

Abstract
All prokaryotes encode a panel of metal sensor or metalloregulatory proteins that govern the expression of genes that allows an organism to quickly adapt to toxicity or deprivation of both biologically essential transition metal ions, e.g., Zn, Cu, Fe, and heavy metal pollutants. As such, metal sensor proteins can be considered arbiters of intracellular transition metal bioavailability and thus potentially control the metallation state of the metalloproteins in the cell. Metal sensor proteins are specialized allosteric proteins that regulate transcription as a result direct binding of one or two cognate metal ions, to the exclusion of all others. In most cases, the binding of the cognate metal ion induces a structural change in a protein oligomer that either activates or inhibits operator DNA binding. A quantitative measure of the degree to which a particular metal drives metalloregulation of operator DNA-binding is the allosteric coupling free energy, ΔGc. In this review, we summarize recent work directed toward understanding metal occupancy and metal selectivity of these allosteric switches in selected families of metal sensor proteins and examine the structural origins of ΔGc in the functional context a thermodynamic “set-point” model of intracellular metal homeostasis.
Keywords: metalloregulation, metal sensor protein, transition metal ions, allosteric coupling free energy, protein-DNA interactions, linkage
Introduction
Transcriptional repressors are allosteric proteins that sense cellular concentrations of metabolites and other small molecular effectors in order to allow for an appropriate response to changing extracellular milieu [1]. These proteins function through a specific interaction with the operator/promoter region DNA just upstream of the regulated gene or operon. Small molecule ligand binding to the protein-DNA complex, typically to a site distinct from the DNA binding site, drives a structural or dynamic change in conformation that modulates the affinity or structure of the regulatory protein-DNA complex.
Metalloregulatory proteins represent a sub-classification of transcriptional regulators that have evolved to balance the expression of cellular metal uptake and efflux/detoxification systems in response to changes in intracellular metal concentration or availability [2,3]. These proteins are often evolutionarily derived from existing transcriptional regulator families and within a single family, the metal selectivity of individual members can vary dramatically. For example, individual members of the ArsR (arsenic repressor) family [4,5] have been described that regulate heavy metal detoxification systems in response to wide range of transition metal ions, organoarsenicals and oxyanions, and in other cases to changes in sulfur metabolism [6,7], via a classical transcriptional derepression mechanism, on what is essentially an identical structural scaffold. Here, the metal inducer is an allosteric inhibitor of DNA operator DNA binding resulting in transcriptional derepression of resistance genes. In contrast, for Fur (ferric uptake repressor) family sensors, now structurally and/or functionally characterized that respond to intracellular Fe(II) levels (Fur), heme availability (Irr), peroxide stress (PerR), Zn(II) (Zur), Mn(II) (Mur) and Ni(II) (Nur), metal binding drives a change in the repressor conformation that allosterically activates DNA operator binding in response to a specific metal or group of closely related metals [8].
Metal sensor proteins as arbiters of the cellular metallome
A particular metal ion can function either as an allosteric activator or inhibitor of DNA operator binding in a way that is consistent with cellular regulatory logic. To illustrate this, we discuss zinc homeostasis in Streptococcus pneumoniae (Fig. 1) [9-13]. Metals that allosterically inhibit DNA binding bind to repressors of genes that encode efflux or detoxification systems. In the absence of metal stress, these genes are transcriptionally repressed, and only become expressed in response to specific metal stress. The tetracycline repressor family [14] regulator SczA is the zinc efflux repressor in S. pneumoniae [9] and is functionally identical to S. aureus CzrA, the paradigm ArsR family zinc regulator [15,16], and E. coli ZntR [17,18], from the MerR family, yet adopts a completely different fold and mechanism of zinc sensing (see below).
Fig. 1.
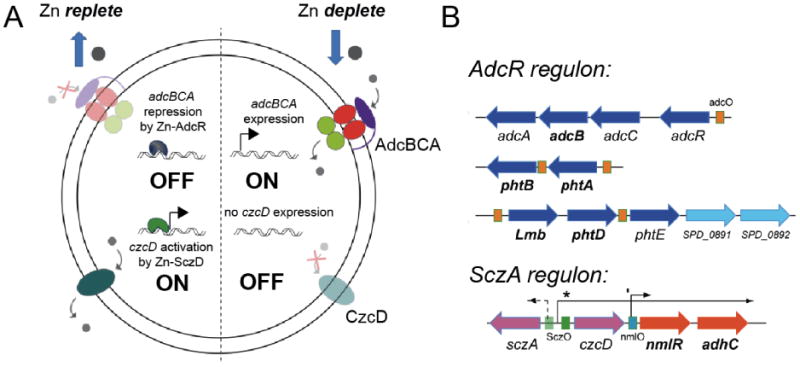
Regulation of zinc homeostasis in Streptococcus pneumonaie. AdcR (adhesin competence repressor) and SczA (streptococcal czcD activator) are uptake and efflux regulators of zinc and thus control zinc availability in the cytoplasm of Sp. AdcR is a member of the MarR family of bacterial repressors [11] and SczA is a TetR (tetracycline repressor) ortholog [9].
On the flip side, metals that allosterically activate DNA binding typically bind to repressors as co-repressors and repress the uptake of that metal [2]. The zinc uptake repressor in S. pneumoniae is adhesin competence repressor (AdcR) [11], also known as ZitR in L. lactis [19,20], and is the functional equivalent of the Fur family regulator Zur found in other organisms [21]. In the absence of metal stress, AdcR has low affinity for its operator, and downstream genes are constitutively expressed in order to bring more Zn(II) into the cell. As intracellular levels rise to a degree that saturates Zn(II) sensing sites on AdcR, this allosterically activates DNA operator binding by AdcR, which then represses expression of the uptake system. Analogously, SczA is allosterically inhibited (and activated) [9] by Zn(II) binding; this in turn induces zinc efflux which ultimately brings intracellular zinc availability (“free ”or weakly chelated zinc) into a range compatible with cell viability (Fig. 1). Thus, SczA and AdcR are hypothesized to collaborate to ensure that intracellular zinc availability is restricted to concentration range compatible with S. pneumoniae viability, despite the presence of total cell-associated Zn(II) in the near millimolar range (Fig. 1) [11,12]. All organisms are known or predicted to possess pairs of efflux and uptake regulators for zinc; the same is likely true for Ni(II), while other more (Cu) or less competitive (Mn, Fe) metals may well be managed at the level of efflux or uptake, respectively, alone [1].
A long-standing hypothesis is that zinc homeostasis is under thermodynamic control, where the corresponding affinities (KZn) of the uptake repressor, AdcR in this example, and the efflux repressor, SczA, for Zn(II) define the set-points for zinc bioavailability in the cell, and effectively “buffer” zinc in this concentration range (Fig. 2) [22]. Zn bioavailability outside of this range on either side signifies either deficiency or toxicity, respectively (Fig. 2). More importantly, this effective range of zinc bioavailability may well effectively dictate the appropriate allocation of metals to metalloproteins in the cell [23,24]. Given that zinc is a highly competitive metal, excursions of weakly chelated, labile zinc to a concentration greater than 1/KZn for the efflux regulator might directly interfere with the ability of metalloproteins and metalloenzymes to acquire a noncompetitive metal (Fe or Mn), as dictated by the Irving-Williams series [23]. The Irving-Williams series of divalent metals ions establishes that Zn(II) and Cu(II) bind with the highest affinity to a model chelate, while Mn(II) and Fe(II) bind with the lowest affinity to the same chelate, Zn(II)≤Cu(II)≫Ni(II)≥Co(II)≥Fe(II)≥Mn(II) [23,25]. Thus, metal sensor proteins may well be the arbiters of metal availability in the cell, and thus may indirectly control the metallation state of metalloproteins, i.e., the metallome.
Fig. 2.

The set-point model of metal homeostasis [1,22,23]. Representation of how the metal affinity (KZn) of a pair of Zn(II)-dependent regulators, AdcR [11] and SczA (G. Campanello and D. Giedroc, unpublished), can effectively buffer the concentration of Zn(II) in the cell, where 1/KZn defines a fractional repression/derepression of 0.5. This set-point model is based on the assumption that metal sensor proteins quickly equilibrate and effectively scan the cytoplasm for bioavailable metal. Dashed curve, Superposition of negative homotropic cooperativity of metal binding to a dimeric regulator on the set-point model, with KZn1=1012 M−1, KZn2 =5×1010 M−1, or a degree of negative cooperativity observed experimentally in BsZur [52] and SaCzrA [48]. As can be seen, negative cooperativity of zinc binding to an uptake regulator has the effect of increasing the range of [Zn]f over which metal-responsive repression is observed [52]. The same would be true for an efflux regulator, e.g., SaCzrA, on the derepression arm (SczA) of this set-point model. The competitiveness of a particular metal follows the Irving-Williams series for divalent cations [24,25,133]; monovalent Cu(I) is known to bind to chelates far more tightly than Zn(II) [1,18].
Do metal sensor proteins operate under thermodynamic or kinetic control in the cell?
The set-point model for thermodynamic control of zinc homeostasis developed in Fig. 2, where the metal sensitivities of metal-specific regulators define the physiological limits of metal availability in the cell, obviously assumes that zinc sensors and intracellular accessible zinc equilibrate rapidly. It is clear that for highly competitive metals, e.g., Cu(I), Zn(II) and perhaps Ni(II), these metals are bound so tightly (KMe≥1010 M−1) by the sensor protein that if on-rates are reasonably fast, as they are anticipated to be (see below), then off-rates of metal into solvent are going to be quite slow, in some extreme cases longer than the lifetime of the cell. Thus, this line of reasoning seem to suggest that the only way to remove the metal from the sensor protein is to simply turn the protein over. Such a model is in direct conflict with the simple set-point model (see Fig. 2), in which free metal, buffered by an overcapacity of the cell to chelate metal ions, is sensed at equilibrium in the range of 1/KMe. Indeed, alternative “kinetic” models have been proposed in yeast in which the high affinity of zinc sensors for zinc simply allows for each to function as “toggle switches” between rapid cycles of zinc deplete and replete states in order to effectively manage both chronic (long-term) and transient (short-term) conditions of metal toxicity or deficiency [26].
One way around this apparent paradox is the recognition that thermodynamically stable transition metal-ligand complexes also tend to be kinetically labile, particularly in the presence of a suitable competitor ligand, which in the cell, would include low molecular weight sulfur, nitrogen and oxygen-rich small molecules and metabolites, many of which will possess non-negligible affinity for divalent ions. A protein-based paradigm for thermodynamically stable, but kinetically labile metal complexes is metallothionein (MT), a small cysteine-rich polypeptide that binds heavy metals Cd(II), Zn(II) and Cu(I) in multinuclear clusters with affinities in the 109-1012 M−1 range for Zn(II) [27]. MTs are known to play a major role in sequestering zinc under conditions of extreme zinc deprivation [28], link cellular zinc levels with redox status of the cell [29], and play important roles in buffering zinc to approximately 10−10-10−12 M “free” Zn in the mammalian cytosol [30-32]. Early 113Cd NMR experiments showed that 113Cd ions are highly dynamic in the metal clusters of MT and readily “move” from one site to another within a cluster, without dissociation into solvent [33,34]. Thus, it seems possible that metal ligand exchange with low molecular weight solutes or with a specific target protein, e.g., an efflux transporter, could potentially catalyze the ability of a transition metal sensor protein to “bind and release” metal cargo rapidly in the cell, particularly under conditions of metal stress in a way that does not require protein turnover.
Nonetheless, it is interesting to note that a number of proteins involved in establishing a metal or oxidative stress response, including the zinc sensor ZntR (Fig, 4A, below) [35], the iron binding protein Dps, and the 4Fe-4S oxygen-sensing regulator FNR [36], are all readily targeted for degradation by the Lon and/or ClpXP proteases, but more so in the absence of metal stress (or in the case of FNR, in the presence of oxygen which leads to destruction of the 4Fe-4S cluster) [37]. In the case of ZntR, zinc and DNA binding strongly stabilize ZntR against proteolytic degradation, suggesting the need to maintain sufficient steady-state levels of Zn-ZntR to provide for continuous activation of the expression of the efflux transporter ZntA under chronic zinc stress. The recent discovery of a new copper resistance system in M. tuberculosis identified in a screen for mutants that would suppress the effects of mutants in the mycobacterial proteosome, further links in some way intracellular copper resistance to protein turnover to effect recovery after exposure to stress [38]. The copper-loaded copper chaperone CopZ in Enterococcus hirae also plays a role in the response of that organism to stress as well [39].
Fig. 4.
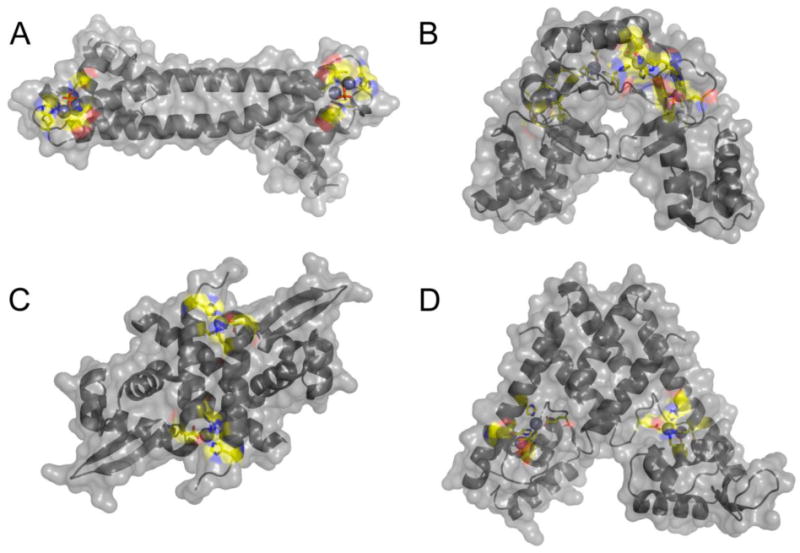
Surface representation of four Zn(II)-dependent transcriptional regulators structurally characterized. These are E. coli ZntR (A) [18], S. coelicolor Zur(B) [66], S. aureus CzrA (C) [68], and S. pneumoniae AdcR (D) [67]. Metal ions are represented as spheres and metal binding ligands are represented as sticks with carbon atoms colored in yellow. Metal binding sites in each protein have high solvent accessibility.
Theory of allosteric linkage by metal ions
Allostery originates with the simple the idea that the binding of a ligand to one site can influence the binding or chemical reactivity of the same or different ligand at a distinct, often distant, site. In the approach taken here, we apply what is essentially a model-free formalism of allostery. We make no effort to distinguish between various models, with the emphasis instead placed on defining the structural and energetic origins of allosteric negative or positive heterotropic cooperativity, by considering only the structures of the allosteric end states (fully ligated with one or both ligands), without explicit consideration of partially ligated intermediate states.
The thermodynamic cycle presented in Fig. 3 represents a closed system (Σi-14 ΔXi=0, where X is any thermodynamic state function) that is inclusive of all four possible “end” states that a dimeric metalloregulatory protein (P2, denoted P here for simplicity) can adopt in equilibrium with a single DNA duplex operator (D) and n total metal ions (M) bound: apo (P), metal-bound (P•Mn), DNA bound (P•D) and the “ternary” metal-protein-DNA complex ((P•Mn)•D) [2]. Note that P2 is in equilibrium with free monomer P as well, defined by Kdimer, and the model assumes that P has negligible affinity for D. Each side of this thermodynamic box represents a measurable transition between two of the four states (K1, K2, βnP and βnP•D). This simplistic view of the macroscopic chemical transitions allows for a generic model-free approach to quantify and normalize the allosteric response of a metalloregulatory protein for its DNA binding partner upon metal binding. Note that this scheme can be expanded across the top and bottom equilibria to expressly consider intermediate ligation states with i ligands bound, e.g., where 1<i<n; in this case, the macroscopic parameters K1 and K2 would be replaced with the appropriate step-wise binding constants [40]. Likewise, this scheme can be expanded to include an additional DNA-binding step, and/or oligomeric assemblies larger than dimers [41].
Fig. 3.
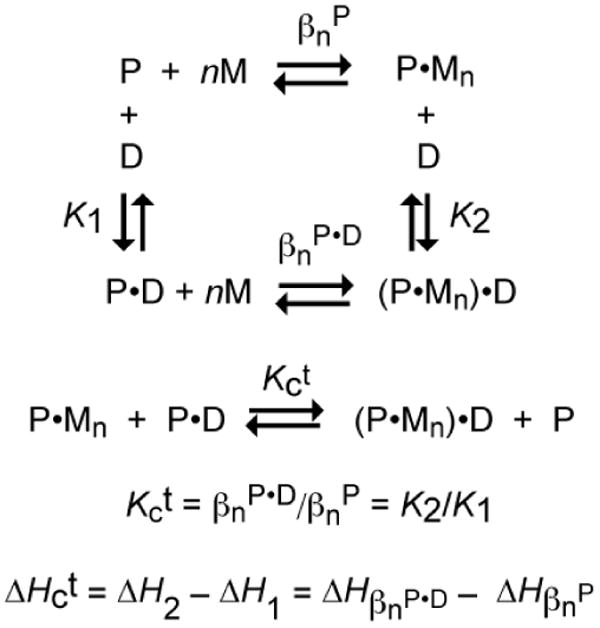
General coupled thermodynamic cycle that describes the relationship between the four allosteric states of a homodimeric or homotetrameric protein P in equilibrium with a total number of n metal ions (M) per oligomer and its DNA operator (D). βn is the overall equilibrium constant for the binding of n metal ions to the oligomer. The coupling constant is given by Kct, and is defined by the disproportion equilibrium shown.
The magnitude of allosteric regulation, Kc, is simply defined as the ligand exchange equilibrium (Fig. 3) defined the unitless disproportionation constant, Kct [42]. In other words, the allosteric response of a protein is dictated by the stability of the P•D and P•Mn states relative to the (P•Mn)•D and P states. Thermodynamically, this can be thought of as the difference in metal affinity between the P•D and P states or the difference in DNA binding affinity between the P•Mn and P states. Therefore, measurement of K1 and K2 or βnP and βnP•D can provide a quantitative determination of the unitless coupling equilibrium constant Kc, which can then be converted to free energy using the standard thermodynamic function (eq 1)
| (1) |
For repressors in which metal binding induces dissociation of the repressor off the DNA operator, the (P•Mn)•D state is substantially destabilized relative to P•Mn and free D, and access to the previously occluded promoter by RNA polymerase results in up regulation of the transcription of downstream genes in the operon. In this case, K2 < K1 (and βnP•D < βnP) and ΔGc > 0; that is the ligand exchange reaction (Fig. 3) is not favorable and the two biologically relevant “end” states are P•Mn and P•D. This is most common for regulation of metal detoxification or efflux mechanisms such as S. pneumoniae SczA (Fig. 2) [9], S. aureus CzrA and S. aureus pI258 CadC in response to Zn(II), Zn(II)/Co(II) and Cd(II)/Pb(II)/Bi(III), respectively [43-45]. Alternatively, when K2 > K1 (and βnP•D > βnP), ΔGc < 0 and free P and (P•Mn)•D are the two biologically relevant states. In this case, excess cellular metal represses downstream gene transcription by formation of the (P2•Mn)•D complex as is the case for the Fe(II), Mn(II) and Zn(II) sensing Fur family members, Fe(II)/Mn(II) sensing DtxR family repressors and S. pneumoniae AdcR (see Fig. 2) [8,11,46,47].
A particularly powerful use of this approach allows one to deconvolute the extent to which individual metal ligand donor atoms are required to simply stabilize the metal complex (βnP in Fig. 3) vs. driving the allosteric switching mechanism (embodied in ΔGc). Using such an approach, we uncovered a “division of labor” among metal ligand donor atoms in the α5 chelate in S. aureus CzrA (see Fig. 5C below): Asp84 and His97 were found to be key allosteric residues while His86 and His100 could be substituted with nonliganding residues with substantial decreases in KZn as expected, but with little or no quantitative effect on in ΔGc [48]. These studies established a 1:1 correlation between the ability to form a metal site of the native coordination geometry, i.e., tetrahedral, with structural switching within the dimer as revealed by NMR studies, and functional coupling to DNA binding. Similar features appear to characterize the Cd(II)/Pb(II) chelates of two other ArsR sensors, CadC [49] and CmtR [50,51].
Fig. 5.

Close-up view of the regulatory metal binding sites of E. coli ZntR (A), S. coelicolor Zur(B), S. aureus CzrA (C), and S. pneumoniae AdcR (D). Note that three of the four Zn(II) regulators harbor two metal binding sites that are within 10 Å of one another, either as part of a binuclear metal center consisting of two shared ligands, as in E. coli ZntR (panel A) [18] or in an amino acid sequence as is the case in S. coelicolor Zur [65,66] and S. pneumoniae AdcR [67] in panels B and D, respectively.
Mass action
In the preceding discussion of the coupled equilibrium shown in Fig. 3, it is usually assumed that the primary way to effect the redistribution of states, and thus regulation, is through a change in the intracellular metal concentration (M) since [D] and [P] are unlikely to change greatly. Although this is likely basically true for D, this may not be the case for the repressor concentration P, since in many cases, a metal-inducible operon or regulon includes the gene encoding the regulator itself (see Fig. 1) [3]. As a result, the transcription and presumably the intracellular concentration of unbound P may also increase (or decrease) on metal stress in a non-negligible fashion relative to M; this in turn, could potentially shift the “set-point” of metal sensitivity in the cell (Fig. 2) by altering the concentration of free M that would trigger allosteric regulation of DNA operator binding by P. Recent experiments with the zinc uptake repressor B. subtilis Zur are consistent with this idea, in that the sensitivity, defined as the concentration of free Zn(II) that is required for half-maximal DNA operator binding, of BsZur for Zn(II) is reduced ≈7-fold with a 10-fold increase in total P from 0.1 to 1.0 μM [52]. When superimposed on negative cooperativity of Zn(II) binding to each of the primary sensing sites in BsZur, this potentially results in a step-wise activation model in which sufficient intracellular Zn(II) present to saturate only the higher affinity of the pair of sensing sites is sufficient to repress transcription at higher concentrations of P, i.e., that which would be present under low Zn(II), whereas at lower concentrations P would require higher Zn(II) and subsequent saturation of both sensing sites to obtain full repression [52]. Clearly, access to mutant proteins that reduce the affinity and/or homotropic cooperativity of M binding to P will become important tools to tease out the physiological significance of the influence of changing P on the linkage scheme shown (Fig. 3) [48,52].
Energetics
This linkage approach also allows direct elucidation of the underlying energetics associated with the magnitude and sign of ΔGc, i.e., the magnitudes and signs of ΔHc and ΔSc which correspond to the enthalpic and entropic contributions to the allosteric coupling free energy, respectively. This is important because resolution of ΔHc and ΔSc allows one to determine the relative contributions that structural changes (ΔHc) vs. changes in dynamics (ΔSc) influence the coupling [53]. Isothermal titration calorimetry provides a measure of the global enthalpy change for a reaction, with ΔHc determined from eq 3 (see Scheme 1, right):
| (5) |
and the global ΔSc determined from the Gibbs relationship ΔSc = (ΔHc − ΔGc)/T. Under favorable cases, residue-specific contributions to the global ΔSc can be investigated from an analysis of the residue-specific dynamics over very short (ps-ns) [54,55], intermediate (μs-ms) [56,57], and long (ms-s) [58] timescales by NMR spectroscopy; these experiments allow direct assessment of the relative contributions that backbone conformational entropy, correlated domain motions, and perturbations in the native state conformational ensemble, respectively, make in stabilizing distinct allosteric states accessible to P. This powerful approach can thus be used to define both global and local origins of allosteric regulation and can provide support for or against mechanistic models that emerge from structural studies. Exactly such an analysis was carried out for S. aureus CzrA [16,40] which was recently reviewed elsewhere [1].
Challenges associated with quantifying allostery in metal sensor proteins
Inspection of the simplified allosteric linkage scheme outlined in Fig. 3 suggests several limitations that can complicate application of this method to the study of metal sensor proteins. First, the general approach is most powerful when knowledge of the structures and energetics associated with all four allosteric “end” states is available, as is the ability to measure all four equilibria indicated. This is a difficult condition in practice since it is sometimes the case that K1 or K2 (see Fig. 3) can not be accurately measured, due to very weak binding to the operator DNA by the apoprotein (for an uptake repressor, e.g., Fur or AdcR) or for the fully metallated protein (for an efflux repressor, e.g., CzrA or SczA), respectively. Very weak binding can sometimes be quite close to the affinity for nonspecific DNA, and there is no provision in this simplified scheme for that. One workaround for this is to employ relative short, fluorescently labeled duplex DNAs harboring a single target operator and/or simply increase the salt concentration, both of which would tend to minimize complications due to nonspecific DNA binding [59]. Alternatively, an excess of nonspecific competitor DNA could be used so that any residual binding that is observed to the specific, fluorescently labeled DNA target is sequence-specific with a stoichiometry of 1:1 for both K1 and K2 (as indicated in Fig. 3) [11]. In fact, this approach has been extensively employed in only one case, S. aureus CzrA, and in that case, the overall thermodynamics are dominated by solvent release from the metal ion and deprotonation of ligand donor atoms on complex formation [40].
In addition, it can sometimes be difficult to identify experimental conditions in which individual equilibria in Fig. 3 can be directly measured without inadvertent complications from coupled equilibria, again often due to the low DNA or metal binding affinity of one or another state in this scheme. For example, in recent work with B. subtlilis PerR, the apparent affinity of PerR for Fe(II) was measured by measuring an increase in the anisotropy of a fluorescently labeled DNA operator upon titration of unbound PerR (P) and DNA (D) with Fe(II) anaerobically (M in Fig 3) [60]. Inspection of Fig. 3 reveals that what was actually measured in this case is not βnP directly but the product, βnP• K2 or equivalently βnP•D •K1. On the other hand, if K1 ≪ βnP•D then βnP•D•K1 ≈ βnP•D if all bound ions contribute to activation of DNA binding. Equivalently, if the concentration of protein and DNA in the binding reaction are such that all P•Mn that is formed binds to the DNA, i.e., stoichiometric binding conditions, then one can obtain an estimate of βnP under these conditions.
A further complication in the quantitative analysis of these coupled equilibria is that transitions metal ions are intrinsically “sticky” and have a significant tendency to bind to adventitious sites on proteins. In fact a bis-imidazole complex with Zn(II) or Ni(II) forms with a stability constant of ≈106 M−1 at neutral pH. Such sites can often be competed away by including small amounts of imidazole in the binding buffer, although this has to be checked carefully to be sure that imidazole offers no significant competition to sites of biological interest [61]. Finally, in a fraction of the crystallographic structures discussed here, Zn(II) is often employed, purposely or not, as a structural surrogate for a redox active and typically weakly bound, less competitive cognate metal, e.g., Fe(II) [62-64]. This has led to considerable confusion concerning the functional role of individual metals in metalloregulatory proteins, particularly pertaining to the Fur family of proteins, and Fe(II)-binding Fur in particular. The coordination geometry of Zn(II) is most often tetrahedral, and thus this geometry, even if non-native, can often be enforced by the metal rather than the protein.
This is further complicated by the fact that many metalloregulatory proteins harbor more than one metal site per protomer (up to 3) in a crystal structure [65], and it can often be difficult to assign a functional role, e.g., primary sensing, structural or other, particularly in cases where individual proteins bind multiple metal ions. In many cases, ligand donor atoms to one metal ion are “next door” in the primary and tertiary structures to donor atoms that make up a second metal site (see Fig. 5, below); as a result, substitution of a ligand to one metal site may not be silent at a neighboring metal site and such studies must be interpreted with caution in absence of structural or biochemical information as to the degree to which metal binding affinities are influenced as a result of individual substitutions. Two examples of “neighboring” metal sites are found in the Fur family member Zur [65,66] and in our recent structure of the zinc uptake regulator, AdcR, from Streptococcus pneumoniae [67], to be discussed further below.
Metal sensing site characteristics and homotropic cooperativity of metal binding
Primary and secondary metal sites in metal sensor proteins introduce characteristics that establish the specificity of the allosteric response and allow a metalloregulator to function as a sensor of intracellular level of one or a few uncomplexed, highly mobile ions in the cell (Fig. 2). In contrast to the active sites of metalloenzymes in which solvent access is restricted in a site that is far from the protein surface, metal coordination sites on metal sensor proteins are positioned at or near the surface. This is illustrated in Fig. 4 with the structures of two Zn(II)-specific efflux regulators, MerR family member E. coli ZntR (Fig. 4A) [18] and ArsR repressor S. aureus CzrA (Fig. 4C) [68], and two uptake repressors, Fur-family member Streptomyces coelicolor Zur (Fig. 4B) [66] and MarR family repressor S. pneumoniae AdcR (Fig. 4D) [67]. Each adopts a distinct homodimeric architecture and each is characterized by distinct metal stoichiometries, from one to three per protomer, zinc nuclearities and coordination complex structures. The common feature is that all metal ligand donor atoms are near or at the protein surface, with nonliganding heteroatoms of donor ligands (oxygen atoms are shaded red; nitrogen atoms, blue; carbon atoms, yellow) often engaging in hydrogen bonds with water molecules. Remarkably, in AdcR, both “sides” of the metal binding “pocket” are exposed to solvent, easily seen when one compares the left and right protomers within the dimer (Fig. 4D) [67]. Positioning a regulatory site at the protein surface likely allows for rapid on-rates of metal binding to a site on the dimer where subsequent coordination bonds can readily drive a change in the quaternary structure of the dimer (or oligomer) which, in turn, allosterically activates or inhibits DNA binding. Association kinetics of metal binding have not been done for any zinc sensor. Kinetic studies have been carried out the Cd/Pb sensor S. aureus pI258 CadC, and these studies reveal biphasic kinetics, in which is rapid protein-concentration dependent on-rate, indicative of formation of an encounter complex, is followed by a much slower intramolecular isomerization, the rate of which is influenced by a key allosteric ligand in the metal sensing site [69]. The simplest interpretation of the second phase is that it represents the rate of quaternary structural switching within the CadC dimer.
S. aureus CzrA (Fig. 4C) is simplest among the structures shown in Fig. 4, in that the metal stoichiometry is 1:1 Zn(II):protomer or two per dimer [70]. Zn(II) is known to bind with strong negative cooperativity to the apo-CzrA dimer (KZn1>KZn2 by ≈20-fold), and the Zn1-CzrA intermediate is incorporates 50-70% of the allosteric coupling energy associated with the fully Zn(II)-saturated dimer [40,48,71]. The functional importance of negative homotropic cooperativity in CzrA has not been firmly established although in simple terms can potentially alter the “two-state” set point model that assumes a single affinity of the repressor for Zn(II) (see Fig. 2, dotted response curve) to a wider range of Zn(II) concentrations over which a full transcriptional response can be mounted (see Fig. 2) [52]. In any case, thermodynamics studies and molecular dynamics simulations of CzrA suggest that negative cooperativity derives from a “stiffening” of the dimer in Zn2 state [1,16] (see below).
ZntR (Fig. 4A) is uniquely characterized by a binuclear zinc cluster in which a bridging phosphate anion from solution completes the binuclear coordination complex [18]. It is unknown as yet if one or both metals are required for allosteric activation of transcription by Zn(II). ScZur (Fig. 4B) like M. tuberculosis and B. subtilis Zurs, contains three zinc binding sites, one of which is clearly a structural tetrathiolate site (site 1), with two additional sites (sites 2 and 3) that play distinct roles [66]. Consensus is clearly emerging that site 2 containing the Cys thiolate ligand is the primary sensing site in Zurs. A recent report with BsZur (of currently unknown structure) suggests that Zn(II) binding to site 3 can not be detected in vitro (KZn≤106 M−1), while zinc binding to the primary sensing site 2 is also strongly negatively cooperative within the dimer (KZn1>KZn2 by ≈20-fold), just the like the case with CzrA [52].
SpAdcR (Fig. 4D) on the other hand, harbors two metal sites per protomer [67]. Biological and biochemical studies establish that site 1 is the primary or only metal sensing site while the function of the other site (site 2) remains to be established [11]. Zn(II) binds to each of the two primary sites with near equal affinity and ≥10,000 fold more tightly than to site 2 (KZn2≈108 M−1 at pH 8.0); NMR structural studies reveal that only site 1 need be occupied to drive the quaternary structural change of the dimer in the absence of DNA operator [11,67]. A C30A substitution in site 2 reduces the binding affinity of site 2, but is functionally dispensable in mediating zinc resistance in the cell [11].
Fur family repressors: A multiplicity of metal sites
The Fur family of metal sensor proteins is named for the founding member E. coli Fe-regulated uptake repressor Fur and is encoded in the genomes of many Gram-negative bacteria [46]. In E. coli, Fur is a global transcriptional regulator of over 90 genes encoding both proteins and noncoding RNAs, and is involved in iron homeostasis as well as oxidative stress and acid tolerance [46]. A handful of Fur orthologs have now been extensively characterized, and include sensors for other transition metal ions, e.g., the Zn(II)-sensor Zur [65,72], the Mn(II)/Fe(II)-sensor Mur [73], the Ni(II)-sensor Nur [74,75], as well as those that sense hydrogen peroxide (H2O2), PerR [76,77] and heme iron (Irr) [78,79]. Fur proteins are typically transcription repressors when bound to specific metal ion effectors and thus bind DNA operator tightly in the presence of the cognate metal.
The last several years have witnessed an explosion in the number of crystallographic structures available for Fur-family regulators in a metal-bound state. The crystallographic structures of four of these, H. pylori Fur [64], S. coelicolor Nur [75], S. coelicolor Zur [66] and B. subtilis PerR [77,80], are shown in Fig. 6. Inspection of these structures allows one to identify similarities and differences in Fur family orthologs. All metallated structures adopt what is clearly a “closed” conformation in which the winged helical DNA-binding domains on opposite protomers are characterized by an interdomain separation of ≈32 Å, ideally suited to bind B-form DNA in successive major grooves. The dimerization domain is a mixed α/β structure that sits atop of the molecule as shown in Fig. 6. There remains just one structure of a nonmetallated Fur protein, apo-PerR, which adopts a “open” or splayed out architecture in which the winged helical domains are not favorably positioned to interact with DNA [81]. The extent to which this is the case in solution, or the degree to which this characterizes other Fur family repressors is not known. However, it is interesting to note that M. tuberculosis Zur adopts a conformation that is more similar to that apo-PerR vs. metal-complexed PerR (Fig. 6D) [65]. In any case, each of these structures appears poised to interact with DNA, thus providing support for a simple “caliper-like” model of allosteric activation by cognate metal ions. On the other hand it is important to recognize that there are currently no available structures of allosterically activated Fur proteins bound to operator DNA.
Fig. 6.
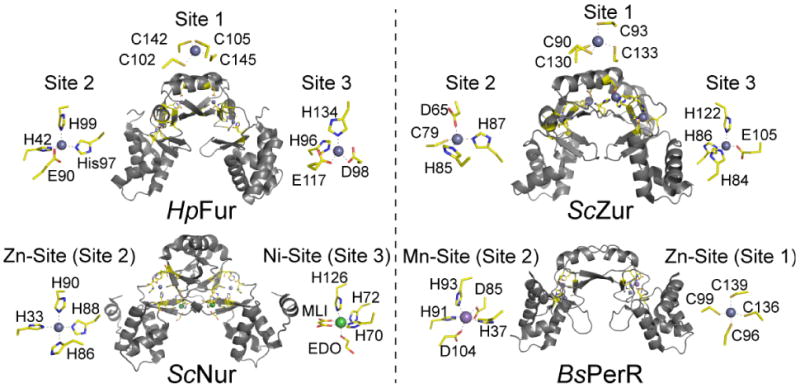
Illustration of multiple binding sites in the Fur family of repressor proteins. HpFur [64], ScZur [66], ScNur [75] and BsPerR [77] illustrate both the total number of different sites as well as the different coordination complexes that have evolved in a single protein family. Metal site designations are internally consistent and labeled sites 1, 2 and 3 according to the convention established for HpFur and MtZur [65] to facilitate comparisons. Sites 1, 2 and 3 in ScZur correspond to the sites previously identified at C, M and D, respectively.
Remarkably, each of these dimeric proteins harbor two or three metal binding sites per protomer, and this of course complicates efforts to assign functional roles to any one site. Although a host of metal site designations exist in the literature, we employ that previously applied to MtZur [65] and HpFur [64] in which metal sites are labeled 1, 2 and 3 to be internally consistent, Many, but not all, Fur family regulators harbor a tetrathiolate Zn(II) site designated site 1; this site is a structural site originally spectroscopically characterized in E. coli Zur [82]. This site plays a role in stabilizing the dimer structure and requires denaturant to remove and thus plays no direct role in metal or oxidative stress sensing [82]. Site 2 lies close or at the interface of the winged helical domain and the dimerization domain within each protomer, and always contains at least one ligand derived from the DNA binding domain, e.g., His42 in HpFur, His33 in ScNur, Asp65 in ScZur and His37 in BsPerR, with the remaining metal ligands from the dimerization domain (Fig. 6). Site 3 contains ligands from the C-terminal domain in all cases (site 3 is not present in BsPerR) and in two cases, HpFur and ScZur, is strongly interdigitated with metal site 2, with neighboring amino acids in the primary structure alternately donating ligands to site 2 and site 3 (see also Fig. 5B), in a manner reminiscent of S. pneumoniae AdcR (Fig. 5D) [67]. In fact, in ScZur, the 10-amino acid sequence from His84-Cys93 donates six metal ligands to each of three metal sites; the same is true in HpFur for residues His96-Cys105 (Fig. 6A).
What are the functional roles of sites 2 and 3 in metalloregulation of DNA binding? This has been most clearly established for ScZur [66] and BsPerR. [77] Site 2 in BsPerR adopts a trigonal bipyramidal coordination complex with Mn(II), used in this case as a surrogate for Fe(II); both metals are equally effective in activating PerR to bind to DNA. This leaves an open coordination site on the Fe(II) which is able to coordinate H2O2, leading to Fe-activation and local generation of OH• which then oxidizes His37 and His91, leading to release of Fe and formation of the “open” architecture that has low affinity for the DNA operator [76,80]. Although BsPerR formally lacks a metal site 3, mutagenesis of predicted residual site 3 ligands in BsPerR (Y92, E114, H128) diminishes the ability of these mutant PerRs to sense H2O2 in vivo, an effect traced to the enhanced ability of mutant PerRs to bind Mn(II) (a poor activator of H2O2 oxidation) over Fe(II). Remarkably, alterations in the availability of intracellular Fe(II) was shown to lead to a restoration of H2O2 sensing function in at least one of these mutant BsPerR proteins (H128A) [60].
In ScZur, likewise, metal site 2 is the primary sensing site which would effect folding of Zur into a closed conformation and subsequent allosteric activation of DNA operator binding. Extensive biological and biochemical experiments in S. coelicolor suggest that site 3 plays a modulatory role and allows ScZur to fully repress a subset of target genes that are not fully repressed on filling only site 2 [66]. The extent to which this is true in other Zurs is not known, although in B. subtilis Zur, site 3 does not bind Zn(II) in solution, and mutagenesis of site 3 ligands simply destabilizes the dimer [52]. Interestingly, reducing the affinity of one of the two site 2 Zn(II) ions via introduction of C84S mutation (analogous to C79 in ScZur) was found to result in enhanced negative cooperativity of metal binding to site 2. This finding potentially supports a step-wise activation model which in turn could potentially widen the intracellular response to Zn(II) [52].
The crystallographic structure of the Ni(II) uptake regulator ScNur bound to two Ni(II) ions reveals metal sites that are formally analogous to site 2 in the other Fur repressors and a second Ni(II) site that is thus far unique among Fur orthologs (labeled site 3 here). Site 3 contains three protein-derived metal ligands, two from the β-wing region (His70, His72) and one from the C-terminal dimerization domain (His126) and is coordinately unsaturated, modeled to contain bound malonate (M) and ethylene glycol (EG) from solution [75]. In contrast, site 2 adopts a square planar coordination geometry around the Ni(II) ion, a common coordination structure for d8 Ni(II) also found in the Ni(II)-uptake regulator NikR [83]. This site is exchangeable with Zn(II) in contrast to site 3, which led An et al. to conclude that site 2 could not function as the Ni(II)-specific sensing site [75]. Mutagenesis experiments were not so revealing in this case since introduction of non-liganding substitutions in either metal site 2 or 3 seemed to influence Ni(II) sensing in vivo [75]. An analogous argument was originally made for P. aureginosa Fur, which was crystallized with two Zn(II) bound (PaFur lacks the Cys-rich site 1) [63]. The metal site analogous to site 2 was found to be exchange-inert with Fe(II), while site 3 was readily exchanged with Fe(II). This suggested to the authors that site 3 was indeed the sensing site. This original functional assignment in PaFur now seems in doubt given the recent characterization of HpFur (see below) [64]. In both Nur and PaFur, site 2 harbors a metal ligand from the DNA binding domain (His33 in Nur), metal occupancy of which could drive formation of the “closed” conformation shown. However, it is not necessary that all Fur family proteins are allosterically activated by their cognate metals in exactly the same way. Detailed biochemical and metal binding experiments with ScNur and metal site mutants will be required to obtain further support for or against what would clearly be a significant departure for cognate metal sensing by a Fur family repressor [75].
Finally, for HpFur the metal binding situation is complicated by the fact that all three sites in the structure are filled with Zn(II), which is obviously a non-cognate metal ion for Fe(II)-dependent activation of DNA binding [64]. Nuclease protection assays with wild-type Fur and mutants designed to selectively abrogate metal binding to site 2 (Δsite2 Fur) or site 3 (Δsite 3 Fur) reveals that Δsite2 Fur could not be activated to bind DNA with any metal in vitro (Co, Ni, Mn and Zn were tested); in contrast, Δsite3 Fur could be activated to bind the operator at roughly twice the protein concentration required to observe wild-type Fur binding [64]. In addition, the structural changes observed by CD spectroscopy are retained in Δsite 3 Fur, but not in the Δsite 2 mutant. These data taken collectively suggest that site 2 is the primary metal sensing site in HpFur, as in ScZur and BsPerR, although the degree to which Fe(II) is capable of stimulating DNA binding remains unknown. It is clear that if site 2 is the primary sensing site for Fe(II), then the coordination number of site 2 will have to increase to five or six from four observed for Zn(II) in the structure. Inspection of the structure suggests that this could be readily accommodated by E90 moving to bidentate coordination (the nonliganding oxygen atom of E90 is 2.46 Å from the Zn(II) ion), and/or via recruitment of E95 from a loop region into the first coordination shell [64]. Either or both appear possible given the solvent-accessible nature of the site.
First vs. second coordination shell-based allostery
Transition metal ions bind to proteins by forming coordinate covalent bonds with protein heteroatoms, with each chelate defined by a coordination number, n, typically ranging from n=2-6, and a defined geometry [1]. Typical coordination geometries range from diagonal, to trigonal, to tetrahedral, to trigonal bipyramidal to octahedral, for n of 2, 3, 4, 5, and 6, respectively. The chemical nature of the ligand donor atoms and the coordination number are characteristic of individual metal ions, and thus provides the essential means to create metal binding sites with a degree of metal specificity by favoring the binding of one metal over another [1,24,84]. These features in toto characterize the first coordination shell. Formation of the first coordination shell sometimes positions metal ligands in such a way that they can donate or accept a hydrogen bond from a neighboring functional groups [3]. These interactions are classically defined as the second coordination shell around the metal ion.
Metalloregulatory proteins exploit either or both the first and second coordination shells in driving conformational changes in an oligomer that allosterically activate or inhibit operator DNA binding. The simplest example of first coordination-based allostery is in the Fur family regulators discussed above [77,85]. Here, a metal ligand derived from the more distal DNA binding domain drives a large “caliper-like” reorganization of the dimer, thereby stabilizing the closed conformation; this in turn positions the “reading heads” of the winged helical domains or each protomer in such a way that they are poised to bind to successive major grooves in a two-fold symmetric operator DNA. Detailed inspection of these structures suggests little in the way of hydrogen bonding interactions that originate with the metal ligands thus suggesting that first shell is necessary and sufficient to effect allosteric positive activation of operator binding.
In contrast, for other regulators, the conformational changes are known or projected to be quite small [68,86]; in these cases, a structural pathway of allosteric linkage between the metal binding site and the DNA binding domain can be used to effect communication between these sites [67,68,87,88]. These pathways often but not always originate with the second coordination shell, followed by a series of additional hydrogen bonds that energetically couple these two domains [2,89]. Three examples of this kind of communication are illustrated in Fig. 7 by the two MerR regulators, the Cu-sensor CueR [18] and the oxidative stress sensor SoxR [90], and the founding member of the CsoR/RcnR family of regulators, M. tuberculosis CsoR [91]. In both CueR and SoxR, a key Arg residue (Arg75 and Arg55, respectively) found at the interface between the C-terminal metal binding loop donates a hydrogen bond to a backbone carbonyl oxygen neighboring one of the metal liganding residues. These hydrogen bonds are buttressed by additional hydrogen bonding interactions that originate from the opposite side of the same helix via a carboxamide side chain (Asn71 in CueR; Gln64 in SoxR) to the face of the recognition helix (shaded orange) opposite to those residues that make sequence-specific contacts to the DNA. It is important to point out that hydrogen bonding interactions are inferred from the structures, and the contribution that each makes to allosteric communication is not yet known.
Fig. 7.
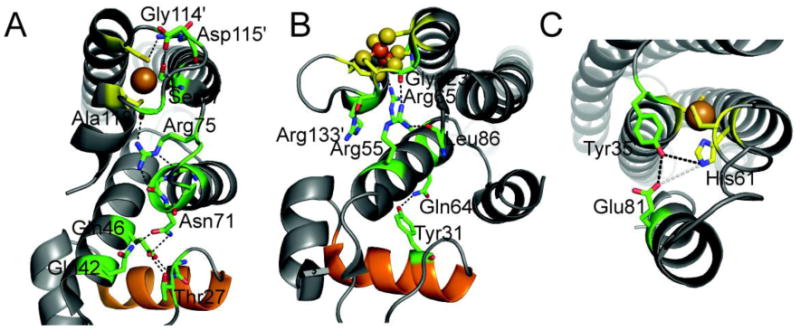
Putative hydrogen bonding network in the allosterically activated states of EcCueR [18] (A), EcSoxR (B) [90], and MtCsoR (C) [91]. Metal binding residues are shown in stick representation with carbon atoms shaded yellow. Key residues in the allosteric hydrogen bonding pathway are also shown as sticks with carbon atoms shaded green. The DNA recognition helix of the HTH DNA binding domain of CueR and SoxR is shaded orange. A native chemical ligation experiment carried out with MtCsoR is consistent with the coupling model shown [41]; the others have not yet been tested.
In the case of histidine ligands, we have used native chemical ligation in an effort to abrogate the second coordination shell, with little or no effect on the first coordination shell, via site-specific introduction of unnatural histidine analogs, e.g., methyl-histidine or thiazole, that perturbs the ε-face of the imidazole ring with no effect on the ε-face which donates a ligand to the Cu(I) ion (Fig. 7C) [41]. These studies reveal that mutant proteins that harbor these substitutions bind Cu(I) with near wild-type Cu(I) binding affinity, but Cu(I) binding is allosterically uncoupled (ΔGc=0) from DNA binding. This finding is consistent with the hypothesis that the ability of H61 to donate a hydrogen bond to the conserved Tyr35, with then donates a hydrogen bond to Glu81 on the α3 helix, is a key aspect of the Cu(I)-induced structural transition in the tetramer which drives CsoR off the DNA [41]. One shortcoming of these studies is that the structure of the apo-CsoR tetramer is not yet known, and thus a comprehensive picture of how this hydrogen bonding pathway impacts regulation in any CsoR/RcnR family sensor is unknown. In addition, a recent study with L. monocytogenes CsoR reveals that the residue corresponding to Glu81 is dispensable for regulation in vivo [92]; this raises the possibility that not all Cu(I)-sensing CsoRs harbor the same obligatory pathway of allosteric communication or an alternative pathway emerges upon substitution via recruitment of a neighboring Glu in the helix.
CsoR is the Cu(I) sensing ortholog of a family of proteins now known to include the Ni(II) efflux regulator RcnR [93,94] and the CsoR-like sulfurtransferase repressor CstR [95]. There are clear differences in the first coordination shells of the Cu(I) sensing site in CsoR relative to the Ni(II)-specific switch in RcnR, which differ yet again from the high reactivity of the two Cys residues in CstR toward the metabolite sulfite to form a mixture of di- and trisulfide linkages across the dimer interface. Mt CsoR does not perform this chemistry, and although it has been shown that BsCsoR binds Ni(II) with high affinity (KNi=3.6×109 M−1), Ni(II) is less capable of driving the allosteric switch, with ΔGc of ≈1.7 kcal mol−1 vs. ≥5.4 kcal mol−1 (25 °C) for Cu(I)-substituted BsCsoR [86]. A similar scenario characterizes the α5 site in ArsR family regulators, discussed below. In any case, a common thread is that a change in metal specificity or reactivity is driven by changes in the first coordination shell which may well alter the second coordination shell or interactions in such a way that a primary communication pathway between metal ion binding and DNA binding is broken. When this pathway is broken, other pathways of communication may well evolve that supersedes the original switch in a progenitor regulator.
ArsR family proteins: Simple “all-in-one” architectures
The ArsR family is one of the most extensively studied and likely the largest and most functionally diverse metalloregulatory protein family [4,96]. The ArsR (or ArsR/SmtB) family is named for its founding members, E. coli As(III)/Sb(III) sensor ArsR [97] and Synechococcus PCC 7942 Zn(II) sensor SmtB [98]. Many bacterial genomes across virtually every bacterial taxonomy encode at least one ArsR-family regulator and the number of unique ArsR/SmtB-encoding genes is conservatively in excess of 500 [96]. Notably, the Actinobacteria Mycobacterium tuberculosis and Streptomyces coelicolor encode ten and thirteen ArsR/SmtB proteins, respectively, many of which are of unknown function [50,99]. One striking aspect of ArsR family proteins is that diverse metal ion binding sites have evolved at structurally distinct places on what is likely the same protein fold, which are designated on the basis of the secondary structural elements that are known to donate metal ligand donor atoms to the metal ion. In addition, in strong contrast to Fur family regulators, ArsR proteins are single-domain, highly compact protein dimers, in which the regulatory and DNA-binding sites are co-located. As a result, allostery must manifest itself in the absence of large-scale interdomain movements induced by metal binding to the apoprotein [68] or redox (thiol-disulfide) switching [7], and sometimes incorporates elements of dynamical stabilization or destabilization of one allosteric state relative to another [16,89].
NmtR vs. CzrA
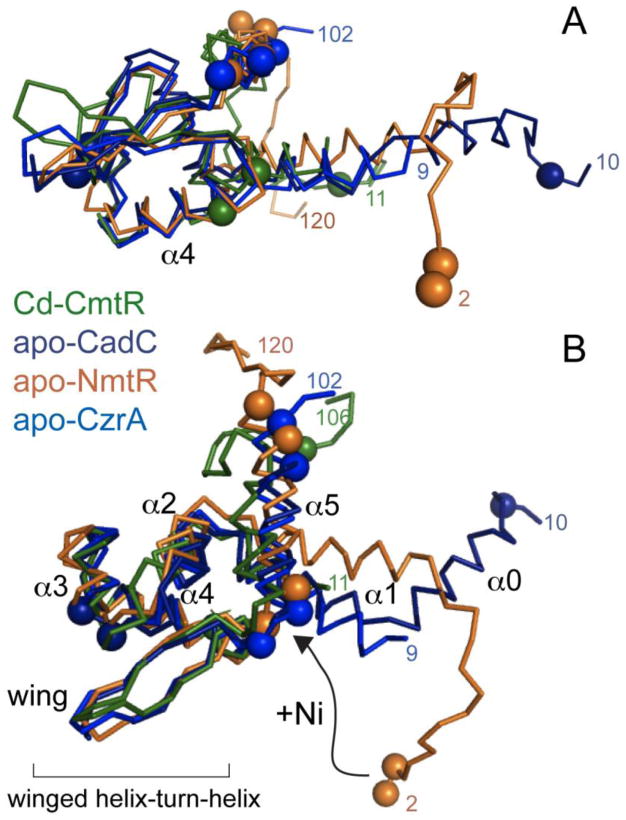
NmtR is a Ni(II)/Co(II)-sensing ArsR family member from M. tuberculosis that exhibits 60% sequence similarity to the Zn(II)-sensor S. aureus CzrA [84,100], with the major difference between the two proteins the presence of flexible “tails” at the N- and C-termini that flank what is anticipated to a CzrA-like homodimeric α1-α2–α3-αR-β1-β2-α5 winged helix scaffold [68]. This prediction is strongly supported by heteronuclear NMR studies and our solution structure of apo-NmtR (Fig. 8) [59,101]. The subunit structure of apo-NmtR compares favorably with the three other known high resolution ArsR-family structures, including CmtR [102], CadC [103] and CzrA [68], except for the distinct disposition of the N-terminal α1 helical region with respect to the rest of the molecule. Since the α1 and α5 helices (and the α0 helix of CadC) [103] make up a large part of the subunit interface, this suggests that the packing of the core region of the dimer may reinforce the specificity of the allosteric response in each case, which is derived from metal sites that form on distinct secondary structural elements in each case [3].
Fig. 8.
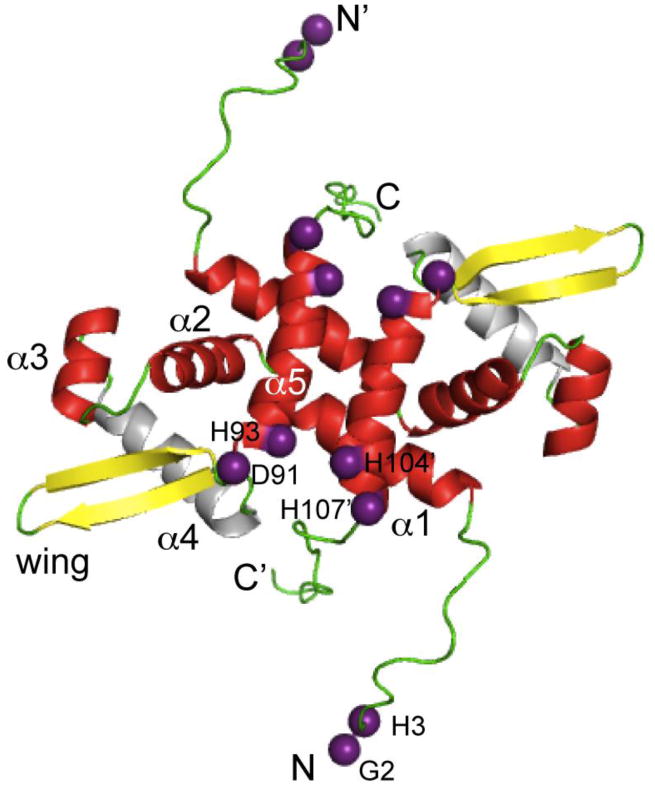
Ribbon representation of the structure of the Ni(II) regulator MtNmtR solved by NMR spectroscopy [101] (PDB code 2LKP). Metal binding ligands are represented as purple spheres on the Cα atom of each residue. Secondary structural elements are shaded red (α-helices), yellow (β-strands), and green (loops and unstructured regions). The N-and C- terminal extensions including residues 2-16 and 109-120 are highly mobile on the ps-ns timescale and defined the limits of the unstructured terminal regions in NmtR [59]; in contrast, residues 17-108 are highly ordered.
Previous comparative studies of the Ni-sensor NmtR vs. CzrA revealed that NmtR forms a six-coordinate octahedral Ni(II) complex vs. the tetrahedral zinc complex of CzrA (see Fig. 5B); in contrast, Zn(II) bound to NmtR to form a non-native tetrahedral complex, which is a poorer allosteric regulator than nickel [84]. Recent biochemical experiments reveal that the N-terminus of NmtR donates 1-2 additional metal ligands, proposed to be His3 and the N-terminal primary amine of Gly2, to the core α5 sensing site in common with CzrA, in contrast to a previous model that implicated His109 and His116 from the C-terminal tail as metal ligands to the Ni(II) [59]. A Ni(II) chelate structure involving coordination of Ni(II) by the extreme N-terminus is quite commonly observed in Ni(II) metalloenzymes and in Ni(II) sensing and trafficking proteins [94,104-106], and is fully consistent with chemical modification and NMR Ni(II)-paramagnetic broadening experiments [101]. Furthermore, the Ni(II)-binding affinity of NmtR is greatly reduced by an H3Q mutation, in much the same manner of Q substitutions of the α5 liganding region, but in strong contrast to Q substitution of H109 and H116 in the C-terminal tail. (Fig. 10, top) [59]. The more striking finding is that application of a coupling model with both cognate and non-cognate metal ions reveals that although His3 plays a nonessential role in maximizing the allosteric coupling free energy [ΔGcNi (H3Q) ≈ (ΔGcNi (wild-type)], it is essential for minimizing the degree to which non-cognate metal Zn(II) is functional in NmtR. Stated another way, substitution of His3 with a non-liganding Q residue changes the rank order of allosteric effectors with non-cognate Zn(II) now more effective than cognate metal Co(II) (Fig. 10) [59]. This suggests that nature incorporates elements of negative design when selecting against a highly competitive metal like Zn(II) in the evolution of metal sensing sites specific for metals other than Zn(II) in a way that reinforces general trends in coordination structure, metal selectivity and metal affinity. It will be interesting to understand the structural basis of these findings.
Fig. 10.
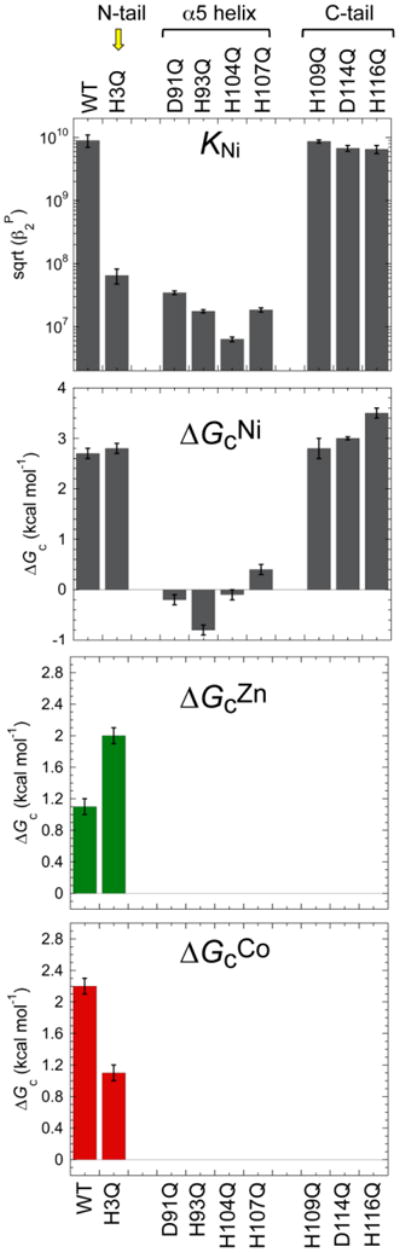
Coupling free energy analysis of MtNmtR [59]. Mutations to Ni(II) binding residues in the α5 helix greatly reduce Ni(II) binding affinity, and abrogate the coupling free energy that connects Ni(II)-binding to negative regulation of DNA-binding. Mutation of H3 in the N-terminal tail, in contrast, lowers the Ni(II) affinity by the nearly the same degree as the α5 mutants, but is characterized by a near wild-type coupling free energy. However, the H3A substitution effectively switches the rank order of allosteric effectors, with this mutant recovering significant allosteric response to the non-cognate Zn(II).
The DNA operator as a non-passive player in allosteric regulation by metal ions
Although the above discussion of allosteric regulation of DNA binding by metal sensor proteins is largely focused on protein-metal interactions and how cognate metal binding drives a change in the structure and/or dynamics of the regulator, several recent reports seem to hint that the nature of the DNA operator, or more precisely, a suite of operators within a regulon, is a potentially important player in metalloregulation of metal homeostasis [66,107]. Much of our current protein-centric view comes from a paucity of high-resolution structures of protein-DNA complexes. Indeed, there are only two such structures, with DNA operator complexes to the oxidized or activated form of E. coli SoxR [90], and Ni(II)-loaded E. coli NikR bound to the nikABDCE operator positioned in front of the genes encoding for the high affinity uptake system [108]. The latter complex has been studied extensively using structural and molecular dynamics methods in an effort to identify the low-affinity Ni(II) binding sites that maximally stabilizes the repressor-operator complex [109,110]. Also available is a molecular dynamics-generated model of a apo-CzrA-czrAB operator consistent with the solution structure of DNA-complexed CzrA [16,89]. Although these studies provide detailed insights into the protein-DNA interface and quaternary structural changes induced in the regulator on DNA binding, they are unable to provide insights into complexes formed with operators derived from other regulated genes, or in the case of SoxR, no direct comparison with the unactivated, reduced form of SoxR bound to DNA. SoxR, of course, is representative to the MerR family of proteins that includes CueR, ZntR [18], PbrR [111,112] and the Hg(II)-sensor MerR itself [113], which are known or presumed to bind DNA tightly in both the activated (or ligand-bound) and unactivated (or ligand-free) states (Fig. 11B) [114]. MerR proteins allosterically activate transcription in response to 2Fe-2S cluster oxidation or metal binding by changing the structure of the operator DNA by inducing underwinding and some local bending [17,114], thereby converting a weak promoter into a strong one [115]. Clearly, DNA structure is not a non-passive player in the MerR-family protein regulation.
Fig. 11.
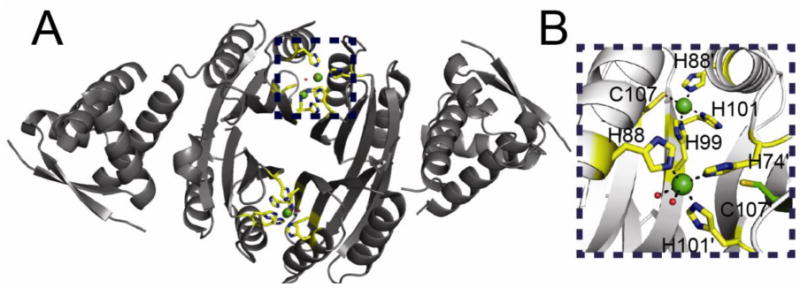
(A) Ribbon representation of the two pairs of Ni(II) binding sites of HpNikR solved at pH 5.6 [107]. Ni(II) ions are represented as green spheres. Metal binding residues are shown in stick representation with carbon atoms shaded yellow. (B) Close up of the two binding sites along the “top” of the NikR tetramer. Metal binding residues are shown as sticks with carbon atoms colored yellow. This view illustrates the two different coordination geometries that are found in this structure. The non-liganding Cys107 which forms part of the canonical square planar site (top site) is also shown in stick representation with carbon atoms colored green.
A recent characterization of S. coelicolor Zur provides evidence in support of the idea that various genes of the Zur regulon are transcriptionally repressed at lower zinc concentrations upon filling the high affinity pair of site 2 metal sites (see Fig. 5), while a different set of genes become fully repressed upon filling both metal sites 2 and 3, at higher Zn(II) intracellular concentrations [66]. These findings suggest that metal binding properties of ScZur orchestrate a “graded response” to zinc stress in the cell. Although the mechanism of this response is not completely understood, one possibility is that it is governed by distinct core or flanking sequences or different DNA structures associated with individual promoters within a regulon, which in turn gives rise to differential affinities of partially vs. fully saturated Zn(II)-bound Zur.
Different DNA operator sequences that respond to structurally distinct metallated repressors may also play a role in nickel homeostasis by H. pylori NikR [116]. H. pylori NikR, unlike E. coli NikR, is a global regulator of a significant number of genes beyond nickel homeostasis, including acid adaptation via urease maturation and catalytic activity, iron uptake and storage and mobility [117-119]. Biochemical studies reveal that HpNikR-regulated promoters can be grouped into those harboring high affinity and low affinity binding sites for NikR, in which all four C-terminal Ni(II) sites are assumed to form classical square planar complexes characteristic of E. coli NikR [116,120]. Strikingly, recent crystallographic studies of HpNikR reveal a number of distinct Ni(II) bound states including 1) Ni1 and Ni2 states in which “intermediate” non-square planar Ni(II) sites were obtained by soaking apo-HpNikR with excess Ni(II) (at pH 5.0) [121]; 2) an asymmetric tetrameric state (solved at pH 5.6), in which two of the four tetramer-bridging sites adopt conventional square planar sites, while the other two adopt five- or six-coordinate sites (5/6 sites) similar to the “I” site of Dian et al. [121], and which share two common metal ligands with the four-coordinate sites (Fig. 11) [107]; or 3) an E. coli NikR-like structure (solved at pH 7.3 from Ni(II)-reconstituted apo-NikR) in which all four Ni(II) ions adopt square planar complexes [122].
It has been speculated that the asymmetry in the tetrameric states significantly influences the mechanism of DNA recognition by HpNikR, [107] alternatively stabilizing cis, open and trans conformations of the repressor via distinct pathways of allosteric communication between functional domains, and/or simply altering the relative mobility of the DNA-binding domains relative the tetrameric nickel binding core to varying degrees [87,88]. While provocative, the functional relevance of the observed “intermediate” and asymmetric tetrameric state structures of HpNikR, both determined at acidic pH, relative to a more conventional square-planar-site bound NikR, to nickel physiology in H. pylori has yet to be firmly established; in this regard, it is worth noting that cytoplasmic pH of H. pylori may well be acidic [123]. In any case, the potential physiological access to multiple allosteric forms of a regulator that differ in the coordination structure and/or occupancy of distinct sites may allow fine tuning of the regulatory response that in turn reinforces quantitative differences in DNA operator affinity.
Conclusions
In this review, we have summarized recent efforts to understand the complexities of allosteric positive and negative regulation of DNA operator binding of metal sensor proteins by transition metal ions at the atomic level, focusing here on the role that metal site coordination chemistry and metal site occupancy in the repressor plays in this process. An emerging theme is that homologous repressors from different organisms that perform the same functional role have evolved different metal site structures and in some cases, even distinct stoichiometries. As a result, even closely related proteins may have evolved subtly altered pathways of allosteric linkage optimized for transcriptional regulation of a simple or complex regulon, ultimately dictated by microenvironmental niche of the particular organism. This of course, complicates efforts to draw general conclusions as to mechanisms of allosteric switching for a family of regulators. In any case, we discuss these findings in the context of a thermodynamic model for metal homeostasis in bacteria in which individual sensors are tuned in such a way that the intracellular bioavailability of transition metals is buffered in a way that minimizes cross-talk and mis-metallation of metalloproteins in the cell [1,23,124]. An increasingly sophisticated, multi-disciplinary approach that employs microbiological, crystallographic, solution NMR and molecular dynamics studies in complementary ways directed toward understanding allosteric regulation by both cognate and non-cognate metal ions will ensure continued progress on this topic.
The time may be ripe to exploit our detailed understanding of these processes at the molecular level and undertake a systems biology approach now being implemented for disease pathologies associated with misregulation of metal homeostasis in humans [125,126]. Such studies could be used to make predictions about key intersections among transition metal homeostasis and oxidative stress sensing systems in microorganisms, and ultimately, identify new players in bacterial transition metal physiology. Indeed, these studies take on added significance in the context of the “double-edged sword” of metals in the biology of human pathogens [127], where host proteins directly compete with bacterial high affinity metal uptake systems and superoxide dismutases for transition metals [128-130], and metal uptake systems for iron, zinc and manganese are important virulence determinants for the survival of microbial pathogens in the human host (for recent reviews, see [131,132]). Successful manipulation of this emerging battleground in nutritional immunity which fundamentally derives from knowledge of the biophysical chemistry of metal homeostasis may well lead to new discoveries in our efforts to ultimately control or eradicate human bacterial infectious disease.
Fig. 9.
Cα-cartoon superimposition of one of the two protomers of dimeric ArsR family metal sensors MtCmtR (shaded green) [102], SaCadC (dark blue) [103], MtNmtR (gold) [101] and SaCzrA (light blue) [68]. All proteins were aligned their HTH-DNA binding motifs with an r.m.s.d. of ∼1 Å for the corresponding 29 Cα atoms. Metal binding residues in each protein are highlighted as spheres on their Cα atoms. A possible movement of the N-terminal extension of MtNmtR is also shown [59].
Highlights.
All cells encode a panel of metalloregulatory proteins that collectively maintain transition metal homeostasis / Regulation is mediated by direct binding of cognate metal ion(s) to these transcriptional regulators / Formation of specific coordination complexes allosterically inhibits or activates DNA operator binding / Recent insights into how metal site occupancy governs structural switching by metal sensor oligomers are discussed.
Acknowledgments
We gratefully acknowledge the US National Institutes of Health (GM042569) for financial support of our studies of bacterial metalloregulation.
Footnotes
For Special Issue: Archives of Biochemistry and Biophysics - Allosteric Regulation, edited by Gregory Grant
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Reyes-Caballero H, Campanello GC, Giedroc DP. Biophys Chem. 2011;156:103–114. doi: 10.1016/j.bpc.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giedroc DP, Arunkumar AI. Dalton Trans. 2007;29:3107–3120. doi: 10.1039/b706769k. [DOI] [PubMed] [Google Scholar]
- 3.Ma Z, Jacobsen FE, Giedroc DP. Chem Rev. 2009;109:4644–4681. doi: 10.1021/cr900077w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Busenlehner LS, Pennella MA, Giedroc DP. FEMS Microbiol Rev. 2003;27:131–143. doi: 10.1016/S0168-6445(03)00054-8. [DOI] [PubMed] [Google Scholar]
- 5.Osman D, Cavet JS. Nat Prod Rep. 2010;27:668–680. doi: 10.1039/b906682a. [DOI] [PubMed] [Google Scholar]
- 6.Mandal S, Chatterjee S, Dam B, Roy P, Das Gupta SK. Microbiology. 2007;153:80–91. doi: 10.1099/mic.0.29197-0. [DOI] [PubMed] [Google Scholar]
- 7.Guimaraes BG, Barbosa RL, Soprano AS, Campos BM, de Souza TA, Tonoli CC, Leme AF, Murakami MT, Benedetti CE. J Biol Chem. 2011;286:26148–26157. doi: 10.1074/jbc.M111.234039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JW, Helmann JD. Biometals. 2007;20:485–499. doi: 10.1007/s10534-006-9070-7. [DOI] [PubMed] [Google Scholar]
- 9.Kloosterman TG, van der Kooi-Pol MM, Bijlsma JJ, Kuipers OP. Mol Microbiol. 2007;65:1049–1063. doi: 10.1111/j.1365-2958.2007.05849.x. [DOI] [PubMed] [Google Scholar]
- 10.Kloosterman TG, Witwicki RM, van der Kooi-Pol MM, Bijlsma JJ, Kuipers OP. J Bacteriol. 2008;190:5382–5393. doi: 10.1128/JB.00307-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reyes-Caballero H, Guerra AJ, Jacobsen FE, Kazmierczak KM, Cowart D, Koppolu UM, Scott RA, Winkler ME, Giedroc DP. J Mol Biol. 2010;403:197–216. doi: 10.1016/j.jmb.2010.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobsen FE, Kazmierczak KM, Lisher JP, Winkler ME, Giedroc DP. Metallomics. 2011;3:38–41. doi: 10.1039/c0mt00050g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shafeeq S, Kloosterman TG, Kuipers OP. Metallomics. 2011;3:609–618. doi: 10.1039/c1mt00030f. [DOI] [PubMed] [Google Scholar]
- 14.Orth P, Schnappinger D, Hillen W, Saenger W, Hinrichs W. Nat Struct Biol. 2000;7:215–219. doi: 10.1038/73324. [DOI] [PubMed] [Google Scholar]
- 15.Kuroda M, Hayashi H, Ohta T. Microbiol Immunol. 1999;43:115–125. doi: 10.1111/j.1348-0421.1999.tb02382.x. [DOI] [PubMed] [Google Scholar]
- 16.Arunkumar AI, Campanello GC, Giedroc DP. Proc Natl Acad Sci U S A. 2009;106:18177–18182. doi: 10.1073/pnas.0905558106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Outten CE, Outten FW, O'Halloran TV. J Biol Chem. 1999;274:37517–37524. doi: 10.1074/jbc.274.53.37517. [DOI] [PubMed] [Google Scholar]
- 18.Changela A, Chen K, Xue Y, Holschen J, Outten CE, O'Halloran TV, Mondragon A. Science. 2003;301:1383–1387. doi: 10.1126/science.1085950. [DOI] [PubMed] [Google Scholar]
- 19.Llull D, Poquet I. Appl Environ Microbiol. 2004;70:5398–5406. doi: 10.1128/AEM.70.9.5398-5406.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Llull D, Son O, Blanie S, Briffotaux J, Morello E, Rogniaux H, Danot O, Poquet I. J Bacteriol. 2011;193:1919–1929. doi: 10.1128/JB.01109-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Panina EM, Mironov AA, Gelfand MS. Proc Natl Acad Sci U S A. 2003;100:9912–9917. doi: 10.1073/pnas.1733691100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Outten CE, O'Halloran TV. Science. 2001;292:2488–2492. doi: 10.1126/science.1060331. [DOI] [PubMed] [Google Scholar]
- 23.Waldron KJ, Robinson NJ. Nat Rev Microbiol. 2009;7:25–35. doi: 10.1038/nrmicro2057. [DOI] [PubMed] [Google Scholar]
- 24.Waldron KJ, Rutherford JC, Ford D, Robinson NJ. Nature. 2009;460:823–830. doi: 10.1038/nature08300. [DOI] [PubMed] [Google Scholar]
- 25.Irving H, Williams RJP. Nature. 1948;162:746–747. [Google Scholar]
- 26.Eide DJ. Biochim Biophys Acta. 2006;1763:711–722. doi: 10.1016/j.bbamcr.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 27.Krezel A, Maret W. J Am Chem Soc. 2007;129:10911–10921. doi: 10.1021/ja071979s. [DOI] [PubMed] [Google Scholar]
- 28.Suhy DA, Simon KD, Linzer DI, O'Halloran TV. J Biol Chem. 1999;274:9183–9192. doi: 10.1074/jbc.274.14.9183. [DOI] [PubMed] [Google Scholar]
- 29.Maret W. J Nutr. 2003;133:1460S–1462S. doi: 10.1093/jn/133.5.1460S. [DOI] [PubMed] [Google Scholar]
- 30.Krezel A, Maret W. J Biol Inorg Chem. 2006;11:1049–1062. doi: 10.1007/s00775-006-0150-5. [DOI] [PubMed] [Google Scholar]
- 31.Bozym RA, Thompson RB, Stoddard AK, Fierke CA. ACS Chem Biol. 2006;1:103–111. doi: 10.1021/cb500043a. [DOI] [PubMed] [Google Scholar]
- 32.Vinkenborg JL, Nicolson TJ, Bellomo EA, Koay MS, Rutter GA, Merkx M. Nat Methods. 2009;6:737–740. doi: 10.1038/nmeth.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otvos JD, Armitage IM. Proc Natl Acad Sci U S A. 1980;77:7094–7098. doi: 10.1073/pnas.77.12.7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zangger K, Oz G, Otvos JD, Armitage IM. Protein Sci. 1999;8:2630–2638. doi: 10.1110/ps.8.12.2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pruteanu M, Neher SB, Baker TA. J Bacteriol. 2007;189:3017–3025. doi: 10.1128/JB.01531-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mettert EL, Kiley PJ. J Mol Biol. 2005;354:220–232. doi: 10.1016/j.jmb.2005.09.066. [DOI] [PubMed] [Google Scholar]
- 37.Pruteanu M, Baker TA. Res Microbiol. 2009;160:677–683. doi: 10.1016/j.resmic.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Festa RA, Jones MB, Butler-Wu S, Sinsimer D, Gerads R, Bishai WR, Peterson SN, Darwin KH. Mol Microbiol. 2011;79:133–148. doi: 10.1111/j.1365-2958.2010.07431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stoyanov JV, Mancini S, Lu ZH, Mourlane F, Poulsen KR, Wimmer R, Solioz M. FEMS Microbiol Lett. 2010;302:69–75. doi: 10.1111/j.1574-6968.2009.01833.x. [DOI] [PubMed] [Google Scholar]
- 40.Grossoehme NE, Giedroc DP. J Am Chem Soc. 2009;131:17860–17870. doi: 10.1021/ja906131b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma Z, Cowart DM, Ward BP, Arnold RJ, DiMarchi RD, Zhang L, George GN, Scott RA, Giedroc DP. J Am Chem Soc. 2009;131:18044–18045. doi: 10.1021/ja908372b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reinhart GD. Methods Enzymol. 2004;380:187–203. doi: 10.1016/S0076-6879(04)80009-0. [DOI] [PubMed] [Google Scholar]
- 43.Singh VK, Xiong A, Usgaard TR, Chakrabarti S, Deora R, Misra TK, Jayaswal RK. Mol Microbiol. 1999;33:200–207. doi: 10.1046/j.1365-2958.1999.01466.x. [DOI] [PubMed] [Google Scholar]
- 44.Xiong A, Jayaswal RK. J Bacteriol. 1998;180:4024–4029. doi: 10.1128/jb.180.16.4024-4029.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun Y, Wong MD, Rosen BP. J Biol Chem. 2001;276:14955–14960. doi: 10.1074/jbc.M010595200. [DOI] [PubMed] [Google Scholar]
- 46.Andrews SC, Robinson AK, Rodriguez-Quinones F. FEMS Microbiol Rev. 2003;27:215–237. doi: 10.1016/S0168-6445(03)00055-X. [DOI] [PubMed] [Google Scholar]
- 47.Stoll KE, Draper WE, Kliegman JI, Golynskiy MV, Brew-Appiah RA, Phillips RK, Brown HK, Breyer WA, Jakubovics NS, Jenkinson HF, Brennan RG, Cohen SM, Glasfeld A. Biochemistry. 2009;48:10308–10320. doi: 10.1021/bi900980g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pennella MA, Arunkumar AI, Giedroc DP. J Mol Biol. 2006;356:1124–1136. doi: 10.1016/j.jmb.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 49.Busenlehner LS, Weng TC, Penner-Hahn JE, Giedroc DP. J Mol Biol. 2002;319:685–701. doi: 10.1016/S0022-2836(02)00299-1. [DOI] [PubMed] [Google Scholar]
- 50.Cavet JS, Graham AI, Meng W, Robinson NJ. J Biol Chem. 2003;278:44560–44566. doi: 10.1074/jbc.M307877200. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y, Hemmingsen L, Giedroc DP. Biochemistry. 2005;44:8976–8988. doi: 10.1021/bi050094v. [DOI] [PubMed] [Google Scholar]
- 52.Ma Z, Gabriel SE, Helmann JD. Nucl Acids Res. 2011 doi: 10.1093/nar/gkr625. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Popovych N, Sun S, Ebright RH, Kalodimos CG. Nat Struct Mol Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lefevre JF, Dayie KT, Peng JW, Wagner G. Biochemistry. 1996;35:2674–2686. doi: 10.1021/bi9526802. [DOI] [PubMed] [Google Scholar]
- 55.Krizova H, Zidek L, Stone MJ, Novotny MV, Sklenar V. J Biomol NMR. 2004;28:369–384. doi: 10.1023/B:JNMR.0000015404.61574.65. [DOI] [PubMed] [Google Scholar]
- 56.Kern D, Zuiderweg ER. Curr Opin Struct Biol. 2003;13:748–757. doi: 10.1016/j.sbi.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 57.Akke M. Curr Opin Struct Biol. 2002;12:642–647. doi: 10.1016/s0959-440x(02)00369-x. [DOI] [PubMed] [Google Scholar]
- 58.Englander JJ, Louie G, McKinnie RE, Englander SW. J Mol Biol. 1998;284:1695–1706. doi: 10.1006/jmbi.1998.2278. [DOI] [PubMed] [Google Scholar]
- 59.Reyes-Caballero H, Lee CW, Giedroc DP. Biochemistry. 2011;50:7941–7952. doi: 10.1021/bi200737a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma Z, Lee JW, Helmann JD. Nucl Acids Res. 2011;39:5036–5044. doi: 10.1093/nar/gkr095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grossoehme NE, Akilesh S, Guerinot ML, Wilcox DE. Inorg Chem. 2006;45:8500–8508. doi: 10.1021/ic0606431. [DOI] [PubMed] [Google Scholar]
- 62.Pohl E, Holmes RK, Hol WG. J Mol Biol. 1999;285:1145–1156. doi: 10.1006/jmbi.1998.2339. [DOI] [PubMed] [Google Scholar]
- 63.Pohl E, Haller JC, Mijovilovich A, Meyer-Klaucke W, Garman E, Vasil ML. Mol Microbiol. 2003;47:903–915. doi: 10.1046/j.1365-2958.2003.03337.x. [DOI] [PubMed] [Google Scholar]
- 64.Dian C, Vitale S, Leonard GA, Bahlawane C, Fauquant C, Leduc D, Muller C, de Reuse H, Michaud-Soret I, Terradot L. Mol Microbiol. 2011;79:1260–1275. doi: 10.1111/j.1365-2958.2010.07517.x. [DOI] [PubMed] [Google Scholar]
- 65.Lucarelli D, Russo S, Garman E, Milano A, Meyer-Klaucke W, Pohl E. J Biol Chem. 2007;282:9914–9922. doi: 10.1074/jbc.M609974200. [DOI] [PubMed] [Google Scholar]
- 66.Shin JH, Jung HJ, An YJ, Cho YB, Cha SS, Roe JH. Proc Natl Acad Sci U S A. 2011;108:5045–5050. doi: 10.1073/pnas.1017744108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guerra AJ, Dann CE, III, Giedroc DP. J Am Chem Soc. 2011 doi: 10.1021/ja2080532. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eicken C, Pennella MA, Chen X, Koshlap KM, VanZile ML, Sacchettini JC, Giedroc DP. J Mol Biol. 2003;333:683–695. doi: 10.1016/j.jmb.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 69.Busenlehner LS, Giedroc DP. J Inorg Biochem. 2006;100:1024–1034. doi: 10.1016/j.jinorgbio.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 70.VanZile ML, Cosper NJ, Scott RA, Giedroc DP. Biochemistry. 2000;39:11818–11829. doi: 10.1021/bi001140o. [DOI] [PubMed] [Google Scholar]
- 71.Lee S, Arunkumar AI, Chen X, Giedroc DP. J Am Chem Soc. 2006;128:1937–1947. doi: 10.1021/ja0546828. [DOI] [PubMed] [Google Scholar]
- 72.Maciag A, Dainese E, Rodriguez GM, Milano A, Provvedi R, Pasca MR, Smith I, Palu G, Riccardi G, Manganelli R. J Bacterio l. 2007;189:730–740. doi: 10.1128/JB.01190-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Diaz-Mireles E, Wexler M, Sawers G, Bellini D, Todd JD, Johnston AW. Microbiology. 2004;150:1447–1456. doi: 10.1099/mic.0.26961-0. [DOI] [PubMed] [Google Scholar]
- 74.Ahn BE, Cha J, Lee EJ, Han AR, Thompson CJ, Roe JH. Mol Microbiol. 2006;59:1848–1858. doi: 10.1111/j.1365-2958.2006.05065.x. [DOI] [PubMed] [Google Scholar]
- 75.An YJ, Ahn BE, Han AR, Kim HM, Chung KM, Shin JH, Cho YB, Roe JH, Cha SS. Nucl Acids Res. 2009:3442–3451. doi: 10.1093/nar/gkp198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee JW, Helmann JD. Nature. 2006;440:363–367. doi: 10.1038/nature04537. [DOI] [PubMed] [Google Scholar]
- 77.Jacquamet L, Traore DA, Ferrer JL, Proux O, Testemale D, Hazemann JL, Nazarenko E, El Ghazouani A, Caux-Thang C, Duarte V, Latour JM. Mol Microbiol. 2009;73:20–31. doi: 10.1111/j.1365-2958.2009.06753.x. [DOI] [PubMed] [Google Scholar]
- 78.Small SK, Puri S, O'Brian MR. Biometals. 2009;22:89–97. doi: 10.1007/s10534-008-9192-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hibbing ME, Fuqua C. J Bacteriol. 2011;193:3461–3472. doi: 10.1128/JB.00317-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Traore DA, El Ghazouani A, Jacquamet L, Borel F, Ferrer JL, Lascoux D, Ravanat JL, Jaquinod M, Blondin G, Caux-Thang C, Duarte V, Latour JM. Nat Chem Biol. 2009;5:53–59. doi: 10.1038/nchembio.133. [DOI] [PubMed] [Google Scholar]
- 81.Traore DA, El Ghazouani A, Ilango S, Dupuy J, Jacquamet L, Ferrer JL, Caux-Thang C, Duarte V, Latour JM. Mol Microbiol. 2006;61:1211–1219. doi: 10.1111/j.1365-2958.2006.05313.x. [DOI] [PubMed] [Google Scholar]
- 82.Outten CE, Tobin DA, Penner-Hahn JE, O'Halloran TV. Biochemistry. 2001;40:10417–10423. doi: 10.1021/bi0155448. [DOI] [PubMed] [Google Scholar]
- 83.Carrington PE, Chivers PT, Al-Mjeni F, Sauer RT, Maroney MJ. Nat Struct Biol. 2003;10:126–130. doi: 10.1038/nsb890. [DOI] [PubMed] [Google Scholar]
- 84.Pennella MA, Shokes JE, Cosper NJ, Scott RA, Giedroc DP. Proc Natl Acad Sci U S A. 2003;100:3713–3718. doi: 10.1073/pnas.0636943100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Giedroc DP. Mol Microbiol. 2009;73:1–4. doi: 10.1111/j.1365-2958.2009.06752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ma Z, Cowart DM, Scott RA, Giedroc DP. Biochemistry. 2009;48:3325–3334. doi: 10.1021/bi900115w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bradley MJ, Chivers PT, Baker NA. J Mol Biol. 2008;378:1155–1173. doi: 10.1016/j.jmb.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sindhikara DJ, Roitberg AE, Merz KM., Jr Biochemistry. 2009;48:12024–12033. doi: 10.1021/bi9013352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chakravorty DK, Wang B, Lee CW, Giedroc DP, Merz KM. J Am Chem Soc. 2011 doi: 10.1021/ja208047b. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Watanabe S, Kita A, Kobayashi K, Miki K. Proc Natl Acad Sci U S A. 2008;105:4121–4126. doi: 10.1073/pnas.0709188105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu T, Ramesh A, Ma Z, Ward SK, Zhang L, George GN, Talaat AM, Sacchettini JC, Giedroc DP. Nat Chem Biol. 2007;3:60–68. doi: 10.1038/nchembio844. [DOI] [PubMed] [Google Scholar]
- 92.Corbett D, Schuler S, Glenn S, Andrew PW, Cavet JS, Roberts IS. Mol Microbiol. 2011 doi: 10.1111/j.1365-2958.2011.07705.x. [DOI] [PubMed] [Google Scholar]
- 93.Iwig JS, Rowe JL, Chivers PT. Mol Microbiol. 2006;62:252–262. doi: 10.1111/j.1365-2958.2006.05369.x. [DOI] [PubMed] [Google Scholar]
- 94.Iwig JS, Leitch S, Herbst RW, Maroney MJ, Chivers PT. J Am Chem Soc. 2008;130:7592–7606. doi: 10.1021/ja710067d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Grossoehme N, Kehl-Fie TE, Ma Z, Adams KW, Cowart DM, Scott RA, Skaar EP, Giedroc DP. J Biol Chem. 2011;286:13522–13531. doi: 10.1074/jbc.M111.220012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Campbell DR, Chapman KE, Waldron KJ, Tottey S, Kendall S, Cavallaro G, Andreini C, Hinds J, Stoker NG, Robinson NJ, Cavet JS. J Biol Chem. 2007;282:32298–32310. doi: 10.1074/jbc.M703451200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wu J, Rosen BP. J Biol Chem. 1993;268:52–58. [PubMed] [Google Scholar]
- 98.Morby AP, Turner JS, Huckle JW, Robinson NJ. Nucl Acids Res. 1993;21:921–925. doi: 10.1093/nar/21.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Harvie DR, Andreini C, Cavallaro G, Meng W, Connolly BA, Yoshida K, Fujita Y, Harwood CR, Radford DS, Tottey S, Cavet JS, Robinson NJ. Mol Microbiol. 2006;59:1341–1356. doi: 10.1111/j.1365-2958.2006.05029.x. [DOI] [PubMed] [Google Scholar]
- 100.Cavet JS, Meng W, Pennella MA, Appelhoff RJ, Giedroc DP, Robinson NJ. J Biol Chem. 2002;277:38441–38448. doi: 10.1074/jbc.M207677200. [DOI] [PubMed] [Google Scholar]
- 101.Lee CW, Reyes-Caballero H, Giedroc DP. unpublished. [Google Scholar]
- 102.Banci L, Bertini I, Cantini F, Ciofi-Baffoni S, Cavet JS, Dennison C, Graham AI, Harvie DR, Robinson NJ. J Biol Chem. 2007;282:30181–30188. doi: 10.1074/jbc.M701119200. [DOI] [PubMed] [Google Scholar]
- 103.Ye J, Kandegedara A, Martin P, Rosen BP. J Bacteriol. 2005;187:4214–4221. doi: 10.1128/JB.187.12.4214-4221.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Barondeau DP, Kassmann CJ, Bruns CK, Tainer JA, Getzoff ED. Biochemistry. 2004;43:8038–8047. doi: 10.1021/bi0496081. [DOI] [PubMed] [Google Scholar]
- 105.Sankararamakrishnan R, Verma S, Kumar S. Proteins. 2005;58:211–221. doi: 10.1002/prot.20265. [DOI] [PubMed] [Google Scholar]
- 106.Chung KC, Cao L, Dias AV, Pickering IJ, George GN, Zamble DB. J Am Chem Soc. 2008;130:14056–14057. doi: 10.1021/ja8055003. [DOI] [PubMed] [Google Scholar]
- 107.West AL, St John F, Lopes PE, MacKerell AD, Jr, Pozharski E, Michel SL. J Am Chem Soc. 2010;132:14447–14456. doi: 10.1021/ja104118r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schreiter ER, Wang SC, Zamble DB, Drennan CL. Proc Natl Acad Sci U S A. 2006;103:13676–13681. doi: 10.1073/pnas.0606247103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Phillips CM, Nerenberg PS, Drennan CL, Stultz CM. J Am Chem Soc. 2009;131:10220–10228. doi: 10.1021/ja9026314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Phillips CM, Schreiter ER, Stultz CM, Drennan CL. Biochemistry. 2010;49:7830–7838. doi: 10.1021/bi100923j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Borremans B, Hobman JL, Provoost A, Brown NL, van Der Lelie D. J Bacteriol. 2001;183:5651–5658. doi: 10.1128/JB.183.19.5651-5658.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chen P, Greenberg B, Taghavi S, Romano C, van der Lelie D, He C. Angew Chem Int Ed Engl. 2005;44:2715–2719. doi: 10.1002/anie.200462443. [DOI] [PubMed] [Google Scholar]
- 113.Lund PA, Ford SJ, Brown NL. J Gen Microbiol. 1986;132(Pt 2):465–480. doi: 10.1099/00221287-132-2-465. [DOI] [PubMed] [Google Scholar]
- 114.Ralston DM, O'Halloran TV. Proc Natl Acad Sci U S A. 1990;87:3846–3850. doi: 10.1073/pnas.87.10.3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hobman JL, Wilkie J, Brown NL. Biometals. 2005;18:429–436. doi: 10.1007/s10534-005-3717-7. [DOI] [PubMed] [Google Scholar]
- 116.Dosanjh NS, West AL, Michel SL. Biochemistry. 2009;48:527–536. doi: 10.1021/bi801481j. [DOI] [PubMed] [Google Scholar]
- 117.van Vliet AH, Ernst FD, Kusters JG. Trends Microbiol. 2004;12:489–494. doi: 10.1016/j.tim.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 118.Dosanjh NS, Michel SL. Curr Opin Chem Biol. 2006;10:123–130. doi: 10.1016/j.cbpa.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 119.Ernst FD, Stoof J, Horrevoets WM, Kuipers EJ, Kusters JG, van Vliet AH. Infect Immun. 2006;74:6821–6828. doi: 10.1128/IAI.01196-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dosanjh NS, Hammerbacher NA, Michel SL. Biochemistry. 2007;46:2520–2529. doi: 10.1021/bi062092w. [DOI] [PubMed] [Google Scholar]
- 121.Dian C, Schauer K, Kapp U, McSweeney SM, Labigne A, Terradot L. J Mol Biol. 2006;361:715–730. doi: 10.1016/j.jmb.2006.06.058. [DOI] [PubMed] [Google Scholar]
- 122.Benini S, Cianci M, Ciurli S. Dalton Trans. 2011;40:7831–7833. doi: 10.1039/c1dt11107h. [DOI] [PubMed] [Google Scholar]
- 123.Wen Y, Feng J, Scott DR, Marcus EA, Sachs G. J Bacteriol. 2009;191:449–460. doi: 10.1128/JB.01219-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tottey S, Harvie DR, Robinson NJ. Acc Chem Res. 2005;38:775–783. doi: 10.1021/ar0300118. [DOI] [PubMed] [Google Scholar]
- 125.Kell DB. Arch Toxicol. 2010;84:825–889. doi: 10.1007/s00204-010-0577-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Burkhead JL, Gray LW, Lutsenko S. Biometals. 2011;24:455–466. doi: 10.1007/s10534-011-9430-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Whitmire JM, Gancz H, Merrell DS. Curr Med Chem. 2007;14:469–478. doi: 10.2174/092986707779941069. [DOI] [PubMed] [Google Scholar]
- 128.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 129.Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, Anderson KL, Dattilo BM, Dunman PM, Gerads R, Caprioli RM, Nacken W, Chazin WJ, Skaar EP. Science. 2008;319:962–965. doi: 10.1126/science.1152449. [DOI] [PubMed] [Google Scholar]
- 130.Kehl-Fie TE, Chitayat S, Hood MI, Damo S, Restrepo N, Garcia C, Munro KA, Chazin WJ, Skaar EP. Cell Host Microbe. 2011;10:158–164. doi: 10.1016/j.chom.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nobles CL, Maresso AW. Metallomics. 2011;3:788–796. doi: 10.1039/c1mt00047k. [DOI] [PubMed] [Google Scholar]
- 132.Kehl-Fie TE, Skaar EP. Curr Opin Chem Biol. 2010;14:218–224. doi: 10.1016/j.cbpa.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang SC, Dias AV, Bloom SL, Zamble DB. Biochemistry. 2004;43:10018–10028. doi: 10.1021/bi049405c. [DOI] [PubMed] [Google Scholar]