

ABSTRACT
We designed, constructed, and evaluated a prototype novel reporter system comprised of two functional cassettes: (i) the SP6 RNA polymerase gene under transcriptional control of a promoter active in mycobacteria and (ii) the consensus SP6 polymerase promoter that directs expression of an otherwise unexpressed sequence. We incorporated the reporter system into a mycobacteriophage for delivery into viable Mycobacterium tuberculosis, and introduction led to synthesis of an SP6 polymerase-dependent surrogate marker RNA that we detected by reverse transcriptase PCR (RT-PCR). The reporter confirmed the susceptibility profile of both drug-susceptible and drug-resistant M. tuberculosis strains exposed to first-line antitubercular drugs and required as little as 16 h of exposure to antibacterial agents targeting bacterial metabolic processes to accurately read the reaction. The reporter system translated the bacterial phenotype into a language interpretable by rapid and sensitive nucleic acid detection. As a phenotypic assay that works only on viable M. tuberculosis, it could be used to rapidly assess resistance to any drug, including drugs for which the mechanism of resistance is unknown or which result from many potential known (and unknown) genetic alterations.
IMPORTANCE
The ability to detect antibiotic resistance of slow-growing bacteria (i.e., Mycobacterium tuberculosis) is hampered by two factors, the time to detection (weeks to months) and the resistance mechanism (unknown for many drugs), delaying the appropriate treatment of patients with drug-resistant or multidrug-resistant tuberculosis (TB). The novel technique described in this article uses a unique surrogate nucleic acid marker produced by phage that infects M. tuberculosis to record phenotypic antibiotic susceptibility in less than a day.
Introduction
Many reporter systems, such as lac promoter-controlled β-galactosidase for blue/white selection of recombinant plasmids (1) or in vivo expression of fluorescent proteins (2, 3), are used as surrogate markers for biological phenomena that are not easy to measure. These reporter systems produce products that are easily and reliably detected. Reporter systems can also be introduced into bacteriophages, obligate intracellular viruses that infect bacteria and use their biosynthetic machinery to manufacture and release phage progeny. These recombinant phages can be used to accurately report metabolic activity of infected cells: a living cell can be co-opted for progeny production, while a dead cell cannot. Detection of specific phage products or phage progeny, therefore, indicates bacterial metabolic activity. Because antimicrobial drugs ultimately affect the metabolic capacity of a susceptible pathogen, antibiotics also have an effect on synthesis of both phage components (i.e., reporter enzymes) and phage progeny.
Due to a notoriously slow doubling time of Mycobacterium tuberculosis, standard antibiotic susceptibility tests using culture take several weeks to months to complete. To solve this problem, investigators generated a recombinant mycobacteriophage that expresses firefly luciferase. After addition of a luciferin substrate to phage-infected cells, light is produced if the cell is sufficiently metabolically active to support the phage infection and gene expression. If antibiotics added prior to phage infection are effective and disturb cell viability or metabolism, the drug susceptibility phenotype is detected as a reduction in the level of light produced compared to that for an untreated control. The assay works well in a research laboratory setting, with spiked and late-stage tuberculosis (TB) patient sputum, but the sputum processing procedure in clinical laboratories is harsh toward M. tuberculosis, killing a large percentage of the bacteria and reducing surface proteins necessary for phage attachment in those that survive. Thus, the optimal time for reading this assay was nearly a week (2, 4). Moreover, we and others found that the luciferase reporter phage assay required >104 bacteria to provide sufficient light for automated detection, and so it could not detect the low number of M. tuberculosis bacteria present in most patient samples (5). A recent adaptation of the luciferase reporter phage assay replaces luciferase with fluorescent proteins that can be detected by fluorescence microscopes used for TB sputum smear microscopy or by fluorescence-activated cell sorters (6). Sensitivity of this assay, when performed directly with clinical samples, is not yet established.
Other efforts to speed antibiotic susceptibility testing of M. tuberculosis have been explored. For example, a phage amplification assay using wild-type mycobacteriophage D29 detects M. tuberculosis in TB patient samples and assesses antibiotic susceptibility by measurement of phage progeny (7–9). This assay relies on two different mycobacteria: (i) M. tuberculosis in the patient’s sample, which is infected with the phage and, if viable, produces progeny, and (b) a fast-growing mycobacterium, grown to a bacterial lawn, on which the M. tuberculosis phage progeny are plated to produce phage plaques. The plaques estimate the number of phage progeny as a surrogate for the number of viable M. tuberculosis bacteria in the sample. This assay employs plaque formation on fast-growing Mycobacterium as the detection methodology, rather than sophisticated luminometers, but the complexity of the assay has limited its commercial application.
Although M. tuberculosis-specific nucleic acid tests can detect the small numbers of M. tuberculosis present in a patient sample, detection of drug resistance is restricted to just a few drugs; for most drugs, the various genes and genetic mutations responsible for clinical drug resistance are either unknown or too numerous to test in a simple assay. Moreover, there is now evidence that some forms of antibiotic evasion are epigenetic (10–12).
To address both sensitivity and timeliness of M. tuberculosis antibiotic susceptibility testing, we combined into a single assay the elements of both phenotypic and genotypic methods: a recombinant phage that reports the phenotypic effects antimicrobials exert on susceptible bacteria, as well as a nucleic acid marker produced by the phage to speed assay time and improve sensitivity. This prototype reporter phage encodes the SP6 RNA polymerase gene expressed by a promoter active in mycobacteria. Once synthesized and folded, SP6 polymerase transcribes a region under exclusive transcriptional control of the SP6 consensus promoter. The generation of this SP6 polymerase-dependent RNA is then detected using nucleic acid detection systems. The SP6 polymerase-dependent RNA is an artificial nucleic acid marker that has a unique sequence not present in nature and is termed a surrogate marker locus (SML); the reporter system incorporated into the phage that directs SML synthesis is the SML generation module (SGM). In this article, we describe the design, construction, and utility of a prototype SGM to identify viable M. tuberculosis and report resistance to first-line TB drugs. We document SGM insertion into a mycobacteriophage to create phSP6-ProPol, a prototype SGM phage, and demonstrate SML synthesis during SGM phage infection of mycobacteria to determine susceptibility to first-line anti-TB antibiotics. This is a report of proof of concept in a development program to create a new and novel type of reporter phage that allows rapid and ultrasensitive nucleic acid amplification technologies to quantify cell viability.
RESULTS
The prototype encoded the SP6 RNA polymerase gene expressed by a promoter active in mycobacteria. Once synthesized and folded, SP6 polymerase transcribed a region under exclusive transcriptional control of the SP6 consensus promoter. The generation of this SP6 polymerase-dependent RNA was then detected using nucleic acid detection systems.
Construction of phSP6-ProPol, a prototype SGM phage.
To create phSP6-ProPol, the prototype SGM phage, we inserted the SGM depicted in Fig. 1A into a targeting plasmid derived from phAE142 as described in Materials and Methods. This insertion resulted in replacement of the Pleft luciferase reporter cassette with the SGM. The targeting plasmid was then used to replace the NotI restriction fragment of phAE142 to create phSP6-ProPol (Fig. 1B). Infectious phage were subsequently created by transformation of phSP6-ProPol phasmid DNA purified from independent clones into the Mycobacterium smegmatis strain mc2 4502. We confirmed that SGM was present in the correct orientation in infectious phSP6-ProPol phage by the presence of a product of the expected size when phage eluted from plaques were amplified by PCR using a primer that hybridized to a site in TM4 outside the transgenic sequences (TM4-50133.52) in combination with the primer PLLF, which hybridized to the Pleft promoter present in the SGM. As a control, we amplified phSP6-ProPol phasmid DNA, with or without MP buffer, and observed a product of the correct size (Fig. 1C). The presence of MP buffer in the phasmid control amplification reaction resulted in reduced reaction efficiency and suggested that MP buffer may have reduced the amount of product obtained from amplification of infectious phage, since it was also used to elute phage from plugs of Noble agar. MP buffer present in the amplification reaction also slowed the migration of products during agarose gel electrophoresis compared to results with the phasmid DNA control. However, the PCR product was clearly distinct in size from that produced by amplification of infectious phAE142 phage (Fig. 1C). Figure 1D demonstrates that no major deletions of the SP6 RNA polymerase gene were present, because a single product consistent with the size of the full-length SP6 RNA polymerase open reading frame was obtained after PCR amplification of infectious phage with primers PLLU and PLLD.
FIG 1 .

Design and characterization of phSP6-ProPol. (A) The SGM was comprised of 2 sections contained within a XbaI-NotI restriction fragment: the SP6 RNA polymerase gene (SP6 RNA Pol) under transcriptional control of the mycobacteriophage L5 Pleft promoter (open star) and the consensus SP6 promoter fused to the SML-encoding sequence (filled star). The SP6-SML section is flanked by 2 transcription terminators: the upstream terminator (filled circle) is E. coli rrnBT2 and precludes basal transcription through the SML-encoding sequence by host RNA polymerase; the downstream terminator (open circle) is the SP6 RNA polymerase terminator from the region downstream of the SP6 phage major capsid subunit described by Dobbins et al. (22). After expression of SP6 RNA polymerase from Pleft, the SML-encoding sequence downstream of the SP6 promoter was transcribed by SP6 RNA polymerase. SP6-dependent transcription of the SML-encoding sequence constituted generation of the SML. SML RNA could then be amplified and detected using primers that bind the SML. (B) The TM4 genome is depicted by the solid black line at the top of the figure. Expression of phage genes occurs on one strand of the genome, and the direction of transcription is indicated by the dashed arrow above the phage genome. Transgenic functions inserted into TM4 are contained on a NotI fragment, which is indicated and expanded. phAE142 encodes an ampicillin resistance cassette (Ampr) and an origin of replication (oriE) for maintenance and selection of the phasmid in E. coli. phAE142 also encodes the luciferase open reading frame fused to Pleft. phSP6-ProPol was derived from phAE142 and replaced the luciferase-encoding XbaI-NotI fragment with the XbaI-NotI SGM. In addition, phSP6-ProPol contained a kanamycin resistance cassette (Kmr) in place of phAE142 Ampr. Pleft transcription occurred on the strand opposite the endogenous phage functions in both phAE142 and phSP6-ProPol. The binding sites and orientation of oligonucleotide primers Ul53-UpSt-113348 and Ul53-DnSt-112112 (UpSt and DnSt, respectively) used for detection of SML generation and those used to characterize transgene structure in phSP6-ProPol are indicated. (C) Phage eluted from primary (1°) plaques originating with transformation of 2 independent phSP6-ProPol-Kan phasmid DNA clones (#4 and #5), as well as phAE142 phasmid DNA, into mc2 4502 were added to a PCR with the primers PLLF and TM4-50133.52. Phasmid DNA with and without the addition of MP buffer was included as controls. PLLF and TM4-50133.52 were predicted to generate a 667-bp product using phSP6-ProPol as a template, compared to a 181-bp product when phAE142 was the substrate. Products were separated on a 2% agarose gel and visualized by ethidium bromide staining. Locations of DNA size markers are indicated. (D) Phage eluted from the 1° plaques in panel C were amplified using primers PLLU and PLLD, which were predicted to mediate amplification of 2,854-bp and 1,882-bp products in phSP6-ProPol and phAE142, respectively. Products were then separated on a 1% agarose gel and visualized by ethidium bromide staining. The locations of DNA size markers are indicated.
Detection of SML RNA in stocks of infectious phSP6-ProPol.
To define conditions that reduce SML RNA to undetectable levels in the phage preparation itself, we added either 5 ng or 1 ng of RNase A to 108 PFU of phSP6-ProPol and incubated the mixture at 37°C for 30 min. Intact RNA was then purified and treated with DNase I, and the SML was amplified by reverse transcriptase PCR (RT-PCR). Both concentrations of RNase A reduced SML RNA to undetectable levels (Fig. 2). In addition, murine RNase inhibitor (MRI) present prior to addition of RNase A completely precluded degradation of SML RNA by RNase A (Fig. 2). These results demonstrated that the amplification product was derived from RNA, and as little as 1 ng RNase A could reduce SML RNA present in the phage preparation to undetectable levels. Furthermore, up to 5 ng RNase A was inactivated by MRI. These conditions sufficiently inactivated RNase A and ensured that newly synthesized SML RNA was not degraded during a test to detect viable bacteria.
FIG 2 .

SML RNA present in phSP6-ProPol stocks. A crude phSP6-ProPol preparation (108 PFU) was either left untreated or treated with MRI to a final concentration of 1 U/μl. Either 5 ng (++) or 1 ng (+) RNase A was then added, and the mixture was incubated for 30 min at 37°C. After purification of RNA and digestion with DNase I, reverse transcription was carried out using the DnSt primer. After reverse transcription but prior to PCR, cDNA from each sample was diluted 1:10 to estimate a 10-fold signal reduction. The primers DnSt and UpSt were then used to amplify a 150-bp section of SML cDNA using PCR. Products were separated on a 2% agarose gel and visualized by ethidium bromide staining. The locations of DNA size markers are indicated.
Detection of SML generation during phSP6-ProPol infection of H37Rv.
To determine if phSP6-ProPol synthesized SML during infection of M. tuberculosis, we infected H37Rv bacteria (described in Materials and Methods) with RNase A-treated phSP6-ProPol. At 30 min postinfection, MRI was added. RNA was purified from infected cells immediately (time 0.5), and at 3 and 4 h postinfection. SML was amplified by RT-PCR. SML generation was below detectable levels at 0.5 h, indicating that residual SML RNA contained in the phSP6-ProPol stock was degraded by the addition of RNase A. At 3 h, new SML generation occurred, and the SML had increased by 4 h (Fig. 3).
FIG 3 .

SML detection occurs at 4 h postinfection (p.i.). H37Rv was infected with phSP6-ProPol. RNase A was added to phSP6-ProPol prior to initiation of infection, and MRI was added at 0.5 h p.i. At 0.5, 3, and 4 h p.i., RNA was purified, digested with DNase I, and amplified using RT-PCR. Products were then separated on a 2% agarose gel and visualized by ethidium bromide staining. The locations of DNA size markers are indicated.
Specificity of SML synthesis.
To ensure that the 150-bp RT-PCR product was not derived from mispriming of cellular RNA during reverse transcription, we mock infected M. tuberculosis H37Rv cells or infected them with RNase A-treated phSP6-ProPol. Mock-infected cells did not receive RNase A to ensure that all RNA in the sample, either intracellular or extracellular, was a candidate for mispriming. At 4 h postinfection, RNA was purified and amplified by RT-PCR using the primers DnSt and UpSt. The 150-bp SML amplification product was observed only in cells infected with phSP6-ProPol (Fig. 4) and was not a result of mispriming. An RT-PCR product was observed in mock-infected (no phage infection) M. tuberculosis, but it was significantly larger than the SML target and was clearly a result of nonspecific amplification of an M. tuberculosis RNA. This slower-migrating product was also observed in phage-infected cells but at significantly reduced levels compared to those in mock-infected cells. We hypothesize that this may be due to RNase A-mediated destruction of extracellular RNA from lysed cells present in the culture and/or destruction of intracellular host RNA by phage-encoded functions.
FIG 4 .
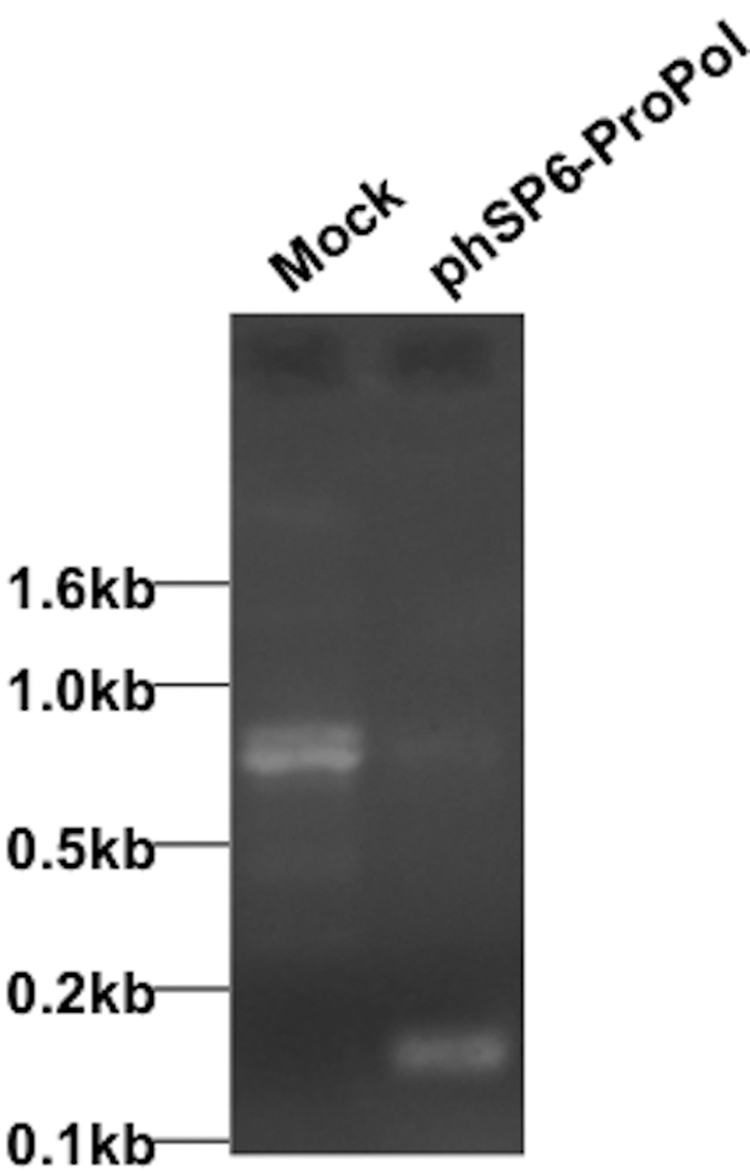
SML generation observed in phSP6-ProPol-infected cells. H37Rv cells were either mock infected or infected with phSP6-ProPol. RNA was purified at 4 h p.i., and SML generation was detected using RT-PCR. Products were then separated on a 2% agarose gel and visualized by ethidium bromide staining. The locations of DNA size markers are indicated.
Detection of RNA derived from promoters upstream of the SP6 promoter.
Although Fig. 4 demonstrates that SML RNA was synthesized during infection of M. tuberculosis, it was not clear if transcription was initiated by SP6 RNA polymerase at the SP6 promoter or by the host cell RNA polymerase from an upstream, cryptic phage promoter. It was important to verify that SML was transcribed by SP6 polymerase, because bacteriostatic antibiotics that inhibit host protein synthesis machinery (translation) leave the cellular transcriptional machinery relatively unaffected (13). With phage infection, this healthy machinery would immediately transcribe the SML if a cryptic host promoter was upstream of the consensus SP6 promoter. This could result in false-positive reports of resistance to a translational inhibitor drug. To determine if transcripts initiated from an upstream cryptic phage promoter synthesized SML, we purified RNA from phSP6-ProPol-infected H37Rv cells harvested at 4 h and amplified it by RT-PCR using one of 2 primer pairs. The first pair, consisting of DnSt and UpSt, mediated amplification of the 150-bp SML sequence. The second pair, comprised of DnSt and SP6-Dep-UpSt, mediated amplification of a 215-bp product. SP6-Dep-UpSt overlapped the SP6 promoter but not the transcription initiation site, so could not amplify an RT-PCR product if SML RNA transcription was initiated at the SP6 promoter. However, if a cryptic phage promoter mediated SML synthesis, a 215-bp RT-PCR product would be observed using this primer pair (Fig. 5A). In Fig. 5B, an RT-PCR product was obtained using the primers DnSt and UpSt. However, although the primers DnSt and SP6-Dep-UpSt mediated PCR amplification of the 215-bp product when the substrate was a plasmid containing the SGM, we observed no product when the substrate was RNA derived from phSP6-ProPol-infected H37Rv cells. These results showed that SML synthesis was initiated at the SP6 promoter and was dependent upon accumulation of SP6 RNA polymerase.
FIG 5 .

Detection of transcription by a cryptic promoter upstream of the SP6 promoter. (A) SP6 promoter-SML transcription unit and the locations of primers UpSt and DnSt, used to amplify SML RNA, are indicated. The primer SP6-Dep-UpSt overlaps the SP6 promoter and terminates one nucleotide 5′ to the transcription initiation site for SP6 polymerase. (B) RNA purified at 4 h p.i. from H37Rv M. tuberculosis infected with phSP6-ProPol was digested with DNase I prior to reverse transcription with the DnSt primer. A portion of cDNA was then left undiluted or diluted 1:10 and was PCR amplified using the primers DnSt and UpSt or DnSt and SP6-Dep-UpSt. The targeting plasmid pSP6-ProPol-Kan was included in separate amplification reactions as a control for successful PCR using each primer set. Products were then separated on a 2% agarose gel and visualized by ethidium bromide staining. The locations of DNA size markers are indicated.
SML phage reported H37Rv susceptibility to first-line anti-TB antibiotics.
To determine if phSP6-ProPol could accurately report susceptibility to first-line anti-TB drugs, we left M. tuberculosis H37Rv untreated or treated the bacteria for either 16 h or 40 h with rifampin (RIF), isoniazid (INH), streptomycin (STR), or ethambutol (EMB) at 37°C. The bacteria were then infected with phSP6-ProPol for 4 h. RNA was purified, followed by amplification of the SML using RT-PCR. Dilutions (10−1 and 10−2) of the untreated control cDNA were amplified to provide an estimation of SML reduction between untreated and drug-treated H37Rv. At both 16 and 40 h of drug exposure, we observed a clear difference in SML generation between the untreated and drug-treated samples (Fig. 6). Bacteria treated with rifampin and streptomycin generated no detectable SML after either 16 or 40 h of treatment; in bacteria treated with isoniazid and ethambutol, both cell wall-active drugs, SML synthesis was present in a small quantity after 16 h of drug treatment, but SML production was completely abolished by 40 h of drug treatment. The level of SML generation in the 16-h samples was approximately 10-fold lower than that for untreated controls, since the amplification product derived from isoniazid- and ethambutol-treated cells was less intense than that with a 10−1 dilution of cDNA from the untreated control. These data showed that the SGM encoded in phSP6-ProPol could report M. tuberculosis drug susceptibility in less than 1 day for metabolically active drugs and less than 2 days for drugs that affect cell wall synthesis.
FIG 6 .

phSP6-ProPol determined H37Rv susceptibility to first-line antitubercular antibiotics with a high signal-to-noise ratio. H37Rv was either left untreated or treated with rifampin, isoniazid, streptomycin, or ethambutol for 16 h or 40 h. Bacteria were then infected with phSP6-ProPol for 4 h. RNA was isolated, purified, and amplified using RT-PCR. The resultant product was separated by 2% agarose gel electrophoresis and was stained with ethidium bromide. Dilutions (1:10 and 1:100) of cDNA from the untreated controls were made prior to PCR amplification to provide a semiquantitative estimation of the effect of each drug on SML synthesis during infection of drug-susceptible M. tuberculosis. The locations and molecular weights of DNA size markers in lane M and the location of the SML signal are indicated.
phSP6-ProPol detects M. tuberculosis resistance to rifampin and ethambutol.
To determine if phSP6-ProPol could report drug-resistant M. tuberculosis, two mono-resistant strains, one RIFr and the other EMBr, were untreated or treated with rifampin and ethambutol for 16 h and then infected with phSP6-ProPol. At 4 h postinfection, RNA was purified and SML was amplified using RT-PCR. A dilution (10−1) of the cDNA derived from both the untreated and ethambutol-treated RIFr strain was amplified to provide an estimation of SML reduction by ethambutol (Fig. 7). Treatment of the RIFr strain with ethambutol (to which it is susceptible) resulted in an approximately 10-fold reduction in SML generation compared to results for the untreated control, whereas there was no observable reduction in SML generation after treatment with rifampin. Treatment of the EMBr strain with rifampin (to which it is susceptible) resulted in no detectable SML generation, reflecting the potent activity of rifampin in this assay; SML generation was not decreased after ethambutol treatment. These data demonstrated that rifampin and ethambutol resistance could be accurately detected by the SGM encoded in phSP6-ProPol after as little as 16 h of drug treatment.
FIG 7 .

phSP6-ProPol can detect M. tuberculosis drug resistance. RIFr M. tuberculosis and EMBr M. tuberculosis were either left untreated or treated with rifampin or ethambutol for 16 h. Bacteria were then infected with phSP6-ProPol for 4 h. RNA from each sample was then purified and amplified using RT-PCR. The resultant product was separated by 2% agarose gel electrophoresis and visualized by staining with ethidium bromide. A 1:10 dilution of cDNA derived from the untreated and ethambutol-treated phSP6-ProPol-infected RIFr strain was made prior to PCR amplification to provide a semiquantitative determination of the reduction of SML synthesis by ethambutol in this strain. The locations of DNA size markers are indicated.
DISCUSSION
For drugs whose resistance genes are known, nucleic acid tests can simultaneously provide microbial identification and resistance detection that can facilitate selection of effective drugs for individual patients. But the resistance mechanisms of most drugs either are very complex, affected by drug efflux rather than genetic changes, or are totally unknown. We designed and constructed a novel phage-based reporter system (the SGM) that could produce an engineered nucleic acid marker of cell viability and susceptibility to any active drug, the SML. When the SGM was incorporated into a mycobacteriophage, phage-infected M. tuberculosis produced SML in sufficient quantity to be easily amplified and detected. Moreover, when we treated M. tuberculosis with drugs to which the bacteria were susceptible, SML production was measurably diminished or undetectable.
Because rifampin and streptomycin interfere directly and immediately with phage gene expression, the reduction in SML production by susceptible M. tuberculosis can be detected as early as 16 h after introduction of the drug. Isoniazid and ethambutol do not have the same drug effect time course as rifampin or streptomycin, since they act on the cell wall. With a replication time of 24 h, exposure of M. tuberculosis to cell-wall-active drugs requires sufficient time to affect the integrity of the wall, so a longer drug exposure is required to observe a complete absence of SML. However, SML production was still substantially (>10-fold) reduced with these drugs at 16 h. We expect that antibiotics used to treat multidrug-resistant TB (MDR-TB) and extensively drug-resistant TB (XDR-TB), specifically fluoroquinolones and the injectable aminoglycosides, will exhibit SML production in a manner similar to that with rifampin and streptomycin, because they interfere directly with the host cell machinery required for phage gene expression. We plan ultimately to create a single test that can simultaneously diagnose MDR- and XDR-TB by interrogating M. tuberculosis in sputum for resistance to rifampin (a surrogate for MDR-TB) and both kanamycin and ciprofloxacin (a surrogate for XDR-TB).
The SGM is a new genetic approach to detect microbial drug susceptibility and resistance: it combines the sensitivity and accuracy of phenotypic antibiotic susceptibility testing (measures the activity of any drug) with the power of nucleic acid tests to detect very few microorganisms in a matter of hours. One study demonstrated that after treatment of M. tuberculosis cultures with isoniazid or rifampin, antigen 85B mRNA, which has a very short half-life, decreases rapidly and accurately reports drug susceptibility and resistance (14). To our knowledge the effectiveness of this approach has not been validated directly with clinical samples. It is possible that the complex mixture of M. tuberculosis nucleic acid from lysed, dying, and viable cells present in sputum may confound this approach. SGM phage may prove to be more suitable for interrogation of the drug susceptibility profile of M. tuberculosis present in clinical samples because the SML is an artificial sequence not present in sputum.
The prototype assay described in this report demonstrates that the SGM functioned as designed and accurately reported the drug susceptibility profile of M. tuberculosis. However, this is still a complex assay, and the limits of detection in clinical samples are unknown. For it to be universally useful as an antibiotic susceptibility test in a clinic, we realize that we must continue to refine the process and design a robust assay. Our goal is to evolve a rapid molecular diagnostic test to identify viable M. tuberculosis and rapidly and accurately report M. tuberculosis susceptibility to any drug to ensure a proper choice of effective drugs for patients. The prototype described above provides us with a road map and a set of tools to make that happen.
MATERIALS AND METHODS
Bacterial strains, phage, and propagation.
M. smegmatis mc2 4502 and phage phAE142 were obtained from William Jacobs at Albert Einstein College of Medicine (Table 1). phAE142 and phSP6-ProPol were propagated using mc2 4502 as described previously (5). H37Rv was maintained in 7H9 broth supplemented with Middlebrook oleic acid-albumin-dextrose-catalase (OADC) and 0.01% Tween 80. Electrocompetent Escherichia coli DH10B and chemically competent E. coli DH5α cells were obtained from Invitrogen Corporation. Both the rifampin-resistant (RIFr) and ethambutol-resistant (EMBr) M. tuberculosis strains used in this study were created and isolated from H37Rv by Sequella, Inc., and were maintained by the same protocols used for H37Rv. The MIC of RIFr M. tuberculosis to rifampin and EMBr M. tuberculosis to ethambutol was >32 µg/ml (each). Both resistant strains were susceptible to all other antituberculosis drugs tested (V. Reddy, personal communication).
TABLE 1 .
Mycobacteriophages, bacterial strains, plasmids, and oligonucleotides used in this study
| Mycobacteriophage, plasmid, strain, or oligonucleotide |
Description or sequence | Reference |
|---|---|---|
| Mycobacteriophage | ||
| phAE142 | Mycobacteriophage TM4-based luciferase reporter phage | 5 |
| phSP6-ProPol | Prototype SGM phage | This study |
| Plasmids | ||
| phMM-001 | Ampr phasmid derived from phAE142; maintained in E. coli | This study |
| pYUB-Bss | Ampr low-copy-number targeting vector encoding E. coli origin of replication and Pleft promoter-driven luciferase gene; derived from BssHII fragment of phMM-001 |
This study |
| pUC57-SP6-ProPol | Ampr high-copy-number E. coli plasmid containing SGM | This study |
| pSP6-ProPol | SGM from pUC57-SP6-ProPol inserted into pYUB-Bss in place of Pleft promoter and luciferase gene |
This study |
| pSP6-ProPol-Kan | pSP6-ProPol with Ampr gene replaced with Kanr gene | This study |
| Bacterial strains | ||
| DH5α | E. coli cloning strain | 23 |
| DH10B | E. coli cloning strain | 24 |
| mc2 4502 |
M. smegmatis strain constitutively expressing mycobacteriophage L5 gp71 polypeptide to downregulate Pleft promoter activity during growth of reporter phage stocks |
5 |
| RIFr M. tuberculosis strain | Rifampin-resistant H37Rv derivative generated at Sequella, Inc. | Unpublished |
| EMBr M. tuberculosis strain | Ethambutol-resistant H37Rv derivative generated at Sequella, Inc. | Unpublished |
| Oligonucleotides | ||
| UL53-DnSt-113348 | 5′ GACCCATGGGCGGGGTCGTT 3′ | This study |
| UL53-UpSt-112112 | 5′ AGTGCTTTGCCGCCAAATGC 3′ | This study |
| PLLF | 5′ GTGGCTGTCAAGCCCTAATC 3′ | This study |
| TM450133-52 | 5′ GTGTCGTGCTCGGTAACCTC 3′ | This study |
Molecular biology reagents.
BssHII, NotI, XbaI, Antarctic phosphatase, MRI, avian myeloblastosis virus reverse transcriptase, DNase I, and T4 DNA ligase were obtained from New England Biolabs. Taq polymerase and deoxyribonucleotides were obtained from GenScript Corporation. Custom oligodeoxyribonucleotides were synthesized by Eurofins MWG/Operon. The RNeasy kit with RNase-free water was obtained from Qiagen. Ampicillin, kanamycin, rifampin, isoniazid, streptomycin, ethambutol, and RNase A were obtained from Sigma-Aldrich.
Generation of phSP6-ProPol.
Mycobacteriophage TM4 is a well-characterized and versatile model for the construction and characterization of reporter mycobacteriophages (15–17). phAE142 is TM4 into which a transgenic cassette has been inserted and encodes firefly luciferase under the transcriptional control of the mycobacteriophage L5 Pleft promoter (5), as well as two elements derived from the cosmid cloning vector SuperCos-1 (5, 18–20) necessary for maintenance of the phage genome in E. coli as a phasmid: an origin of replication and an ampicillin resistance cassette. To create the phasmid phMM-001, phAE412 genomic DNA was prepared as described previously (21), circularized by intramolecular ligation with T4 DNA ligase, and transformed into electrocompetent DH10B according to the manufacturer’s directions. Transformants were selected on Luria broth agar supplemented with 100 µg/ml ampicillin. To create the targeting vector pYUB-Bss, which contains the transgenic fragments present in phAE142, phMM-001 DNA was isolated and digested with BssHII, which does not cut in the transgenic fragment. This digestion mixture was then diluted, and circular DNA molecules were generated by addition of T4 DNA ligase. The transgenic fragment was recovered by transformation into competent DH5α according to the manufacturer’s directions, followed by selection on Luria broth agar supplemented with ampicillin.
SGM synthesized de novo by GenScript Corporation was maintained in the plasmid pUC57 and designated pUC57-SP6-ProPol. To create phSP6-ProPol, we digested pUC57-SP6-ProPol with NotI and XbaI, followed by gel purification of the SGM. The SGM was then ligated into NotI- and XbaI-digested pYUB-Bss to create pSp6-ProPol. The ampicillin resistance gene in pSp6-ProPol was then replaced with the kanamycin resistance gene by lambda red recombineering (4) to create pSp6-ProPol-Kan. pSp6-ProPol-Kan was then digested with NotI, dephosphorylated by incubation with Antarctic phosphatase, gel purified, ligated with NotI-digested phMM-001, and electroporated into DH10B. Kanamycin-resistant colonies were expanded and assayed by PCR for correct insertion of the SGM into phMM-001 using the primers pLeft-LocusForward and TM450133-52. Phasmid DNA was then isolated and electroporated into electrocompetent mc2 4502 to create infectious phSP6-ProPol, as described elsewhere (21). Individual plaques were harvested and crude stocks of phSP6-ProPol were obtained by elution from confluently lysed agar Noble overlays using MP buffer (50 mM Tris, 150 mM NaCl, 10 mM MgCl2, 2 mM CaCl2 [pH = 7.6]). Phage were maintained at 2 × 109 PFU/ml in MP buffer at 4°C.
Preparation of M. tuberculosis prior to phage infection.
Prior to phage infection, cultures of H37Rv were washed 3 times with 7H9 broth supplemented with OADC, diluted to an optical density at 600 nm (OD600) of 0.1 to 0.2, and incubated overnight at 37°C with shaking. For susceptibility testing, antibiotics were added immediately after washing to a final concentration of 2 µg/ml rifampin, 0.2 µg/ml isoniazid, 2 µg/ml streptomycin, and 7.5 µg/ml ethambutol.
phSP6-ProPol infection.
At phage infection, we added 5 ng of RNase A to 100 µl phSP6-ProPol, and 10 µl of phSP6-ProPol containing RNase A was added to 100 µl washed H37Rv, RIFr M. tuberculosis, or EMBr M. tuberculosis and incubated at 37°C. MRI was subsequently added between 0.5 h and 2 h postinfection.
RNA purification.
RNA released into the culture supernatant after lysis of phage-infected cells by phage-encoded lytic functions was purified using the RNeasy kit according to the manufacturer’s directions. Briefly, buffer RLT was added to samples, followed by the addition of 100% ethanol, and RNA was purified using RNeasy spin columns. RNA was eluted in 50 µl RNase-free water, and MRI was added to a final concentration of 1 U/μl. After the addition of buffer RLT but before that of ethanol, samples containing M. tuberculosis strains were sterile filtered using 0.22-µm Acrodisc syringe filters (Pall Corporation).
RT-PCR.
Sixteen microliters of purified RNA was treated with 4 U DNase I for 30 min at 37°C in a total reaction volume of 20 µl. The solution was then adjusted to 5 mM EDTA and heated at 75°C for 10 min. Five microliters of DNase I-treated RNA was then reverse transcribed using 10 U reverse transcriptase for 30 min at 55°C with the primer UL53-DnSt-113348 at a final concentration of 1 µM in a total reaction volume of 20 µl. Two microliters of cDNA, or a dilution of the cDNA in water, was then added to a 25-µl hot-start PCR mixture using the primers UL53-DnSt-113348 and UL53-UpSt-112112 at final concentrations of 120 µM and 40 µM, respectively. Amplification was performed for 35 cycles using an annealing temperature of 55°C. Ten microliters of the amplification product was then separated by electrophoresis through a 2% agarose gel impregnated with 500 ng/ml ethidium bromide. An internal amplification control was not performed because each drug differentially affects the level of intracellular RNA in susceptible mycobacteria. Furthermore, each has variable effects on cell growth, leading to differences in bacterial numbers between the drug-treated samples. Normalizing the amount of RNA between all samples would therefore underreport the dynamic range of the assay.
ACKNOWLEDGMENTS
This work was supported by grant award 1R43AI077167-01 from NIAID and Sequella corporate funds.
Footnotes
Citation Mulvey MC, Sacksteder KA, Einck L, Nacy CA. 2012. Generation of a novel nucleic acid-based reporter system to detect phenotypic susceptibility to antibiotics in Mycobacterium tuberculosis. mBio 3(2):e00312-11. doi:10.1128/mBio.00312-11.
REFERENCES
- 1. Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor: Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 2. Chudakov DM, Lukyanov S, Lukyanov KA. 2005. Fluorescent proteins as a toolkit for in vivo imaging. Trends Biotechnol. 23:605–613 [DOI] [PubMed] [Google Scholar]
- 3. Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. 2002. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295:868–872 [DOI] [PubMed] [Google Scholar]
- 4. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bardarov S, Jr, et al. 2003. Detection and drug-susceptibility testing of M. tuberculosis from sputum samples using luciferase reporter phage: comparison with the Mycobacteria Growth Indicator Tube (MGIT) system. Diagn. Microbiol. Infect. Dis. 45:53–61 [DOI] [PubMed] [Google Scholar]
- 6. Piuri M, Jacobs WR, Jr, Hatfull GF. 2009. Fluoromycobacteriophages for rapid, specific, and sensitive antibiotic susceptibility testing of Mycobacterium tuberculosis. PLoS One 4:e4870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eltringham IJ, Wilson SM, Drobniewski FA. 1999. Evaluation of a bacteriophage-based assay (phage amplified biologically assay) as a rapid screen for resistance to isoniazid, ethambutol, streptomycin, pyrazinamide, and ciprofloxacin among clinical isolates of Mycobacterium tuberculosis. J. Clin. Microbiol. 37:3528–3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Park DJ, Drobniewski FA, Meyer A, Wilson SM. 2003. Use of a phage-based assay for phenotypic detection of Mycobacteria directly from sputum. J. Clin. Microbiol. 41:680–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rees CE, Loessner MJ. 2005. Phage for the detection of pathogenic Bacteria, p 267–285 In Kutter E, Sulakvelidze A, Bacteriophages: biology and applications. CRC Press, Boca Raton, FL [Google Scholar]
- 10. Adam M, Murali B, Glenn NO, Potter SS. 2008. Epigenetic inheritance based evolution of antibiotic resistance in bacteria. BMC Evol. Biol. 8:52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gumbo T, et al. 2007. Isoniazid’s bactericidal activity ceases because of the emergence of resistance, not depletion of Mycobacterium tuberculosis in the log phase of growth. J. Infect. Dis. 195:194–201 [DOI] [PubMed] [Google Scholar]
- 12. Srivastava S, et al. 2010. Efflux-pump-derived multiple drug resistance to ethambutol monotherapy in Mycobacterium tuberculosis and the pharmacokinetics and pharmacodynamics of ethambutol. J. Infect. Dis. 201:1225–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lembach KJ, Buchanan JM. 1970. The relationship of protein synthesis to early transcriptive events in bacteriophage T4-infected Escherichia coli B. J. Biol. Chem. 245:1575–1587 [PubMed] [Google Scholar]
- 14. Hellyer TJ, DesJardin LE, Hehman GL, Cave MD, Eisenach KD. 1999. Quantitative analysis of mRNA as a marker for viability of Mycobacterium tuberculosis. J. Clin. Microbiol. 37:290–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bardarov S, et al. 1997. Conditionally replicating mycobacteriophages: a system for transposon delivery to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 94:10961–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carrière C, et al. 1997. Conditionally replicating luciferase reporter phages: improved sensitivity for rapid detection and assessment of drug susceptibility of Mycobacterium tuberculosis. J. Clin. Microbiol. 35:3232–3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ford ME, Stenstrom C, Hendrix RW, Hatfull GF. 1998. Mycobacteriophage TM4: genome structure and gene expression. Tuber. Lung Dis. 79:63–73 [DOI] [PubMed] [Google Scholar]
- 18. Balasubramanian V, et al. 1996. Allelic exchange in Mycobacterium tuberculosis with long linear recombination substrates. J. Bacteriol. 178:273–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bange FC, Collins FM, Jacobs WR., Jr 1999. Survival of mice infected with Mycobacterium smegmatis containing large DNA fragments from Mycobacterium tuberculosis. Tuber. Lung Dis. 79:171–180 [DOI] [PubMed] [Google Scholar]
- 20. Brown KL, Sarkis GJ, Wadsworth C, Hatfull GF. 1997. Transcriptional silencing by the mycobacteriophage L5 repressor. EMBO J. 16:5914–5921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hatfull G, Jacobs WR., Jr 2000. Molecular genetics of mycobacteria. American Society for Microbiology, Washington, DC [Google Scholar]
- 22. Dobbins AT, et al. 2004. Complete genomic sequence of the virulent Salmonella bacteriophage SP6. J. Bacteriol. 186:1933–1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bethesda-Research-Laboratories 1986. BRL pUC host: E. coli DH5α competent cells. Focus 8:9 [Google Scholar]
- 24. Grant SG, Jessee J, Bloom FR, Hanahan D. 1990. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc. Natl. Acad. Sci. U. S. A. 87:4645–4649 [DOI] [PMC free article] [PubMed] [Google Scholar]