

Abstract
Maintenance of the cellular redox balance has vital importance for correcting organism functioning. Methionine sulfoxide reductases (Msrs) are among the key members of the cellular antioxidant defence system. To work properly, methionine sulfoxide reductases need to be reduced by their biological partner, thioredoxin (Trx). This process, according to the available kinetic data, represents the slowest step in the Msrs catalytic cycle. In the present paper, we investigated structural aspects of the intermolecular complex formation between mammalian MsrB1 and Trx. NMR spectroscopy and biocomputing were the two mostly used through the research approaches. The formation of NMR detectable MsrB1/Trx complex was monitored and studied in attempt to understand MsrB1 reduction mechanism. Using NMR data, molecular mechanics, protein docking, and molecular dynamics simulations, it was found that intermediate MsrB1/Trx complex is stabilized by interprotein β-layer. The complex formation accompanied by distortion of disulfide bond within MsrB1 facilitates the reduction of oxidized MsrB1 as it is evidenced by the obtained data.
1. Introduction
Oxygen is vital for all aerobic biological processes. However, about 5% of it is converted into reactive oxygen species (ROS) [1]. Methionine residues in proteins are susceptible to oxidation by reactive oxygen and nitrogen species leading to formation of methionine sulfoxide (MetSO). This modification can result in loss of proteins' function [2]. Organisms developed a complex antioxidant defence system that includes enzymatic and nonenzymatic antioxidants [3–5]. Methionine sulfoxide reductases (Msrs) are redox repairing enzymes which reduce MetSO back to methionine (Met). A number of published reports describe the role of methionine sulfoxide reductases in antioxidant defence and the regulation of protein function [6–10]. Methionine sulfoxide reductases reduce both free and protein-bound MetSO, back to Met in the presence of thioredoxin (Trx). Two distinct families constitute Msrs: MsrA, which specifically reduces S-epimer, and MsrB, which is stereospecific for R-epimer of methionine sulfoxide [11–14].
Mammalian MsrB enzymes constitute three different subclasses: MsrB1, MsrB2, and MsrB3 [15–20]. All of them contain Zn2+ ion, coordinated by two motifs CxxC (two cysteines separated by two residues), which stabilize their structure. Whereas mammalian MsrB1 contains a resolving cysteine (Cys4) and catalytic selenocysteine (Sec95) in its active site, the other two subclasses, MsrB2 and MsrB3, in analogy with bacterial MsrBs, contain only catalytic cysteine [21–24]. NMR solution [25] and X-ray crystal (PDB entry 3MAO) structures of mammalian MsrB1 have recently been determined.
Reduction of methionine sulfoxide by selenocysteine containing MsrB1 is a multistep process [24, 25]. First, a catalytic Sec95 attacks a sulfoxide moiety of the substrate, MetSO, resulting in the formation of selenenic acid intermediate and concomitant release of methionine. Second, a resolving Cys4 attacks the selenenic acid intermediate to form an intramolecular selenide-sulfide bond, and finally, a fully reduced enzyme is regenerated by reduction of the latter bond with thioredoxin, a natural electron donor (Scheme 1). It has been established that thioredoxin is the natural reducing agent for the members of Msr family, though DTT can be used as the reductant in vitro [14, 24, 26]. Reduction of oxidized MsrB2 and MsrB3 as well as of the most bacterial MsrBs occurs through a similar mechanism without formation of the intramolecular disulfide bridge. However, it was reported that bacterial MsrB from Neisseria meningitides upon oxidation form intramolecular disulfide bridge [27]. It was shown that its reduction by Trx is overall rate-limiting step of MsrB cycle and it is described as an apparent irreversible process.
Scheme 1.
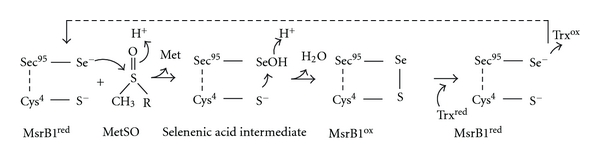
The biochemical cycle of selenoprotein MsrB1.
Thioredoxin is a ubiquitous protein that plays an important role in maintaining redox balance in cells [28]. In most of its reactions, Trx reduces substrate disulfide bond. The proposed mechanism of Trx-catalysed substrate disulfide reduction [29–31] includes nucleophilic attack by thiolate of Cys32 supported by hydrophobic interactions resulting in transient mixed disulfide formation followed by nucleophilic attack of the resolving Cys35 generating Trx-S2 and reduced protein.
Summarising, reduction of MsrBs (mammalian and bacterial) disulfide bond by Trx, can be described by the following steps. First, thioredoxin and MsrB form an intermolecular complex. In the second step, the just-formed intermolecular bond gets resolved by the thioredoxin resolving cysteine 35, followed by the formation of reduced MsrB and oxidized Trx. Subsequently, the S–S bridge of oxidized thioredoxin is further reduced by NADPH and the specific FAD-containing enzyme thioredoxin reductase. Some studies suggest that specific structural recognitions exist between oxidized substrate disulfide bond and reduced Trx [14, 32]. The knowledge of the structure aspects of the MsrB/Trx complex formation would help to understand full catalytic mechanism of methionine sulfoxide reductases.
Here, we have investigated the structural aspects of the interaction between mammalian oxidized MsrB1 and reduced thioredoxin. Mutants of MsrB1 and Trx were produced in order to get an intermolecular complex suitable for its investigation using NMR spectroscopy. Firstly, since Sec-containing MsrB1 is not possible to express in bacterial cells, Sec95 of MsrB1 was replaced by Cys giving Sec95Cys mutant (hereafter MsrB1). Further, thioredoxin containing a Cys35Ser mutation (hereafter Trx) was obtained as this modification removes the second resolving Cys35 leading to the formation of a detectable intermediate MsrB1-Trx (Scheme 2). The resultant MsrB1-Trx complex is stable and could, therefore, be structurally characterized by NMR spectroscopy and with the aid of computational approach.
Scheme 2.
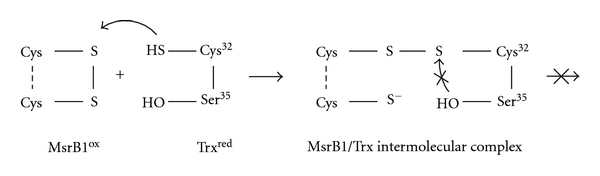
MsrB1ox-Trxred complex formation. Here, Cys35 of Trx is replaced with catalytically (redox) inactive Ser residue.
2. Materials and Methods
2.1. Microorganisms and Plasmids
The genes of C-terminal his-tagged mouse MsrB1 (Sec95Cys) and human Trx (Cys35Ser) mutants, cloned into pET21 expression vectors were kindly provided by Professor V. Gladyshev's group. Escherichia coli strains ER2566 (New England Biolabs) and BL21 (DE3) (Novagen) cells were respectively transformed with the constructs using the standard protocol [33]. The transformed cells were spread on several LB agar plates containing 100 mg/L of ampicillin and were further stored at 4°C.
2.2. Protein Expression, Purification, and Sample Preparation
E. coli BL21(DE3) cells carrying plasmid pET21-Trx and E. coli ER2566 cells containing pET21-MsrB1 were grown in LB-media with 100 mg/L ampicillin. To uniformly label proteins either with 15N or with 15N-13C, cells were grown in M9 minimal media containing 1 g/L 99%-enriched 15NH4Cl, 4 g/L 13C6-glucose and 100 mg/L of ampicillin. MsrB1 and Trx were expressed, by growing host cells at 37°C until an OD600 reached 0.8, followed by induction of protein synthesis with 1 mM IPTG and subsequent incubation for 3 h at 37°C. Cells were harvested by centrifugation and disrupted by sonication in 50 mM phosphate buffer, pH 8.0, containing 400 mM NaCl, 0.01% Tween 20 (Sigma Aldrich); 5 mM β-mercaptoethanol as well as complete protease inhibitor cocktail (Roche) (1 tab for 50 mL).
Cell extract was clarified by centrifugation and filtering through 0,45 μm filter. Clear supernatant was loaded onto the preequilibrated in binding buffer (50 mM Na phosphate buffer, pH 8.0, 400 mM NaCl, 5 mM β-mercaptoethanol, and 5 mM imidazole) Ni-NTA column (Novagen). The column was step-washed with the increasing concentration of imidazole (5, 10, and 20 mM) in binding buffer followed by elution of protein with 250 mM imidazole in binding buffer. The purity of the samples was examined with SDS-PAGE gel (Invitrogen). The yield of the proteins per liter of growth medium was 20 mg and 10 mg for MsrB1 and Trx, respectively.
The obtained reduced MsrB1 was further subjected to oxidation by excess of dabsyl-Met-R-SO (kindly provided by Professor H-Y. Kim) for 3 h at 25°C in 20 mM phosphate buffer, 20 mM NaCl, and pH 7.5. The final NMR MsrB1ox and Trxred samples contained 1,5–2 mM of protein in 10 mM NaCl, 10 mM phosphate, pH 5.5, 90%H2O, 10% D2O buffer. The sample for the backbone assignment of reduced double enriched Trx (15N- and 13C) also contained 5 mM DTT.
2.3. NMR Spectroscopy
All NMR spectra were recorded at 298 K either on Bruker Avance 600 MHz spectrometer, equipped with 5 mm z-gradients TXI (H/C/N) cryoprobe at the NMR centre of NT faculty, NTNU or Varian DirectDrive NMR System 700 MHz spectrometer, equipped with 5 mm z-gradients salt tolerant H/C/N probe at the SPbSPU. Proton chemical shifts were referenced to external 3-(trimethylsilyl)-propane-sulfonic acid sodium salt (DSS), while 15N and 13C chemical shifts were referenced indirectly to a liquid ammonia and DSS, respectively, based on the absolute frequency ratios [34].
The comparison between reduced and oxidized forms of MsrB1 was performed by analyzing 2D 15N-1H HSQC spectra of both reduced and oxidized MsrB1. The spectra were acquired using pulse sequence from the standard pulse sequence library. N-H coupling constant was set to 90 ms, the relaxation delay in HSQC experiments was of 1 s. 2048 complex points were collected in F2 dimension, while 256 were collected in F1 dimension. 32 scans per each transient have been recorded.
1H, 13C, and 15N backbone resonance assignments for Trx protein were achieved using 15N HSQC, HNCA, CBCA(CO)NH, CBCANH, HBHANH, HBHA(CO)NH, and 15N NOESY NMR spectra from the standard pulse sequence library.
The NMR data were processed with the BRUKER XWinNMR version 3.5 and Varian VNMRJ version 2.2C software. Spectral analysis was performed using CARA version 1.8.4.2 [35].
In order to map the interacting sites of both Trx and MsrB1 proteins, that is, to determine which aminoacids are involved in the formation of the interprotein complex, NMR titration of each protein was performed. 15N-1H HSQC of 15N labeled oxidized MsrB1 was recorded followed by acquisition of the series of HSQC spectra of oxidized MsrB1 in the presence of increasing amounts of 15N-unlabeled Trx. Likewise, the 15N-1H HSQC spectrum of 15N-enriched pure Trx was recorded first, followed by the set of HSQC spectra of Trx containing an increasing amount of unlabeled oxidized MsrB1. All titration experiments were carried out three times at 298 K at two different pH values 5.5 and 6.5 in order to have statistically significant results.
2.4. Molecular Modelling
Structural calculations for MsrB1 and Trx and their complex were performed using molecular modelling techniques, including molecular mechanics and protein docking (Molsoft ICM Pro 3.6 program package [36], ECEPP/3 force field [37]) as well as molecular dynamics simulations (GROMACS program package version 4.0.7 [38], G53a6 force field [39]). VMD [40] and ICM Pro 3.6 program packages were used to visualize and analyze molecular dynamics (MD) trajectories and the resulting averaged spatial structures of the proteins and their complexes. The initial sets of atomic coordinates of the proteins (reduced forms) were taken from PDB (2kv1 and 3trx for MsrB1 and Trx, resp.).
MD simulations were carried out using a standard protocol including the following steps: energy minimization, water box equilibration, and productive run. Both structures of MsrB1 protein and four complexes of its oxidized form with reduced Trx were placed in dodecahedral water box. The box dimensions were chosen in such a way that water shell around the protein structures was no less than 12 Å. Water molecules were represented by SPC model [41]. The intrinsic MsrB1 positive charge was neutralized with two Cl− ions, and the negative charge of the protein complex was neutralized by three Na+ ions. The energy minimization procedure was executed with steepest descent method. During 300 ps equilibration of the water molecules surrounding protein, spatial position of its atoms was constrained. The equilibration procedure was followed by two productive runs with 5 ns and 20 ns in order to separate proteins and their complexes, respectively. Distances between Zn2+ ion and Sγ atoms of the coordinating cysteines of MsrB1 (Cys23, Cys26, Cys71, and Cys74) were restricted with help of harmonic potential having a minimum at a value of 2.35 Å. The averaged molecular structures were obtained by the cluster analysis [42] after each productive run of MD.
MD integration step was equal to 2 fs. LINCS algorithm [43] was used to constrain the covalent bond distances and the valence angle values. The temperature of the system was held at 300 K with velocity-rescale algorithm [44]. To hold system pressure at 1 atm value, Berendsen barostate [45] with the time constant for coupling, τ = 0.5 ps was used. Long-range electrostatics was calculated with PME method [46]. The cut-off distance of nonbonded interactions was set to 10 Å for van der Waals interactions and to 14 Å for electrostatic interactions.
To obtain the disulfide bond between Cys95 and Cys4 residues in oxidized MsrB1, MD simulations in explicit water box with the described above protocol were used, except that the distance between Sγ atoms of two cysteines was restrained at 2.8 Å with the force exceeding van der Waals repulsion of these atoms. The disulfide bond itself was formed and its geometry was optimized by molecular mechanic procedures in ECEPP/3 force field implemented in ICM Pro 3.6 program package. To construct the mutant form Cys35Ser of Trx, the side chain of the mutated aminoacid together with its neighbours situated within 5 Å radius, were subjected to the energy minimization procedure in ECEPP/3 potential. The average structures of oxidized MsrB1 and reduced Trx have been subsequently subjected to the protein-protein docking in ICM Pro 3.6. This algorithm uses optimal docking area parameter representing the protein surface regions with the maximum dehydration energy arising upon formation of the tight protein-protein contact. To reduce conformational sampling calculations, only the “hot points” that were situated near MsrB1 and Trx active sites were used within the study. Interaction energies were calculated using potential grids with 0.5 Å cells, and the truncated van der Waals potential with maximum 1.0 kcal/mol was used.
The conformations of MsrB1-Trx complex, obtained from docking, having a representative distance between Sγ Cys95 of MsrB1 and Sγ C32 of Trx less than 10 Å, were taken for further analysis and sorted into four groups depending on the mutual orientations of the two proteins. Then, the representative conformations from each of four groups were refined by the above-described molecular dynamics simulation methods.
3. Results and Discussion
3.1. Assignment of Oxidized MsrB1
Upon oxidation, MsrB1 undergoes structural changes caused by the intramolecular Cys95/Cys4 disulfide bond formation [25]. In order to assign 1H and 15N NMR spectra of the oxidized MsrB1, the HSQC spectra of MsrB1 protein in both oxidized and reduced states were compared [25, 47].
This analysis revealed close similarity between the HSQC spectra of both redoxed forms: the majority of the cross-peaks assigned in the case of reduced MsrB1 protein remained either at the same position in 1H-15N HSQC spectra or their positions were slightly altered. This overall spectral correspondence between reduced and oxidized forms of MsrB1 allowed us to unambiguously identify and assign the cross-peaks related to the most of the aminoacids of the oxidized MsrB1. Nevertheless, the signal broadening and the shift of some of the cross-peaks in the oxidized protein corresponding to the residues either belonging to the protein active site or situated in its vicinity were observed (Cys4, Phe82, Cys95, and Ile96). The performed comparison provided us with the assignment of the 1H and 15N chemical shifts of oxidized MsrB1 protein and indicated that no major structural changes occur in MsrB1 protein upon oxidation.
3.2. Assignment of Reduced Trx
The protein's backbone assignment was performed using a standard procedure. The availability of the assignment of native human Trx (313 K, pH = 5.5) [48] assisted in the obtainment of the present assignment. As a result, 95% of all Trx aminoacids were identified and assigned in the present study. The backbone assignments of Trx's 1H, 13C, and 15N nuclei were deposited in BioMagResBank under accession number BMRB-16850. The obtained within the study assignments for both oxidized MsrB1 and reduced Trx provided a basis for our further investigations on interaction between Trx and MsrB1.
3.3. Monitoring of Interaction of Oxidized MsrB1-Trx
NMR spectroscopy is a commonly used technique for mapping the interacting site of a protein upon complex formation with its ligands. Within the study, it was performed the NMR titration of oxidized MsrB1 with reduced Trx and vice versa. To determine whether the interaction of these proteins can be observed on NMR time scale and, in the positive case, to determine which aminoacids from those proteins are involved in the interaction, the following approach was used. 15N-enriched protein (either MsrB1 or Trx) was titrated with nonenriched partner, and their interaction was monitored by comparative analysis of 1H-15N HSQC spectra of a single protein and the protein in the presence of its partner. Since only one protein was enriched with 15N NMR active isotope in each single experiment, the presence in solution of another protein did not hamper the observation of 1H-15N spectrum under analysis, and it was possible to clearly identify aminoacids changing their NMR parameters (either chemical shifts (shifting of the cross-peak) or relaxation rates (broadening of the cross-peak)), which indicated that these residues are involved in interaction with another protein. This analysis allowed us to determine which aminoacids from each protein are directly involved in the complex formation.
3.4. The NMR Titration of Oxidized MsrB1 with Reduced Trx
The titration of MsrB1 revealed its aminoacids involved in binding with Trx. The integral chemical shift differences calculated for all MsrB1 residues using the relationship Δ1H + Δ15N/7 (Δ1H + Δ15N/5 for glycine residues) [49] and plotted as a function of the aminoacid number, are shown in Figure 1(a). These data show that the residues belonging to the MsrB1 active site as well as their neighbouring residues directly participate in interaction with reduced Trx (C4-F7, E65, and C95-F97). It was also observed that the MsrB1 HSQC cross peaks corresponding to the residues revealing the maximal shifts upon interaction with Trx became broader (increase of transverse relaxation rates), thus supporting our finding in pinpointing of the interaction relevant aminoacids. Altogether, our studies indicate that these MsrB1 residues are involved in protein complex formation with Trx.
Figure 1.
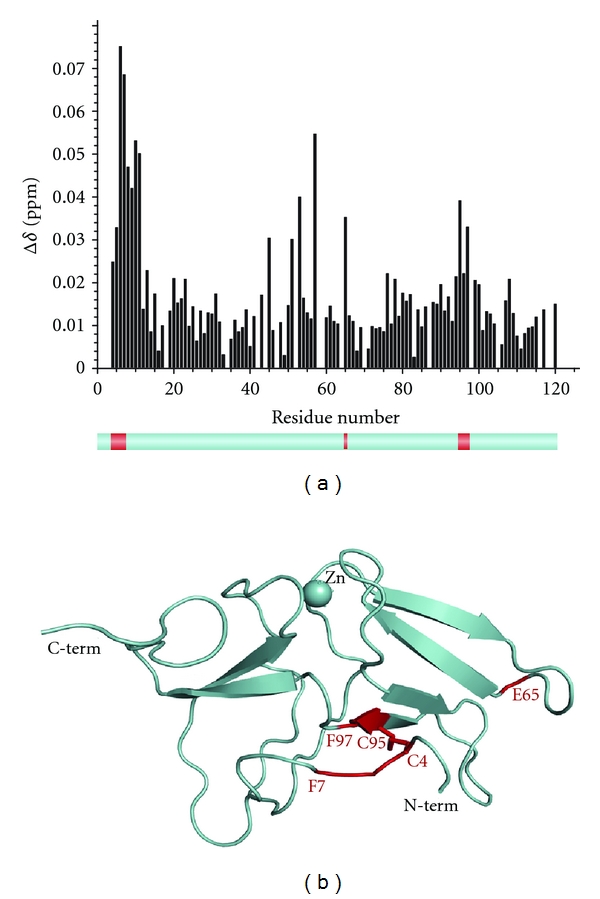
(a) Chemical shift difference observed for each residue of MsrB1 upon NMR titration by Trx. Lower panel shows in red the residues belonging to the protein active site revealing maximal changes upon titration. (b) The 3D structure of oxidized mammalian MsrB1. “Hot points” used for molecular docking are evidenced in red.
3.5. The NMR Titration of Reduced Trx with Oxidized MsrB1
The integral 1H-15N HSQC chemical shift differences for all Trx residues plotted as a function of the aminoacid number were obtained as described above and are shown in Figure 2(a). The titration of Trx has revealed three aminoacid areas which are involved in interaction with MsrB1: D26-S35, V57-E68, and K72-M74. These segments are evidenced in red on the lower panel of Figure 2(a). Our results indicate that: (i) Trx active site (as expected) is involved in the interaction; (ii) the monitored segments are situated mostly on the Trx external loops; (iii) these segments are rather close in space, thus supporting and validating our findings.
Figure 2.
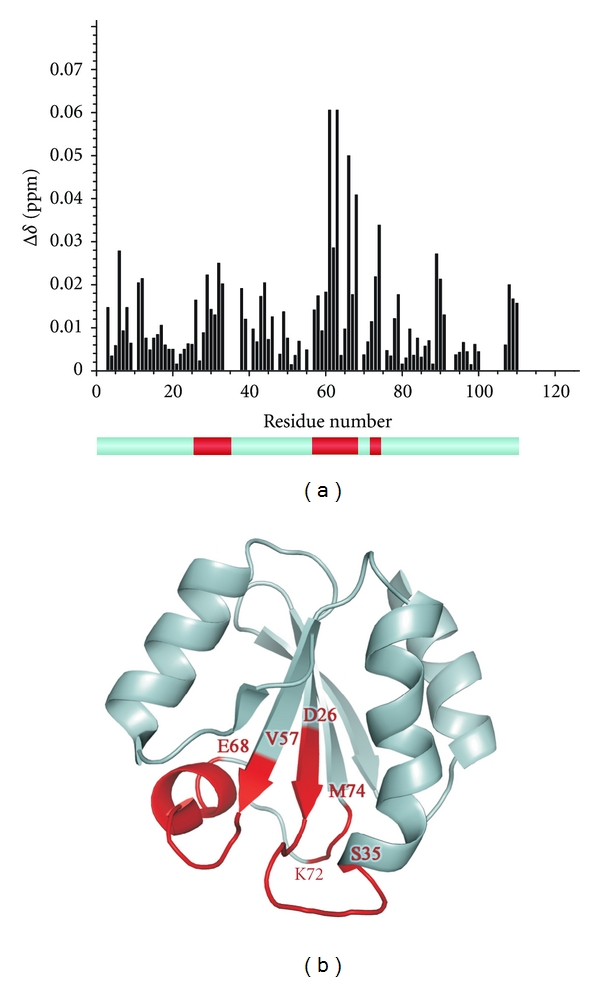
(a) Chemical shift difference observed for each residue of Trx upon NMR titration by MsrB1. Lower panel shows in red the residues revealing maximal changes upon titration. (b) The 3D structure of oxidized mammalian Trx. “Hot points” used for molecular docking are evidenced in red.
3.6. Structural Modelling of Oxidized MsrB1 and Reduced Trx
The average structures of oxidized MsrB1 and reduced Trx were obtained from the molecular dynamics simulations starting from coordinates of the reduced proteins taken from PDB (2kv1 and 3trx for MsrB1 and Trx, resp.). The calculated structures are shown in Figures 1(b) and 2(b).
3.7. Docking of Oxidized MsrB1 and Trx
Upon formation of an intermolecular protein complex, catalytic Cys32 (active residue) and Cys35 (resolving residue) of Trx should become close in space to the disulfide bond, connecting Cys4 and Cys95 residues of oxidized MsrB1. Such arrangement makes possible that Cys32 of Trx attacks the MsrB1 disulfide bond. A protein docking procedure was carried out in order to identify possible options for mutual arrangement of these proteins. Based on the above-mentioned distance restrains and experimental NMR data, the “hot points of docking”, were found (evidenced for both proteins in Figures 1(b) and 2(b) in red), and more than 1000 spatial structure orientations were generated. Our experimental data on residues involved in complex formation and application of 10 Å distance restrains between the Sγ atoms of Cys32 of Trx and Cys4-Cys95 disulfide bond of MsrB1 were used to sort these structures into four groups. The neighbouring contacts between residues and the distances between sulphur atom of Cys32 (Trx) and the nearest sulphur atom of oxidized MsrB1 disulfide bond, differentiating each group, are shown in Table 1.
Table 1.
Structural parameters for MsrB1-Trx complexes (four groups) revealed by protein docking. The distances between sulphur atom of Cys32 of Trx and the nearest sulphur atom of MsrB1 disulfide bond as well as three pairs of the nearest aminoacids approaching in space for each group (to outline the spatial orientation of the proteins within a complex) are presented.
| Group A | Group B | Group C | Group D | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Distance, Å (Trx Cys32) S/S–S bond (MsrB1) | 7,1 | 7,8 | 7,8 | 5,3 | ||||||||
|
| ||||||||||||
| Nearest neighbouring contacts between residues | ||||||||||||
|
| ||||||||||||
| MsrB1 residues | W43 | H39 | P87 | S2 | W43 | P87 | W43 | N62 | P87 | F6 | P42 | R93 |
| Trx residues | M37 | E95 | S90 | W31 | A92 | E70 | M37 | K72 | K96 | K72 | D60 | K36 |
Among the four represented complexes, the fourth (D) is the most plausible since the distance in this structure between Cys32 of Trx, and two sulphur atoms of disulfide bond in MsrB1 were found to be minimal (5.3 Å). Further, each of the four representative structures has been subjected to 5 ns molecular dynamics simulations.
3.8. Molecular Dynamics Procedure
The aim of the molecular dynamics simulation was to refine the possible structure of the Trx-MsrB1 complex taking into account the obtained NMR data, the effects of water environment, and flexibility of proteins backbone and side chains. The analysis of the averaged structures of the complexes obtained after 5 ns of MD simulations reveal that only for complex D the distance SγCys32 Trx–SγCys4 MsrB1 remains the smallest among all possible complexes. Therefore, the MD trajectory for this complex was extended to 20 ns (Figures 3(a) and 3(c)). It is necessary to emphasize that even if these data suggest that the SγCys32 Trx is closer to SγCys4 of oxidized MsrB1, the analogous distance to Cys95 is rather comparable. This observation precludes us to determine which Cys residue from intramolecular MsrB1 disulfide bridge will be subsequently targeted to form intermolecular disulfide bond.
Figure 3.
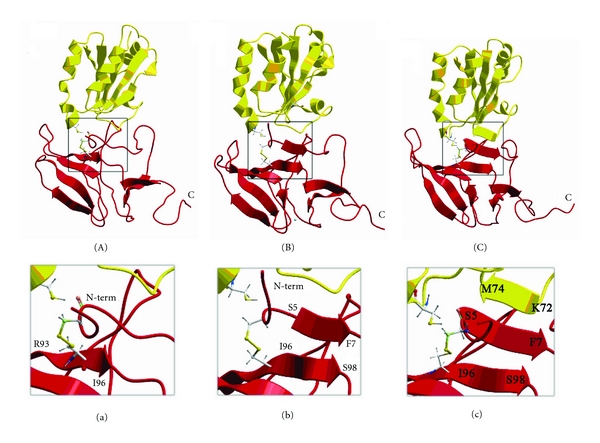
The snapshots (A, B, and C) of the protein complex formation along the molecular dynamics trajectory. The panels (a), (b), and (c) show zoomed views (interactions interfaces) of the snapshots (A) and (C), respectively.
As it was calculated by our MD, the N-terminal tail of MsrB1 upon oxidation gets closer to the β-strand of the protein formed by Arg93-Ile96 (Figures 3(A) and 3(a)). Further, elongation of the mentioned MsrB1's β-strand occurs additionally involving residues I96-S98 accompanied by formation of three additional hydrogen bonds with residues S5-F7 (Figures 3(B) and 3(b)). This newly constituted strand belongs to the N-terminus of MsrB1, which now is stabilized by the intramolecular disulfide bond formed upon oxidation. At the final stage of the interaction, Trx adjusts its unstructured loop to the newly formed N-terminal β-strand of MsrB1, leading to formation of interprotein β-sheet, composed of six β-strands, where five of them belong to MsrB1 and the sixth to Trx (residues K72-M74) (Figures 3(C) and 3(c)). Thus, in developing of two new β-strands (one intramolecular MsrB1 and one intermolecular), nine aminoacids participate. This observation is in good agreement with our NMR titration results (Figures 1 and 2), thus further validating our calculations. In addition, β-strand of MsrB1 formed by G77-F82 extends periodically to residues G75-G77 allowing residues G75-L78 to form hydrogen bonds with the second antiparallel β-strand of MsrB1 constituted by A66-G72. The distance between SγCys32 of thioredoxin and SγCys95 of MsrB1, a reference distance, oscillates during the interaction from the smallest value of about 3.2 Å (first 100 ps of the trajectory) to 9.5 Å and then down to 4.8 Å. Such approaching may cause a catalytic act, upon which the disulfide bond becomes resolved followed by reduction of MsrB1.
Upon the formation of the protein intermolecular complex, six new hydrogen bonds evolve (three of them are interproteins), thus stabilizing the intermolecular complex. Analysis of MD trajectories for MsrB1-Trx complex (prior formation of the intermolecular disulfide bond) indicates an existence of two conformations of C4-C95 MsrB1 bond. The disulfide bond in both conformations is strained in different extent. The degree of strain could be estimated by comparison of the energies for the van der Waals (vw) and torsion (to) terms of disulfide bond for oxidized MsrB1 in the step preceding the complex formation and in the free state. In accordance with ECEPP/3 force field, the following energies have been obtained (kcal/mol): 3.1 and 3.2 (vw), −0.5 and −1.7 (to), 2.6 and 1.5 (total) for “strained” and “relaxed” S–S bond, respectively. It is worth to mention that during the MD simulations this MsrB1 intramolecular S–S bond prefers its “strained” conformation. This bond thus becomes totally destabilized by 2.6 kcal/mol for “strained” or by 1.5 kcal/mol for “relaxed” conformation. However, the presence of six hydrogen bonds inside the developed interprotein β-sheet obviously overcompensates the straining of Cys4-Cys95 bond in MsrB1, as energy profit from one hydrogen bond formation in β-sheet can be estimated by −0.78 kcal/mol [50]. Therefore, the formation of the interprotein MsrB1-Trx complex becomes energetically preferable.
4. Conclusions
In this study, both the experimental and the computational approaches were combined to investigate MsrB1-Trx protein complex formation. Through the analysis of the NMR data, the aminoacids involved in protein complex formation have been determined. The average 3D structures of oxidized MsrB1 and reduced Trx were generated and subsequently subjected to protein docking and molecular dynamics. Complementary use of the mentioned approaches indicates that the formation of the interprotein β-sheet, stabilized by new hydrogen bonds, distorting the MsrB1 intramolecular disulfide bond. This disulfide bond straining favours the formation of intermolecular disulfide bridge of MsrB1-Trx complex. The study evidences structural and energetic driving forces of MsrB1-Trx complex formation.
Authors' Contribution
O. Dobrovolska, G. Rychkov, and E. Shumilina have made an equal contribution to the Paper.
Acknowledgments
AD acknowledges the support from the NT Faculty, NTNU. ES acknowledges the NT Faculty, NTNU, for financial support through Post-doctoral fellowship. Dr. F. L. Aachmann is acknowledged for his help in early paper steps. The work was partially supported by the Ministry of Education and Science of Russian Federation (the State Contracts 02.740.11.5014 and 16.552.11.7037) and was performed with use of scientific equipment of “The analytical center of nano- and biotechnologies of SPbSPU.”
References
- 1.Bandyopadhyay U, Das D, Banerjee RK. Reactive oxygen species: oxidative damage and pathogenesis. Current Science. 1999;77(5):658–666. [Google Scholar]
- 2.Vogt W. Oxidation of methionyl residues in proteins: tools, targets, and reversal. Free Radical Biology and Medicine. 1995;18(1):93–105. doi: 10.1016/0891-5849(94)00158-g. [DOI] [PubMed] [Google Scholar]
- 3.Stadtman ER, Van Remmen H, Richardson A, Wehr NB, Levine RL. Methionine oxidation and aging. Biochimica et Biophysica Acta. 2005;1703(2):135–140. doi: 10.1016/j.bbapap.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 4.Friguet B. Oxidized protein degradation and repair in ageing and oxidative stress. FEBS Letters. 2006;580(12):2910–2916. doi: 10.1016/j.febslet.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 5.Petropoulos I, Friguet B. Maintenance of proteins and aging: the role of oxidized protein repair. Free Radical Research. 2006;40(12):1269–1276. doi: 10.1080/10715760600917144. [DOI] [PubMed] [Google Scholar]
- 6.Moskovitz J, Flescher E, Berlett BS, Azare J, Poston JM, Stadtman ER. Overexpression of peptide-methionine sulfoxide reductase in saccharomyces cerevisiae and human T cells provides them with high resistance to oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(24):14071–14075. doi: 10.1073/pnas.95.24.14071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruan H, Tang XD, Chen ML, et al. High-quality life extension by the enzyme peptide methionine sulfoxide reductase. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(5):2748–2753. doi: 10.1073/pnas.032671199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moskovitz J. Roles of methionine suldfoxide reductases in antioxidant defense, protein regulation and survival. Current Pharmaceutical Design. 2005;11(11):1451–1457. doi: 10.2174/1381612053507846. [DOI] [PubMed] [Google Scholar]
- 9.Weissbach H, Resnick L, Brot N. Methionine sulfoxide reductases: history and cellular role in protecting against oxidative damage. Biochimica et Biophysica Acta. 2005;1703(2):203–212. doi: 10.1016/j.bbapap.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Kim HY, Gladyshev VN. Methionine sulfoxide reductases: selenoprotein forms and roles in antioxidant protein repair in mammals. Biochemical Journal. 2007;407(3):321–329. doi: 10.1042/BJ20070929. [DOI] [PubMed] [Google Scholar]
- 11.Grimaud R, Ezraty B, Mitchell JK, et al. Repair of oxidized proteins: identification of a new methionine sulfoxide reductase. Journal of Biological Chemistry. 2001;276(52):48915–48920. doi: 10.1074/jbc.M105509200. [DOI] [PubMed] [Google Scholar]
- 12.Neiers F, Kriznik A, Boschi-Muller S, Branlant G. Evidence for a new sub-class of methionine sulfoxide reductases B with an alternative thioredoxin recognition signature. Journal of Biological Chemistry. 2004;279(41):42462–42468. doi: 10.1074/jbc.M407464200. [DOI] [PubMed] [Google Scholar]
- 13.Weissbach H, Etienne F, Hoshi T, et al. Peptide methionine sulfoxide reductase: structure, mechanism of action, and biological function. Archives of Biochemistry and Biophysics. 2002;397(2):172–178. doi: 10.1006/abbi.2001.2664. [DOI] [PubMed] [Google Scholar]
- 14.Boschi-Muller S, Gand A, Branlant G. The methionine sulfoxide reductases: catalysis and substrate specificities. Archives of Biochemistry and Biophysics. 2008;474(2):266–273. doi: 10.1016/j.abb.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 15.Lescure A, Gautheret D, Carbon P, Krol A. Novel selenoproteins identified in silico and in vivo by using a conserved RNA structural motif. Journal of Biological Chemistry. 1999;274(53):38147–38154. doi: 10.1074/jbc.274.53.38147. [DOI] [PubMed] [Google Scholar]
- 16.Huang W, Escribano J, Sarfarazi M, Coca-Prados M. Identification, expression and chromosome localization of a human gene encoding a novel protein with similarity to the pilB family of transcriptional factors (pilin) and to bacterial peptide methionine sulfoxide reductases. Gene. 1999;233(1-2):233–240. doi: 10.1016/s0378-1119(99)00131-6. [DOI] [PubMed] [Google Scholar]
- 17.Jung S, Hansel A, Kasperczyk H, Hoshi T, Heinemann SH. Activity, tissue distribution and site-directed mutagenesis of a human peptide methionine sulfoxide reductase of type B: hCBS1. FEBS Letters. 2002;527(1–3):91–94. doi: 10.1016/s0014-5793(02)03171-x. [DOI] [PubMed] [Google Scholar]
- 18.Hansel A, Heinemann SH, Hoshi T. Heterogeneity and function of mammalian MSRs: enzymes for repair, protection and regulation. Biochimica et Biophysica Acta. 2005;1703(2):239–247. doi: 10.1016/j.bbapap.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 19.Kim HY, Gladyshev VN. Methionine sulfoxide reduction in mammals: characterization of methionine-R-Sulfoxide reductases. Molecular Biology of the Cell. 2004;15(3):1055–1064. doi: 10.1091/mbc.E03-08-0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim HY, Gladyshev VN. Characterization of mouse endoplasmic reticulum methionine-R-sulfoxide reductase. Biochemical and Biophysical Research Communications. 2004;320(4):1277–1283. doi: 10.1016/j.bbrc.2004.06.078. [DOI] [PubMed] [Google Scholar]
- 21.Boschi-Muller S, Olry A, Antoine M, Branlant G. The enzymology and biochemistry of methionine sulfoxide reductases. Biochimica et Biophysica Acta. 2005;1703(2):231–238. doi: 10.1016/j.bbapap.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 22.Moskovitz J. Methionine sulfoxide reductases: ubiquitous enzymes involved in antioxidant defense, protein regulation, and prevention of aging-associated diseases. Biochimica et Biophysica Acta. 2005;1703(2):213–219. doi: 10.1016/j.bbapap.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 23.Kryukov GV, Kumar RA, Koc A, Sun Z, Gladyshev VN. Selenoprotein R is a zinc-containing stereo-specific methionine sulfoxide reductase. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(7):4245–4250. doi: 10.1073/pnas.072603099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim HY, Gladyshev VN. Different catalytic mechanisms in mammalian selenocysteine- and cysteine-containing methionine-R-sulfoxide reductases. PLoS Biology. 2005;3(12, article e375):1–10. doi: 10.1371/journal.pbio.0030375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aachmann FL, Sal LS, Kim HY, Marino SM, Gladyshev VN, Dikiy A. Insights into function, catalytic mechanism, and fold evolution of selenoprotein methionine sulfoxide reductase B1 through structural analysis. Journal of Biological Chemistry. 2010;285(43):33315–33323. doi: 10.1074/jbc.M110.132308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim HY, Kim JR. Thioredoxin as a reducing agent for mammalian methionine sulfoxide reductases B lacking resolving cysteine. Biochemical and Biophysical Research Communications. 2008;371(3):490–494. doi: 10.1016/j.bbrc.2008.04.101. [DOI] [PubMed] [Google Scholar]
- 27.Antoine M, Boschi-Muller S, Branlant G. Kinetic characterization of the chemical steps Involved in the catalytic mechanism of methionine sulfoxide reductase A from Neisseria meningitidis. Journal of Biological Chemistry. 2003;278(46):45352–45357. doi: 10.1074/jbc.M307471200. [DOI] [PubMed] [Google Scholar]
- 28.Holmgren A. Thioredoxin and glutaredoxin systems. Journal of Biological Chemistry. 1989;264(24):13963–13966. [PubMed] [Google Scholar]
- 29.Holmgren A. Thioredoxin. Annual Review of Biochemistry. 1985;54:237–271. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 30.Kallis GB, Holmgren A. Differential reactivity of the functional sulfhydryl groups of cysteine-32 and cysteine-35 present in the reduced form of thioredoxin from Escherichia coli. Journal of Biological Chemistry. 1980;255(21):10261–10265. [PubMed] [Google Scholar]
- 31.Holmgren A. Thioredoxin structure and mechanism: conformational changes on oxidation of the active-site sulfhydryls to a disulfide. Structure. 1995;3(3):239–243. doi: 10.1016/s0969-2126(01)00153-8. [DOI] [PubMed] [Google Scholar]
- 32.Qin J, Clore GM, Kennedy WMP, Huth JR, Gronenborn AM. Solution structure of human thioredoxin in a mixed disulfide intermediate complex with its target peptide from the transcription factor NFκB. Structure. 1995;3(3):289–297. doi: 10.1016/s0969-2126(01)00159-9. [DOI] [PubMed] [Google Scholar]
- 33.Ausubel FM. Short Protocols in Molecular Biology: A Compendium of Methods from Current Protocols in Molecular Biology. 5th edition. Vol. 1. Hoboken, NJ, USA: John Wiley & Sons; 2002. [Google Scholar]
- 34.Zhang H, Neal S, Wishart DS. RefDB: a database of uniformly referenced protein chemical shifts. Journal of Biomolecular NMR. 2003;25(3):173–195. doi: 10.1023/a:1022836027055. [DOI] [PubMed] [Google Scholar]
- 35.Keller RLJ. Optimizing the Process of Nuclear Magnetic Resonance Spectrum Analysis and Computer Aided Resonance Assignment. Zürich; 2005. [Google Scholar]
- 36.Abagyan R, Totrov M, Kuznetsov D. ICM—a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. Journal of Computational Chemistry. 1994;15(5):488–506. [Google Scholar]
- 37.Nemethy G, Gibson KD, Palmer KA, et al. Energy parameters in polypeptides. 10. Improved geometrical parameters and nonbonded interactions for use in the ECEPP/3 algorithm, with application to proline-containing peptides. Journal of Physical Chemistry. 1992;96(15):6472–6484. [Google Scholar]
- 38.Hess B, Kutzner C, Van Der Spoel D, Lindahl E. GRGMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of Chemical Theory and Computation. 2008;4(3):435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 39.Oostenbrink C, Villa A, Mark AE, Van Gunsteren WF. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. Journal of Computational Chemistry. 2004;25(13):1656–1676. doi: 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- 40.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. Journal of Molecular Graphics. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 41.Berweger CD, van Gunsteren WF, Müller-Plathe F. Force field parametrization by weak coupling. Re-engineering SPC water. Chemical Physics Letters. 1995;232(5-6):429–436. [Google Scholar]
- 42.Daura X, Gademann K, Jaun B, Seebach D, Van Gunsteren WF, Mark AE. Peptide folding: when simulation meets experiment. Angewandte Chemie International Edition. 1999;38(1-2):236–240. [Google Scholar]
- 43.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: a Linear constraint solver for molecular simulations. Journal of Computational Chemistry. 1997;18(12):1463–1472. [Google Scholar]
- 44.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. Journal of Chemical Physics. 2007;126(1) doi: 10.1063/1.2408420. Article ID 014101. [DOI] [PubMed] [Google Scholar]
- 45.Berendsen HJC, Postma JPM, Van Gunsteren WF, Dinola A, Haak JR. Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics. 1984;81(8):3684–3690. [Google Scholar]
- 46.Darden T, York D, Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. The Journal of Chemical Physics. 1993;98(12):10089–10092. [Google Scholar]
- 47.Sal LS, Aachmann FL, Kim HY, Gladyshev VN, Dikiy A. NMR assignments of 1H, 13C and 15N spectra of methionine sulfoxide reductase B1 from Mus musculus. Biomolecular NMR assignments. 2007;1(1):131–133. doi: 10.1007/s12104-007-9039-7. [DOI] [PubMed] [Google Scholar]
- 48.Forman-Kay JD, Clore GM, Driscoll PC, Wingfield P, Richards FM, Gronenborn AM. A proton nuclear magnetic resonance assignment and secondary structure determination of recombinant human thioredoxin. Biochemistry. 1989;28(17):7088–7097. doi: 10.1021/bi00443a045. [DOI] [PubMed] [Google Scholar]
- 49.Williamson RA, Carr MD, Frenkiel TA, Feeney J, Freedman RB. Mapping the binding site for matrix metalloproteinase on the N-terminal domain of the tissue inhibitor of metalloproteinases-2 by NMR chemical shift perturbation. Biochemistry. 1997;36(45):13882–13889. doi: 10.1021/bi9712091. [DOI] [PubMed] [Google Scholar]
- 50.Munoz V, Serrano L. Elucidating the folding problem of helical peptides using empirical parameters. II. Helix macrodipole effects and rational modification of the helical content of natural peptides. Journal of Molecular Biology. 1995;245(3):275–296. doi: 10.1006/jmbi.1994.0023. [DOI] [PubMed] [Google Scholar]