

Abstract
Transforming growth factor (TGF)-β family members are multifunctional cytokines regulating diverse cellular functions such as growth, adhesion, migration, apoptosis, and differentiation. TGF-βs elicit their effects via specific type I and type II serine/threonine kinase receptors and intracellular Smad transcription factors. Knockout mouse models for the different components of the TGF-β signaling pathway have revealed their critical roles in smooth muscle cell (SMC) differentiation. Genetic studies in humans have linked mutations in these signaling components to specific cardiovascular disorders such as aorta aneurysm and congenital heart diseases due to SMC defects. In this review, the current understanding of TGF-β function in SMC differentiation is highlighted, and the role of TGF-β signaling in SMC-related diseases is discussed.
Keywords: Transforming growth factor β, Smad, Smooth muscle cell, Differentiation, Cardiovascular diseases
TGF-β SIGNALING TRANSDUCTION
Transforming growth factor-β (TGF-β) is the founding member of the TGF-β superfamily that comprise TGF-βs, activins, bone morphogenetic proteins (BMPs), and growth and differentiation factors (GDFs)[1]. Three TGF-β isoforms (TGF-β1, TGF-β2, and TGF-β3) have been identified in mammals. In most cases, these isoforms exhibit similar functional properties and regulate various cellular activities including cell growth, differentiation, apoptosis and extracellular matrix synthesis in endothelial cells and vascular smooth muscle cells (SMCs)[2-8].
TGF-β ligands are synthesized as latent precursor molecules (LTGF-β), which are activated via proteolytic cleavage by endoproteases such as furin[9]. Active TGF-β signaling is transmitted through two types of transmembrane serine/threonine protein kinase receptors: TGF-β type I (TβRI) and type II (TβRII)[1,10-12]. TGF-β first binds to TβRII with the assistance of the membrane-anchored proteoglycan betaglycan TGF-β receptor III (TβRIII)[13], which leads to heterotetrameric complex formation with TβRI, resulting in TβRI phosphorylation[14,15]. TβRI (also known as activin receptor-like kinase 5; ALK5) transduces TGF-β signaling in most cell types although the signaling can also be mediated by ALK1 or other type I receptors in certain cell types[16,17]. Activated TβRI propagates signaling by recruiting and phosphorylating receptor-regulated Smad (R-Smad) proteins. ALK5 phosphorylates Smad2 and Smad3, while ALK1 phosphorylates Smad1, Smad5, and Smad8. Activated Smads form a complex with the common Smad (Smad4) and then are translocated into the nucleus, where they regulate target gene expression by binding to regulatory promoter DNA alone or interacting with other transcription factors[18,19].
Smad3 homomer can form DNA-binding complexes through its MH1 domain independent of Smad4. But Smad2 cannot bind to DNA without Smad4 because of the lack of the additional 30 amino acids present in Smad3 MH1 domain. Smad4 and phosphorylated Smad3 bind multiple 5’-AGAC-3’ sequences called Smad binding elements (SBEs) and GC-rich sequences[20]. Smad2 and Smad3 interact with a number of common and distinct transcription factors for SBE selectivity and specific gene transcription[21]. In most cases, Smad-binding transcription factors can function independent of Smads in controlling a specific gene transcription. However, Smad interacts with these transcription factors to modulate their transcriptional activity by recruiting co-activators or co-repressors[20,22,23]. For example, Smads recruit transcription coactivator p300/CBP, which has histone acetyltransferase activity, to facilitate the initiation of transcription[20]. In addition to p300/CBP, various other transcription factors such as Forkhead, homeobox, zinc-finger, AP1, Ets, and basic helix-loop-helix families have also been shown to act in concert with Smad proteins[24,25]. The diversity of Smad/co-factor combinations enables the regulation of the transcription of a vast amount of target genes. The differential expression of these factors in different cells are thought to contribute, at least in part, to the cell type-specific responses observed upon TGF-β stimulation[19].
TGF-β/Smad signaling pathway is regulated in multiple steps by different factors. SARA (SMAD anchor for receptor activation) presents R-Smads to the activated receptor complexes[26], while TMEPAI (transmembrane prostate androgen-induced protein) sequesters R-Smad proteins from active participation in TGF-β signaling[27]. Inhibitory Smad (I-Smad), Smad6 or Smad7, inhibits R-Smad binding to TGF-β receptor[28-30]. Smad Phosphorylation is reversed by phosphatases such as PPM1A and PDP in order to create a rapid activation-deactivation cycle[31-33]. Moreover, activated Smad proteins may be ubiquitinized by E3 ligases for proteasomal degradation[34,35]. In addition, transcriptional repressors Ski and SnoN also regulate TGF-β signaling by interacting with Smad proteins[36,37].
In addition to the canonical Smad signaling pathway that directly regulates the transcription of Smad-dependent target genes, TGF-β function can also be mediated by Smad-independent pathways including MAPK signaling pathways, such as p38 MAPK and c-Jun NH2-terminal kinase, phosphatidylinositol 3-kinase/Akt pathway, and Wnt signaling[38].
TGF-β SIGNALING IN SMOOTH MUSCLE DIFFERENTIATION DURING EMBRYONIC DEVELOPMENT
SMC differentiation is an integral part of embryonic vascular development. Vascular development in the embryo starts with the formation of a primitive vascular network from endothelial precursors through a process known as vasculogenesis. This primary vessel network undergoes angiogenesis to grow into a complex vascular system through branching and remodeling[39]. Recruitment and differentiation of SMC progenitor cells are essential process for both vasculogenesis and angiogenesis. The function of SMCs is to stabilize nascent vessels by inhibiting excessive endothelial cell proliferation and migration. In addition, SMCs express vasoactive peptides, growth factors and cytokines which are important for the overall function of vasculature. After birth, the principal function of SMCs is to regulate pulse pressure and blood flow through contraction[40]. SMCs are capable of reversibly modulating their phenotype during postnatal development and can de-differentiate into proliferative, matrix synthetic cells in response to vascular injury[41,42]. Abnormal SMC differentiation or function contributes to a number of cardiovascular disorders including congenital heart diseases, aortic aneurysm, atherosclerosis, hypertension, and restenosis[42-51].
TGF-β signaling plays pivotal roles in SMC differentiation during vascular development as well as phenotypic switching in disease states[52]. The importance of TGF-β signaling pathway in SMC differentiation during embryonic development has been demonstrated by numerous studies[53]. Gene-targeting studies in mice have shown that a loss of TGF-β signaling components generally leads to abnormal differentiation and maturation of the primitive vascular network, resulting in defective vessels losing integrity of the vessel wall. One of the defects is the failure of smooth muscle cell recruitment and/or differentiation[54]. 50% of mice with both alleles of TGF-β gene deleted die in utero around 10.5 dpc due to abnormalities in yolk sac vessel development. The vascular defects are caused, at least in part, by the failed differentiation of mesenchymal precursors into vascular SMCs[55]. In young mice with one allele of the TGF-β gene deleted, the levels of both TGF-β and smooth muscle differentiation markers are reduced as compared with that of wild-type mice. This regulation of smooth muscle differentiation by TGF-β also occurs dynamically in the adult animals[56]. Quantitative immunofluorescence data in rat arteries demonstrate that levels of smooth muscle differentiation markers correlate with the levels of TGF-β expression[57].
TβRII is unique and essential for TGF-β signaling[1]. SMC-specific deletion of TβRII gene is the best method to generate mice with ablation of TGF-β signaling in SMCs. Langlois and colleagues have generated mice with conditional deletion of the gene in cells expressing SMC-specific marker SM22α. Their results have shown that all SM22α-Cre/TβRII-floxed embryos die between E14.5 and the end of pregnancy. All mutant embryos display profound vascular abnormalities in the descending thoracic aorta including irregular thickness, occasional aneurysms and elastic fiber disarray. Importantly, VSMC differentiation is impaired in the descending thoracic aorta in these embryos. TβRII gene deletion in the VSMCs of the descending thoracic aorta diminishes the number of smooth muscle α-actin (α-SMA)-positive VSMCs in the media at E11.5. These results suggest that TGF-β plays an irreplaceable role in the differentiation of VSMCs in the descending thoracic aorta during mouse development[58].
The role of TβRII in SMC differentiation has also been demonstrated by tissue-specific knockout of TβRII gene in neural crest cells. During embryonic development, neural crest cells migrate to various locations within the embryo and differentiate into non-neural tissues. One subpopulation named cardiac neural crest can differentiate to SMCs of ascending aorta and great arteries by a number of growth factors including TGF-β[59,60]. TGF-β function in this process is demonstrated by neural crest-specific ablation of TβRII using Cre-loxP system. TβRII protein is specifically deleted in neural crest and neural crest-derived cells by mating TβRII-floxed mice with Wnt1-Cre mice. Mouse hearts with TβRII deletion display truncus arteriosus together with ventricular septal defects. In addition, the mutant mice exhibit abnormal patterning of the arteries arising from the aortic arch, the main cause of mortality in human DiGeorge patients. Importantly, although the mutant neural crest cells are able to migrate and form aorto-pulmonary septum at E10.5, they do not contribute to the development of the smooth musculature and fail to adopt a smooth muscle cell fate[61]. The absence of neural crest-derived smooth muscle cells in mutants explains the defective separation of the aorta from the pulmonary trunk, leading inevitably to a truncus arteriosus. Although a later report using the same strategy has failed to identify SMC defect, which is likely due to, as discussed by the authors, an in vivo compensatory mechanism or the use of a different TβRII-floxed mouse line[62], TGF-β signaling appears to be crucial in SMC differentiation from neural crest cell during embryonic development.
ALK5, a type I receptor, has been shown to be involved in the induction of epicardial to mesenchymal cells, one of the processes by which differentiated smooth muscle cells are produced. Ablation of Alk5 in epicardial lineages using Gata5-Cre mouse lines results in the failure of TGF-β-induced epicardial to mesenchymal cell transition. Late-term mutant embryos lacking epicardial Alk5 display defective formation of the SMC layer around coronary arteries and aberrant formation of capillary vessels in the myocardium[63]. In addition to ALK5, ALK1 is also required for the differentiation and recruitment of vascular SMCs to the vascular endothelium cells because ALK1 knockout embryos contain no VSMCs[64-68]. Mice mutant for ALK1 develops arterio-venous malformations (AVMs), a serious condition characterized by shunting between the arterial and venous circulations.
Endoglin (also known as CD105) is a homodimeric membrane glycoprotein located on cell surface of vascular endothelial cells, hematopoietic cells, neural crest stem cells, etc[69-71]. Endoglin has been identified as a part of the TGF-β receptor complexes and can be co-precipitated with TβRII and TβRI in endothelial and leukemic cells[72-74]. Endoglin has a pivotal function in the development of the cardiovascular system and in vascular remodeling. Mice lacking endoglin gene die during embryonic development due to cardiovascular abnormalities[68,75]. In contrast to the mice lacking TGF-β or its signaling receptors, the process of vasculogenesis occurs normally in endoglin mutant embryos. However, the second stage of vascular development, angiogenesis, is affected as shown by the absence of organized vessels in yolk sacs. Therefore, endoglin is important in angiogenesis rather than in vasculogenesis. Importantly, disrupted development of SMC in the yolk sac is observed in endoglin null mice. One important mechanism underlying the limited number of SMC in the vessel walls is the reduced availability of TGF-β protein levels. In endoglin knockout mice, the lacking of TGF-β pathway in endothelial cells of the yolk sac leads to the decreased phosphorylation of Smad2 in the mesothelial layer, which eventually inhibits the recruitment and differentiation of mesenchymal cells into VSMCs[76].
Smad proteins are important components of TGF-β signaling pathway[1,4,22,23]. Smad5 is expressed predominantly in mesenchyme and somites during embryogenesis and in many tissues of the adult. The Smad5 homozygous mutant embryos (Smad5ex6/ex6) exhibit phenotypes similar to those of TGF-β and TβRII knockouts. Smad5ex6/ex6 embryos die at E10.5-11.5 due to defects in angiogenesis which requires extensive interactions of endothelial cells with pericytes or smooth muscle cells. Smad5ex6/ex6 embryos have dilated blood vessels, and the layer of endothelial cells is dissociated from mesenchymal cells, suggesting that the interaction between the endothelial and mesenchymal cells is affected. Many Smad5ex6/ex6 embryos suffer massive apoptosis of mesenchymal cells. The abnormal blood vessels display a decrease in the thickness of SMC layer, indicating that the differentiation of mesenchymal cells into SMC is impaired in Smad5ex6/ex6 mutants[65].
MOLECULAR MECHANISMS OF TGF-β-INDUCED SMC DIFFERENTIATION
SMCs are defined by specific molecular markers and contractile functions. Smooth muscle α-actin (α-SMA) and SM22α are early markers of developing SMCs while calponin, caldesmon, and smooth muscle myosin heavy chain (SMMHC) are late markers. The principal function of SMCs is to regulate pulse pressure and blood flow through contraction[40]. In order to understand the underlying mechanisms of TGF-β-induced SMC differentiation, several in vitro models have been developed including primary cultured VSMCs[77], C3H10T1/2 (10T1/2, a multipotent mouse embryonic mesenchymal cell line)[78], and neural crest Monc-1 cells (pluripotent neural crest stem cells)[79], etc. TGF-β has been shown to induce these cells to change into a polarized and elongated SMC morphology accompanied by an up-regulation of SMC contractile proteins[78-84]. These models have significantly contributed to the understanding of transcriptional regulation of genes essential for SMC function. Three TGF-β responsive elements have been identified: the TGF-β control element (TCE), the SBE, and the CArG box. Mutation of any of these elements abolishes TGF-β induction. In addition, crosstalk between TGF-β and Notch signaling is found to be involved in SMC differentiation. Moreover, microRNA is recently emerging as an important regulator for TGF-β-induced SMC differentiation.
TCE/KLF4/KLF5
TCE is a cis-element in SMC promoter region and highly conserved across species in multiple SMC marker genes including α-SMA, SM22α, SMMHC and calponin[85]. Mutation of TCE in α-SMA or SM22α promoter region abolishes TGF-β-induced α-SMA and SM22α promoter activity in cultured SMCs[85-87]. The importance of TCE in the regulation of α-SMA and SM22α promoter activity was further studied in transgenic mice with wild-type and TCE-mutant promoters coupled to a LacZ reporter gene. TCE mutations completely block the promoter activity in directing LacZ transcription in arterial SMCs[86,87]. Both α-SMA and SM22α TCE form a TGF-β-dependent complex with nuclear proteins in electrophoretic mobility shift assays (EMSAs)[86,88]. Mutation of SM22α TCE completely abolishes this complex formation[86]. GKLF/KLF4, a Kruppel-like transcription factor (KLF) containing three C2H2 zinc fingers, specifically binds to SM22α or α-SMA TCE. Interestingly, KLF4 represses rather than activates TCE activity. Overexpression of KLF4 inhibits TGF-β-stimulated increase in SM22α or α-SMA promoter activity in 10T1/2 cells. KLF4-mediated repression of the promoter activity is TCE-dependent because in rat aortic SMCs, KLF4 overexpression inhibits the activity of wild type α-SMA promoter but has no effect on the activity of TCE mutant α-SMA promoter[87]. In addition, inhibition of KLF4 with antisense morpholinos increases α-SMA and SMMHC expression[87]. TGF-β inhibits KLF4 expression in cultured SMCs through induction of microRNA-143 (miR-143) and miR-145, leading to a reduction of KLF4 transcripts and decreased KLF4 protein expression[86,89].
Studies of KLF4 lead to the finding of another Kruppel like factor, a GKLF-related basic transcriptional element-binding protein (KLF5). KLF5 binds specifically to SM22α TCE[86]. Overexpression of KLF5 enhances TGF-β-dependent SM22-LacZ promoter activity in 10T1/2 cells, while reversing KLF4-mediated repression of α-SMA promoter activity induced by SRF in NIH3T3 cells. These studies suggest that TCE may act as an activator or a repressor of SMC marker genes depending on the stoichiometry of specific binding factors.
SBE/Smad signaling
As mentioned earlier, Smads are major intracellular mediators of TGF-β signaling pathway. When Smad2/Smad3 is phosphorylated, they are translocated into nuclear to regulate gene transcription. Smads bind to SBE (CAGA or GTCT) to regulate gene transcription. SBE is an important TGF-β responsive element in the promoter region of SMC marker genes and thus regulates SMC differentiation. Mutation of SBE in SM22α promoter inhibits TGF-β-induced SM22α promoter activity in 10T1/2, Balb3T3 and Monc-1 cells[90,91]. Transgenic embryos with SBE-mutated SM22α promoter show diminished transcription activation potential of the promoter in the arteries[90]. TGF-β induces a nuclear complex bound to the SBE sequence, and mutation of the SBE blocks this inducible interaction, indicating that SBE is required for the formation of the TGF-β-inducible complex. Smad3 and Smad4 but not Smad2 are present in these inducible complexes. The Smad3 binding to the SBE of SM22α promoter in vivo is demonstrated by chromatin immunoprecipitation assay[90]. It appears that Smad3, but not Smad2 or Smad4, activates SBE activity. Smad3 increases the transactivation of SBE reporter but not the mutant SBE reporter. Therefore, Smad3 is the major mediator of TGF-β-induced SM22α transcription, and SBE in the SM22α promoter is a direct target of Smad3.
In addition to Smad, several other pathways such as RhoA also mediate TGF-β signaling. RhoA is a member of Rho GTPase family that has intrinsic GTPase activity and can shuttle between an inactive GDP-bound state and an active GTP-bound state[92]. RhoA is highly expressed in mature VSMCs. RhoA and p160 Rho kinase (ROCK), a downstream effector of RhoA, regulate the expression of α-SMA and SM22α[93]. Overexpression of RhoA or activation of RhoA in cultured VSMCs causes a contractile phenotype and organized arrangement of actin and myosin. On the other hand, inhibition of RhoA leads to a loss of actin and myosin filaments, indicating that RhoA plays a key role in regulating SMC contractile function[94]. RhoA/ROCK regulates the expression and nuclear translocation of SRF in SMCs, and ROCK inhibitor decreases SRF enrichment to CArG regions of α-SMA and SMMHC promoters[95,96]. It appears that RhoA activates SMC marker gene expression via both Rock-dependent and independent pathways in rat pulmonary artery SMCs[97]. Our studies show that RhoA regulates TGF-β-induced SMC differentiation via modulating Smad signaling. RhoA is activated as early as 5 minutes following TGF-β induction. Inhibition of RhoA blocks TGF-β-induced expression of α-SMA, SM22α and calponin and reverses TGF-β-induced morphology alteration and contractility, indicating that RhoA is essential in TGF-β-stimulated SMC differentiation. Dominant negative RhoA blocks Smad2 and Smad3 phosphorylation, resulting in an impaired nuclear translocation and transcriptional activity, which eventually inhibits SMC marker gene expression. Conversely, constitutively active RhoA significantly enhances Smad-dependent promoter activity[98]. These results suggest that RhoA cross-talks with Smad to regulate TGF-β-induced SMC differentiation.
CArG/SRF and myocardin
Almost all SMC-specific genes have conserved CArG elements with a consensus sequence CC(A/T-rich)6GG in their promoter regions. CArG box, also called serum response element, is involved in TGF-β-induced SMC marker gene expression via binding of SRF[42,85,99,100]. Overexpression of SRF increases SMC marker expression in 10T1/2 cells. Moreover, cell morphology changes from flat to elongated shape in SRF-transfected 10T1/2 cells[100]. TGF-β induces SRF protein expression and enhances its binding activity to the CArG boxes in inducing SMC phenotype[85,100,101]. CArG box mutation disrupts SRF binding and completely abolishes TGF-β-induced transcriptional activation of SMC marker genes[100]. SRF appears to interact with Smad3 upon TGF-β induction and regulates Smad3 transactivation of SM22α promoter.
Myocardin is a transcriptional cofactor of SRF and is highly expressed in aortic medial SMCs[102]. Overexpression of myocardin leads to a high induction of calponin and α-SMA with a cell morphological alteration from flat to spindle shape in several cell lines[103-106]. In addition, overexpression of myocardin stimulates SM22α, α-SMA and SMMHC promoter activities in mouse ES cells[104]. siRNA knockdown of myocardin significantly reduces transcriptional activity of α-SMA, SM22α and SMMHC in aortic SMCs[106]. Myocardin-null embryos die at E10.5 and lack the differentiation of vascular SMCs[107]. Myocardin induces SMC marker transcription in a CArG-dependent manner[106]. Moreover, myocardin alone is sufficient to induce a SMC-like contractile phenotype[108].
Myocardin is also involved in TGF-β-induced SMC differentiation. Myocardin appears to directly interact with Smad3 in a CArG box-independent manner. Myocardin enhances TGF-β-induced alteration of cell morphology and SM22α transcription in 10T1/2 cells[90]. Overexpression of myocardin and Smad3, but not Smad2, leads to a synergistic increase of SBE promoter activity. Moreover, myocardin enhances Smad3-mediated activation of SM22α, SMMHC and α-SMA promoter activities[90]. Taken together, both SRF and its transcription cofactor myocardin play important roles in TGF-β-induced SMC differentiation.
Although myocardin is considered to be a master regulator of SMC differentiation, the expression of some SMC-associated genes such as smoothelin-B is independent of myocardin[109,110]. In addition, some progenitor cells such as A404 expressing a low level of myocardin are not converted to SMC phenotype without retinoic acid (RA) induction[106]; Conversely, other SMC progenitors such as 10T1/2 cells can be converted to SMC phenotype by overexpression of myocardin[106], suggesting that a threshold level of myocardin is required for SMC differentiation. In vivo studies show that the expression of early SMC marker genes such as SM22α and α-SMA emerges prior to detectable myocardin mRNA in the embryonic dorsal aorta, indicating that myocardin has a minor role in the initiation of SMC differentiation in some vascular tissues[104,111-113]. A recent report shows that myocardin null embryonic stem cells can readily form vascular SMCs in the setting of chimeric knockout mice. The results from this study provide novel evidence that myocardin is essential for development of visceral SMCs and ventricular myocytes but is dispensable for development of atrial myocytes and vascular SMCs[113]. Our in vitro studies demonstrate that myocardin may not participate in the initiation of TGF-β-induced SMC differentiation because the early SMC markers are induced preceding the induction of myocardin. It appears that Smad3 activation by TGF-β has blocked the expression of myocardin. Smad3 blocks myocardin transcription by interacting with Nkx2.5, which prevents Nkx2.5 from activating myocardin promoter[114]. Our data suggest that Smad may mediate the initiation of TGF-β-induced SMC differentiation, while myocardin is likely to contribute to the maturation of SMCs during a later stage (Figure 1).
Figure 1.
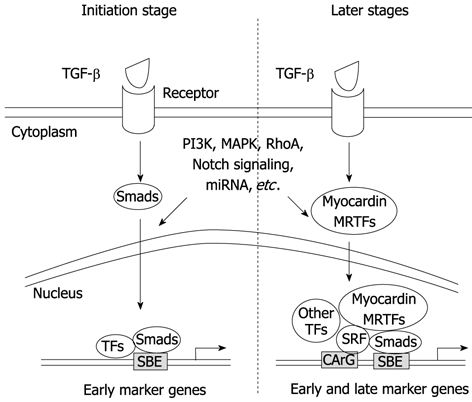
Transforming growth factor-β in smooth muscle cell differentiation. In the initiation stage of smooth muscle cell (SMC) differentiation, transforming growth factor (TGF)-β rapidly activates Smad signaling, leading to the activation of SMC early marker genes and cell fate determination by interacting with other transcription factors (TFs). At later stages, myocardin or MRTFs, via interacting with SRF, Smads and other TFs, enforce and accelerate SMC differentiation and maturation. In both the initial or later stages, other signaling pathways including PI3K, MAPK, RhoA, Notch, and miRNA may participate in the regulation of the differentiation process by interacting with TGF-β signaling molecules or downstream targets.
TGF-β and notch signaling
Like TGF-β, Notch signaling induces SMC differentiation[115-118]. Once ligands (such as Delta-like or Jagged) bind to Notch receptor (Notch1, Notch2, Notch3 and Notch4), the Notch intracellular domain (NICD) is cleaved and translocated into the nucleus to interact with the DNA-binding protein CSL (CBF-1, suppressor of hairless, and Lag-1, also known as RBP-Jκ), mastermind-like (MAML), and other transcriptional coactivators to modulate the expression of Notch target genes that regulate cell fate decisions[118]. Numerous data show that Notch induces SMC specific marker expression including α-SMA, SM22α, calponin and SMMHC in a number of cell lines[116,117,119]. Notch signaling specific inactivation in the neural crest causes cardiac outflow tract defects with decreased expression of SMC markers[116]. Although there are four types of Notch receptor, only Notch1 and Notch3 are expressed in VSMCs. In adult Notch3-/- mice, VSMCs show deficiency in postnatal maturation stage. The expression of late stage SMC marker smoothelin B is significantly inhibited in mutant arteries, suggesting a pivotal role of Notch3 in the maturation of VSMCs[120].
It appears that TGF-β cross-talks with Notch signaling in the regulation of SMC differentiation. TGF-β and Notch have cooperative effect on SMC differentiation[121]. In human SMCs, both Jagged1 and Notch induce SMC marker expression. SMCs embedded within collagen matrix exhibit a greater contractile response with both TGF-β and NICD comparing to individual treatment. CBF1 interacts with Smad2/3, which leads to an increased Smad2/3 transcriptional activity. In addition, Notch increases TGF-β-induced binding of Smad2/3 to SMC marker promoter. As most of SMC marker gene promoters contain CBF1 and Smad consensus binding sites, it is possible that NICD/CBF1 complex binding to adjacent promoter region, which provides a cis regulatory signal to promote Smad binding. In addition to the mature SMCs, TGF-β and Notch also show cooperative activity in SMC differentiation of huMSCs and embryonic stem cells. TGF-β induces Jagged1 expression in huMSCs, suggesting that Notch activation mediates TGF-β signaling during huMSC differentiation into SMC. Knockdown of Jagged1 using shRNA inhibits TGF-β-induced SMC marker expression in huMSCs[122]. Although TGF-β and Notch cooperate in most of cases, TGF-β appears to inhibit Notch 3 in SMC differentiation of 10T1/2 cells[123], suggesting that TGF-β and Notch signaling pathways interacts in a cell-specific manner.
MicroRNA
MicroRNA (miRNA) are small non-coding RNAs that function as negative regulators of gene expression by associating with the complementary sequences in the 3′ untranslated regions (UTRs) of mRNAs, resulting in mRNA degradation and/or translational inhibition[124,125]. A number of studies have shown that miRNAs plays a role in VSMC phenotype switch[126-130]. TGF-β/BMP regulate around 20 miRNAs[131], which control expression of protein-coding genes associated with epithelial-mesenchymal transition, skeletal muscle cell differentiation, and cell proliferation, etc[39,132,133]. miR-143 and miR-145, which are encoded as a gene cluster, target KLF4 and play a critical role in regulating VSMC phenotype[127,134,135]. miR-143 or miR-145 VSMC knock-out mice exhibit abnormal vascular tone and reduced contractile gene expression[134]. The expression of miR-143 and miR-145 is repressed during platelet-derived growth factor (PDGF)-induced VSMC dedifferentiation and during neointimal formation[127]. Recent studies indicate that miR143/145 plays a role in TGF-β-induced SMC differentiation[89,136]. TGF-β stimulates miR143/145 expression in a dose- and time-dependent manner in VSMC. TGF-β-induced miR143/145 expression is myocardin /SRF-, p38-, and Smad4-dependent[136]. Both CArG box and SBE are essential for TGF-β-dependent activation of miR143/145 enhancer[136]. BMP-4 also induces miR143/145 expression. TGF-β and BMP-4 induction of miR143/145 results in down-regulation of KLF4[89]. Interestingly, BMP-4 induces miR143/145 through myocardin-related factor A (MRTFA), but not myocardin, suggesting that TGF-β and BMP4 signaling regulate KLF4 expression through different mechanisms[89].
TGF-β IN SMC-RELATED DISEASES
The principal postnatal function of SMCs is to regulate pulse pressure and blood flow through contraction[40]. SMCs are capable of reversibly modulating their phenotype during postnatal development and can de-differentiate into proliferative, matrix synthetic cells in response to vascular injury[41,42]. TGF-β regulates both SMC differentiation during embryonic development and postnatal phenotypic switching[74,75,137]. Overexpression of TGF-β increases the neointimal formation and smooth muscle proliferation and differentiation in balloon injury models[138,139]. Therefore, it is conceivable that TGF-β plays an important role in the re-differentiation phenomena[140]. TGF-β has been shown to be involved in the development of many cardiovascular diseases including atherosclerosis, congenital heart diseases, aortic aneurysm, hypertension and hereditary hemorrhagic telangiectasia, etc[141-143]. Many of these diseases are due to the failed regulation of SMC function or differentiation.
Atherosclerosis
Atherosclerosis is triggered in response to chronic injury to the vascular endothelium by various risk factors. It is a progressive disease characterized by the formation of a plaque in the inner lining of large arteries. VSMC proliferation, migration, and hypertrophy are involved in the development of atherosclerosis. VSMCs play a maladaptive role in the lesion development and the progression of the disease[144]. TGF-β directs the response of SMC to the injury. In animals, deletion of a single allele of the TGF-β gene increases its susceptibility to endothelial cell activation and vascular lipid lesion formation in response to pro-atherogenic stimuli such as a lipid-rich diet[145]. TGF-β stimulates SMC proliferation at low concentrations via both PDGF-dependent and -independent manner. Ribozyme oligonucleotides against TGF-β increase vascular inflammation, accelerate lipid lesion formation, and shift the plaque morphology towards an unstable phenotype[146]. Inhibition of TGF-β signaling in ApoE deficient mice, an animal model for atherosclerosis, suggest that the cytokine is critical for the production of extracellular matrix and the maintenance of a stable plaque phenotype through SMC phenotypic regulation[146,147]. Indeed, SMC in stable lesions express greater amounts of TGF-β than unstable lesions. MacCaffrey et al[148] demonstrate that SMCs isolated from atherosclerotic plaque tissue expressed less TβRII than SMCs from healthy vessel wall. These data directly or indirectly show that TGF-β plays a pivotal role in the maintenance of normal blood vessel wall architecture.
Congenital heart diseases
Defective TGF-β signaling causes congenital heart diseases (CHD) during embryonic development[73]. CHD are the most commonly occurring birth defect in humans. Moderate and severe forms of congenital heart disease, including outflow tract defects and aortic arch anomalies, occur in 6 per 1000 live births[149]. Several studies have shown that cardiac neural crest contribute SMCs to the ascending and arch portions of the aorta and the ductus arteriosus. A number of congenital human diseases such as heart and outflow tract malformations are now attributed to failure of cardiac neural crests to differentiate into aortic arch complex[150]. Patients with Alagille syndrome always have CHD with right-sided outflow tract defects and tetralogy of Fallot influenced by cardiac neural crests. Patients with DiGeorge syndrome (DGS) always have congenital defects with heart and outflow tract malformations influenced by cardiac neural crest[151]. Mice with TβRII mutation in neural crest develop all the morphological features of DGS. The hearts of TβRII-mutant mice display a truncus arteriosus together with a ventricular septum defects (VSD) at E18. Both control and TβRII-mutant neural crest cells are able to populate the pharyngeal apparatus and form aorto-pulmonary septum at E10.5. However, TβRII-mutant neural crest cells in the aorto-pulmonary septum do not develop into smooth muscle cells[61]. The absence of neural crest derived SMCs in mutants explains the defective separation of the aorta from the pulmonary trunk, leading inevitably to a truncus arteriosus. Thus, TGF-β regulation of neural crest differentiation rather than migration plays a crucial role in the etiology of DiGeorge syndrome.
Thoracic aortic aneurysms and dissections
Combination of human molecular genetics and animal modeling has demonstrated the involvement of TGF-β signaling in aortic aneurysm[72]. Mutations in TβRI and TβRII result in a spectrum of genetic conditions, associating with thoracic aortic aneurysms and dissections (TAAD)[152]. Mutations in TβRII are initially identified in individuals with a Marfan-like connective tissue syndrome with TAAD and skeletal features of Marfan syndrome (MFS). TβRI and TβRII mutations are subsequently described in individuals with Loeys-Dietz syndrome (LDS), a syndrome characterized by TAAD in children and young adults, arterial tortuosity, aneurysms and dissections of peripheral arteries. TβRII mutations also lead to descending aortic disease and aneurysms of other arteries. An arginine residue of TβRII at position 460 has been identified as a mutation “hot spot” for TAAD. Structural analysis has revealed that the amino acid substitutions may interfere with the receptor’s ability to transduce signals[153]. In aortic SMCs explanted from patients with TβRII mutations, the expression of SMC contractile proteins is decreased compared with controls. In vivo expression of contractile proteins is also decreased in aortas from patients with TβRII mutations relative to unaffected aortas. The failed expression of SMC contractile proteins in TβRII-mutant SMCs may influence the contractile function of SMCs, which contributes to the pathogenesis of TAAD[154].
Hypertension
Hypertension is defined as a sustained diastolic pressure of > 90 mmHg or a systolic blood pressure > 140 mmHg. Hypertension is another disease that related to SMC phenotypic switching. Although the etiology is extremely complex and varies among individuals, a common feature in the majority of cases of hypertension is an increase in peripheral resistance as a result of increased SMC contractility and vascular remodeling that are related with the phenotypic switching of SMC[155]. Primary pulmonary hypertension (PPH) is a rare disease with symptoms of fatigue, anorexia, an increase in pulmonary arterial pressure, right ventricular failure and death[156]. PPH is caused by mutations in either of two genes: the BMP type II receptor gene (BMPR-II) and ALK-1[157-159]. BMPR-II mutations increase the incidence of PPH. BMP-2, -4 and -7 have been shown to inhibit SMC proliferation but increase the SMC marker expression in cultured pulmonary artery SMCs (PASMCs). PASMCs derived from the pulmonary arteries of patients with PPH exhibit abnormal growth responses to TGF-β. TGF- β inhibits serum-induced proliferation of PASMC from healthy individuals while stimulates cell proliferation of PASMCs from patients with PPH[160].
Hereditary hemorrhagic telangiectasia
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant vascular disorder which always happened in nasal, mucocutaneous, gastrointestinal, pulmonary, cerebral, and hepatic vascular beds. The common syndromes are nose bleeding, skin telangiectases, gastrointestinal bleeding[161]. Pulmonary arteriovenous malformations (PAVM) are always present in 20% of HHT populations. Genetic analyses reveal that Endoglin are responsible for type I HHT[162,163]. As aforementioned, Endoglin form TGF-β receptor complexes with TβRI and TβRII to modulate the phosphorylation of TβRI and TβRII and plays a pivotal role in angiogenesis as demonstrated in the Eng-/- mice. Eng-/- embryos show a defective development of VSMCs because of the reduced availability of active TGF-β protein. The lack of TGF-β inhibits the recruitment and the differentiation of mesenchymal cells into VSMCs, leading to weak vessel walls, which may contribute to the development of HHT[76]. In addition to Endoglin, ALK-1 and Smad4 are also involved in HHT[164]. ALK-1 gene heterozygous mutation causes type II HHT-2[164]. Recently, a remarkable ALK1 germinal and somatic mosaicism characterized by the presence of two distinct mutant alleles and a non-mutant ALK1 allele are identified in a woman with HHT and PAH[70]. It is proposed that genetic background and/or environmental factors (second hits), in addition to the mutations in Endoglin and ALK-1 genes, may also play an important role in the development of vascular malformations in HHT patients. Park and colleagues demonstrate using ALK-1-knockout mice that excisional skin wounding, as a second hit, is essential for the development of AVMs in HHT. These results provide new insights for understanding the pathogenesis of HHT[71].
CONCLUSION
SMC differentiation and phenotypic modulation play critical roles in embryonic cardiovascular development as well as pathological conditions in adults. TGF-β and its downstream signaling molecules including receptors, coreceptors and intermediate Smad proteins are all indispensible for the SMC differentiation or phenotypic modulation. As aforementioned, TGF-β itself can activate multiple signaling pathways such as MAPK, PI3K and RhoA. In addition, TGF-β signaling crosstalks with other pathways including Notch and SRF/myocardin. These diversified interactions ensure a precise cell fate determination and maturation of SMCs. Among the various SMC regulators, TGF-β/Smad signaling appears to be critical in regulating the initiation of SMC differentiation. Defective TGF-β signaling leads to development of several prominent cardiovascular diseases including congenital heart diseases, aortic aneurysm, hypertension, neointimal hyperplasia observed in vascular injury and atherosclerosis, etc.
Footnotes
Supported by Grants from National Institutes of Health, No. HL093429 and No. HL107526 to Dr. Chen
Peer reviewers: Emil Martin, PhD, Assistant Professor, Center for Cell Signaling, Brown Foundation Institute of Molecular Medicine, University of Texas Health Science Center in Houston, 1825 Pressler street, room 530A, Houston, TX 77030, United States; Carlo Ventura, MD, PhD, Full Professor of Molecular Biology, Chief, Laboratory of Molecular Biology and Stem Cell Engineering-National Institute of Biostructures and Biosystems, University of Bologna, S. Orsola-Malpighi Hospital, Cardiovascular Department, Pavilion 21 Via Massarenti 9, 40138 Bologna, Italy; Jianyu Liu, Dr., Markey Cancer Center, University of Kentucky, 741 S Limestone Rd, Lexington, KY 40536, United States
S- Editor Cheng JX L- Editor A E- Editor Zhang DN
References
- 1.Massagué J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 2.Yang L, Moses HL. Transforming growth factor beta: tumor suppressor or promoter? Are host immune cells the answer? Cancer Res. 2008;68:9107–9111. doi: 10.1158/0008-5472.CAN-08-2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akhurst RJ, Derynck R. TGF-beta signaling in cancer--a double-edged sword. Trends Cell Biol. 2001;11:S44–S51. doi: 10.1016/s0962-8924(01)02130-4. [DOI] [PubMed] [Google Scholar]
- 4.Dennler S, Goumans MJ, ten Dijke P. Transforming growth factor beta signal transduction. J Leukoc Biol. 2002;71:731–740. [PubMed] [Google Scholar]
- 5.Moustakas A, Pardali K, Gaal A, Heldin CH. Mechanisms of TGF-beta signaling in regulation of cell growth and differentiation. Immunol Lett. 2002;82:85–91. doi: 10.1016/s0165-2478(02)00023-8. [DOI] [PubMed] [Google Scholar]
- 6.Schuster N, Krieglstein K. Mechanisms of TGF-beta-mediated apoptosis. Cell Tissue Res. 2002;307:1–14. doi: 10.1007/s00441-001-0479-6. [DOI] [PubMed] [Google Scholar]
- 7.Gong Z, Calkins G, Cheng EC, Krause D, Niklason LE. Influence of culture medium on smooth muscle cell differentiation from human bone marrow-derived mesenchymal stem cells. Tissue Eng Part A. 2009;15:319–330. doi: 10.1089/ten.tea.2008.0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Massagué J, Gomis RR. The logic of TGFbeta signaling. FEBS Lett. 2006;580:2811–2820. doi: 10.1016/j.febslet.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 9.Dubois CM, Laprise MH, Blanchette F, Gentry LE, Leduc R. Processing of transforming growth factor beta 1 precursor by human furin convertase. J Biol Chem. 1995;270:10618–10624. doi: 10.1074/jbc.270.18.10618. [DOI] [PubMed] [Google Scholar]
- 10.Feng XH, Derynck R. A kinase subdomain of transforming growth factor-beta (TGF-beta) type I receptor determines the TGF-beta intracellular signaling specificity. EMBO J. 1997;16:3912–3923. doi: 10.1093/emboj/16.13.3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Feng XH, Derynck R. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced transcription. Nature. 1998;394:909–913. doi: 10.1038/29814. [DOI] [PubMed] [Google Scholar]
- 13.López-Casillas F, Wrana JL, Massagué J. Betaglycan presents ligand to the TGF beta signaling receptor. Cell. 1993;73:1435–1444. doi: 10.1016/0092-8674(93)90368-z. [DOI] [PubMed] [Google Scholar]
- 14.Wieser R, Wrana JL, Massagué J. GS domain mutations that constitutively activate T beta R-I, the downstream signaling component in the TGF-beta receptor complex. EMBO J. 1995;14:2199–2208. doi: 10.1002/j.1460-2075.1995.tb07214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamashita H, ten Dijke P, Franzén P, Miyazono K, Heldin CH. Formation of hetero-oligomeric complexes of type I and type II receptors for transforming growth factor-beta. J Biol Chem. 1994;269:20172–20178. [PubMed] [Google Scholar]
- 16.Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, ten Dijke P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell. 2003;12:817–828. doi: 10.1016/s1097-2765(03)00386-1. [DOI] [PubMed] [Google Scholar]
- 17.Daly AC, Randall RA, Hill CS. Transforming growth factor beta-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol Cell Biol. 2008;28:6889–6902. doi: 10.1128/MCB.01192-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 19.Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 20.Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8:970–982. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 21.Massagué J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19:1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Derynck R, Zhang Y, Feng XH. Smads: transcriptional activators of TGF-beta responses. Cell. 1998;95:737–740. doi: 10.1016/s0092-8674(00)81696-7. [DOI] [PubMed] [Google Scholar]
- 23.Miyazono K, ten Dijke P, Heldin CH. TGF-beta signaling by Smad proteins. Adv Immunol. 2000;75:115–157. doi: 10.1016/s0065-2776(00)75003-6. [DOI] [PubMed] [Google Scholar]
- 24.Koinuma D, Tsutsumi S, Kamimura N, Imamura T, Aburatani H, Miyazono K. Promoter-wide analysis of Smad4 binding sites in human epithelial cells. Cancer Sci. 2009;100:2133–2142. doi: 10.1111/j.1349-7006.2009.01299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koinuma D, Tsutsumi S, Kamimura N, Taniguchi H, Miyazawa K, Sunamura M, Imamura T, Miyazono K, Aburatani H. Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor beta signaling. Mol Cell Biol. 2009;29:172–186. doi: 10.1128/MCB.01038-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell. 1998;95:779–791. doi: 10.1016/s0092-8674(00)81701-8. [DOI] [PubMed] [Google Scholar]
- 27.Watanabe Y, Itoh S, Goto T, Ohnishi E, Inamitsu M, Itoh F, Satoh K, Wiercinska E, Yang W, Shi L, et al. TMEPAI, a transmembrane TGF-beta-inducible protein, sequesters Smad proteins from active participation in TGF-beta signaling. Mol Cell. 2010;37:123–134. doi: 10.1016/j.molcel.2009.10.028. [DOI] [PubMed] [Google Scholar]
- 28.Imamura T, Takase M, Nishihara A, Oeda E, Hanai J, Kawabata M, Miyazono K. Smad6 inhibits signalling by the TGF-beta superfamily. Nature. 1997;389:622–626. doi: 10.1038/39355. [DOI] [PubMed] [Google Scholar]
- 29.Nakao A, Afrakhte M, Morén A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389:631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 30.Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, Richardson MA, Topper JN, Gimbrone MA, Wrana JL, et al. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell. 1997;89:1165–1173. doi: 10.1016/s0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- 31.Chen HB, Shen J, Ip YT, Xu L. Identification of phosphatases for Smad in the BMP/DPP pathway. Genes Dev. 2006;20:648–653. doi: 10.1101/gad.1384706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sapkota G, Knockaert M, Alarcón C, Montalvo E, Brivanlou AH, Massagué J. Dephosphorylation of the linker regions of Smad1 and Smad2/3 by small C-terminal domain phosphatases has distinct outcomes for bone morphogenetic protein and transforming growth factor-beta pathways. J Biol Chem. 2006;281:40412–40419. doi: 10.1074/jbc.M610172200. [DOI] [PubMed] [Google Scholar]
- 33.Lin X, Duan X, Liang YY, Su Y, Wrighton KH, Long J, Hu M, Davis CM, Wang J, Brunicardi FC, et al. PPM1A functions as a Smad phosphatase to terminate TGFbeta signaling. Cell. 2006;125:915–928. doi: 10.1016/j.cell.2006.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lo RS, Massagué J. Ubiquitin-dependent degradation of TGF-beta-activated smad2. Nat Cell Biol. 1999;1:472–478. doi: 10.1038/70258. [DOI] [PubMed] [Google Scholar]
- 35.Gao S, Alarcón C, Sapkota G, Rahman S, Chen PY, Goerner N, Macias MJ, Erdjument-Bromage H, Tempst P, Massagué J. Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-beta signaling. Mol Cell. 2009;36:457–468. doi: 10.1016/j.molcel.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stroschein SL, Wang W, Zhou S, Zhou Q, Luo K. Negative feedback regulation of TGF-beta signaling by the SnoN oncoprotein. Science. 1999;286:771–774. doi: 10.1126/science.286.5440.771. [DOI] [PubMed] [Google Scholar]
- 37.Luo K, Stroschein SL, Wang W, Chen D, Martens E, Zhou S, Zhou Q. The Ski oncoprotein interacts with the Smad proteins to repress TGFbeta signaling. Genes Dev. 1999;13:2196–2206. doi: 10.1101/gad.13.17.2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo X, Wang XF. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009;19:71–88. doi: 10.1038/cr.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kong W, Yang H, He L, Zhao JJ, Coppola D, Dalton WS, Cheng JQ. MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol. 2008;28:6773–6784. doi: 10.1128/MCB.00941-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev. 1995;75:487–517. doi: 10.1152/physrev.1995.75.3.487. [DOI] [PubMed] [Google Scholar]
- 41.Mosse PR, Campbell GR, Campbell JH. Smooth muscle phenotypic expression in human carotid arteries. II. Atherosclerosis-free diffuse intimal thickenings compared with the media. Arteriosclerosis. 1986;6:664–669. doi: 10.1161/01.atv.6.6.664. [DOI] [PubMed] [Google Scholar]
- 42.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 43.Aikawa M, Sivam PN, Kuro-o M, Kimura K, Nakahara K, Takewaki S, Ueda M, Yamaguchi H, Yazaki Y, Periasamy M. Human smooth muscle myosin heavy chain isoforms as molecular markers for vascular development and atherosclerosis. Circ Res. 1993;73:1000–1012. doi: 10.1161/01.res.73.6.1000. [DOI] [PubMed] [Google Scholar]
- 44.Kocher O, Gabbiani G. Cytoskeletal features of normal and atheromatous human arterial smooth muscle cells. Hum Pathol. 1986;17:875–880. doi: 10.1016/s0046-8177(86)80637-2. [DOI] [PubMed] [Google Scholar]
- 45.Glukhova MA, Kabakov AE, Frid MG, Ornatsky OI, Belkin AM, Mukhin DN, Orekhov AN, Koteliansky VE, Smirnov VN. Modulation of human aorta smooth muscle cell phenotype: a study of muscle-specific variants of vinculin, caldesmon, and actin expression. Proc Natl Acad Sci USA. 1988;85:9542–9546. doi: 10.1073/pnas.85.24.9542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwartz SM. Cellular proliferation in atherosclerosis and hypertension. Proc Soc Exp Biol Med. 1983;173:1–13. doi: 10.3181/00379727-173-41601. [DOI] [PubMed] [Google Scholar]
- 47.Libby P. Current concepts of the pathogenesis of the acute coronary syndromes. Circulation. 2001;104:365–372. doi: 10.1161/01.cir.104.3.365. [DOI] [PubMed] [Google Scholar]
- 48.Schwartz SM. Smooth muscle migration in atherosclerosis and restenosis. J Clin Invest. 1997;100:S87–S89. [PubMed] [Google Scholar]
- 49.Milewicz DM, Guo DC, Tran-Fadulu V, Lafont AL, Papke CL, Inamoto S, Kwartler CS, Pannu H. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet. 2008;9:283–302. doi: 10.1146/annurev.genom.8.080706.092303. [DOI] [PubMed] [Google Scholar]
- 50.Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, Wegman M, Glancy L, Gasc JM, Brunotte F, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38:343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 51.Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 52.Sinha S, Hoofnagle MH, Kingston PA, McCanna ME, Owens GK. Transforming growth factor-beta1 signaling contributes to development of smooth muscle cells from embryonic stem cells. Am J Physiol Cell Physiol. 2004;287:C1560–C1568. doi: 10.1152/ajpcell.00221.2004. [DOI] [PubMed] [Google Scholar]
- 53.Perrella MA, Jain MK, Lee ME. Role of TGF-beta in vascular development and vascular reactivity. Miner Electrolyte Metab. 1998;24:136–143. doi: 10.1159/000057361. [DOI] [PubMed] [Google Scholar]
- 54.Goumans MJ, Mummery C. Functional analysis of the TGFbeta receptor/Smad pathway through gene ablation in mice. Int J Dev Biol. 2000;44:253–265. [PubMed] [Google Scholar]
- 55.Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development. 1995;121:1845–1854. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- 56.Kocher O, Gabbiani F, Gabbiani G, Reidy MA, Cokay MS, Peters H, Hüttner I. Phenotypic features of smooth muscle cells during the evolution of experimental carotid artery intimal thickening. Biochemical and morphologic studies. Lab Invest. 1991;65:459–470. [PubMed] [Google Scholar]
- 57.Grainger DJ, Metcalfe JC, Grace AA, Mosedale DE. Transforming growth factor-beta dynamically regulates vascular smooth muscle differentiation in vivo. J Cell Sci. 1998;111(Pt 19):2977–2988. doi: 10.1242/jcs.111.19.2977. [DOI] [PubMed] [Google Scholar]
- 58.Langlois D, Hneino M, Bouazza L, Parlakian A, Sasaki T, Bricca G, Li JY. Conditional inactivation of TGF-β type II receptor in smooth muscle cells and epicardium causes lethal aortic and cardiac defects. Transgenic Res. 2010;19:1069–1082. doi: 10.1007/s11248-010-9379-4. [DOI] [PubMed] [Google Scholar]
- 59.Kirby ML, Gale TF, Stewart DE. Neural crest cells contribute to normal aorticopulmonary septation. Science. 1983;220:1059–1061. doi: 10.1126/science.6844926. [DOI] [PubMed] [Google Scholar]
- 60.Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- 61.Wurdak H, Ittner LM, Lang KS, Leveen P, Suter U, Fischer JA, Karlsson S, Born W, Sommer L. Inactivation of TGFbeta signaling in neural crest stem cells leads to multiple defects reminiscent of DiGeorge syndrome. Genes Dev. 2005;19:530–535. doi: 10.1101/gad.317405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Choudhary B, Ito Y, Makita T, Sasaki T, Chai Y, Sucov HM. Cardiovascular malformations with normal smooth muscle differentiation in neural crest-specific type II TGFbeta receptor (Tgfbr2) mutant mice. Dev Biol. 2006;289:420–429. doi: 10.1016/j.ydbio.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 63.Sridurongrit S, Larsson J, Schwartz R, Ruiz-Lozano P, Kaartinen V. Signaling via the Tgf-beta type I receptor Alk5 in heart development. Dev Biol. 2008;322:208–218. doi: 10.1016/j.ydbio.2008.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pepper MS. Transforming growth factor-beta: vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev. 1997;8:21–43. doi: 10.1016/s1359-6101(96)00048-2. [DOI] [PubMed] [Google Scholar]
- 65.Yang X, Castilla LH, Xu X, Li C, Gotay J, Weinstein M, Liu PP, Deng CX. Angiogenesis defects and mesenchymal apoptosis in mice lacking SMAD5. Development. 1999;126:1571–1580. doi: 10.1242/dev.126.8.1571. [DOI] [PubMed] [Google Scholar]
- 66.Urness LD, Sorensen LK, Li DY. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat Genet. 2000;26:328–331. doi: 10.1038/81634. [DOI] [PubMed] [Google Scholar]
- 67.Chang H, Huylebroeck D, Verschueren K, Guo Q, Matzuk MM, Zwijsen A. Smad5 knockout mice die at mid-gestation due to multiple embryonic and extraembryonic defects. Development. 1999;126:1631–1642. doi: 10.1242/dev.126.8.1631. [DOI] [PubMed] [Google Scholar]
- 68.Li DY, Sorensen LK, Brooke BS, Urness LD, Davis EC, Taylor DG, Boak BB, Wendel DP. Defective angiogenesis in mice lacking endoglin. Science. 1999;284:1534–1537. doi: 10.1126/science.284.5419.1534. [DOI] [PubMed] [Google Scholar]
- 69.Conley BA, Smith JD, Guerrero-Esteo M, Bernabeu C, Vary CP. Endoglin, a TGF-beta receptor-associated protein, is expressed by smooth muscle cells in human atherosclerotic plaques. Atherosclerosis. 2000;153:323–335. doi: 10.1016/s0021-9150(00)00422-6. [DOI] [PubMed] [Google Scholar]
- 70.Eyries M, Coulet F, Girerd B, Montani D, Humbert M, Lacombe P, Chinet T, Gouya L, Roume J, Axford M, et al. ACVRL1 germinal mosaic with two mutant alleles in hereditary hemorrhagic telangiectasia associated with pulmonary arterial hypertension. Clin Genet. 2011:Epub ahead of print. doi: 10.1111/j.1399-0004.2011.01727.x. [DOI] [PubMed] [Google Scholar]
- 71.Park SO, Wankhede M, Lee YJ, Choi EJ, Fliess N, Choe SW, Oh SH, Walter G, Raizada MK, Sorg BS, et al. Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J Clin Invest. 2009;119:3487–3496. doi: 10.1172/JCI39482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lindsay ME, Dietz HC. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature. 2011;473:308–316. doi: 10.1038/nature10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Arthur HM, Bamforth SD. TGFβ signaling and congenital heart disease: Insights from mouse studies. Birth Defects Res A Clin Mol Teratol. 2011;91:423–434. doi: 10.1002/bdra.20794. [DOI] [PubMed] [Google Scholar]
- 74.Mack CP. Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler Thromb Vasc Biol. 2011;31:1495–1505. doi: 10.1161/ATVBAHA.110.221135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen YE. Vascular cell lineage determination and differentiation. Arterioscler Thromb Vasc Biol. 2011;31:1467–1468. doi: 10.1161/ATVBAHA.111.230813. [DOI] [PubMed] [Google Scholar]
- 76.Carvalho RL, Jonker L, Goumans MJ, Larsson J, Bouwman P, Karlsson S, Dijke PT, Arthur HM, Mummery CL. Defective paracrine signalling by TGFbeta in yolk sac vasculature of endoglin mutant mice: a paradigm for hereditary haemorrhagic telangiectasia. Development. 2004;131:6237–6247. doi: 10.1242/dev.01529. [DOI] [PubMed] [Google Scholar]
- 77.Owens GK, Geisterfer AA, Yang YW, Komoriya A. Transforming growth factor-beta-induced growth inhibition and cellular hypertrophy in cultured vascular smooth muscle cells. J Cell Biol. 1988;107:771–780. doi: 10.1083/jcb.107.2.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hirschi KK, Rohovsky SA, D’Amore PA. PDGF, TGF-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J Cell Biol. 1998;141:805–814. doi: 10.1083/jcb.141.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen S, Lechleider RJ. Transforming growth factor-beta-induced differentiation of smooth muscle from a neural crest stem cell line. Circ Res. 2004;94:1195–1202. doi: 10.1161/01.RES.0000126897.41658.81. [DOI] [PubMed] [Google Scholar]
- 80.Herbst-Kralovetz MM, Quayle AJ, Ficarra M, Greene S, Rose WA, Chesson R, Spagnuolo RA, Pyles RB. Quantification and comparison of toll-like receptor expression and responsiveness in primary and immortalized human female lower genital tract epithelia. Am J Reprod Immunol. 2008;59:212–224. doi: 10.1111/j.1600-0897.2007.00566.x. [DOI] [PubMed] [Google Scholar]
- 81.Morishita R, Nagata K, Ito H, Ueda H, Asano M, Shinohara H, Kato K, Asano T. Expression of smooth muscle cell-specific proteins in neural progenitor cells induced by agonists of G protein-coupled receptors and transforming growth factor-beta. J Neurochem. 2007;101:1031–1040. doi: 10.1111/j.1471-4159.2006.04405.x. [DOI] [PubMed] [Google Scholar]
- 82.Shah NM, Groves AK, Anderson DJ. Alternative neural crest cell fates are instructively promoted by TGFbeta superfamily members. Cell. 1996;85:331–343. doi: 10.1016/s0092-8674(00)81112-5. [DOI] [PubMed] [Google Scholar]
- 83.Manabe I, Owens GK. Recruitment of serum response factor and hyperacetylation of histones at smooth muscle-specific regulatory regions during differentiation of a novel P19-derived in vitro smooth muscle differentiation system. Circ Res. 2001;88:1127–1134. doi: 10.1161/hh1101.091339. [DOI] [PubMed] [Google Scholar]
- 84.Fei T, Chen YG. Regulation of embryonic stem cell self-renewal and differentiation by TGF-beta family signaling. Sci China Life Sci. 2010;53:497–503. doi: 10.1007/s11427-010-0096-2. [DOI] [PubMed] [Google Scholar]
- 85.Hautmann MB, Madsen CS, Owens GK. A transforming growth factor beta (TGFbeta) control element drives TGFbeta-induced stimulation of smooth muscle alpha-actin gene expression in concert with two CArG elements. J Biol Chem. 1997;272:10948–10956. doi: 10.1074/jbc.272.16.10948. [DOI] [PubMed] [Google Scholar]
- 86.Adam PJ, Regan CP, Hautmann MB, Owens GK. Positive- and negative-acting Kruppel-like transcription factors bind a transforming growth factor beta control element required for expression of the smooth muscle cell differentiation marker SM22alpha in vivo. J Biol Chem. 2000;275:37798–37806. doi: 10.1074/jbc.M006323200. [DOI] [PubMed] [Google Scholar]
- 87.Liu Y, Sinha S, Owens G. A transforming growth factor-beta control element required for SM alpha-actin expression in vivo also partially mediates GKLF-dependent transcriptional repression. J Biol Chem. 2003;278:48004–48011. doi: 10.1074/jbc.M301902200. [DOI] [PubMed] [Google Scholar]
- 88.King KE, Iyemere VP, Weissberg PL, Shanahan CM. Krüppel-like factor 4 (KLF4/GKLF) is a target of bone morphogenetic proteins and transforming growth factor beta 1 in the regulation of vascular smooth muscle cell phenotype. J Biol Chem. 2003;278:11661–11669. doi: 10.1074/jbc.M211337200. [DOI] [PubMed] [Google Scholar]
- 89.Davis-Dusenbery BN, Chan MC, Reno KE, Weisman AS, Layne MD, Lagna G, Hata A. down-regulation of Kruppel-like factor-4 (KLF4) by microRNA-143/145 is critical for modulation of vascular smooth muscle cell phenotype by transforming growth factor-beta and bone morphogenetic protein 4. J Biol Chem. 2011;286:28097–28110. doi: 10.1074/jbc.M111.236950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Qiu P, Ritchie RP, Fu Z, Cao D, Cumming J, Miano JM, Wang DZ, Li HJ, Li L. Myocardin enhances Smad3-mediated transforming growth factor-beta1 signaling in a CArG box-independent manner: Smad-binding element is an important cis element for SM22alpha transcription in vivo. Circ Res. 2005;97:983–991. doi: 10.1161/01.RES.0000190604.90049.71. [DOI] [PubMed] [Google Scholar]
- 91.Chen S, Kulik M, Lechleider RJ. Smad proteins regulate transcriptional induction of the SM22alpha gene by TGF-beta. Nucleic Acids Res. 2003;31:1302–1310. doi: 10.1093/nar/gkg224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans. 2005;33:891–895. doi: 10.1042/BST20050891. [DOI] [PubMed] [Google Scholar]
- 93.Mack CP, Somlyo AV, Hautmann M, Somlyo AP, Owens GK. Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization. J Biol Chem. 2001;276:341–347. doi: 10.1074/jbc.M005505200. [DOI] [PubMed] [Google Scholar]
- 94.Worth NF, Campbell GR, Rolfe BE. A role for rho in smooth muscle phenotypic regulation. Ann N Y Acad Sci. 2001;947:316–322. doi: 10.1111/j.1749-6632.2001.tb03955.x. [DOI] [PubMed] [Google Scholar]
- 95.Liu HW, Halayko AJ, Fernandes DJ, Harmon GS, McCauley JA, Kocieniewski P, McConville J, Fu Y, Forsythe SM, Kogut P, et al. The RhoA/Rho kinase pathway regulates nuclear localization of serum response factor. Am J Respir Cell Mol Biol. 2003;29:39–47. doi: 10.1165/rcmb.2002-0206OC. [DOI] [PubMed] [Google Scholar]
- 96.Wamhoff BR, Bowles DK, McDonald OG, Sinha S, Somlyo AP, Somlyo AV, Owens GK. L-type voltage-gated Ca2+ channels modulate expression of smooth muscle differentiation marker genes via a rho kinase/myocardin/SRF-dependent mechanism. Circ Res. 2004;95:406–414. doi: 10.1161/01.RES.0000138582.36921.9e. [DOI] [PubMed] [Google Scholar]
- 97.Deaton RA, Su C, Valencia TG, Grant SR. Transforming growth factor-beta1-induced expression of smooth muscle marker genes involves activation of PKN and p38 MAPK. J Biol Chem. 2005;280:31172–31181. doi: 10.1074/jbc.M504774200. [DOI] [PubMed] [Google Scholar]
- 98.Chen S, Crawford M, Day RM, Briones VR, Leader JE, Jose PA, Lechleider RJ. RhoA modulates Smad signaling during transforming growth factor-beta-induced smooth muscle differentiation. J Biol Chem. 2006;281:1765–1770. doi: 10.1074/jbc.M507771200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Miano JM. Serum response factor: toggling between disparate programs of gene expression. J Mol Cell Cardiol. 2003;35:577–593. doi: 10.1016/s0022-2828(03)00110-x. [DOI] [PubMed] [Google Scholar]
- 100.Hirschi KK, Lai L, Belaguli NS, Dean DA, Schwartz RJ, Zimmer WE. Transforming growth factor-beta induction of smooth muscle cell phenotpye requires transcriptional and post-transcriptional control of serum response factor. J Biol Chem. 2002;277:6287–6295. doi: 10.1074/jbc.M106649200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Qiu P, Feng XH, Li L. Interaction of Smad3 and SRF-associated complex mediates TGF-beta1 signals to regulate SM22 transcription during myofibroblast differentiation. J Mol Cell Cardiol. 2003;35:1407–1420. doi: 10.1016/j.yjmcc.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 102.Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA, Olson EN. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell. 2001;105:851–862. doi: 10.1016/s0092-8674(01)00404-4. [DOI] [PubMed] [Google Scholar]
- 103.Chen J, Kitchen CM, Streb JW, Miano JM. Myocardin: a component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol. 2002;34:1345–1356. doi: 10.1006/jmcc.2002.2086. [DOI] [PubMed] [Google Scholar]
- 104.Du KL, Ip HS, Li J, Chen M, Dandre F, Yu W, Lu MM, Owens GK, Parmacek MS. Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol Cell Biol. 2003;23:2425–2437. doi: 10.1128/MCB.23.7.2425-2437.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang Z, Wang DZ, Pipes GC, Olson EN. Myocardin is a master regulator of smooth muscle gene expression. Proc Natl Acad Sci USA. 2003;100:7129–7134. doi: 10.1073/pnas.1232341100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yoshida T, Sinha S, Dandré F, Wamhoff BR, Hoofnagle MH, Kremer BE, Wang DZ, Olson EN, Owens GK. Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ Res. 2003;92:856–864. doi: 10.1161/01.RES.0000068405.49081.09. [DOI] [PubMed] [Google Scholar]
- 107.Li S, Wang DZ, Wang Z, Richardson JA, Olson EN. The serum response factor coactivator myocardin is required for vascular smooth muscle development. Proc Natl Acad Sci USA. 2003;100:9366–9370. doi: 10.1073/pnas.1233635100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Long X, Bell RD, Gerthoffer WT, Zlokovic BV, Miano JM. Myocardin is sufficient for a smooth muscle-like contractile phenotype. Arterioscler Thromb Vasc Biol. 2008;28:1505–1510. doi: 10.1161/ATVBAHA.108.166066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yoshida T, Kawai-Kowase K, Owens GK. Forced expression of myocardin is not sufficient for induction of smooth muscle differentiation in multipotential embryonic cells. Arterioscler Thromb Vasc Biol. 2004;24:1596–1601. doi: 10.1161/01.ATV.0000137190.63214.c5. [DOI] [PubMed] [Google Scholar]
- 110.Yoshida T, Owens GK. Molecular determinants of vascular smooth muscle cell diversity. Circ Res. 2005;96:280–291. doi: 10.1161/01.RES.0000155951.62152.2e. [DOI] [PubMed] [Google Scholar]
- 111.Li L, Miano JM, Cserjesi P, Olson EN. SM22 alpha, a marker of adult smooth muscle, is expressed in multiple myogenic lineages during embryogenesis. Circ Res. 1996;78:188–195. doi: 10.1161/01.res.78.2.188. [DOI] [PubMed] [Google Scholar]
- 112.Zhang JC, Kim S, Helmke BP, Yu WW, Du KL, Lu MM, Strobeck M, Yu Q, Parmacek MS. Analysis of SM22alpha-deficient mice reveals unanticipated insights into smooth muscle cell differentiation and function. Mol Cell Biol. 2001;21:1336–1344. doi: 10.1128/MCB.2001.21.4.1336-1344.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hoofnagle MH, Neppl RL, Berzin EL, Teg Pipes GC, Olson EN, Wamhoff BW, Somlyo AV, Owens GK. Myocardin is differentially required for the development of smooth muscle cells and cardiomyocytes. Am J Physiol Heart Circ Physiol. 2011;300:H1707–H1721. doi: 10.1152/ajpheart.01192.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xie WB, Li Z, Miano JM, Long X, Chen SY. Smad3-mediated myocardin silencing: a novel mechanism governing the initiation of smooth muscle differentiation. J Biol Chem. 2011;286:15050–15057. doi: 10.1074/jbc.M110.202747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Doi H, Iso T, Shiba Y, Sato H, Yamazaki M, Oyama Y, Akiyama H, Tanaka T, Tomita T, Arai M, et al. Notch signaling regulates the differentiation of bone marrow-derived cells into smooth muscle-like cells during arterial lesion formation. Biochem Biophys Res Commun. 2009;381:654–659. doi: 10.1016/j.bbrc.2009.02.116. [DOI] [PubMed] [Google Scholar]
- 116.High FA, Zhang M, Proweller A, Tu L, Parmacek MS, Pear WS, Epstein JA. An essential role for Notch in neural crest during cardiovascular development and smooth muscle differentiation. J Clin Invest. 2007;117:353–363. doi: 10.1172/JCI30070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Doi H, Iso T, Sato H, Yamazaki M, Matsui H, Tanaka T, Manabe I, Arai M, Nagai R, Kurabayashi M. Jagged1-selective notch signaling induces smooth muscle differentiation via a RBP-Jkappa-dependent pathway. J Biol Chem. 2006;281:28555–28564. doi: 10.1074/jbc.M602749200. [DOI] [PubMed] [Google Scholar]
- 118.Iso T, Hamamori Y, Kedes L. Notch signaling in vascular development. Arterioscler Thromb Vasc Biol. 2003;23:543–553. doi: 10.1161/01.ATV.0000060892.81529.8F. [DOI] [PubMed] [Google Scholar]
- 119.Noseda M, Fu Y, Niessen K, Wong F, Chang L, McLean G, Karsan A. Smooth Muscle alpha-actin is a direct target of Notch/CSL. Circ Res. 2006;98:1468–1470. doi: 10.1161/01.RES.0000229683.81357.26. [DOI] [PubMed] [Google Scholar]
- 120.Domenga V, Fardoux P, Lacombe P, Monet M, Maciazek J, Krebs LT, Klonjkowski B, Berrou E, Mericskay M, Li Z, et al. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev. 2004;18:2730–2735. doi: 10.1101/gad.308904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tang Y, Urs S, Boucher J, Bernaiche T, Venkatesh D, Spicer DB, Vary CP, Liaw L. Notch and transforming growth factor-beta (TGFbeta) signaling pathways cooperatively regulate vascular smooth muscle cell differentiation. J Biol Chem. 2010;285:17556–17563. doi: 10.1074/jbc.M109.076414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kurpinski K, Lam H, Chu J, Wang A, Kim A, Tsay E, Agrawal S, Schaffer DV, Li S. Transforming growth factor-beta and notch signaling mediate stem cell differentiation into smooth muscle cells. Stem Cells. 2010;28:734–742. doi: 10.1002/stem.319. [DOI] [PubMed] [Google Scholar]
- 123.Kennard S, Liu H, Lilly B. Transforming growth factor-beta (TGF- 1) down-regulates Notch3 in fibroblasts to promote smooth muscle gene expression. J Biol Chem. 2008;283:1324–1333. doi: 10.1074/jbc.M706651200. [DOI] [PubMed] [Google Scholar]
- 124.Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10:126–139. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- 125.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chan MC, Hilyard AC, Wu C, Davis BN, Hill NS, Lal A, Lieberman J, Lagna G, Hata A. Molecular basis for antagonism between PDGF and the TGFbeta family of signalling pathways by control of miR-24 expression. EMBO J. 2010;29:559–573. doi: 10.1038/emboj.2009.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, Deitch EA, Huo Y, Delphin ES, Zhang C. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res. 2009;105:158–166. doi: 10.1161/CIRCRESAHA.109.197517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circ Res. 2007;100:1579–1588. doi: 10.1161/CIRCRESAHA.106.141986. [DOI] [PubMed] [Google Scholar]
- 129.Liu X, Cheng Y, Zhang S, Lin Y, Yang J, Zhang C. A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res. 2009;104:476–487. doi: 10.1161/CIRCRESAHA.108.185363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A. Induction of microRNA-221 by platelet-derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype. J Biol Chem. 2009;284:3728–3738. doi: 10.1074/jbc.M808788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A. Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol Cell. 2010;39:373–384. doi: 10.1016/j.molcel.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kato M, Putta S, Wang M, Yuan H, Lanting L, Nair I, Gunn A, Nakagawa Y, Shimano H, Todorov I, et al. TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat Cell Biol. 2009;11:881–889. doi: 10.1038/ncb1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sun Q, Zhang Y, Yang G, Chen X, Zhang Y, Cao G, Wang J, Sun Y, Zhang P, Fan M, et al. Transforming growth factor-beta-regulated miR-24 promotes skeletal muscle differentiation. Nucleic Acids Res. 2008;36:2690–2699. doi: 10.1093/nar/gkn032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, Richardson JA, Bassel-Duby R, Olson EN. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009;23:2166–2178. doi: 10.1101/gad.1842409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, Srivastava D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705–710. doi: 10.1038/nature08195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Long X, Miano JM. Transforming growth factor-beta1 (TGF-beta1) utilizes distinct pathways for the transcriptional activation of microRNA 143/145 in human coronary artery smooth muscle cells. J Biol Chem. 2011;286:30119–30129. doi: 10.1074/jbc.M111.258814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Xie C, Ritchie RP, Huang H, Zhang J, Chen YE. Smooth muscle cell differentiation in vitro: models and underlying molecular mechanisms. Arterioscler Thromb Vasc Biol. 2011;31:1485–1494. doi: 10.1161/ATVBAHA.110.221101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yamamoto K, Morishita R, Tomita N, Shimozato T, Nakagami H, Kikuchi A, Aoki M, Higaki J, Kaneda Y, Ogihara T. Ribozyme oligonucleotides against transforming growth factor-beta inhibited neointimal formation after vascular injury in rat model: potential application of ribozyme strategy to treat cardiovascular disease. Circulation. 2000;102:1308–1314. doi: 10.1161/01.cir.102.11.1308. [DOI] [PubMed] [Google Scholar]
- 139.Kanzaki T, Tamura K, Takahashi K, Saito Y, Akikusa B, Oohashi H, Kasayuki N, Ueda M, Morisaki N. In vivo effect of TGF- beta 1. Enhanced intimal thickening by administration of TGF- beta 1 in rabbit arteries injured with a balloon catheter. Arterioscler Thromb Vasc Biol. 1995;15:1951–1957. doi: 10.1161/01.atv.15.11.1951. [DOI] [PubMed] [Google Scholar]
- 140.Orlandi A, Ropraz P, Gabbiani G. Proliferative activity and alpha-smooth muscle actin expression in cultured rat aortic smooth muscle cells are differently modulated by transforming growth factor-beta 1 and heparin. Exp Cell Res. 1994;214:528–536. doi: 10.1006/excr.1994.1290. [DOI] [PubMed] [Google Scholar]
- 141.Deng HB, Jiang CQ, Tomlinson B, Liu B, Lin JM, Wong KS, Cheung BM, Lam TH, Thomas GN. A polymorphism in transforming growth factor-β1 is associated with carotid plaques and increased carotid intima-media thickness in older Chinese men: the Guangzhou Biobank Cohort Study-Cardiovascular Disease Subcohort. Atherosclerosis. 2011;214:391–396. doi: 10.1016/j.atherosclerosis.2010.11.025. [DOI] [PubMed] [Google Scholar]
- 142.Rao M, Guo D, Jaber BL, Tighiouart H, Pereira BJ, Balakrishnan VS. Transforming growth factor-beta 1 gene polymorphisms and cardiovascular disease in hemodialysis patients. Kidney Int. 2004;66:419–427. doi: 10.1111/j.1523-1755.2004.00748.x. [DOI] [PubMed] [Google Scholar]
- 143.Saltis J, Agrotis A, Bobik A. Regulation and interactions of transforming growth factor-beta with cardiovascular cells: implications for development and disease. Clin Exp Pharmacol Physiol. 1996;23:193–200. doi: 10.1111/j.1440-1681.1996.tb02595.x. [DOI] [PubMed] [Google Scholar]
- 144.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Grainger DJ, Mosedale DE, Metcalfe JC, Böttinger EP. Dietary fat and reduced levels of TGFbeta1 act synergistically to promote activation of the vascular endothelium and formation of lipid lesions. J Cell Sci. 2000;113(Pt 13):2355–2361. doi: 10.1242/jcs.113.13.2355. [DOI] [PubMed] [Google Scholar]
- 146.Mallat Z, Gojova A, Marchiol-Fournigault C, Esposito B, Kamaté C, Merval R, Fradelizi D, Tedgui A. Inhibition of transforming growth factor-beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ Res. 2001;89:930–934. doi: 10.1161/hh2201.099415. [DOI] [PubMed] [Google Scholar]
- 147.Cipollone F, Fazia M, Mincione G, Iezzi A, Pini B, Cuccurullo C, Ucchino S, Spigonardo F, Di Nisio M, Cuccurullo F, et al. Increased expression of transforming growth factor-beta1 as a stabilizing factor in human atherosclerotic plaques. Stroke. 2004;35:2253–2257. doi: 10.1161/01.STR.0000140739.45472.9c. [DOI] [PubMed] [Google Scholar]
- 148.McCaffrey TA, Consigli S, Du B, Falcone DJ, Sanborn TA, Spokojny AM, Bush HL. Decreased type II/type I TGF-beta receptor ratio in cells derived from human atherosclerotic lesions. Conversion from an antiproliferative to profibrotic response to TGF-beta1. J Clin Invest. 1995;96:2667–2675. doi: 10.1172/JCI118333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890–1900. doi: 10.1016/s0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- 150.Kirby ML, Waldo KL. Role of neural crest in congenital heart disease. Circulation. 1990;82:332–340. doi: 10.1161/01.cir.82.2.332. [DOI] [PubMed] [Google Scholar]
- 151.Kochilas L, Merscher-Gomez S, Lu MM, Potluri V, Liao J, Kucherlapati R, Morrow B, Epstein JA. The role of neural crest during cardiac development in a mouse model of DiGeorge syndrome. Dev Biol. 2002;251:157–166. doi: 10.1006/dbio.2002.0819. [DOI] [PubMed] [Google Scholar]
- 152.Tran-Fadulu V, Pannu H, Kim DH, Vick GW, Lonsford CM, Lafont AL, Boccalandro C, Smart S, Peterson KL, Hain JZ, et al. Analysis of multigenerational families with thoracic aortic aneurysms and dissections due to TGFBR1 or TGFBR2 mutations. J Med Genet. 2009;46:607–613. doi: 10.1136/jmg.2008.062844. [DOI] [PubMed] [Google Scholar]
- 153.Pannu H, Fadulu VT, Chang J, Lafont A, Hasham SN, Sparks E, Giampietro PF, Zaleski C, Estrera AL, Safi HJ, et al. Mutations in transforming growth factor-beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation. 2005;112:513–520. doi: 10.1161/CIRCULATIONAHA.105.537340. [DOI] [PubMed] [Google Scholar]
- 154.Inamoto S, Kwartler CS, Lafont AL, Liang YY, Fadulu VT, Duraisamy S, Willing M, Estrera A, Safi H, Hannibal MC, et al. TGFBR2 mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovasc Res. 2010;88:520–529. doi: 10.1093/cvr/cvq230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Folkow B. Physiological aspects of primary hypertension. Physiol Rev. 1982;62:347–504. doi: 10.1152/physrev.1982.62.2.347. [DOI] [PubMed] [Google Scholar]
- 156.Rubin LJ. Primary pulmonary hypertension. N Engl J Med. 1997;336:111–117. doi: 10.1056/NEJM199701093360207. [DOI] [PubMed] [Google Scholar]
- 157.Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67:737–744. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 159.Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I, Simonneau G, Galie N, Loyd JE, Humbert M, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2001;345:325–334. doi: 10.1056/NEJM200108023450503. [DOI] [PubMed] [Google Scholar]
- 160.Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, Sheares KK, Trembath RC. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation. 2001;104:790–795. doi: 10.1161/hc3201.094152. [DOI] [PubMed] [Google Scholar]
- 161.Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med. 1995;333:918–924. doi: 10.1056/NEJM199510053331407. [DOI] [PubMed] [Google Scholar]
- 162.McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- 163.Pece N, Vera S, Cymerman U, White RI, Wrana JL, Letarte M. Mutant endoglin in hereditary hemorrhagic telangiectasia type 1 is transiently expressed intracellularly and is not a dominant negative. J Clin Invest. 1997;100:2568–2579. doi: 10.1172/JCI119800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.ten Dijke P, Arthur HM. Extracellular control of TGFbeta signalling in vascular development and disease. Nat Rev Mol Cell Biol. 2007;8:857–869. doi: 10.1038/nrm2262. [DOI] [PubMed] [Google Scholar]