although we observed a significant decrease in DNA methylation associated with exon II, we found no significant changes in exon II mRNA levels after DNMT inhibition. In addition, we used BSP to examine site-specific methylation of 12 CpG dinucleotides within the promoter region of exon IV (Fig. 3D). The results showed significant decreases in cytosine residues methylated at CpG sites 10, 11, and 12 after Zebularine infusion (Fig. 3D). Together, these data demonstrate that DNA methylation in the hippocampus is sufficient to drive altered exon-specific bdnf transcription.
Although exon IV transcription increased with zebularine treatment (Fig. 3B), the percentage of cytosine residues methylated at CpG site 7 of exon IV DNA significantly increased with zebularine treatment with a trend of hypermethylation at CpG sites 4, 5, and 6 (Fig. 3D). This result was unexpected and suggests the possibility of compensatory increases in DNA methylation at CpG sites not sensitive to zebularine.
We also evaluated the effect of blocking DNMT activation on memory consolidation by infusing one of two distinct DNMT inhibitors, zebularine or RG108, directly into area CA1 of hippocampus 1 h before training (Fig. 4A). When memory was assessed 24 h later (test day 1), animals infused with the indirect DNMT inhibitor, zebularine, displayed significantly less freezing than did their vehicle-infused counterparts (Fig. 4B). We also observed a significant decrease in freezing from animals treated with RG108 (Fig. 4B), which is a non-nucleoside compound that directly inhibits DNMT enzyme activity (Brueckner et al., 2005; Schirrmacher et al., 2006). As a control for nonspecific drug effects, we also performed a short-term memory test 1 h after training and found that vehicle- and Zebularine-treated animals displayed similar freezing behavior (t(10) = 0.2527, p = 0.8056, n = 6). Thus, our results confirm that ongoing regulation of DNA methylation of genes is required for proper long-term memory consolidation in the adult CNS.
Figure 4.

Inhibition of DNMT interferes with contextual fear memory. A, Diagram outlines the experimental design used with data presented in B. B, On test day 1, zebularine- or RG108-treated animals exhibited lower amounts of freezing behavior compared with vehicle-treated animals (zebularine, t(9) = 2.52, p = 0.0328; RG108, t(8) = 3.72, p = 0.0059, n = 9–10; Student's t test, *p < 0.05, **p < 0.01). Error bars indicate SEM.
Histone modifications at bdnf promoters after contextual fear conditioning
In conjunction with DNA methylation, histone modifications play a crucial role in gene activation through chromatin remodeling. We examined whether histone modifications at specific bdnf promoters are altered in the hippocampus after contextual fear conditioning, using ChIP. Given the relatively low sensitivity of ChIP, we focused on the changes associated with fear conditioning because these were the most robust transcriptional changes we observed in our earlier studies (Fig. 1C). At 2 h after training, area CA1 was extracted and processed for ChIP assays with antibodies against histone H3 acetylation (AcH3), histone H4 polyacetylation (AcH4), and histone H3 phosphoacetylation (PH3/AcH3). The immunoprecipitated DNA was analyzed for bdnf promoter activity using real-time PCR. Both histone H3 acetylation and phosphoacetylation increased at promoter 4 in area CA1 of hippocampus after fear conditioning relative to naive controls (Fig. 5A). In addition, at 2 h after training, a decrease in histone H4 acetylation was detected at promoter 2 (Fig. 5A). No statistically significant changes were found in histone modifications (AcH3, AcH4, or AcH3/PH3) at promoters 1 and 6 at 2 h after fear conditioning. These results support the hypothesis that chromatin remodeling (histone modification) is directly involved in initiation start site-specific bdnf promoter regulation in hippocampus during consolidation of fear memory (Levenson et al., 2004; Chwang et al., 2006; Lubin and Sweatt, 2007). The selective increase in histone acetylation and phosphoacetylation at promoter 4 with fear conditioning is in good agreement with fear-conditioning-associated increased transcription at this site as well (Fig. 1D).
Figure 5.

Histone modifications at exon-specific bdnf promoters during memory consolidation. A, At the bdnf gene, PH3/AcH3 and AcH3 levels were increased at promoter 4 during fear memory consolidation relative to naive controls (PH3/AcH3, t(2) = 4.185, p = 0.05; AcH3, t(2) = 6.21, p = 0.0250, n = 3). AcH4 levels were significantly reduced at promoter 2 relative to naive controls (t(2) = 12.26, p = 0.0066, n = 3). The top shows the schematic location of the bdnf promoters preceding each exon (P1, P2, P4, and P6) as indicated by the star. In the graph, the areas with no bars indicate that the anti-histone antibody precipitated negligible levels of the bdnf promoter regions that were not detectable. B, At the bdnf promoter 4, AcH3 levels were increased during memory consolidation and significantly attenuated with DNMT inhibition [zebularine (ZEB)] relative to naive controls (t(8) = 3.047, p = 0.0318, n = 4–6). C, There were no significant histone modifications (AcH3) around the β-actin promoter during memory consolidation (n = 3; one-way ANOVA, *p < 0.05, compared with naive controls; Student's t test, #p < 0.05, compared with context + shock alone). Error bars indicate SEM.
Given our finding that histone acetylation is increased at bdnf promoter 4 after fear conditioning (Fig. 5A), we next determined whether or not DNMT inhibition would affect histone modifications at this promoter during memory consolidation. In theory, histone deacetylases (HDACs) are recruited to methylated DNA to produce transcriptional silencing of genes. To this end, we performed additional ChIP assays and examined the effects of DNMT inhibition on AcH3 levels at the bdnf gene in response to fear conditioning. At 2 h after fear conditioning, zebularine treatment significantly attenuated the increase in AcH3 levels at bdnf promoter 4 relative to vehicle-treated fear-conditioned counterparts (Fig. 5B). As a control, we also included analysis of DNMT inhibition on AcH3 levels around the β-actin promoter during memory consolidation and found no significant changes (Fig. 5C). Although somewhat surprising, these results are consistent with our previous results suggesting cross talk between DNA methylation and histone acetylation (Miller et al., 2008), specifically suggesting that these mechanisms regulate histone acetylation at bdnf promoter 4 during memory consolidation.
Next, we reasoned that if DNMT inhibition decreased histone acetylation at bdnf promoter 4 during memory consolidation, we might observe altered methylation at exon IV with DNMT inhibition that is characteristic of an inactive promoter. Thus, we assessed the effect of blocking DNMT activity with zebularine on the levels of bdnf gene methylation in area CA1 2 h after training in fear-conditioned animals. BSP analysis revealed that the observed decrease in percentage methylation at cytosine residues at exon IV CpG sites with fear conditioning was significantly altered with DNMT inhibition (Fig. 6A). Consequently, we also found that DNMT inhibition significantly attenuated exon IV mRNA levels with fear conditioning (Fig. 6B). These results are consistent with the finding that DNMT inhibition decreased histone acetylation at bdnf promoter 4 during memory consolidation. Moreover, these findings provide a parsimonious explanation for the mechanism by which DNMT inhibition blocks memory formation: through inhibition of transcription of a gene that promotes synaptic plasticity and memory, bdnf. It is important to note that although DNMT inhibition reduced total bdnf mRNA levels (exon IX) produced by fear conditioning, total bdnf expression remained significantly elevated relative to naive controls (data not shown).
Figure 6.

Inhibition of DNMT activation during memory consolidation prevents bdnf DNA demethylation and mRNA expression. A, Sequence map of bdnf exon IV DNA is shown including the 12 CpG sites (bold). Methylation analysis of the 12 CpG dinucleotides of exon IV show that DNMT inhibition altered the percentage of cytosine residues that were demethylated with fear conditioning. B, At 2 h after fear conditioning, DNMT inhibition with zebularine prevented an increase in bdnf exon IV mRNA (t(10) = 2.493, p = 0.0318, n = 6; one-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001, compared with naive controls; Student's t test, #p < 0.05, compared with context + shock). Error bars indicate SEM.
NMDA receptor activation is necessary for regulation of bdnf DNA methylation by contextual fear conditioning
Given the documented role of NMDA receptor activation in long-term memory formation and hippocampal chromatin remodeling (Levenson et al., 2004; Chwang et al., 2006), we determined whether or not NMDA receptor activation was necessary for bdnf DNA methylation and altered gene expression in contextual fear conditioning. Again, we focused on associative fear conditioning because the most robust increases in bdnf DNA methylation in hippocampus were observed with associative contextual fear learning.
We examined the effect of inhibiting NMDA receptor activation on memory consolidation by injecting rats with the noncompetitive antagonist MK801 1 h before training (Fig. 7A). Twenty-four hours later (test day 1), MK801-treated animals displayed significantly less freezing than did vehicle-treated animals (t(14) = 14.90, p = 0.0001, n = 8), as has been previously reported (Levenson et al., 2004; Chwang et al., 2006). We next isolated mRNA from either vehicle- or MK801-treated fear-conditioned animals at 2 h after training and assessed exon IX mRNA levels (all transcripts) in area CA1 of hippocampus. Vehicle-treated animals displayed a significant increase in total bdnf mRNA in area CA1 of hippocampus relative to naive controls, whereas MK801 treatment blocked this increase (Fig. 7B). These findings demonstrate that NMDA receptor activation is necessary for increased bdnf gene transcription during consolidation of contextual fear memory. In control experiments we confirmed that the effects of NMDA receptor blockade on bdnf mRNA expression were reversible after 24 h (data not shown).
Figure 7.

Inhibition of NMDA receptor activation prevents bdnf DNA demethylation and mRNA expression. A, Diagram outlines the experimental design used with data presented in B–D. B, At 2 h after fear conditioning, NMDA receptor inactivation with MK801 prevented an increase in bdnf total mRNA (exon IX, t(9) = 4.18, p = 0.0024, n = 7–8). C, NMDA receptor inactivation prevented the demethylation of exon IV induced by fear conditioning (methylated exon IV, t(10) = 2.99, p = 0.0308; unmethylated exon IV, t(11) = 4.441, p = 0.1108, n = 6–7). D, NDMA receptor inactivation prevented changes in exon IV mRNA (exon IV, t(10) = 6.85, p = 0.0156, n = 6). The solid line across the bars represents normalized naive control levels (one-sample t test, *p < 0.05, **p < 0.01, ***p < 0.001, compared with naive controls; Student's t test, #p < 0.05, ##p < 0.01, ###p < 0.001, compared with context + shock). Error bars indicate SEM.
To ascertain whether NMDA receptor activation mediates altered DNA methylation at the bdnf locus during consolidation of contextual fear conditioning, we examined the effect of MK801 on the methylation status of DNA associated with exon IV. We observed no change in exon IV methylation in area CA1 of hippocampus from MK801-treated animals, indicating that NMDA receptor blockade during fear-conditioning training prevented decreased bdnf methylation (Fig. 7C). Moreover, the increase in unmethylated exon IV levels normally seen with fear conditioning was also blocked with MK801 treatment (supplemental Fig. 1, available at www.jneurosci.org as supplemental material; Fig. 7C). Importantly, the lack of altered exon IV methylation after MK801 treatment was paralleled by a block in exon IV mRNA levels after fear conditioning when compared with vehicle treatment (Fig. 7D). BSP analysis confirmed that cytosine residues at exon IV CpG sites were demethylated with fear conditioning, and that MK801 treatment significantly attenuated demethylation (Fig. 8). Interestingly, we found that MK801 treatment did not alter demethylation at CpG site 3 of exon IV normally observed with fear conditioning, thus suggesting complex CpG site-specific regulation of DNA methylation within the bdnf gene. In parallel experiments, we examined the effect of MK801 on the methylation status of another site within the bdnf gene, the bdnf coding exon (exon IX) via two approaches, MSP and methylated DNA immunoprecipitation. Using these two independent methods, we confirmed that exon IX methylation also decreased with fear conditioning and that MK801 treatment blocked this effect at exon IX (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Together, these findings confirm that NMDA receptor activation contributes to the regulation of bdnf gene methylation during contextual fear conditioning.
Figure 8.

Inhibition of NMDA receptor activation (top) alters demethylation of cytosine residues at exon IV bdnf DNA induced by fear conditioning. Sequence map of bdnf exon IV DNA is shown (middle), including the 12 CpG sites (bold). Methylation analysis of the 12 CpG dinucleotides of exon IV (bottom) show that NMDA receptor blockade altered the percentage of cytosine residues that were demethylated with fear conditioning (Student's t test, *p < 0.05, **p < 0.01, ***p < 0.001, compared with naive controls). A two-way ANOVA revealed a highly significant effect of behavior group (F = 35.71, p < 0.0001) and region (F = 3.428, p < 0.0002), as well as a significant behavior group × region interaction effect (F = 1.676, p < 1.676). Error bars indicate SEM.
Discussion
In this study we produced four main findings. First, we found differential regulation of bdnf exons in hippocampus after context exposure versus fear conditioning, and found that both types of environmental stimuli triggered altered DNA methylation at the bdnf gene locus. Second, we observed that DNA methylation is dynamic in the adult CNS and appears to regulate bdnf gene readout during memory consolidation in the adult hippocampus. Third, we found that DNMT inhibition causes bdnf DNA demethylation and that demethylation is sufficient to lead to increased bdnf mRNA levels in hippocampus in vivo. Finally, we found that NMDA receptor activation plays a pivotal role in the regulation of bdnf DNA methylation and altered bdnf gene expression during memory consolidation. Overall, these observations concerning DNA methylation in the adult CNS are in stark contrast to findings from developmental studies that led to the historical view that DNA methylation is a static process after cell differentiation and neural development. Thus, based on these findings, we propose that bdnf DNA methylation is subject to ongoing regulation in the adult nervous system, likely occurring in nondividing cells and subject to control by environmental influences. This interpretation is in good agreement with recent findings in non-neuronal cells indicating that rapid cycling of DNA methylation is a potent transcription control mechanism in nondividing cells (Kangaspeska et al., 2008; Métivier et al., 2008).
Using a mammalian contextual fear- conditioning model, we observed differential regulation of specific bdnf exons in hippocampus after contextual fear conditioning. Although the purpose for this differential control and usage of specific bdnf exons in the CNS is unknown, these findings are in agreement with recent studies examining differential usage of bdnf noncoding exons in amygdala during consolidation of fear learning (Rattiner et al., 2004; Ou and Gean, 2007). In these earlier studies, bdnf exons I and IV were transcriptionally upregulated in amygdala during the consolidation of fear learning. In our current study, we found that only bdnf exon IV is transcriptionally upregulated in hippocampus during consolidation of fear learning (2 h after training), suggesting that bdnf exon usage is brain region specific and responds differently to environmental cues. These results also suggest that exon IV-containing bdnf transcripts are responsible for total bdnf gene expression in hippocampus during consolidation of contextual conditioned fear memory. In addition, we observed that mRNAs for bdnf exons I and VI are upregulated in hippocampus 2 h after context exposure alone. Thus, it is possible that differential exon usage in hippocampus contributes to highly regulated transcriptional control of the bdnf gene in response to unique events such as learning about a novel environment versus an association about that same environment. Overall, the present results together with previous findings in the amygdala suggest the intriguing possibility that site-specific initiation of transcription of the bdnf gene might participate in the encoding of salient features of environmental cues.
The complexity in the regulation of the bdnf gene is further demonstrated by our DNA methylation results. We hypothesized that DNA methylation within bdnf promoter or exonic regions is an epigenetic mechanism regulating bdnf gene expression in hippocampus after contextual fear conditioning. In support of this, methylation-specific real-time PCR revealed that bdnf exon VI DNA methylation increased after fear conditioning. This correlated with no change in exon VI mRNA levels in hippocampus during memory consolidation. These findings suggest active transcriptional silencing of promoter-specific bdnf transcripts in hippocampus after fear learning. Using methylation-specific real-time PCR and bisulfite sequencing, we also found a decrease in DNA methylation of exons IV, which corresponded to an increase in exon IV-containing transcripts and an overall increase in total bdnf gene mRNA levels (exon IX) in hippocampus during fear memory consolidation. These important observations support the idea that demethylation of CpG sites within CpG islands promotes transcriptional activity of the bdnf gene during memory consolidation.
In light of our DNA methylation studies, we triggered bdnf gene demethylation using hippocampal infusion of the DNMT inhibitor zebularine and confirmed that altered methylation of CpG island sites within the bdnf gene is sufficient to mediate bdnf gene expression in the adult hippocampus. As a complement to these studies, we confirmed that both zebularine and a structurally distinct, direct DNMT inhibitor, RG108, blocked memory consolidation (Brueckner et al., 2005; Schirrmacher et al., 2006). Inhibition of DNMT activity with zebularine increased basal levels of bdnf exon I, IV, and VI mRNA in area CA1 in vivo. This paralleled increases in total bdnf gene expression as assessed by exon IX mRNA levels. In contrast, DNMT inhibition produced no changes in exon II mRNA levels in hippocampus, which did not parallel the overall decrease in bdnf exon II methylation. One possible explanation for this contrasting effect at exon II is that other modifications at this promoter, such as altered histone modifications, suppressed changes in exon II mRNA levels by a DNMT-insensitive transcriptional complex. Nonetheless, these results confirmed that DNMT inhibition with zebularine decreased basal bdnf DNA methylation in hippocampus at bdnf exons I, IV, and VI. Overall, these findings indicate that DNA demethylation at sites I, IV, and VI is sufficient to drive altered transcription of the bdnf gene and suggest that this mechanism may underlie bdnf gene expression changes in hippocampus during the consolidation of conditioned fear memories.
Because many previous reports have demonstrated that after cellular differentiation, DNA methylation is stable to control tissue-specific gene expression for the maintenance of cell identity, dynamic regulation of CpG methylation in the adult hippocampus was unanticipated. However, our methylation studies indicate that dynamic regulation of CpG methylation at the bdnf gene does indeed occur in the adult hippocampus in response to environmental influences. Thus, our findings provide an additional level of regulation of gene transcription in the adult nervous system and are in agreement with recent studies that indicate that similar active, rapid cycling of CpG methylation occurs in other experimental systems (Kangaspeska et al., 2008; Métivier et al., 2008). Furthermore, identifying a role for DNMTs in the methylation of the bdnf gene in the adult hippocampus expands the current understanding of the regulation of epigenetic marks after cellular differentiation in this brain region (Levenson et al., 2006; Miller and Sweatt, 2007).
Our findings also indicate the involvement of histone modifications at the bdnf gene during consolidation of contextual fear learning. We discovered that histone H3 phosphoacetylation and acetylation increased at promoter 4 in hippocampus, which paralleled changes in exon IV mRNA transcription and total bdnf gene expression (exon IX). We also observed decreases in histone H4 acetylation at promoter 2 with no change in exon II mRNA levels in hippocampus during consolidation of fear learning. We speculate that decreased histone H4 acetylation at promoter 2 may help serve to suppress exon II mRNA transcription and to allow promoter 4 transcriptional control of the bdnf gene during consolidation of fear memory. It is also possible that although histone H4 acetylation is modulated at promoter 2 in hippocampus, histone H3 phosphoacetylation and acetylation at promoter 4 play the dominant role in bdnf gene expression in hippocampus regardless of other signals. Whichever scenario is correct, these findings support the general hypothesis that specific histone modifications, such as histone H3 acetylation, are associated with regulation of DNA methylation and altered gene transcription during memory consolidation.
In addition, we report that DNMT inhibition prevents histone H3 acetylation at bdnf promoter 4 during memory consolidation. We also discovered that blocking DNMT activity prevented demethylation of the bdnf gene associated with exon IV and blocked changes in bdnf exon IV mRNA levels with fear conditioning. These results were not predicted, because DNMT inhibition should lead to a decrease in methylation and an increase in gene expression. Perhaps DNMT inhibition may have suppressed bdnf gene transcription through secondary effects on memory suppressor genes, such as protein phosphatase 1 (PP1), as the laboratory has reported previously (Miller and Sweatt, 2007). Alternatively, DNMTs have recently been discovered to be regulators of DNA demethylation as well as DNA methylation, and this mechanism might account for an effect of DNMT inhibition to affect both increases and decreases in DNA methylation (Kangaspeska et al., 2008; Métivier et al., 2008). Given the compensatory functions of memory suppressor genes, such as PP1, and the emerging importance of chromatin modifying enzymes, such methyl-CpG-binding protein 2 (MeCP2) and HDACs, in establishing epigenetic marks (Nan et al., 1998), combinatorial epigenetic mechanisms should be considered for future study of multiple gene transcription-regulating mechanisms underlying memory formation.
It is well appreciated that NMDA receptor activation is important for the acquisition and consolidation of memories (Morris et al., 2003). Thus, we hypothesized that NMDA receptor activation might be involved in the regulation of DNA methylation as a means of transcriptional control during the early stages of long-term memory formation. We observed that NMDA receptor blockade prevented the increase in total bdnf gene expression (exon IX) normally seen in area CA1. This finding complements previous investigations concerning the effects of blocking NMDA receptor activation on bdnf gene expression in the rat brain (Castrén et al., 1993). Moreover, we observed that inhibiting NMDA receptor activation blocked memory-associated bdnf DNA demethylation at exon IV. Furthermore, inhibition of NMDA receptor activation prevented increases in exon IV bdnf mRNA levels in hippocampus with fear conditioning. These findings are significant because they indicate that signaling through the NMDA receptor is critical for DNA demethylation of the bdnf gene in this learning paradigm, and also because they implicate DNA methylation as a nuclear target of the NMDA receptor.
The signaling mechanisms involved in NMDA receptor-mediated bdnf DNA demethylation are mysterious at present but clearly deserve further examination. In particular, the methylcytosine-binding proteins, including MeCP2, are abundantly expressed in the nervous system, and these proteins mediate the gene-silencing effect of DNA methylation (Lewis et al., 1992; Ng and Bird, 1999). In addition, the disassociation of the repression complex, MeCP2–HDAC–mSin3A, from bdnf promoter 4 has been shown to increase bdnf gene transcription in an activity-dependent manner in cultured cortical cells (Martinowich et al., 2003). Thus, one appealing but very speculative hypothesis at this point is that NMDA receptor-mediated signaling may target MeCP2 to specific bdnf gene promoter or exonic regions to regulate bdnf gene methylation states during associative memory formation (Chen et al., 2003; Zhou et al., 2006).
In summary, we have shown using direct molecular techniques that differential bdnf DNA methylation is dynamically regulated in the adult hippocampus during consolidation of fear memories. These results provide the first evidence of differential DNA methylation of the bdnf gene in the adult brain in response to a behaviorally relevant learning paradigm. Importantly, our results underscore the complexity of mechanisms regulating de novo bdnf methylation and their possible roles in subserving adult cognition. Because aberrant bdnf gene expression continues to be implicated in psychiatric disorders associated with cognitive dysfunction (schizophrenia, depression, and bipolar disorder), our results provide support for the hypothesis that manipulating chromatin structure may be a viable therapeutic mechanism to restore cognitive function in these disorders. Thus, understanding the complex epigenetic regulation of the bdnf gene appears to be integral to understanding the possible role of bdnf dysregulation in psychiatric illness. In a more general context, exploration of changes in DNA methylation as a mechanism for regulating gene transcription in memory formation will shed light on the potential role of epigenetic molecular mechanisms in the functioning of the adult CNS.
Footnotes
This work was supported by the National Institutes of Health (NS048811, MH082106 to F.D.L.; MH57014, NS37444, NS013546 to J.D.S.), the Epilepsy Foundation, the National Alliance for Research on Schizophrenia and Depression, the Rotary Clubs Coins for Alzheimer's Research Trust Fund, and the Evelyn F. McKnight Brain Research Foundation. We thank G. Calfa and L. Pozzo-Miller for critical reading of this manuscript and F. Hester for her much appreciated assistance. We thank Drs. Y. Li, T. O. Tollefsbol, M. Han, and the University of Alabama at Birmingham Genomics Core Facility for their assistance in bisulfite sequencing.
References
- Abdolmaleky HM, Cheng KH, Russo A, Smith CL, Faraone SV, Wilcox M, Shafa R, Glatt SJ, Nguyen G, Ponte JF, Thiagalingam S, Tsuang MT. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet. 2005;134B:60–66. doi: 10.1002/ajmg.b.30140. [DOI] [PubMed] [Google Scholar]
- Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res. 2007;85:525–535. doi: 10.1002/jnr.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcón JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Angelucci F, Brenè S, Mathé AA. BDNF in schizophrenia, depression and corresponding animal models. Mol Psychiatry. 2005;10:345–352. doi: 10.1038/sj.mp.4001637. [DOI] [PubMed] [Google Scholar]
- Aranda-Abreu GE, Hernández ME, Soto A, Manzo J. Possible Cis-acting signal that could be involved in the localization of different mRNAs in neuronal axons. Theor Biol Med Model. 2005;2:33. doi: 10.1186/1742-4682-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blichenberg A, Schwanke B, Rehbein M, Garner CC, Richter D, Kindler S. Identification of a cis-acting dendritic targeting element in MAP2 mRNAs. J Neurosci. 1999;19:8818–8829. doi: 10.1523/JNEUROSCI.19-20-08818.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Bredy TW, Wu H, Crego C, Zellhoefer J, Sun YE, Barad M. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn Mem. 2007;14:268–276. doi: 10.1101/lm.500907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueckner B, Boy RG, Siedlecki P, Musch T, Kliem HC, Zielenkiewicz P, Suhai S, Wiessler M, Lyko F. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005;65:6305–6311. doi: 10.1158/0008-5472.CAN-04-2957. [DOI] [PubMed] [Google Scholar]
- Castrén E, da Penha Berzaghi M, Lindholm D, Thoenen H. Differential effects of MK-801 on brain-derived neurotrophic factor mRNA levels in different regions of the rat brain. Exp Neurol. 1993;122:244–252. doi: 10.1006/exnr.1993.1124. [DOI] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Chwang WB, O'Riordan KJ, Levenson JM, Sweatt JD. ERK/MAPK regulates hippocampal histone phosphorylation following contextual fear conditioning. Learn Mem. 2006;13:322–328. doi: 10.1101/lm.152906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Kubota T, Song M, Daniel R, Berry-Kravis EM, Prior TW, Popovich B, Rosser L, Arinami T, Ledbetter DH. Methylation analysis of the fragile X syndrome by PCR. Genet Test. 1997;1:151–155. doi: 10.1089/gte.1997.1.151. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- Grayson DR, Jia X, Chen Y, Sharma RP, Mitchell CP, Guidotti A, Costa E. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci U S A. 2005;102:9341–9346. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Giustetto M, Lomvardas S, Kim JH, Miniaci MC, Schwartz JH, Thanos D, Kandel ER. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;111:483–493. doi: 10.1016/s0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- Hall J, Thomas KL, Everitt BJ. Rapid and selective induction of BDNF expression in the hippocampus during contextual learning. Nat Neurosci. 2000;3:533–535. doi: 10.1038/75698. [DOI] [PubMed] [Google Scholar]
- Kangaspeska S, Stride B, Métivier R, Polycarpou-Schwarz M, Ibberson D, Carmouche RP, Benes V, Gannon F, Reid G. Transient cyclical methylation of promoter DNA. Nature. 2008;452:112–115. doi: 10.1038/nature06640. [DOI] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DE, Truong HT, Russo SJ, Laplant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48:303–314. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Lauterborn JC, Rivera S, Stinis CT, Hayes VY, Isackson PJ, Gall CM. Differential effects of protein synthesis inhibition on the activity-dependent expression of BDNF transcripts: evidence for immediate-early gene responses from specific promoters. J Neurosci. 1996;16:7428–7436. doi: 10.1523/JNEUROSCI.16-23-07428.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, Malone LM, Sweatt JD. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem. 2006;281:15763–15773. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- Linnarsson S, Björklund A, Ernfors P. Learning deficit in BDNF mutant mice. Eur J Neurosci. 1997;9:2581–2587. doi: 10.1111/j.1460-9568.1997.tb01687.x. [DOI] [PubMed] [Google Scholar]
- Liu QR, Lu L, Zhu XG, Gong JP, Shaham Y, Uhl GR. Rodent BDNF genes, novel promoters, novel splice variants, and regulation by cocaine. Brain Res. 2006;1067:1–12. doi: 10.1016/j.brainres.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lubin FD, Sweatt JD. The IkappaB kinase regulates chromatin structure during reconsolidation of conditioned fear memories. Neuron. 2007;55:942–957. doi: 10.1016/j.neuron.2007.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. Memory—a century of consolidation. Science. 2000;287:248–251. doi: 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- Métivier R, Gallais R, Tiffoche C, Le Péron C, Jurkowska RZ, Carmouche RP, Ibberson D, Barath P, Demay F, Reid G, Benes V, Jeltsch A, Gannon F, Salbert G. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452:45–50. doi: 10.1038/nature06544. [DOI] [PubMed] [Google Scholar]
- Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Miller CA, Campbell SL, Sweatt JD. DNA methylation and histone acetylation work in concert to regulate memory formation and synaptic plasticity. Neurobiol Learn Mem. 2008;89:599–603. doi: 10.1016/j.nlm.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Moser EI, Riedel G, Martin SJ, Sandin J, Day M, O'Carroll C. Elements of a neurobiological theory of the hippocampus: the role of activity-dependent synaptic plasticity in memory. Philos Trans R Soc Lond B Biol Sci. 2003;358:773–786. doi: 10.1098/rstb.2002.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- Nanda S, Mack KJ. Multiple promoters direct stimulus and temporal specific expression of brain-derived neurotrophic factor in the somatosensory cortex. Brain Res Mol Brain Res. 1998;62:216–219. doi: 10.1016/s0169-328x(98)00242-3. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J Neurosci. 2008;28:395–406. doi: 10.1523/JNEUROSCI.3796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Bird A. DNA methylation and chromatin modification. Curr Opin Genet Dev. 1999;9:158–163. doi: 10.1016/s0959-437x(99)80024-0. [DOI] [PubMed] [Google Scholar]
- Ou LC, Gean PW. Transcriptional regulation of brain-derived neurotrophic factor in the amygdala during consolidation of fear memory. Mol Pharmacol. 2007;72:350–358. doi: 10.1124/mol.107.034934. [DOI] [PubMed] [Google Scholar]
- Pal R, Agbas A, Bao X, Hui D, Leary C, Hunt J, Naniwadekar A, Michaelis ML, Kumar KN, Michaelis EK. Selective dendrite-targeting of mRNAs of NR1 splice variants without exon 5: identification of a cis-acting sequence and isolation of sequence-binding proteins. Brain Res. 2003;994:1–18. doi: 10.1016/j.brainres.2003.08.046. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. San Diego: Academic; 1998. The rat brain in stereotaxic coordinates. [DOI] [PubMed] [Google Scholar]
- Rattiner LM, Davis M, Ressler KJ. Differential regulation of brain-derived neurotrophic factor transcripts during the consolidation of fear learning. Learn Mem. 2004;11:727–731. doi: 10.1101/lm.83304. [DOI] [PubMed] [Google Scholar]
- Schirrmacher E, Beck C, Brueckner B, Schmitges F, Siedlecki P, Bartenstein P, Lyko F, Schirrmacher R. Synthesis and in vitro evaluation of biotinylated RG108: a high affinity compound for studying binding interactions with human DNA methyltransferases. Bioconjug Chem. 2006;17:261–266. doi: 10.1021/bc050300b. [DOI] [PubMed] [Google Scholar]
- Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- Soulé J, Messaoudi E, Bramham CR. Brain-derived neurotrophic factor and control of synaptic consolidation in the adult brain. Biochem Soc Trans. 2006;34:600–604. doi: 10.1042/BST0340600. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Weickert CS, Hyde TM, Lipska BK, Herman MM, Weinberger DR, Kleinman JE. Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol Psychiatry. 2003;8:592–610. doi: 10.1038/sj.mp.4001308. [DOI] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]