

Abstract
Information on the secondary structure and interactions of RNA is important to understand the biological function of RNA as well as in applying RNA as a tool for therapeutic purposes. Recently, the isoenergetic microarray mapping method was developed to improve the prediction of RNA secondary structure. Herein, for the first time, isoenergetic microarrays were used to study the binding of RNA to protein or other RNAs, as well as the interactions of two different RNAs and protein in a three-component complex. The RNAs used as models were the regulatory DsrA and OxyS RNAs from Escherichia coli, the fragments of their target mRNAs (fhlA and rpoS), and their complexes with Hfq protein. The collected results showed the advantages and some limitations of microarray mapping.
Knowledge about RNA structure as well as RNA interactions is important for understanding biological processes and for the practical applications of RNA. Recently, a method to study the structure of native RNA using isoenergetic microarrays has been developed (1, 2). This approach is based on the observation that microarray oligonucleotide probes bind to single, but not to double stranded, fragments of target RNAs. To enhance thermodynamic stability and to make binding of the target RNA to 2′-O-methylated probes sequence independent, locked nucleic acid (LNA) nucleotides, including 2,6-diaminopurine riboside (D) derivatives were introduced at selected positions (3-6). Microarray mapping was successfully applied to solve the secondary structure of the 5′ region of the R2 retrotransposon from Bombyx mori and four another closely related R2 RNAs (2, 7). Based on studies of tRNAPhe from Saccharomyces cerevisiae and its unmodified transcript as well as tRNAiMet and tRNA Met m from Lupinus luteus, it has been shown that isoenergetic microarry probing can be used to study the influence of modified nucleotides and magnesium cations on RNA folding (8, 9). Recently, several RNA structural motifs (hairpins, internal and bulged loops, 3′- and 5′-dangling ends, and pseudoknots) were studied in detail using isoenergetic microarrays (10).
In this paper, the application of isoenergetic microarrays to study natural interactions of RNAs in two- and three-component complexes is shown. The targets for experiments were DsrA and OxyS RNAs, and their interactions with Hfq protein, various length fragments of rpoS and fhlA mRNA, and also with both: fragment of target mRNA and Hfq protein.
DsrA and OxyS RNA are non-coding RNAs (ncRNA) whose regulatory functions, among the many ncRNA identified in Escherichia coli, are particularly well understood (11-14). Non-coding RNAs have been identified in both prokaryotes and eukaryotes. In bacteria, which must rapidly adapt their metabolism in response to environmental changes or to establish virulence, ncRNAs regulate the expression of the multiple genes and have profound effects on cell physiology (13, 15, 16). The 87 nucleotide DsrA RNA is synthesized at low temperatures and is involved in the regulation of several target genes: such as rpoS (17-19). It has been postulated that DsrA RNA binds to the 5′UTR of rpoS mRNA; this binding leads to the formation of an alternative secondary structure of rpoS mRNA that is translationaly active. The OxyS RNA ncRNA is 109 nucleotides long and is induced in response to oxidative stress in E. coli (20). OxyS RNA regulates the expression of multiple genes. It has been shown that OxyS RNA represses the translation of two genes: fhlA and rpoS. An interaction with fhlA mRNA is achieved probably by the formation of two kissing loops (21, 22) or by kissing loop as well as loop and bulge interactions (23),while the mechanism of rpoS mRNA repression is less clear. In the formation of the complexes between DsrA or OxyS RNAs and their target mRNAs, the protein Hfq is involved (18, 19, 23-26). This 11.2 kDa protein is active as homohexamer and has been reported to bind preferentially to A and U rich single stranded RNA regions (25, 27-30).
Here we show that isoenergetic microarrays applied to study complexed RNA can give new and alternative insight into RNA interactions.
EXPERIMENTAL PROCEDURES
Materials
A, C, G, and U or T RNA, DNA and 2′-O-methyl RNA phosphoramidites and C6-aminolinker for oligonucleotide synthesis were purchased from Glen Research and Proligo. Phosphoramidites of LNA 2,6-diaminopurine riboside and 2′-O-methyl-2,6-diaminopurine riboside were synthesized as described previously (31).
Taq polymerase, T4 polynucleotide kinase, dNTPs and ddNTPs were from Fermentas. Reverse transcriptase SuperScript III was from Invitrogen, T7-MEGAshortscript transcription kit was from Ambion. 1-Cyclohexyl-3-(2-morpholinoethyl)-carbodiimide metho-p-toluenesulfonate (CMCT) was from Aldrich and kethoxal was from ICN Biomedicals. N-methylisatoic anhydride (NMIA) was from Molecular Probes. Silanized slides and probe-clip press seal incubation chambers for hybridization experiments were purchased from Sigma.
Chemical synthesis of oligonucleotides
Oligonucleotides and oligonucleotide probes were synthesized by the phosphoramidite approach on an ABI 392 synthesizer or MerMade 12 (BioAutomatization) synthesizers (32). The oligonucleotides probes for microarrays were synthesized with a C6-aminohexyl linker on the 5′-end. Oligonucleotides were deprotected and purified according to published procedures (2) Molecular weights were confirmed by mass spectrometry (MALDI TOF, model Autoflex, Bruker). Concentrations of all oligonucleotides were determined from predicted extinction coefficients for RNA and measured absorbance at 260 nm at 80°C (33, 34). It was assumed that 2′-O-methyl RNA-LNA chimeras and RNA strands with identical sequences have identical extinction coefficients.
DNA template construction and RNA synthesis
Generation of DNA templates for OxyS and DsrA RNAs synthesis and their in vitro transcription were performed as described previously using oligonucleotides DsrA1, DsrA2, OxyS1 and OxyS2 (35) (Table S7). DNA templates for transcription of rpoS140, rpoS325, rpoS578, fhlA115, fhlA282 were amplified by PCR from chromosomal DNA in E. coli DH5α cells (sequences of primers in Table S7). RNAs were synthesized by in vitro transcription using T7-MEGAshortscript kit (Qiagen).
Hfq expression and purification
Expression and purification of Hfq protein was performed according to published procedures with some modifications (36). Cell pellet (obtained from 1 liter of cell culture) was resuspended in lysis buffer and lysed by ultrasonification. The lysate was treated with DNase I (200 U) and RNase A (200 μg) and incubated 7 h at room temperature. Centrifugally clarified lysate was passed over HiTrap chelating column (GE Healthcare) preloaded with NiSO4. The column was washed with 10 volumes of lysis buffer, then 20 volumes of wash buffer. Hfq was eluted with 10 volumes of elution buffer (50 mM HEPES pH 7.5, 500 mM NH4Cl, 250 mM imidazole, 5% (w/v) glicerol). Protein was concentrated and dialyzed against storage buffer.
Native gel electrophoresis
Native gel electrophoresis indicated that all RNAs used form a single structure under various buffer conditions. RNA was radioactively labeled at the 5′-end with γ32P-ATP (20,000 cpm) and was folded in 10 μl of one of the following buffers: (I) 1 M NaCl, 4 mM MgCl2, 10 mM Tris-HCl, pH 7.5, (II) 200 mM NaCl, 4 mM MgCl2, 10 mM Tris-HCl, pH 7.5, (III) 1 M NaCl, 0.2 mM MgCl2, 10 mM Tris-HCl, pH 7.5, (IV) 200 mM NaCl, 4 mM MgCl2, 0.2 mM Tris-HCl, pH 7.5, (V) 1 M NaCl, 10 mM MgCl2, 10 mM Tris-HCl, pH 7.5, (VI) 200 mM NaCl, 10 mM MgCl2, 10 mM Tris-HCl, pH 7.5, (VII) 200 mM NaCl, 30 mM MgCl2, 10 mM Tris-HCl, pH 7.5, (VIII) 1 M NaCl, 0.5 mM Na2EDTA, 10 mM Tris-HCl, pH 7.5, (IX) 200 mM NaCl, 0.5 mM Na2EDTA, 10 mM Tris-HCl, pH 7.5, with addition of 1 μl 50% glycerol and traces of tracking dyes. The mixture was incubated for 3 min at 75°C except 95°C for DsrA RNA and renatured for 20 min at 25°C. After folding, the RNA sample was analyzed by non-denaturing polyacrylamide gel electrophoresis (PAGE) containing TBE buffer with a TBE running buffer or containing TBM buffer (100 mM Tris-HCl pH 8.3, 100 mM boric acid, 10 mM MgCl2) with a TBM running buffer. The gel was pre-electrophoresed for 0.5 h. Electrophoresis on 27 cm long plates were run at 20W at 4°C maintained by water-coat and dried gels were analyzed by exposing to a phosphorimager screen.
For RNA/Hfq complexes RNA (20,000 cpm) was renaturated as described earlier and a 10 μl sample was prepared by combining the pre-annealed RNA, in selected buffer, and increasing concentration of Hfq. Reactions were incubated for 20 min at 25°C. After incubation, samples were analyzed by non-denaturing PAGE at 4°C using TBE buffer.
For RNA/RNA and RNA/RNA/Hfq complexes each RNA was renaturated separately as described earlier and 10 μl samples were prepared by combining pre-annealed RNAs in selected buffer: 20,000 cpm labeled RNA with increasing concentration of complementary RNA. Reactions were incubated for 20 min at 25°C. For three-component complexes, after addition of appropriate amount of Hfq, the reaction was incubated another 20 min at 25°C. After incubation, samples were analyzed by non-denaturing PAGE with either TBE or TBM buffer.
Preparation of isoenergetic microarrays
Isoenergetic microarray probes were designed and synthesized as described earlier (2). They were built with 2′-O-methyl oligonucleotide pentamers and hexamers with incorporated LNA nucleotides and 2,6-diaminopurine riboside (LNA or 2′-O-methyl). Modifications were chosen to provide predicted free energies (ΔG°37) for binding at 37°C to unstructured RNA between −8.0 and −10.5 kcal/mol. The sequence UUUUU and spotting buffer were used as negative controls. Microarrays were prepared according to the method described earlier (2, 37). Silanized slides were coated with 2% agarose activated by NaIO4 and probes were spotted in 4-8 repeats by a microarray printer (SpotArray 24, Perkin Elmer Life Sciences) with a spot distance of 750 μm. Microarrays contained complementary probes that fully covered the DsrA and OxyS sequences. For some A and U rich regions of DsrA and OxyS there were no pentamer probes with sufficient calculated binding free energy. For those positions additional heptanucleotide probes were prepared (Table 1 and 2). Microarrays were incubated for 12 h at 37°C in 100% humidity chambers. The remaining aldehyde groups on microarrays were reduced with 35 mM NaBH4 solution in PBS buffer (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4, pH 7.5) and ethanol (3:1 v/v). Then slides were washed in water at room temperature (3 washes for 30 min each) and in 1% SDS solution at 55°C for 1 h, and finally in water at room temperature (3 washes for 30 min each) and dried at room temperature overnight.
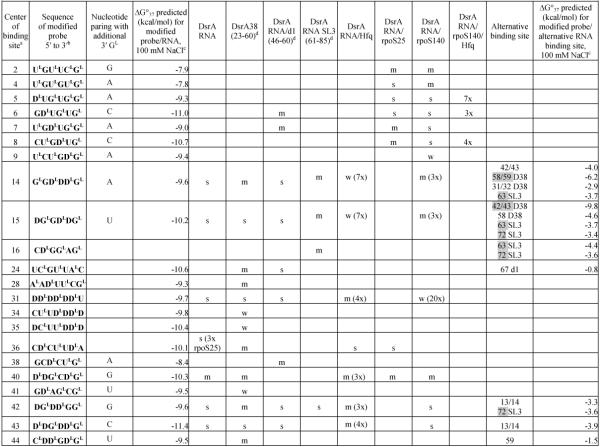
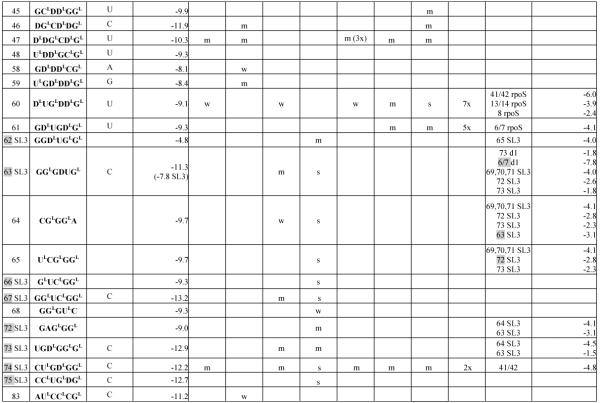
Table 1.
Hybridization results for DsrA RNA and itscomplexes
|
|
Center of binding site is the target RNA nucleotide complementary to the third nucleotide from the 5′-end of the probe.
In sequence of modified probe: LNA nucleotides are marked with superscript L; D represents 2,6-diaminopurineriboside; nucleotides without a superscript are 2′-O-Me-nucleotides.
Calculated according to a published equation (2).
Numbers in brackets show positions in DsrA RNA. Symbols: s - strong binding, m - medium binding, w - weak binding. Numbers with X mean how many times binding signals are diminished: for DsrA/Hfq and DsrA/rpoS140 relative to DsrA alone, for DsrA/rpoS140/Hfq relative to DsrA/rpoS140. The most probably complementary or alternative binding sites are marked grey. If in alternative binding site there are two numbers separated by a slash, it means that probe may bind to an even number of nucleotides.
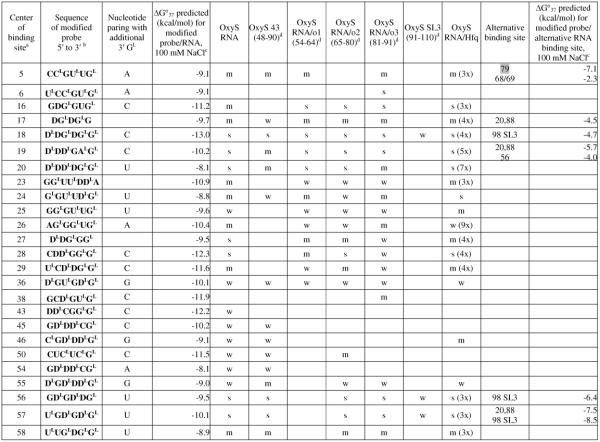
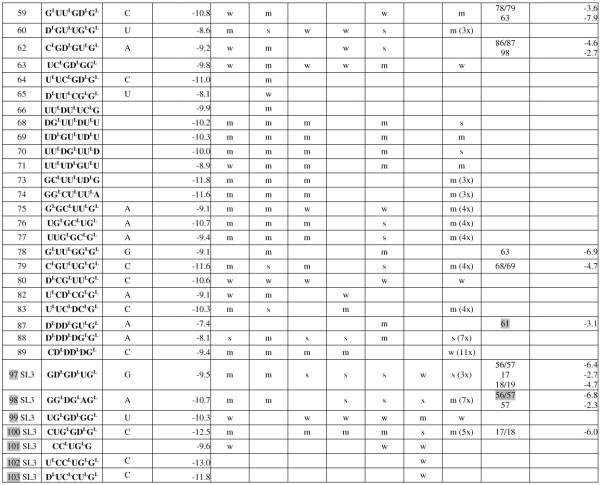
Table 2.
Hybridization results for OxyS RNA and its complexes (without some alternative binding sites eliminated after experiments with OxyS43, o1, o2, o3 and OxyS SL3; complete table is accessible in supplementary materials)
|
|
|
|
Center of binding site is the target RNA nucleotide complementary to the third nucleotide from the 5′-end of the probe.
In sequence of modified probe: LNA nucleotides are marked with superscript L; D represents 2,6-diaminopurineriboside; nucleotides withouta superscript are 2′-O-Me-nucleotides.
Calculated according to a published equation (2).
Numbers in brackets show positions in OxyS RNA. Symbols: s- strong binding, m- medium binding, w-weak binding. Numbers with X mean how many times binding signals are diminished: for OxyS/Hfq relativeto OxyS alone. The most probably complementary or alternative binding sites are marked in grey, the rest alternative binding sites are possible. Ifin an alternative binding site there are two numbers separated by slash it means that probe may bind to an even number of nucleotides.
Hybridization on isoenergetic microarrays
Prior to hybridization, RNA was 5′ radioactively labeled and purified by denaturing PAGE. RNAs were folded in one of the above buffers I-IX by incubation for 3 min at 75°C (except 95°C for DsrA RNA) and renatured for 20 min at 25°C. For hybridization, ca. 10 nM of 32P labeled RNA in 200 μl of folding buffer was placed on a microarray slide and covered with a HybriSlip. During hybridization of RNA/Hfq pre-formed complexes, RNA renaturation was done in reduced volume and then incubated with Hfq for 20 min at 25°C in 200 μl volume. For hybridization of RNA/RNA complexes each RNA was folded separately in 100 μl volume and then RNAs were mixed and incubated for 20 min at 25°C. Three-component complexes were prepared by folding RNAs separately and then mixing them with Hfq for 20 min at 25°C in 200 μl final volume. The microarray slide was placed in a humidity-temperature chamber with 100% humidity and incubated for 18 h at 4°C or 22°C. After hybridization, buffers with RNA were poured out and slides were washed in buffer with the same salt concentrations for 3 min at 0°C. Different hybridization times (4 h, 8 h) and temperatures (4°C, room temperature, 37°C) and also various washing times (1, 3, 10 and 15 min at 0°C) were also tested. Slides were dried by centrifugation in a clinical centrifuge (2000 rpm, 2 min) and covered with plastic wrap. Hybridization was visualized by exposure to a phosphorimager screen which was scanned on a Molecular Dynamics or Fuji Phosphorimager. Quantitative analysis was performed with ImageQuant 5.2. Binding was considered strong, medium, or weak when the integrated spot intensity was ≥ 1/3, ≥ 1/9, or ≥ 1/27 of the strongest integrated spot intensity, respectively. The site of probe binding is marked (and named) by the number of middle nucleotide complementary to probe sequence. Experiments were repeated at least three times and the average of the data is presented. Only strong and medium binding signals were considered as constraints in structure predictions by RNAstructure 4.6 program (38). Probes that bound were checked for alternative binding sites with bimolecular mode of RNAstructure.
Chemical mapping
Chemical mapping of DsrA and OxyS RNAs was performed according to procedures described earlier (35). Kethoxal was used to modify guanosine, CMCT to modify uridine, and NMIA to modify flexible riboses.
Modification sites were identified by primer extension with DNA-LNA modified primers (Table S8) according to a procedure described earlier (35).
RESULTS
Determination of secondary structure of DsrA RNA by isoenergetic microarray mapping
Native PAGE showed that DsrA RNA folds into one unique structure in various buffers and folding conditions used. Therefore 5′-labelled DsrA RNA was hybridized to microarrays at 4 or 22°C, in buffers containing various amount of magnesium chloride (0, 0.2, 4 and 30 mM) and sodium chloride (0.2 or 1 M). As was the case for other RNAs, the most intense signals were observed at 4°C and buffers with the highest ionic strength (2, 10). The binding pattern, however, was similar in each buffer used. The results showed that the presence of 1 M NaCl in hybridization buffer is sufficient to detect binding and the presence of MgCl2 did not affect probe binding. Decreasing NaCl to 0.2 M, however, must be compensated by at least 4 mM MgCl2.
Averaged hybridization results were used as constraints for the in-silico folding of DsrA RNA with RNAstructure 4.6 (Figure 1, structure A4). Structure A1 on Figure 1 is based on energy minimization alone and is currently disfavored, while structures A2 and A3 were proposed based on enzymatic mapping data (39-41). The microarray binding results support the presence of previously reported hairpins I and III. Strong binding to positions 14 and 15 (hairpin I) and medium binding to position 74 (hairpin III) were detected. No binding to stem regions of hairpins I and III were observed. The structure is less well-determined in the region of hairpin II, where alternative secondary structure models have been previously proposed. Results do not allow for the unambiguous determination of the secondary structure of this fragment of DsrA RNA. Strong binding was observed to positions 31, 36, 42, 43 and medium binding to positions 40 or 47 (the same probe may bind to both positions, Table 1). The newly predicted secondary structure for DsrA RNA (Figure 1, A4), based on free energy minimization constrained by microarray binding data, is unlike those previously suggested, which are not well-supported by the microarray results.
Figure 1.

Isoenergetic microarray mapping and chemical mapping results for DsrA RNA, marked on (A1) structure proposed by Sledejski based only on computational analysis, (A2) structure proposed by Lease and Belfort, (A3) structure proposed by Rolle, (A4) structure predicted by RNAstructure constrained by microarray mapping data. Red squares represent sites of binding to probe (middle nucleotide of target binding region), orange squares represent two sites which can bind to the same probe. Chemical modification data are superimposed: kethoxal with triangles, CMCT with squares, and NMIA with circles. Free energy of each structure is predicted by RNAstructure 4.6. Isoenergetic microarray mapping results for (B) DsrA 38, (C) DsrA SL3, (D) complex DsrA RNA/oligonucleotide d1. The navy line shows complementary region of binding for d1. Red squares represent binding sites to probes which have the same intensity relative to DsrA RNA alone. Orange squares represent two sites which can bind to the same probe and have the same intensity relative to DsrA RNA alone. Blue squares represent additional binding sites to probes, not observed in DsrA RNA. Light-blue squares represent additional binding sites to probes, not observed in DsrA RNA, which have the possibility of binding to alternative binding sites. Yellow squares represent lack of binding to probes in the presence of d1.
To get additional information on folding of the middle region of the DsrA RNA, a 38 nucleotide fragment of the DsrA RNA, corresponding to nucleotides 23-60 of the full-length RNA (named DsrA38), was chemically synthesized and used in hybridization experiments. The results showed stronger binding to microarrays for DsrA38 than the entire DsrA RNA. The same pattern of binding was observed in DsrA38 as was seen in the DsrA RNA. In DsrA38, however, several additional medium and weak bindings were observed for probes complementary to positions 24, 28, 44, 46 and 59. Moreover, probes complementary to positions 14 and 15, absent in DsrA38, also bound, probably by mismatch interactions with positions 58/59 and 42/43, respectively (Figure 1B, Table 1). Isoenergetic microarray hybridization experiments on isolated RNA structural motifs demonstrated that single stranded regions of RNA and terminal helixes adjacent to single stranded regions are particularly accessible for probe binding (10). This could explain the observed differences in binding of DsrA38 and entire DsrA RNA. Overall, the hybridization experiments of DsrA38 support the secondary structure labeled A4 in Figure 1.
To test the structure of the central fragment of DsrA RNA the oligonucleotide d1 (GMDLDMDMCMUMUMGLCMUMUMDMDMGLC, where D is 2,6-diaminopurine riboside; superscripts M and L mark 2′-O-methylated and LNA nucleotides, respectively) was hybridized to the complementary fragment of DsrA RNA (nucleotides 46-60) and then mapped with microarrays. Because the central fragment of DsrA RNA is A and U rich, LNA and 2′-O-methylated and 2,6-diaminopurine nucleotide residues were used to enhance the thermodynamic stability of the oligonucleotide d1/DsrA RNA hybrid (5, 31): the calculated free energy of the hybrid is −27.9 kcal/mol (2). In consequence, the binding of d1 to the complementary region of DsrA RNA may result in linearization of this fragment and structural rearrangements in other regions. Newly released single stranded regions of the DsrA RNA could appear and bind to complementary probes on the isoenergetic microarray. The control experiments were the hybridizations of DsrA RNA alone and d1 alone (Table S2). Hybridization experiments in various buffers confirm this hypothesis: in addition to previously observed binding sites (14, 15, 31, 42, 43 and 74), new strong and medium binding for probes complementary to sites 6, 7, 24, 38, 63, 67 and 73 were detected (Figure 1D, Table 1). Bindings at positions 24 and 38 are possible only if d1 binds and unfolds the native structure of DsrA RNA. Unexpectedly, no binding to position 36 was observed. It could be the result of structural rearrangement in the d1/DsrA RNA complex. Lack of binding to positions 40 or 47 confirms the binding of d1 to the intended fully complementary site. Additional unexpected bindings of probes complementary to positions 63 and 67 as well as 6 and 7 were observed. Detailed hybridization analysis suggests that the probe complementary to site 63 could also bind to position 6/7 (Table 1); only this alternative binding may have been present during the hybridization. Nevertheless, the results of microarray mapping for the d1/DsrA RNA complex suggest some rearrangement of DsrA RNA secondary structure, even in the well defined regions of hairpins I and III.
To confirm the possibility of rearrangement of the stable hairpin in DsrA RNA, the structure of oligoribonucleotide DsrA SL3, corresponding to hairpin III, was also studied. Hybridization of model hairpin III (nucleotides 61-85) in the same conditions as DsrA RNA show strong binding to positions 63-67, 74 and 75 and medium binding to positions 62, 72 and 73 (Figure 1C, Table 1). These results demonstrate substantial differences in the hybridization of model hairpin III and of DsrA RNA. Some sites (63 and 67) were also available for binding in the d1/DsrA RNA complex. Results show that the loop was more accessible for hybridization in DsrA SL3, than in the entire DsrA RNA. Additionally, these results support the observation reported by Rolle that hydroxyl radicals modified the 5′-side of hairpin III in DsrA RNA (41). The significantly enhanced binding ability of DsrA SL3 was observed not only in the loop region but also the 5′-side of stem. This correlates with the previous observation that in hairpins the 5′-side of the hairpin stem is more accessible to hybridization than the 3′ one (10).
Due to the novel nature of using microarray mapping to study RNA structure and interactions in multi-component complexes, traditional chemical mapping was applied as well. Due to thermodynamic stability of hairpin III of DsrA RNA, a chimeric LNA-DNA primer was used for primer extension (rather than the less stable DNA primer) (35). This approach allows for the formation of the thermodynamically stable LNA-DNA primer/DsrA RNA duplex which can be extend by reversed transcriptase. The application of an LNA-DNA primer allowed for the visualization of the chemical mapping of the 3′-side of DsrA RNA which was impossible to achieve using standard DNA primers. Most of the chemical modifications were observed in central part of DsrA RNA. In hairpin I, only modification of U12 was found. In the central region of DsrA RNA, chemical modifications were observed at positions 23-27, 29, 35-39, 41, 46, 48 and 52. The majority of the modified positions (with the exception of positions 23-27 and 52) are consistent with microarray mapping results (Figure 1).
Determination of interactions of DsrA RNA in complex with Hfq by isoenergetic microarray mapping
One of the main goals of this work was to study a model RNA/protein complex using microarray mapping; the DsrA RNA and Hfq protein complex provides an interesting object of study. Gel shift assays demonstrate that Hfq can form two complexes with DsrA RNA in various hybridization buffers, which is consistent with previous observations (18, 36, 42). Based on the analysis of the stabilities of DsrA RNA/Hfq complexes, the following hybridization conditions were selected: hybridization buffer VIII and 3 μM concentration of Hfq (first complex, with lower mobility on gel). As a control, the hybridization of DsrA RNA alone was used. The hybridization experiments of DsrA RNA/Hfq showed 3 to 7 fold diminishing of binding signal of some probes in comparison with DsrA RNA (Figure 2, Table 1). The binding of any of the probes was inhibited completely, but also no new binding sites were observed. This suggests a lack of structural rearrangement in DsrA RNA in complex with Hfq, which is in agreement with previous work (18, 30). Diminishing of binding was observed at positions: 14, 15, 31, 42, 43 and 40 (47). These data are consistent with the claim of other authors that the middle region of DsrA RNA is the main Hfq binding site (18, 30, 41).
Figure 2.

Isoenergetic microarray mapping results for DsrA RNA in complex with Hfq. Left side: two microarrays - the first with hybridized DsrA RNA, the second with hybridized complex DsrA RNA/Hfq. Right side: bar graph of relative binding intensity to the probes and hybridization results for DsrA RNA/Hfq marked on proposed structure of DsrA RNA. Red squares represent sites of binding to probe which have the same intensity relative to DsrA RNA alone. Yellow circles represent decreased binding intensities in the presence of Hfq. Light-yellow circles represent decreased binding intensities in the presence of Hfq for sites 40 and/or 47 to which can bind the same probe.
There is the possibility that released Hfq protein could bind to oligonucleotide probes and block binding to the Hfq/DsrA RNA complex. To correct for any such artifacts, control experiments were performed. It is well established that A and U rich polynucleotide strongly bind to Hfq (25, 28-30). Several 2′-O-methyl and LNA modified probes, especially rich in U and 2,6-diaminopurine riboside, which potentially could strongly bind to Hfq, were tested (Table S5). Native PAGE showed no or negligible amounts of binding of modified probes to Hfq, even with 100 fold excess of tested probes: Hfq could not block probes for interactions with DsrA RNA.
Determination of the secondary structure and interactions of DsrA RNA in complex with rpoS mRNA using isoenergetic microarray mapping
Bacterial ncRNA are involved in the regulation of biological activity of some mRNAs. DsrA RNA regulates the activity of rpoS mRNA. It was shown that DsrA RNA and rpoS mRNA hybridized via 22 residues. In DsrA RNA, interactions involve nucleotides 10 to 32, whereas in rpoS mRNA, the region between −97 and −119 is interacting (18). To study those interactions by isoenergetic microarrays, DsrA RNA and two fragments of rpoS mRNA were used for hybridization. One of them, corresponding to a 25 nucleotide fragment of rpoS mRNA (rpoS25), is complementary to DsrA RNA and is involved in base pairing. Second, a 140 nucleotide fragment (rpoS140) contains a complementary fragment and also has the structure responsible for inhibition of translation, which is translationally activated through base paring with DsrA RNA. Besides hybridization of complexes in buffer VIII the control hybridizations were performed using DsrA RNA alone and rpoS140 alone under the same conditions. During annealing, a 30 fold molar excess of rpoS25 or rpoS140 was used to form complexes with DsrA RNA.
The hybridization of the complex DsrA RNA/rpoS25 showed significant differences in binding to isoenergetic microarrays compared to DsrA RNA (Figure 3, Table 1). Strong binding to positions 4-6 and medium binding to positions 2, 7 and 8 were observed. In DsrA RNA, nucleotides at those positions are involved in the formation of hairpin I and no binding was detected. Moreover, there was a lack of binding to positions 14, 15 and 31 of DsrA RNA in complex with rpoS25; whereas in DsrA RNA alone, hybridizations to those positions were observed. As expected, the binding of rpoS25 to DsrA RNA caused refolding of hairpin I. The interacting region within DsrA RNA is protected from binding while the 5′ end becomes single stranded and accessible for hybridization. Moreover, some changes in the hybridization pattern were observed in the remaining regions of DsrA RNA (for probes complementary to positions 36, 40 (47), 42, 60 and 61). This suggests that interactions with rpoS mRNA can influence the structure of these regions of DsrA RNA.
Figure 3.

Isoenergetic microarray mapping results for DsrA RNA in complex with (A) rpoS25 (B) rpoS140 (C) rpoS140 and Hfq. (D) Bar graph of relative binding intensity to the probes. Red squares represent sites of binding to probes which have the same intensity relative to DsrA RNA alone. Orange squares represent two sites which can bind to the same probe and have the same intensity relative to DsrA RNA alone. Blue squares represent additional binding sites to probes in the presence of rpoS or rpoS and Hfq. Grey circles represent lack of binding relative to DsrA RNA alone. Yellow circles represent decreased binding in the presence of rpoS140. Green circles represent decreased binding in the presence of rpoS25. Purple circles represent lack of binding relative to complex DsrA RNA/rpoS140.
The hybridization results with a 140 nucleotide (rpoS140) fragment of rpoS mRNA bound with DsrA RNA showed some differences relative to those observed for the rpoS25/DsrA RNA complex (Figure 3, Table 1). As for rpoS25/DsrA RNA also in complex rpoS140/DsrA RNA binding of probes at positions 2, 4-8 were observed. Signal intensity of binding to positions 14, 15 and 31 was diminished by 3, 3 and 20 times for respective positions, relative to DsrA RNA. Unexpectedly, there was no binding to position 36 (observed in rpoS25/DsrA RNA complex). Also additional binding of probes complementary to sites 45, 46, 60 and 61 were observed. Results of the hybridization experiments suggest that rpoS140 binding to DsrA RNA is extended to nucleotide 41 in DsrA RNA.
Determination of the secondary structure and interactions of DsrA RNA in complex with rpoS mRNA and Hfq by isoenergetic microarray mapping
It was found that in vivo the activation of the translation of rpoS mRNA by DsrA RNA requires Hfq protein (43). It is postulated that Hfq is involved in interactions of DsrA RNA and rpoS mRNA. Hfq can bind to both RNAs separately but also can form three-compounds complex containing DsrA RNA/Hfq/rpoS mRNA. It was reported that the presence of Hfq increases the rate of RNA association and the stability of DsrA RNA/rpoS mRNA complex. However, the rate of association depends on length of rpoS mRNA leader (18, 19).
To form a model three-molecular complex for microarray mapping: DsrA RNA, rpoS140 and Hfq were chosen. For rpoS140 Hfq increases base paring with DsrA RNA twofold (18), and it is the simple model for our research. Additionally, these experiments allowed for the correlation of the hybridization results with results on the complex of DsrA RNA/rpoS140 presented here. To form the three-molecular complex DsrA RNA (radioactively labeled) was mixed and annealed with 30 fold molar excess of rpoS140 in presence of 1.5 μM of Hfq hexamer in buffer VIII. The formation of DsrA RNA/Hfq/rpoS140 complex was confirmed by native polyacrylamide gel electrophoresis. For each set of three-component hybridization experiments DsrA RNA and complex DsrA RNA/rpoS140 were used as control. During hybridization of complex DrsA RNA/Hfq/Rpos140 to the microarray, binding was observed for probes complementary to sites 5, 6, 8, 60, 61 and 74 (Figure 3, Table 1). Those signals were 2-7 times weaker than in control DsrA RNA/rpoS140 experiments. Hybridization of DsrA RNA/rpoS140 gave 3-20 fold decreased binding in the region of interactions (sites 14, 15 and 31), relative to DsrA RNA alone, whereas, in the three-molecular complex, no binding in those positions was observed (see Table 1). This suggests that Hfq stabilizes binding of complementary fragments of DsrA RNA and rpoS140. Disappearance of binding signals at positions 42, 43 and 47 was noted, this suggests that Hfq remains bound to DsrA RNA in the three-component complex, because the same hybridization pattern was observed in DsrA RNA/Hfq complex but not in DsrA RNA/rpoS140 complex. Also, no binding at positions 45 and 46 was observed; those positions were not accessible in DsrA RNA in contrast to DsrA RNA/rpoS140 complex. This may suggest that in the tested three-component complex (where the main Hfq binding site is covered) binding sites of Hfq in DsrA RNA are transferred to region 41-47.
Determination of secondary structure of OxyS RNA with isoenergetic microarray mapping
The analysis of OxyS RNA by native gel electrophoresis showed that it forms one structure which remains the same in various buffers used during hybridization experiments. OxyS was hybridized to microarray at 4 or 22°C in buffers containing various concentrations of NaCl (0.2 and 1 M) and MgCl2 (0, 2, 4 and 30 mM). The pattern of hybridization was similar in all buffers, which confirmed that buffers do not influence the structure of OxyS RNA. However, the most intense signals were observed in 4°C and buffers with the highest ionic strength.
OxyS RNA was more accessible for probe binding than DsrA RNA. Hybridization was observed for probes complementary to positions 5, 16-20, 23, 24, 26-29, 56-58, 60, 68-70, 73-77, 79, 83, 88, 89, 97, 98 and 100. Unfortunately, most probes (except binding at positions 68-70, 72-77 and 83) could bind to alternative (with mismatched duplex formation) binding sites (Figure 4A, Table S1). Despite this, the microarray mapping confirmed the presence of hairpins I and III (20) and the region postulated as single stranded was accessible for probes. To get insight into details of the secondary structure of OxyS RNA several additional hybridization experiments were performed. This included microarray mapping of the central region of OxyS RNA (OxyS43), hairpin III (OxyS SL3), as well as a complex of OxyS RNA and three modified oligonucleotides complementary to various region of OxyS RNA (o1, o2 and o3). Those experiments allowed discriminating between real and alternative binding sites of the probes.
Figure 4.

Isoenergetic microarray mapping results for (A) OxyS RNA. (B) OxyS 43 (C) OxyS SL3 (D) complex OxyS RNA/ o1 (E) complex OxyS RNA/ o2 (F) complex OxyS RNA/ o3. Red squares represent sites of binding to probes without alternative possibility of binding. Orange squares represent sites of binding to probes with possibility of binding to alternative binding sites. Red squares represent sites of binding to probes without alternative possibility of binding which have the same intensity relative to OxyS RNA alone. Orange squares represent sites of binding to probes with possibility of binding to alternative binding sites which have the same intensity relative to OxyS alone. Blue squares represent increased intensity or additional binding sites to probes relative to OxyS RNA without alternative possibility of binding. Light-blue squares represent increased intensity or additional binding sites to probes relative to OxyS RNA which have possibility of binding to alternative binding sites. Open squares represent sites of weak binding intensity for OxyS SL3. Yellow squares represent binding with decreased intensity in the presence of oligonucleotide. Grey squares represent lack of binding in the presence of oligonucleotide. Blue lines represent the region of paring with oligonucleotide (G) Isoenergetic microarray mapping results after elimination of alternative binding sites of probes and chemical mapping results for OxyS RNA. Chemical modification data are superimposed: kethoxal with triangles, CMCT with squares, and NMIA with circles.
The chemically synthesized OxyS43, corresponding to region 48-90 in OxyS RNA was hybridized to an isoenergetic microarray designed to cover the entire OxyS RNA in buffers I, II and VIII. These data showed that the isolated fragment is much more accessible to binding than OxyS RNA. In the absence of hairpins I and III almost the entire OxyS43 oligonucleotide binds to microarray, including the stem region of hairpin II (Figure 4B, Table 2). That experiment confirmed that the central region of OxyS RNA is relatively unstructured. Moreover, these results eliminate some alternative binding sites (Table S1). For example, position 5 is absent in OxyS43, so the probe complementary to this position must bind to an alternative one (e.g. position 79); and likewise for probes complementary to positions 18 and 19. Similarly the probe complementary to position 97 must bind to position 56/57 with mismatched duplex formation.
More informative, especially with the elimination of alternative binding sites, were the hybridization experiments in which three modified oligonucleotides: CMGLDMGMUMULGMDMGMDLDM (o1, complementary to region of 54-64 in OxyS), GMULUMGMGMCMUMULUMDLGMUMUMDMULUM (o2, complementary to region of 65-80 in OxyS) and GMDLDMDLCMUMUMGMCMUMULDMDMGLCM (o3, complementary to region of 81-91 in OxyS) were hybridized to OxyS RNA in buffers I and II. Modifications of oligonucleotides result in the increase of thermodynamic stabilities of formed hybrids (5, 31). The calculated free energies of hybrids were −21.6, −27.7 and −19.6 kcal/mol, respectively for o1, o2 and o3 (2). The hybridization was performed in buffer containing 1 M NaCl and 4 mM MgCl2 with 50 fold molar excess of oligonucleotide. Annealing of OxyS RNA was performed in presence or absence of oligonucleotide and the procedure of annealing does not influence the hybridization results. As part of a control procedure the hybridization of oligonucleotides o1-o3 to an isoenergetic microarray designed to cover the entire OxyS RNA was also performed (Table S3). In these control experiments, the binding of modified oligonucleotides o1-o3 to several probes was observed. The probes on microarray were in sufficient excess that even if oligonucleotides strongly bind to several of them (for example o3 to probes dedicated to position 77 or 79) they could still strongly bind to OxyS RNA (see Tables 2 and S3).
The hybridization of complexes OxyS RNA/o1, OxyS RNA/o2 and OxyS RNA/o3 demonstrated the binding of o1-o3 to complementary regions of OxyS RNA (Figure 4D-F, Table 2). In the case of OxyS RNA/o1, hairpin II was protected from binding. This hybridization allowed for the confirmation that probes complementary to positions 56-58 in OxyS RNA alone are not binding at alternative sites. Moreover, the probe complementary to site 98 was verified as a bona fide alternative binding site, probably to position 56/57. Unexpectedly, lack of binding was observed at position 83. Positions 23, 26-29, 60 and 75 were less accessible to probes, but 16 and 71 were more accessible. Also, the probe complementary to position 97 bound stronger. In complex OxyS RNA/o2 oligonucleotide o2 was complementary to region 65-80. A lack of binding was observed at positions base pairing with oligonucleotide o2. Again, the experiments allowed for verification of alternative binding of the probe dedicated to position 5. Additionally, decreased intensity of binding was observed for probes complementary to positions 23, 24, 26, 27 and 60, but increased for 16, 50, 97 and 98. In the case of OxyS RNA/o3, lack of binding at positions 82, 83 and 89 was observed as consequence of complementary oligonucleotide o3 binding, excluding alternative binding for probes dedicated for those positions. Additionally, a lack of binding was observed for positions 73 and 74, and less signal intensity for 20, 23, 27-29, 75, and 88. Additional binding was observed for probes complementary to positions 6, 38, 78 and 87. However, higher intensities were noticed for 60, 62, 63, 71, 76, 77, 79, 97 and 98. As previously stated, the comparison of hybridization of OxyS RNA and complex OxyS RNA/o3 allowed for the determination of probe binding as being a full match or mismatch (Table S1). The hybridization of complexes OxyS RNA and oligonucleotides o1-o3 demonstrated the enhancement or the diminishment of probe binding abilities not only in the region of complementarity but also at additional positions. Presumably, the binding of oligonucleotides o1-o3 to OxyS RNA results also in OxyS RNA secondary and tertiary structure rearrangement.
In the case of DsrA RNA the hybridization patterns of hairpin III (DsrA SL3) alone and as part of entire DsrA RNA were different. Also for OxyS RNA a similar comparison with hairpin III (OxyS SL3) was performed. During the hybridization of OxyS SL3 in buffers I and II the strong binding of probes complementary to positions 98-100 were observed which are similar to binding in entire OxyS RNA. This experiment is also consistent with previous observations for DsrA RNA; the isolated hairpin loop is more accessible for probes than the same loop in the entire molecule. Additionally, medium binding was observed for the probe complementary to position 107, but it is probably mismatch binding to position 99; the energy of this binding, however, is unfavourable (Figure 4C, Table 2).
Finally, to get additional structure information and relate it to microarray mapping, chemical mapping of OxyS RNA was performed. For the same structural reason as in the case of DsrA RNA LNA-modified DNA primer was used to primer extension procedure (35). The chemical modifications take place at positions: 8, 23, 24, 31, 33, 39, 49, 55, 57, 64-69, 71-75, 78, 79 and 81-84 (Figure 4G). The results of chemical and microarray mapping were consistent. They show the presence of hairpins I-III and a long unstructured region within the OxyS RNA. However, results achieved with oligonucleotides o1-o3 could suggest some intramolecular interactions within OxyS RNA.
OxyS RNA in complex with Hfq, and fhlA mRNA or rpoS mRNA
Hfq protein binds to OxyS RNA as well as to DsrA RNA (25). Interactions between these two molecules have also been used for testing the microarray mapping method. Gel shift experiments showed that Hfq can interact with OxyS RNA in the buffer used to perform microarray mapping. For the formation of a stable complex, the hybridization buffer VI and 0,6 μM Hfq (first complex) were selected. Hybridization experiments on the complex OxyS RNA/Hfq were performed similarly as for DsrA RNA. Diminished binding intensity was observed for probes complementary to the following positions: 89 (11 times), 26 (9 times), 20, 88, 98 (probe 98 presumably binds with mismatch to site 56/57) (7 times), 5, 16-19, 23, 27-29, 56-58, 60, 73-77, 79, 83, 97 and 100 (3-5 times) (Figure 5, Table 2). Hybridization results suggest that regions with the highest reduction of binding signals are major Hfq binding sites in OxyS RNA.
Figure 5.

Isoenergetic microarray mapping results for OxyS RNA in complex with Hfq. Left side: two microarrays - first with hybridized OxyS RNA, second with hybridized complex OxyS RNA/Hfq. Right side: bar graph of relative binding intensity to the probes. Hybridization results for OxyS RNA/Hfq marked on proposed structure of OxyS RNA. Red squares represent sites of binding to probes which have the same intensity relative to OxyS RNA alone. Yellow circles represent decreased binding in the presence of Hfq. Light-yellow circles represent decreased binding to probes in the presence of Hfq which have possibility of binding to alternative binding sites. Green circles represent positions with significantly decreased binding to probes in the presence of Hfq without the possibility of binding to alternative binding sites. Light-green circles represent positions with significantly decreased binding to probes in the presence of Hfq which have possibility of binding to alternative binding sites.
As a control, the binding ability of Hfq to several oligonucleotide probes (Table S5) was checked. Non-denaturing PAGE showed basically no Hfq/probe complex formation in 100 time molar excess of protein.
Complexes of OxyS RNA with other RNA and the three-component complexes with Hfq were also studied using microarray mapping. OxyS RNA regulates the activity of fhlA mRNA via interactions with two short separate regions: first at the RBS, and second within the coding region (21-23). Initially, to simplify the experiments, two model oligonucleotides fhlA9 and fhlA11 (5′UUCCUGGAA and 5′ACAAGGGUUGU, respectively) which mimic natural interactions, were tested in their ability to form complexes with OxyS RNA. Model oligonucleotides fhlA9 and fhlA11 were longer than complementary loops in OxyS RNA (Figure S1). During annealing in buffer I (1 M NaCl, 4 mM MgCl2) 50 fold molar excess of both oligonucleotides was used to form complexes with OxyS RNA. OxyS RNA alone was hybridized in the same conditions as a control experiment. The hybridization experiments of OxyS RNA/fhlA11 complex showed lack of probe binding at positions 26-29 and significant diminishing of binding at positions 23 and 24 in OxyS RNA. Changes of hybridizations were observed in the loop of hairpin I of OxyS RNA complementary to the fhlA11 oligonucleotide. OxyS RNA interactions with fhlA11 do not change the structure of OxyS RNA. The second model oligonucleotide fhlA9 was complementary to hairpin III of OxyS RNA. The hybridization of OxyS RNA/fhlA9 complex resulted in a lack of probe binding at position 100 of OxyS RNA (covered by complementary fhlA9), whereas other binding sites were intact (Table S4). It suggests no structure changes of OxyS RNA due to hybridization to fhlA9 and fhlA11. Additionally, experiments showed that a short oligoribonucleotide (where calculated free energy of the hybrid is about -12 kcal/mol) can be stably bound to RNA during hybridization to microarray.
Hybridization of the 115 and 282 nucleotide long fragments of fhlA mRNA (fhlA115, fragment between −54 and 60 nucleotide residues, fhlA282 between −221 to 60) containing both fragments involved in complex formation with OxyS RNA was also studied (22, 23). It was reported that fhlA115 and fhlA282 has different structures in regions involved in interactions (22, 23). Native gel electrophoresis of fhlA115 and OxyS RNA demonstrated that all of OxyS RNA is bound in complex when 84 times molar excess of fhlA115 was used, while for fhlA282 10 fold molar excess was sufficient. To observe the formation of both complexes it was necessary to use 10 mM MgCl2 and 0.2 or 1 M NaCl (buffer V or VI). Unfortunately, the microarray experiments performed at various conditions did not allow for the observation of differences in the hybridization pattern of complex OxyS RNA/fhlA115 and also OxyS RNA/fhlA282 relative to OxyS RNA and fhlA RNAs alone. It was postulated that Hfq increases interactions between OxyS and its target mRNAs (23, 25). Regrettably, no difference in hybridization pattern was obtained for three-components complex of fhlA282/Hfq/OxyS RNA comparing to OxyS RNA. Presumably, the interactions of those complexes are still thermodynamically too weak to be investigated by microarray method.
OxyS RNA also regulates the biological activity of rpoS mRNA (21, 24). Mechanism of this regulatory function is not clear. One of the hypotheses suggests that formation of three-components complex OxyS RNA/Hfq/rpoS mRNA is necessary for translation inhibition. The presence of Hfq is required for the repression of rpoS mRNA. It was shown that hairpin III and the single stranded region between hairpins II and III of OxyS RNA are necessary for this repression. However, the partners of those interactions which could be Hfq or/and rpoS mRNA remain unclear. To get insight into those interactions and check if direct interaction between OxyS RNA and rpoS mRNA is possible, various length fragments of rpoS mRNA leader, containing 25, 140, 325 and 578 nucleotides (marked as rpoS25, rpoS140, rpoS325 and rpoS578, respectively) were investigated (where rpoS578 is the full-length rpoS leader, which was shortened from the 5′ end to give rpoS325 and rpoS140, whereas rpoS25 is a fragment of the leader complementary to DsrA RNA). The ability of OxyS RNA to form complexes with various fragments of rpoS mRNA showed Mg+2 concentration dependence; best results were obtained in buffers containing 4 or 10 mM MgCl2 and 0.2 M NaCl (buffer II and VI). Lengths of model rpoS influenced the binding. Also, increase of molar excess of model rpoS, and magnesium concentration facilitate complex formation. The most effective interactions were found for 20 fold excess of rpoS325 in 10 mM MgCl2 and 0.2 M NaCl (Figure S2). These conditions were used for hybridization on microarrays of complex rpoS325/OxyS RNA. The hybridization pattern for complex rpoS325/OxyS RNA was similar as OxyS RNA alone. This suggests that OxyS RNA/rpoS325 complex is too weak to be seen using microarray mapping. It was reported that the presence of Hfq stabilizes OxyS RNA/rpoS mRNA complex (25). The three-component complex OxyS RNA/Hfq/rpoS325 was formed and hybridized to isoenergetic microarrays. Also in this case, there was no difference in accessibility for probes comparing to OxyS RNA, suggesting that the complex was not strong enough to be seen by tested method.
DISCUSSION
Knowledge about the secondary and tertiary structure as well as the character of interactions with other biomolecules is important for understanding the biological function of RNAs (44). That information is also essential in the use of RNAs as tools and targets for therapeutic treatment (45). Several methods are available to study the secondary structure of RNAs (46-49). The most precise are NMR and X-ray crystallography, which give atomic resolution detail, but they are time consuming and have limitations (50, 51).
Enzymatic mapping using ribonucleases (46-48), and chemical mapping using reagents such as DMS, CMCT or kethoxal are widely applied to facilitate prediction of RNA secondary structure (46, 48). Another useful approach is the SHAPE method developed by Weeks which is based on using single reagent capable of distinguishing the chemical reactivity of the 2′-hydroxyl in single and double stranded regions of RNA (49). Recently, the method was used also to investigate RNA/protein complexes (52). The microarray mapping method, based on penta/hexanucleotide isoenergetic probes, was used successfully to improve prediction of secondary structure of several native RNAs (2, 7).
The secondary structure is more readily accessible, and several computational methods have been developed for making structural predictions (38, 53, 54). Computational predictions may be improved when experimental constraints are used to limit the folding of RNA. For example, the computer program RNAstructure allows one to predict the lowest free energy structure of RNA consistent with experimental data. Constraints can be used from the results of enzymatic and chemical mappings (including SHAPE) as well as microarray mapping (2, 7, 53, 55).
Knowledge about interactions with cellular components such as others RNAs and proteins brings better understanding of the role of RNAs within functioning cells. The study of RNA structures complexed with other molecules is more complicated than RNA secondary structure alone due to the large size of complexes and number of interactions within them. In this work microarray mapping was used to investigate the structure and interactions of two regulatory RNAs, DsrA RNA and OxyS RNA, in complexes with Hfq protein and various length fragments of their target mRNAs.
DsrA RNA and its complexes. Isoenergetic microarray mapping shows different RNA accessibility in complexes
Microarray mapping of DsrA RNA generally confirmed the secondary structure reported in the literature (40, 41). DsrA RNA hairpins I and III are undoubtedly present in the natively folded molecule. Experiments confirmed that isoenergetic probes bind only to single stranded fragments of the loops and not to double stranded fragments of the stems. The same holds true for isolated model structural motifs of RNA (10). The structure of the region in the middle of the molecule is controversial. In the literature there are different proposed folds for this region (39-41). The calculations of the free energies of hairpins I-III (ΔG°37 equal to −7.0, −4.5 and −18.6 kcal/mol, respectively) show that the hairpin II structure is the least stable of the three; the structure of DsrA RNA proposed here (including hairpin II; Figure 1, structure A4), however, is the most thermodynamically stable from all proposed previously based on experiments data (40, 41). Moreover, postulated here is the presence of tandem 5′UU/3′UU mismatch in hairpin II which could stabilize hairpin stem as shown by Wu et al (56). The different folding of DsrA RNA proposed by various authors could be consequences of different methods of analysis. It is also possible that the central fragment is thermodynamically flexible and can form several conformers, not detectable by electrophoresis that may switch depending on experimental conditions.
The interactions with specific enzymes (during the enzymatic mapping) or long oligonucleotides (during RNase H and DNazyme cleavage) could result in structural rearrangements of DsrA RNA during analysis. This may also occur during microarray mapping; if a rearrangement occurs after binding of microarray probe, however, it will not influence the results for other probes, because the hybridization duplex (and rearranged RNA) is already fixed on the microarray surface. Also, the hybridization approach allows performing probing at a wider range of buffer, salt, and temperature conditions.
The proposed secondary structure of DsrA RNA was also confirmed by microarray mapping of DsrA38 and by hybridization of DsrA RNA in complex with oligonucleotide d1. An unusual feature of the proposed structure is the limited number of binding sites in the region between nucleotides 23 and 38. One explanation may be that there are additional factors at work, such as tertiary interactions, which may not be explained simply by the base pairing pattern within this region. This hypothesis is supported by experiments with DsrA38 and with complex DsrA/d1.
The results of DsrA RNA chemical modifications also support the structure proposed in this work (Figure 1 A4), but are also consistent with the structure proposed by Lease and Belfort (structure A2) (40) as well as Rolle (structure A3) (41). The chemical mapping data does not allow for the unambiguous selection of the correct DsrA RNA structure. The structures proposed in Figure 1 (structures A2-A4, ΔG°37 in range between −25.8 to −31.4 kcal/mol) fit well both microarray and chemical mapping results. Based on the results discussed above, DsrA RNA structure marked as A4 seems to be the most likely. The thermodynamic stability of this structure calculate by RNAstructure is −31.4 kcal/mol and it is one of the suboptimal structure of DsrA RNA.
It is postulated that the middle region of DsrA RNA is involved in interactions with Hfq as part of its regulatory activity (18, 30). Native gel electrophoresis showed the formation of two types of complexes of DsrA RNA and Hfq: the first is formed by DsrA RNA and one Hfq hexamer, whereas the second is presumably with two Hfq hexamers. The first type of complex was used for hybridization to microarrays. It is proposed that Hfq does not bind with high sequence specificity which is consistent with the lack of sequence homology between the known Hfq substrates (25). Probably, for Hfq binding, RNA secondary and higher-ordered structure is more important than sequence.
The microarray method gave results consistent with previous observations, suggesting that there is no structure rearrangement after binding Hfq and that the middle region of DsrA RNA is the main Hfq binding site (Figure 1, Table 1)(18, 30). The microarray mapping indicated 3-7 fold diminishment of probe binding mainly in the middle region and loop of hairpin I of DsrA RNA and achieved results were consistent with previously published data. Lease and Woodson observed lower ability of cleavage of DsrA RNA at 14 and 15 nucleotide residues by RNase T2 in complex with Hfq (18). Position 31 is postulated by various authors as a Hfq binding site (18, 30). Brescia reported that presence of Hfq enhances cleave of region 42-45 by V1 ribonuclease which in opinion of the authors could mean enhancement of helical character of this DsrA RNA region in complex with Hfq or this helical region could be sterically protected in DsrA/Hfq complex (30). It is consistent with reported here microarrays mapping results suggesting very unstable character of internal region of naked DsrA RNA, whereas complex with Hfq stabilize this fragment and make it unavailable for probes binding. Additionally, Rolle observed that presence of Hfq protected region 43-52 of DsrA RNA from cleavage by lead ions (41).
As an additional control experiment, the binding of short model oligonucleotides to Hfq were tested, which eliminated the possibility that inhibition of probes binding is a consequence of binding Hfq released from complex DsrA RNA/Hfq.
In the cell DsrA RNA is involved in regulation the translation of many mRNAs, including rpoS mRNA. It was postulated that for this mRNA the regulation is achieved by interactions of DsrA RNA (region 10-32) and the 5′UTR of rpoS mRNA (region −97 and −119). As a consequence of this binding there is rearrangement of the secondary structure in the rpoS mRNA so the translation can start (18, 36). As the model of rpoS mRNA, oligonucleotides 25- (rpoS25) and 140-nucleotides long (rpoS140) were used. The hybridization of DsrA RNA/rpoS25 and DsrA RNA/rpoS140 to the isoenergetic microarray showed lack of binding to probes in the region of DsrA RNA, which base paired with rpoS25 and rpoS140 and new binding sites in the region which becomes single stranded after complexation (Figure 3,Table 1). The hybridization of DsrA RNA/rpoS140 (rpoS140 better mimic rpoS mRNA) demonstrated that base pairing include nucleotides between 10 and 41 within DsrA. Also Lease and Woodson observed resistance to cleavage by nucleases T1 and T2 not only within the region proposed as a region of interaction (region 10-32) but also in addition positions (18). Combination of these results may suggest that region of complementarities is longer than proposed earlier.
Finally, the three-component complex DsrA RNA/Hfq/rpoS140 was also studied by microarray mapping. Intensities of probes binding to DsrA RNA/Hfq/rpoS140 were significantly diminished (by 2-7 folds relative to DsrA RNA/rpoS140) or some probe binding was inhibited completely (Figure 3,Table 1). Microarray mapping confirmed that the presence of Hfq additionally stabilizes base pairing of DsrA RNA/rpoS140 complex and that Hfq remains bound to DsrA RNA during interactions with rpoS140. Stabilization of interactions between DsrA RNA and rpoS140 by Hfq is consistent with data reported by Lease and Woodson (18). Hybridization data reveals that in the three-molecular complex, the main Hfq binding site is probably displaced to another site on DsrA RNA (region 41-47). Lease and Woodson also consider that possibility. Their finding indicates that in the three-molecular complex Hfq interact with rpoS outside of the region of complementary to DsrA RNA and simultaneously it is displaced from high-affinity binding site on DsrA RNA (18).
The experiments described above show that the microarray mapping method can be applied as a tool to study not only the structure of RNA but also the accessibility of RNA in complex with other molecules such as RNA or protein.
OxyS RNA and its complexes. Formation of stable complex between molecules is necessary for using microarray mapping
Regulatory functions of OxyS RNA are less understood than DsrA RNA, however, it is postulated that it participates in the regulation of over 40 genes (20). Previously, it was shown that OxyS RNA represses the translation of fhlA mRNA and rpoS mRNA, but (especially for rpoS mRNA) the mechanism of action is not clear (21, 24). In the literature, a secondary structure of OxyS RNA was published based only on chemical mapping with DMS (20). Microarray mapping of OxyS RNA in various conditions has shown that its secondary structure is consistent with the one proposed earlier (20). OxyS RNA contains three hairpins, labeled I-III, and a 27 nucleotide long single stranded region between hairpins II and III. Calculated free energies (ΔG°37) of the hairpins I-III were −14.4, −1.5 and −11.5 kcal/mol, respectively. Hairpin I contains internal loops and a bulge, hairpin II contains only a short and thermodynamically unstable stem, whereas terminal hairpin III has a very stable GC rich stem. The relatively unstructured region between hairpins I and III was confirmed by microarray mapping of oligonucleotide OxyS43. Moreover, microarray mapping of model hairpin III (OxyS SL3) as well as OxyS RNA base paired to oligonucleotides o1-o3, and finally chemical mapping (NMIA, CMCT, kethoxal), confirmed the proposed folding of OxyS RNA, but suggested that within this molecule exists significant higher-ordered structural interactions (Figure 4,Table 2).
The regulatory function of OxyS RNA also involves Hfq protein (24, 25). Hybridization of OxyS RNA and Hfq reveals the formation of two types of complexes. This complexation is dependent on the concentration of Hfq. Microarray mapping was used on the first type of OxyS RNA/Hfq complex. The hybridization of OxyS RNA/Hfq complex to isoenergetic microarrays results in 3-11 fold diminishment of binding of most of oligonucleotide probes. The most intense reduction of probe binding was observed at the loop regions of hairpins I (positions 20, 26) and II (position 98 (56/57)) as well as in the single stranded region near hairpin III (positions 80, 89) (Figure 5,Table 2). Based on hybridization results it could be postulated that the single stranded region between hairpin II and III is a major Hfq binding site; however, other regions contribute to the Hfq binding as well, or Hfq sterically prevents probes for binding to those positions. These results are consistent with the observation of Zhang et al. where for binding of Hfq to OxyS RNA, beside single stranded region, hairpins I or III are crucial (25).
Among the many regulatory functions of OxyS RNA, one is to regulate the biological activity of the fhlA mRNA. It was proposed that the regulatory interactions involve the hairpin loops I and III, which based paired with complementary fragments of fhlA mRNA (22). Model oligonucleotides fhlA11 and fhlA9, which mimic complementary regions of fhlA mRNA, were used to study these interactions by isoenergetic microarrays. Microarray mapping has shown these complexes are stable during hybridization. Lack or very significant reduction of probe binding was observed at hairpin loops I and III, which base pair to fhlA11 and fhlA9 (Figure S1,Table S4). The other binding sites remain the same as in OxyS RNA alone. Next, there were used 115 (fhlA115) and 282 (fhlA282) nucleotide long model fragments of mRNA. Also the three-component complex OxyS/fhlA282/Hfq was tested (Hfq enhanced OxyS RNA binding to their target mRNA) (23, 25). Unfortunately, the hybridization of the OxyS RNA/fhlA115, OxyS RNA/fhlA282 and OxyS/fhlA282/Hfq complexes, did not demonstrate significant differences in hybridization pattern relative to control. Perhaps, the complexes could be too thermodynamically unstable during the hybridization. This shows the limitation of such an approach, but also illustrates the labile nature of studied complexes.
OxyS RNA also regulates the expression of rpoS mRNA and Hfq is involved in this process as well (21, 24). The details of this regulatory process are not known definitively. The formation of OxyS RNA/rpoS mRNA complexes was probed using 25 (rpoS25), 140 (rpoS140), 325 (rpoS325) and 578 (rpoS578) nucleotide long model fragments of rpoS mRNA. The results indicate that OxyS RNA has the ability to interact with rpoS mRNA, which was shown for the first time. The ability of rpoS mRNA model oligomers to form the complexes with OxyS RNA increases with their length and magnesium concentration. The most effective interactions occurred for rpoS325 (Figure S2). However the microarray hybridization pattern of the OxyS RNA/rpoS325 and OxyS RNA/Hfq/rpoS325 complexes were very similar to OxyS RNA or rpoS325 alone. Results may suggest that the nature of the interaction between OxyS RNA and their both target mRNAs is thermodynamically weak and cannot be analyzed by microarray mapping.
Isoenergetic microarrays as an alternative method to study two- and three-component complexes
The experiments which have been described here show that microarray mapping can be applied as a tool to study the structure of RNA in complex with another molecule, such as RNA or protein. Microarray mapping provides data about the ability of RNA to interact with short oligonucleotides, which is very important for using oligonucleotides as modulators of RNA biological function. This method can also give a significant amount of information about the nature of interactions between molecules within complexes. Microarray mapping of RNA complexes allows for studies to be performed under a broader spectrum of conditions, without satisfying the requirements necessary for enzymatic or chemical reactivity. In consequence, microarray mapping should not change the native structure of RNA or its complexes with other biomolecules. Microarray mapping is fast, easy and reproducible. The only limitation of the method is having sufficient thermodynamic stability between interacting biomolecules and/or structural elements of each target molecule.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Andrew L. Feig (Wayne State University) for plasmid encoding Hfq protein and Walter Moss (University of Rochester) for comments and suggestions on this manuscript.
Footnotes
This work was supported by National Institute of Health grant 1R03TW008739-01 (E.K. and D.H. Turner) and National Science Center grant N N301 788 440 (E.K.). R.K. is the recipient of a Foundation of Polish Science fellowship.
SUPPORTING INFORMATION Supporting information contains tables (with additional hybridization results and a list of sequences for the oligonucleotides used) and figures (with microarray mapping results for OxyS RNA in complex with fhlA11 and fhlA9 and gel of complexes between OxyS RNA and rpoS fragments) is available at http://pubs.acs.org.
REFERENCES
- (1).Kierzek E, Kierzek R, Turner DH, Catrina IE. Facilitating RNA structure prediction with microarrays. Biochemistry. 2006;45:581–593. doi: 10.1021/bi051409+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kierzek E, Kierzek R, Moss WN, Christensen SM, Eickbush TH, Turner DH. Isoenergetic penta- and hexanucleotide microarray probing and chemical mapping provide a secondary structure model for an RNA element orchestrating R2 retrotransposon protein function. Nucleic Acids Res. 2008;36:1770–1782. doi: 10.1093/nar/gkm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kierzek E, Mathews DH, Ciesielska A, Turner DH, Kierzek R. Nearest neighbor parameters for Watson-Crick complementary heteroduplexes formed between 2′-O-methyl RNA and RNA oligonucleotides. Nucleic Acids Res. 2006;34:3609–3614. doi: 10.1093/nar/gkl232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Pasternak A, Kierzek E, Pasternak K, Turner DH, Kierzek R. A chemical synthesis of LNA-2,6-diaminopurine riboside, and the influence of 2′-O-methyl-2,6-diaminopurine and LNA-2,6-diaminopurine ribosides on the thermodynamic properties of 2′-O-methyl RNA/RNA heteroduplexes. Nucleic Acids Res. 2007;35:4055–4063. doi: 10.1093/nar/gkm421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kierzek E, Ciesielska A, Pasternak K, Mathews DH, Turner DH, Kierzek R. The influence of locked nucleic acid residues on the thermodynamic properties of 2′-O-methyl RNA/RNA heteroduplexes. Nucleic Acids Res. 2005;33:5082–5093. doi: 10.1093/nar/gki789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Pasternak A, Kierzek E, Pasternak K, Fratczak A, Turner DH, Kierzek R. The thermodynamics of 3′-terminal pyrene and guanosine for the design of isoenergetic 2′-O-methyl-RNA-LNA chimeric oligonucleotide probes of RNA structure. Biochemistry. 2008;47:1249–1258. doi: 10.1021/bi701758z. [DOI] [PubMed] [Google Scholar]
- (7).Kierzek E, Christensen SM, Eickbush TH, Kierzek R, Turner DH, Moss WN. Secondary structures for 5′ regions of R2 retrotransposon RNAs reveal a novel conserved pseudoknot and regions that evolve under different constraints. J. Mol. Biol. 2009;390:428–442. doi: 10.1016/j.jmb.2009.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kierzek E, Barciszewska MZ, Barciszewski J. Isoenergetic microarray mapping reveals differences in structure between tRNAiMet and tRNAmMet from Lupinus luteus. Nucleic Acids Symp Ser (Oxf) 2008:215–6. doi: 10.1093/nass/nrn109. [DOI] [PubMed] [Google Scholar]
- (9).Jenek M, Kierzek E. Isoenergetic microarray mapping-the advantages of this method in studying the structure of Saccharomyces cerevisiae tRNAPhe. Nucleic Acids Symp Ser (Oxf) 2008:219–20. doi: 10.1093/nass/nrn111. [DOI] [PubMed] [Google Scholar]
- (10).Kierzek E. Binding of short oligonucleotides to RNA: studies of the binding of common RNA structural motifs to isoenergetic microarrays. Biochemistry. 2009;48:11344–11356. doi: 10.1021/bi901264v. [DOI] [PubMed] [Google Scholar]
- (11).Livny J, Waldor MK. Identification of small RNAs in diverse bacterial species. Current Opinion in Microbiology. 2007;10:96–101. doi: 10.1016/j.mib.2007.03.005. [DOI] [PubMed] [Google Scholar]
- (12).Vogel J, Sharma CM. How to find small non-coding RNAs in bacteria. Biological Chemistry. 2005;386:1219–1238. doi: 10.1515/BC.2005.140. [DOI] [PubMed] [Google Scholar]
- (13).Storz G, Gottesman S. RNA World. 3rd Ed Cold Spring Harbor Labolatory Press; New York: 2006. Versatle roles of small RNA regulators in bacteria; pp. 567–594. [Google Scholar]
- (14).Gottesman S. The small RNA regulators of Escherichia coli: Roles and mechanisms. Annual Review of Microbiology. 2004;58:303–328. doi: 10.1146/annurev.micro.58.030603.123841. [DOI] [PubMed] [Google Scholar]
- (15).Storz G. An expanding universe of noncoding RNAs. Science. 2002;296:1260–1263. doi: 10.1126/science.1072249. [DOI] [PubMed] [Google Scholar]
- (16).Waters LS, Storz G. Regulatory RNAs in Bacteria. Cell. 2009;136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sledjeski DD, Gupta A, Gottesman S. The small RNA, DsrA, is essential for the low temperature expression of RpoS during exponential growth in Escherichia coli. EMBO J. 1996;15:3993–4000. [PMC free article] [PubMed] [Google Scholar]
- (18).Lease RA, Woodson SA. Cycling of the Sm-like protein Hfq on the DsrA small regulatory RNA. J. Mol. Biol. 2004;344:1211–1223. doi: 10.1016/j.jmb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- (19).Soper TJ, Woodson SA. The rpoS mRNA leader recruits Hfq to facilitate annealing with DsrA sRNA. RNA-Publ. RNA Soc. 2008;14:1907–1917. doi: 10.1261/rna.1110608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Altuvia S, WeinsteinFischer D, Zhang AX, Postow L, Storz G. A small, stable RNA induced by oxidative stress: Role as a pleiotropic regulator and antimutator. Cell. 1997;90:43–53. doi: 10.1016/s0092-8674(00)80312-8. [DOI] [PubMed] [Google Scholar]
- (21).Altuvia S, Zhang AX, Argaman L, Tiwari A, Storz G. The Escherichia coli OxyS regulatory RNA represses fhlA translation by blocking ribosome binding. EMBO J. 1998;17:6069–6075. doi: 10.1093/emboj/17.20.6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Argaman L, Altuvia S. fhlA repression by OxyS RNA: Kissing complex formation at two sites results in a stable antisense-target RNA complex. J. Mol. Biol. 2000;300:1101–1112. doi: 10.1006/jmbi.2000.3942. [DOI] [PubMed] [Google Scholar]
- (23).Salim NN, Feig AL. An upstream Hfq binding site in the fhlA mRNA leader region facilitates the OxyS-fhlA Interaction. Plos One. 2010;5:e13028. doi: 10.1371/journal.pone.0013028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhang AX, Altuvia S, Tiwari A, Argaman L, Hengge-Aronis R, Storz G. The OxyS regulatory RNA represses rpoS translation and binds the Hfq (HF-I) protein. EMBO J. 1998;17:6061–6068. doi: 10.1093/emboj/17.20.6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zhang AX, Wassarman KM, Ortega J, Steven AC, Storz G. The Sm-like Hfq protein increases OxyS RNA interaction with target mRNAs. Mol. Cell. 2002;9:11–22. doi: 10.1016/s1097-2765(01)00437-3. [DOI] [PubMed] [Google Scholar]
- (26).Soper T, Mandin P, Majdalani N, Gottesman S, Woodson SA. Positive regulation by small RNAs and the role of Hfq. Proc. Natl. Acad. Sci. U. S. A. 2010;107:9602–9607. doi: 10.1073/pnas.1004435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).de Fernandez M. T. Franze, Hayward WS, August JT. Bacterial proteins required for replication of phage Q ribonucleic acid. Pruification and properties of host factor I, a ribonucleic acid-binding protein. J Biol Chem. 1972;247:824–31. [PubMed] [Google Scholar]
- (28).Moller T, Franch T, Hojrup P, Keene DR, Bachinger HP, Brennan RG, Valentin-Hansen P. Hfq: a bacterial Sm-like protein that mediates RNA-RNA interaction. Mol. Cell. 2002;9:23–30. doi: 10.1016/s1097-2765(01)00436-1. [DOI] [PubMed] [Google Scholar]
- (29).Geissmann TA, Touati D. Hfq, a new chaperoning role: binding to messenger RNA determines access for small RNA regulator. EMBO J. 2004;23:396–405. doi: 10.1038/sj.emboj.7600058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Brescia CC, Mikulecky PJ, Feig AL, Sledjeski DD. Identification of the Hfq-binding site on DsrA RNA: Hfq binds without altering DsrA secondary structure. RNA-Publ. RNA Soc. 2003;9:33–43. doi: 10.1261/rna.2570803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Pasternak A, Kierzek E, Pasternak K, Turner DH, Kierzek R. The chemical synthesis of 2′-O-methyl-2,6-diaminopurine riboside and LNA-2,6-diaminopurine riboside and their influence on the thermodynamic properties of 2′-O-methyl RNA/RNA heteroduplexes. Nucleic Acids Res. 2007;35:4055–4063. doi: 10.1093/nar/gkm421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).McBride LJ, Caruthers MH. An investigation of several deoxyribonucleoside phosphoramidites useful for synthesizing deoxyoligonucleotides. Tetrahedron Lett. 1983;24:245–248. [Google Scholar]
- (33).Borer PN. Optical properties of nucleic acids, absorption and circular dichroism spectra. In: Fasman GD, editor. CRC Handbook of Biochemistry and Molecular Biology: Nucleic Acids. 3rd edn CRC Press; Cleveland, OH: 1975. pp. 589–595. [Google Scholar]
- (34).Richards EG. Use of tables in calculations of absorption, optical rotatory dispersion and circular dichroism of polyribonucleotides. In: Fasman GD, editor. CRC Handbook of Biochemistry and Molecular Biology: Nucleic Acids. 3rd edn CRC Press; Cleveland, OH: 1975. pp. 596–603. [Google Scholar]
- (35).Fratczak A, Kierzek R, Kierzek E. LNA-modified primers drastically improve hybridization to target RNA and reverse transcription. Biochemistry. 2009;48:514–516. doi: 10.1021/bi8021069. [DOI] [PubMed] [Google Scholar]
- (36).Mikulecky PJ, Kaw MK, Brescia CC, Takach JC, Sledjeski DD, Feig AL. Escherichia coli Hfq has distinct interaction surfaces for DsrA, rpoS and poly(A) RNAs. Nature Structural & Molecular Biology. 2004;11:1206–1214. doi: 10.1038/nsmb858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Afanassiev V, Hanemann V, Wölfl S. Preparation of DNA and protein micro arrays on glass slides coated with an agarose film. Nucleic Acids Res. 2000;28:e66. doi: 10.1093/nar/28.12.e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- (39).Sledjeski DD, Gotessman S. A small RNA acts as an antisilencer of the H-NS- silencer rcsA gene of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 1995;92:2003–2007. doi: 10.1073/pnas.92.6.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Lease RA, Belfort M. A trans-acting RNA as a control switch in Escherichia coli: DsrA modulates function by forming alternative structures. Proc. Natl. Acad. Sci. U. S. A. 2000;97:9919–9924. doi: 10.1073/pnas.170281497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Rolle K, Zywicki M, Wyszko E, Barciszewska MZ, Barciszewski J. Evaluation of the dynamic structure of DsrA RNA from E. coli and its functional consequences. J. Biochem. 2006;139:431–438. doi: 10.1093/jb/mvj045. [DOI] [PubMed] [Google Scholar]
- (42).Sun XG, Wartell RM. Escherichia coli Hfq binds A(18) and DsrA domain II with similar 2:1 Hfq(6)/RNA stoichiometry using different surface sites. Biochemistry. 2006;45:4875–4887. doi: 10.1021/bi0523613. [DOI] [PubMed] [Google Scholar]
- (43).Sledjeski DD, Whitman C, Zhang AX. Hfq is necessary for regulation by the untranslated RNA DsrA. Journal of Bacteriology. 2001;183:1997–2005. doi: 10.1128/JB.183.6.1997-2005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Simons W, Grunberg-Mango M. RNA structure and function. Cold Spring Harbor Labolatory Press; 1998. [Google Scholar]
- (45).Kurreck J. Antisense technologies - Improvement through novel chemical modifications. Eur. J. Biochem. 2003;270:1628–1644. doi: 10.1046/j.1432-1033.2003.03555.x. [DOI] [PubMed] [Google Scholar]
- (46).Ehresmann C, Baudin F, Mougel M, Romby P, Ebel JP, Ehresmann B. Probing the structure of RNAs in solution. Nucleic Acids Res. 1987;15:9109–9128. doi: 10.1093/nar/15.22.9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Knapp G. Enzymatic approaches to probing of RNA secondary and tertiary structure. Method Enzymol. 1989;180:192–212. doi: 10.1016/0076-6879(89)80102-8. [DOI] [PubMed] [Google Scholar]
- (48).Ziehler WA, Engelke DR. Probing RNA structure with chemical reagents and enzymes. Current Protocols in Nucleic Acid Chemistry. 2000;2:6.1.1–6.1.21. doi: 10.1002/0471142700.nc0601s00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Wilkinson KA, Merino EJ, Weeks KM. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 2006;1:1610–1616. doi: 10.1038/nprot.2006.249. [DOI] [PubMed] [Google Scholar]
- (50).Felden B. RNA structure: experimental analysis. Current Opinion in Microbiology. 2007;10:286–291. doi: 10.1016/j.mib.2007.05.001. [DOI] [PubMed] [Google Scholar]
- (51).Selmer M, Dunham CM, Murphy FV, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- (52).McGinnis JL, Duncan CDS, Weeks KM. High-throughput SHAPE and hydroxyl radical analysis of RNA structure and ribonucleoprotein assembly. Methods in Enzymology. 2009. pp. 67–89. [DOI] [PMC free article] [PubMed]
- (53).Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. U. S. A. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Zuker M. On finding all suboptimal foldings of an RNA molecule. Science. 1989;244:48–52. doi: 10.1126/science.2468181. [DOI] [PubMed] [Google Scholar]
- (55).Deigan KE, Li TW, Mathews DH, Weeks KM. Accurate SHAPE-directed RNA structure determination. Proc. Natl. Acad. Sci. U. S. A. 2009;106:97–102. doi: 10.1073/pnas.0806929106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Wu M, McDowell JA, Turner DH. A periodic-table of symmetrical tandem mismatches in RNA. Biochemistry. 1995;34:3204–3211. doi: 10.1021/bi00010a009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.