

Abstract

A flexible synthesis of the C1–C12 fragment of Tedanolide C has been accomplished in eight steps from 2-methyl-2,4-pentadienal. Asymmetric hydroformylation of a 1,3-diene allows for the late-stage generation of either C10 epimer with complete catalyst control. Diastereoselective addition of an isobutyryl β-ketoester dianion to an α,β-disubstituted chiral aldehyde sets the C5 stereochemistry while installing the geminal dimethyl unit. Differential protection of a syn-1,3-diol is performed as a highly efficient single-pot operation.
Tedanolide C (1) is a marine natural product isolated from a species of Ircina sponge off the coast of Papua New Guinea. Characterized by Ireland in 2006,1 tedanolide C is one of the more recently reported members of an intriguing family of marine macrolides that include tedanolide (2),2 13-deoxytedanolide (3),3 and the candidaspongiolides (4)4—which exhibit in vitro cytotoxicities in the nanomolar to picomolar range (Figure 1).5 The diversity of genera and geographies yielding these structural congeners, along with their low natural abundance, suggests that these toxins are derived from symbiotic microorganisms.4
Figure 1.
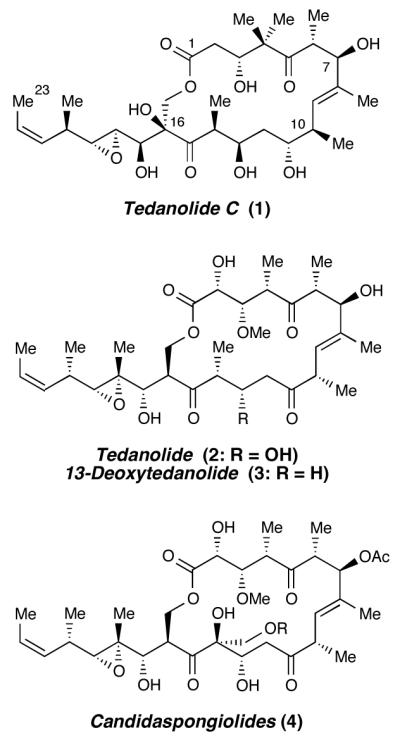
Tedanolide family of natural products.
Tedanolide C resembles other members of the tedanolide family in the fundamental structure of its complex 18-membered macrolide core and epoxidebearing side chain, yet differs in its substitution pattern and proposed stereochemistry; every analogous stereocenter beyond C7 (other than the epoxide positions) is assigned a configuration epimeric to the other tedanolides. Relative stereochemistry between the different subunits of the molecule was deduced by comparison of measured and calculated NMR coupling constants.1 The absolute configuration of tedanolide C has not yet been ascertained, nor has the relative stereochemistry between the subunits been definitively proven.
The tedanolides’ exceedingly potent cytotoxicity might someday be harnessed for chemotherapeutic use if the molecular basis of their biological activity can be elucidated.6 Initial biological testing indicated that both tedanolide and tedanolide C cause accumulation of cells in the S phase of the cell cycle without further progression and division.1,2 13-Deoxytedanolide selectively inhibits eukaryotic protein synthesis by binding to the 60S large ribosomal subunit,7 likely as a competitive inhibitor of tRNA at the E site via binding of the C17 hydroxyl and the side-chain epoxide.8 Indeed, limited SAR studies on 13-deoxytedanolide derivatives implicate these interactions and bolster the supposition that macrolide conformation is also important for effective binding.9
With the hope of further explicating these pharmacological and stereochemical issues, we initiated synthetic studies on tedanolide C. While tedanolide and 13-deoxytedanolide have attracted considerable attention among synthetic chemists,10 with notable total syntheses reported by Smith,11 Roush,12 and Kalesse,13 only two studies on tedanolide C have been reported to date—both of which are concerned with the “southwest” hemisphere of the natural product.14 In planning an asymmetric synthesis of the C1–C12 “northeast” fragment of tedanolide C, we kept several design concepts in mind: We desired equally easy access to both enantiomers and we valued flexibility in reliably setting relative stereochemistry throughout the fragment. In particular, because of plausible stereochemical ambiguity at C10, we wished to establish this stereocenter late in the synthesis to maximize common intermediates on the route to both C10 epimers.
We viewed this last challenge of installing the C10 stereochemistry as the most difficult. Due to the isolated location of this stereocenter relative to others, substrate-based asymmetric induction was unlikely to be advantageous. Survey of the literature confirmed that most approaches to this problem relied upon chiral pool starting materials that already contained this C10 stereochemical information. These solutions were not attractive. Particularly intriguing here is that the structural motif represented by the C7–C11 fragment—also including a critical E-trisubstituted olefin—is common to an array of complex natural products and a solution to this problem could have broader synthetic relevance.15
In 2001, Jacobsen installed the corresponding (C14–C18) subunit within ambruticin by way of an asymmetric diene hydroformylation (Scheme 1).16 Nozaki had shown that simple 1,3-dienes undergo regio- and enantioselective hydroformylations in the presence of a chiral rhodium catalyst.17 Yet, despite the potential utility of this transformation to selectively modify structurally elaborate dienes, Jacobsen’s work remains the only application within the core of a complex synthesis. We anticipated that early installation of a relatively unreactive diene unit followed by late-stage unmasking of a latent chiral β,γ-unsaturated aldehyde would inspire a highly efficient and flexible tedanolide C fragment synthesis.
Scheme 1.
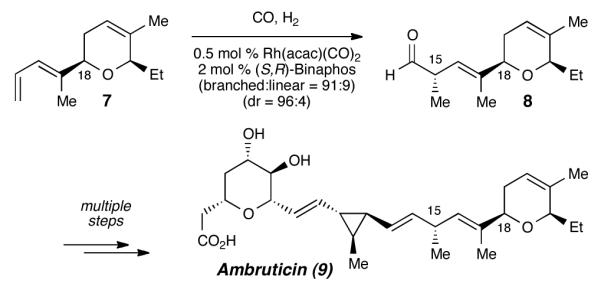
Diene hydroformylation as a key step in Jacobsen’s synthesis of ambruticin
To test the validity of our strategy as quickly as possible, we set out to prepare model diene 13 and investigate its rhodium-catalyzed hydroformylation with a variety of ligands (Table 1). Enacting these plans, a thizolidinethione-directed aldol reaction of known dienal 1018 was used to set the stereochemistry at C6 and C7 and install the diene component at the outset of the synthesis. Protection of the C7 hydroxyl group and reductive cleavage of the chiral auxiliary provided rapid access to model system 13.19
Table 1.
Synthesis and Hydroformylation of a model diene
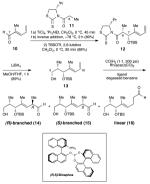 | ||||||
|---|---|---|---|---|---|---|
| entry | ligand | temp (°C) |
time (h) |
conv. (%) |
branched/ linear |
R/S |
| 1 | PPh3 | 35 | 24 | 95 | 86:14 | 48:52 |
| 2 | (R,R)-Chiraphite | 35 | 72 | 85 | 100:0 | 57:43 |
| 3 | (R,R)-Kelliphite | 35 | 24 | 87 | 95:5 | 59:41 |
| 4 | (R,R)-Ph-Bpe | 35 | 24 | trace | — | — |
| 5 | (R,R)-Ph-Bpe | 80 | 18 | 100 | 96:4 | 52:48 |
| 6 | (R,S)-Binaphos | 35 | 113 | 100 | 96:4 | 93:7 |
| 7 | (S,R)-Binaphos | 35 | 113 | 100 | 92:8 | 6:94 |
Hydroformylation using an achiral control ligand (entry 1) verified that there was negligible substrate control over the reaction diastereoselectivity. Commercially available chiral ligands (entries 2–5), with documented success for the asymmetric hydroformylation of simple olefins,20 demonstrated high branched/linear regioselectivity but unexceptional R/S diastereoselectivity. Ultimately, Nozaki’s original conditions proved to be the best of those evaluated;17 (R,S)-Binaphos (entry 6) led to a 93:7 ratio of C10 epimers 14 and 15, while the (S,R)-Binaphos (entry 7) provided complete reversal of the selectivity.21,19
With corroboration that the C10 stereochemistry of tedanolide C fragments could be catalyst controlled, construction of our actual hydroformylation substrate was resumed in earnest (Scheme 2). We envisaged that the entire C1–C4 subunit could be installed efficiently via nucleophilic addition of a β-ketoester dianion. For direct addition to an α,β-aldehyde such as 17, however, prediction of the (C5) stereochemical outcome was complicated by conflicting precedents. Evans’ polar model predicts that the 1,3-anti induction from the β-silyloxy substituent and the Felkin 1,2-syn directing effects from the α-methyl group should be stereononreinforcing and therefore lead to poor selectivity, yet several notable exceptions have been observed in complex systems.22 In the event, addition of 18 proceeded with ≥10:1 selectivity for the desired all-syn product (19)23. The large steric demand of the nucleophile likely contributes to the dominance of Felkin selectivity.
Scheme 2.
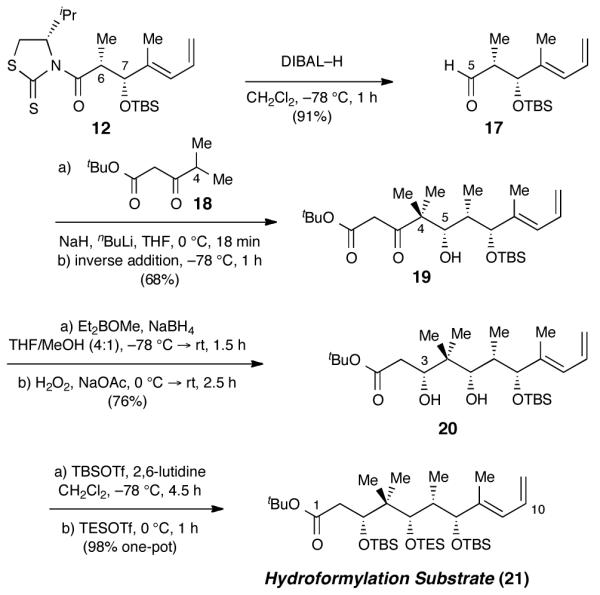
Synthesis of hydroformylation substrate.
Stereoselective C3 reduction of β-hydroxy ketone 19 required extensive optimization. The only other example of a 1,3-syn reduction on a substrate bearing an intervening gem-dimethyl group was reported by Jamison in his synthesis of acutiphycin.24 The catecholborane protocol used therein gave incomplete conversion and poor yields with our system. Prasad conditions25 gave excellent syn selectivity for the reduction,23,19 but nondestructive liberation of the product diol (20) from its unusually stable boron complex proved challenging. Ultimately, a sodium acetate-buffered hydrogen peroxide workup was found to give reproducible yields.26 Sequential addition of TBSOTf and TESOTf enabled the differential protection of diol 20 as a highly efficient single-pot operation27 and completed the synthesis of hydroformylation substrate 21.28
Gratifyingly, hydroformylations on this real system proceeded smoothly (Scheme 3). Using conditions optimized for the model system, each C10 epimer was generated with >95:5 diastereoselectivity depending upon the enantiomer of Binaphos ligand employed. Although it was possible to isolate the aldehyde products (22 and 25) directly—or derive the primary alcohols by quenching the reactions with sodium borohydride—addition of the mild methylating agent MeTi(iPrO)3 provided the corresponding methyl carbinols (23 and 26) as an inconsequential mixture of C11 epimers.29 Dess-Martin oxidation then delivered the desired methyl ketones (24 and 27), which were isolated as single stereoisomers.
Scheme 3.

Hydroformylation and completion of fragment.
In summary, the C1–C12 fragment of tedanolide C has been prepared in protected form. The synthesis is short, only eight steps from 2-methyl-2,4-pentadienal (10), and enables efficient access to both C10 epimers via late-stage hydroformylation of a common diene intermediate. Additional synthetic and stereochemical studies on tedanolide C and its analogs are underway. Further development of our general diene elaboration/hydroformylation strategy and application toward other natural product challenges will be forthcoming.
Supplementary Material
Acknowledgment
This work was supported by a Grant (R15CA132072) from the National Cancer Institute. A Pfizer Summer Undergraduate Research Fellowship to SJF is also gratefully acknowledged. Special thanks to Wesley Yoshida of the University of Hawaii, Manoa NMR facility for many helpful discussions.
Footnotes
Supporting Information Available Experimental procedures and full spectroscopic data for all new compounds. A stereochemical proof for model diene hydroformylation is also included. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Chevallier C, Bugni TS, Feng X, Harper MK, Orendt AM, Ireland CM. J. Org. Chem. 2006;71:2510–2513. doi: 10.1021/jo052285+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Schmitz FJ, Gunasekera SP, Yalamanchili G, Hossain MB, Van der Helm D. J. Am. Chem. Soc. 1984;106:7251–7252. [Google Scholar]
- (3).Fusetani N, Sugawara T, Matsunaga S. J. Org. Chem. 1991;56:4971–4974. [Google Scholar]
- (4).(a) Meragelman TL, Willis RH, Woldemichael GM, Heaton A, Murphy PT, Snader KM, Newman DJ, van Soest R, Boyd MR, Cardellina JH, II, McKee TC. J. Nat. Prod. 2007;70:1133–1138. doi: 10.1021/np0700974. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Whitson EL, Pluchino KM, Hall MD, McMahon JB, McKee TC. Org. Lett. 2011;13:3518–3521. doi: 10.1021/ol201329p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Tedanolide: ED50 = 16 pg/mL (26 pM) against cultured leukemia lymphocytes (ref. 2); 13-Deoxytedanolide: IC50 = 94 pg/mL (158 pM) against P388 murine leukemia cells (ref. 3); Candidaspongiolide core: mean GI50 = 14 ng/mL (20 nM) against NCI 60 tumor cell line (ref. 4a); Tedanolide C: IC50 = 57 ng/mL (95 nM) against HCT-116 cells (ref. 1).
- (6).(a) Taylor RE. Nat. Prod. Rep. 2008;25:854–861. doi: 10.1039/b805700c. [DOI] [PubMed] [Google Scholar]; (b) Daniela T, Uranchimeg B, Cardellina JH, Meragelman TL, Matsunaga S, Fusetani N, Del Bufalo D, Shoemaker RH, Melillo G. J. Nat. Cancer Inst. 2008;100:1233–1246. doi: 10.1093/jnci/djn239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Nishimura S, Matsunaga S, Yoshida M, Hirota H, Yokoyama S, Fusetani N. Bioorg. Med. Chem. 2005;13:449–454. doi: 10.1016/j.bmc.2004.10.012. [DOI] [PubMed] [Google Scholar]
- (8).Schroeder SJ, Blaha G, Tirado-Rives J, Steitz T, Moore PB. J. Mol. Biol. 2007;367:1471–1479. doi: 10.1016/j.jmb.2007.01.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Nishimura S, Matsunaga S, Yoshida S, Nakao Y, Hirota H, Fusetani N. Bioorg. Med. Chem. 2005;13:455–462. doi: 10.1016/j.bmc.2004.10.014. [DOI] [PubMed] [Google Scholar]
- (10).For a review of synthetic studies through 2007 see: Roy M, Kalesse M. Nat. Prod. Rep. 2008;25:862–870. doi: 10.1039/b719607e. and references cited therein. For more recent synthetic studies see. Park SH, Min JK, Park SH, Lee HW. Bull. Korean Chem. Soc. 2009;30:537–538. Jung ME, Yoo D. Tetrahedron. 2011;67:10281–10286.
- (11).(a) Smith AB, III, Adams CM, Barbosa SAL, Degnan AP. J. Am. Chem. Soc. 2003;125:350–351. doi: 10.1021/ja0289649. [DOI] [PubMed] [Google Scholar]; (b) Smith AB, III, Adams CM, Barbosa SAL, Degnan AP. Proc. Natl. Acad. Sci. U. S. A. 2004;101:12042–12047. doi: 10.1073/pnas.0402084101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Smith AB, III, Lee D. J. Am. Chem. Soc. 2007;129:10957–10962. doi: 10.1021/ja073329u. [DOI] [PubMed] [Google Scholar]
- (12).(a) Julian LD, Newcom JS, Roush WR. J. Am. Chem. Soc. 2005;127:6186–6187. doi: 10.1021/ja050729d. [DOI] [PubMed] [Google Scholar]; (b) Dunetz JR, Julian LD, Newcom JS, Roush WR. J. Am. Chem. Soc. 2008;130:16407–16416. doi: 10.1021/ja8063205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Ehrlich G, Hassfeld J, Eggert U, Kalesse M. J. Am. Chem. Soc. 2006;128:14038–14039. doi: 10.1021/ja0659572. [DOI] [PubMed] [Google Scholar]; (b) Ehrlich G, Hassfeld J, Egert U, Kalesse M. Chem.–Eur. J. 2008;14:2232–2247. doi: 10.1002/chem.200701529. [DOI] [PubMed] [Google Scholar]
- (14).(a) Barth R, Roush WR. Org. Lett. 2010;12:2342–2345. doi: 10.1021/ol1006955. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bülow L, Mani A, Fohrer J, Kalesse M. Org. Lett. 2011;13:6038–6041. doi: 10.1021/ol202515x. [DOI] [PubMed] [Google Scholar]
- (15).Notable examples include rapamycin, geldanamycin, calyculin, bafilomycin, and dictyostatin.
- (16).Liu P, Jacobsen E. J. Am. Chem. Soc. 2001;123:10772–10773. doi: 10.1021/ja016893s. [DOI] [PubMed] [Google Scholar]
- (17).Horiuchi T, Ohta T, Shirakawa E, Nozaki K, Takaya H. Tetrahedron. 1997;53:7795–7804. doi: 10.1021/jo9624051. Watkins AL, Landis CR. Org. Lett. 2011;13:164–167. doi: 10.1021/ol102797t. Landis has recently shown that diazaphospholane ligands are also highly effective for the hydroformylation of simple dienes:
- (18).Prepared in one step from 3-ethoxymethacrolein and vinylmagesium bromide: Spangler CW, McCoy RK, Karavakis AA. J. Chem. Soc., Perkin Trans. 1. 1986;7:1203–1207.
- (19).See Supporting Information for details.
- (20).Klosin J, Landis CR. Acc. Chem. Res. 2007;40:1251–1259. doi: 10.1021/ar7001039. [DOI] [PubMed] [Google Scholar]
- (21).The absolute stereochemistry at C10 was proved by chemical correlation and is consistent with previous reports.
- (22).(a) Evans DA, Dart MJ, Duffy JL, Yang MG. J. Am. Chem. Soc. 1996;118:4322–4343. and references cited therein. [Google Scholar]; (b) Balog A, Bertinato P, Su D-S, Meng D, Sorensen E, Danishefsky, Zheng Y-H, Chou T-C, He L, Horwitz SB. Tetrahedron Lett. 1997;38:4529–4532. [Google Scholar]; (c) Liu J, Wong C-H. Angew. Chem. Int. Ed. 2002;41:1404–1407. doi: 10.1002/1521-3773(20020415)41:8<1404::aid-anie1404>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- (23).Confirmed by NMR analysis of the corresponding acetonide.
- (24).Moslin RM, Jamison TF. J. Org. Chem. 2007;72:9736–9745. doi: 10.1021/jo701821h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chen KM, Hardtmann GE, Prasad K, Repic O, Shapiro MJ. Tetrahedron Lett. 1987;28:155–158. [Google Scholar]
- (26).Galobardes M, Mena M, Romea P, Urpi F, Vilarrasa J. Tetrahedron Lett. 2002;43:6145–6148. [Google Scholar]
- (27).No TBS protection was observed at the C5-OH at −78 °C, even in the presence of excess TBSOTf.
- (28).Although C5 is eventually needed in its ketone oxidation state, we elected to prepare the fragment in reduced and protected form.
- (29).This reagent may be used in excess with no concern for overaddition to the ester: Weidmann B, Seebach D. Helv. Chim. Acta. 1980;63:2451–2454.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.