

Abstract
The diet-induced atherosclerotic rabbit is an ideal model for atherosclerosis study, but temporal changes in atherosclerotic development in hypercholesterolemic rabbits are poorly understood. Japanese white rabbits were fed a high-cholesterol diet to induce sustained hypercholesterolemia, and each group of 10–12 animals was then sacrificed at 6, 12, 16, or 28 weeks. The rabbit aortas were harvested, and the sizes of the gross and intima atherosclerotic lesions were quantified. The cellular component of macrophages (Mφs) and smooth muscle cells (SMCs) in aortic intimal lesions was also quantified by immunohistochemical staining, and the correlation between plasma cholesterol levels and the progress of atherosclerotic lesions was studied. The ultrastructure of the atherosclerotic lesions was observed by transmission electron microscopy (TEM). Widely variable atherosclerotic plaques were found from 6 weeks to 28 weeks, and the lesional progress was closely correlated with cholesterol exposure. Interestingly, a relatively reduced accumulation of Mφ, an increased numbers of SMCs, and a damaged endothelial layer were presented in advanced lesions. Moreover, SMCs were closely correlated with cholesterol exposure and lesional progress for the whole period. Cholesterol exposure directly determines atherosclerotic progress in a rabbit model, and the changes in the cellular component of advanced lesions may affect plaque stability in an atherosclerotic rabbit model.
1. Introduction
Atherosclerosis is characterized by chronic inflammation in arteries that involves the accumulation of oxidized lipoproteins, an increased number of inflammatory cells and hypertrophic degeneration of the arterial intima, and it is closely associated with hypercholesterolemia [1–3]. Atherosclerosis complications often occur suddenly, and they can seriously jeopardize patients' lives [3]. Pathologically, plaque rupture is primarily responsible for the occurrence of dramatic clinical events, such as unstable angina, acute myocardial infarction, and stroke [2, 4]. Although the mechanism of plaque rupture is unclear, numerous studies have suggested that altered plaque contents result in destabilization, and these studies indicate that macrophages (Mφs) and smooth muscle cells (SMCs) in the lesion play pivotal roles in plaque rupture [5–9].
The laboratory rabbit is a good model in which to study atherosclerosis because its lipoprotein profile is more similar to humans than that of mouse or rat [10]. However, the temporal process of Mφs and SMCs accumulation in atherosclerotic lesions in rabbit is unclear, and the relationship between cholesterol exposure and the changes of cellular component is not completely determined. Moreover, the pathological mechanism underlying Mφ death and defective resolution of inflammation remains uncertainly. To address these questions, we performed this study to investigate the temporal changes in development of atherosclerosis at four time points (6, 12, 16, and 28 weeks) in rabbits, as well as the potential relationship between the changes of cellular component and cholesterol exposure. We observed the SMCs proliferation, a reduction in Mφs accumulation and damaged endothelium copresenting in advanced lesions. Furthermore, we proposed a possible explanation for why the changes of cellular component occurred in the advanced lesions of hypercholesterolemic rabbits.
2. Materials and Methods
2.1. Experimental Design
Male Japanese white rabbits (1.5–2.0 kg; 16–18 weeks old) were provided by the Laboratory Animal Center of Xi'an Jiaotong University and were randomly divided into two groups. The control group (n = 10) was fed a standard chow diet. The other rabbits (n = 44) were fed a high-cholesterol diet that contained 0.3–0.5% cholesterol and 3% corn oil; this group was designated as the high-cholesterol diet (HCD) group. Animals were individually maintained in a temperature-controlled (20–24°C) facility with a 12 h light/dark cycle, given free access to food and water. As Figure 1 shown, at 6 (n = 10), 12 (n = 11), 16 (n = 12), and 28 (n = 11) weeks, rabbits from the HCD group were randomly sacrificed by administering an overdose of thiamylal sodium, and a pathological analysis of aortic atherosclerosis was performed. All of the experimental protocols were approved by the Xi'an Jiaotong University Laboratory Animal Administration Committee and performed according to the Xi'an Jiaotong University Guidelines for Animal Experimentation.
Figure 1.
A schematic of experimental design.
2.2. Analysis of the Plasma Lipoprotein Profile
Each week, after a period of overnight fasting, blood samples were collected via the auricular artery. The blood samples were stored on ice and centrifuged (3000 rpm, 15 min, 4°C) to obtain plasma. The plasma total cholesterol (TC) and triglyceride levels and high-density lipoprotein-cholesterol (HDL-C) levels were measured using commercial assay kits (Dongou Bioengineering, Beijing, China). To evaluate cholesterol exposure, the area under the curve (AUC) was calculated according to the trapezium rule before the animal was sacrificed [11].
2.3. Measure of Plasma C-Reactive Protein
The plasma C-reactive protein (CRP) was measured using high-sensitivity rabbit CRP ELISA kits (Immunology Consultants Laboratory, USA) as previously described [12].
2.4. Quantification of Gross Lesions
At the end of each time point, the rabbits were sacrificed, and their aortic trees were carefully isolated and opened. After the samples were fixed in formalin, they were stained with Sudan IV and analyzed as previously described [13]. The area that was stained positively with Sudan IV was quantified using the Image Pro-PlusV2.0 image analysis software program (Media Cybernetics Inc., America).
2.5. Histological and Immunohistochemical Staining
For the histological analysis, the aortic arch of the cholesterol-fed rabbits was used and cut into 10 sections as previously described [14]. All of the sections were embedded in paraffin and cut into 5-μm-thick serial sections. The gross atherosclerotic lesions were evaluated as previously described [14]. For the microscopic quantification of the lesion areas, all of the aortic sections were stained with hematoxylin and eosin (H&E) and elastica van Gieson (EVG). Serial paraffin sections of the thoracic aorta were immunohistochemically stained with antibodies against Mφ (RAM11, Dako Inc.) and smooth muscle a-actin (alpha-actin, Thermo Fisher Scientific Inc.). The primary antibodies were diluted in PBS as follows: RAM11 (1 : 100) and alpha-actin (1 : 200). For the preparations, the sections were washed 3 times with PBS for 10 minutes per wash. Endogenous peroxidases were blocked by incubation for 10 minutes in 0.3% hydrogen peroxide. The sections were washed 3 times with PBS for 10 minutes each. To reduce the background staining, the sections were then incubated in 10% normal goat serum for 60 minutes. The sections were incubated in the primary antibody at 4°C overnight, and they were then washed 3 times for 10 minutes with PBS. The secondary antibody (antimurine IgG, Beijing Zhong Shan Biotechnology CO., China) was applied to the sections for 60 minutes. The sections were washed 3 times with PBS for 10 minutes each and were detected with an AEC kit (AEC kit, Beijing Zhong Shan Biotechnology CO., China). All of the sections (EVG and immunostained) that were used for microscopic quantification were captured under an Olympus BX51 light microscope that was equipped with a DP70 digital camera (Olympus, Tokyo, Japan) and were measured using the image pro-plusV2.0 image analysis software program (Media Cybernetics Inc., America).
2.6. Transmission Electron Microscopic (TEM) Analysis
The atherosclerotic lesions in the aortic arch (n = 3; each group) were further analyzed using TEM. The tissues were cut into 5 m pieces and fixed with a phosphate buffer that contained 2.5% glutaraldehyde and 4% paraformaldehyde at 4°C overnight. After the phosphate buffer was washed, the tissues were fixed secondarily in osmium tetroxide for 2 hours. The tissues were dehydrated in serial alcohol solutions and then polymerized at 60°C for 48 hours. Ultrathin sections (50 nm) were cut (LKB ultratome, Sweden) and stained with uranyl acetate, which was followed by lead citrate. They were observed under a transmission electron microscope (H-600, Hitachi, Japan) [15].
2.7. Statistical Analysis
All of the data were expressed as the means ± SEM. The two groups were compared using Student's t-test. Multiple groups were compared using a 1-way ANOVA, which was followed by a Bonferrioni test. The Pearson correlation coefficient was used to the check correlation between different atherosclerotic parameters. A probability value of less than 0.05 was considered to be significant.
3. Results
3.1. Plasma Lipid Profile and CRP Levels
The high-cholesterol diet intake of the HCD group resulted in severe hypercholesterolemia in these rabbits; their plasma cholesterol levels reached 800 mg/dL at 6 weeks and remained between 800 and 1200 mg/dL during the entire experimental period (Table 1). As shown in Table 1, the HDL-C levels decreased in the cholesterol-fed rabbits following a cholesterol feeding. However, the plasma triglyceride levels did not change in the cholesterol-fed rabbits compared to the normal rabbits (Table 1).
Table 1.
Body weight, plasma lipid profiles, and inflammatory marker.
| 0 weeks | 6 weeks | 12 weeks | 16 weeks | 28 weeks | ||
|---|---|---|---|---|---|---|
| Body weight (kg) | HCD | 2.51 ± 0.06 | 2.78 ± 0.06 | 2.95 ± 0.07 | 3.09 ± 0.06 | 2.86 ± 0.08** |
| Chow diet | 2.63 ± 0.05 | 2.98 ± 0.06 | 3.05 ± 0.05 | 3.15 ± 0.05 | 3.54 ± 0.06 | |
| Total cholesterol (mg/dL) | HCD | 50.44 ± 3.93 | 1032.56 ± 53.2** | 810.74 ± 64.22** | 1008.92 ± 118.39** | 1143.82 ± 129.62** |
| Chow diet | 48.29 ± 3.37 | 65.89 ± 3.32 | 65.89 ± 3.32 | 45.63 ± 6.87 | 77.32 ± 8.37 | |
| Triglyceride (mg/dL) | HCD | 53.52 ± 11.13 | 83.11 ± 7.46 | 72.15 ± 9.62 | 76.51 ± 16.82 | 70.76 ± 10.11 |
| Chow diet | 80.40 ± 18.25 | 99.26 ± 23.30 | 92.73 ± 24.68 | 98.73 ± 29.87 | 86.51 ± 10.64 | |
| HDL-C (mg/dL) | HCD | 20.00 ± 4.08 | 7.17 ± 1.39** | 4.89 ± 1.09** | 2.65 ± 0.42** | 5.31 ± 1.22** |
| Chow diet | 21.91 ± 1.91 | 17.40 ± 2.10 | 17.98 ± 1.75 | 17.81 ± 2.09 | 16.97 ± 1.54 | |
| pCRP (mg/dL) | HCD | 16.76 ± 3.53 | 38.47 ± 10.95 | 81.51 ± 27.90 | 155.41 ± 20.55** | 226.10 ± 28.55** |
| Chow diet | 16.66 ± 3.20 | 21.06 ± 4.72 | 24.66 ± 11.66 | 26.27 ± 5.90 | 13.59 ± 1.90 |
HCD, the group for high-cholesterol diet, 0 weeks (n = 10), 6 weeks (n = 10), 12 weeks (n = 11), 16 weeks (n = 12), 28 weeks (n = 11); Chow diet, the control group for normal diet, n = 10; HDL-C, high-density lipoprotein-cholesterol; pCRP, plasma C-reactive protein levels. Values are expressed as mean ± SEM, *P < 0.05 and **P < 0.01 to control group.
To assess the inflammatory levels in the HCD group, we performed ELISA experiment to monitor the plasma high-sensitivity CRP levels. After a cholesterol feeding, the plasma CRP levels increased linearly from 6 weeks to 28 weeks. At the end of the experiment, the plasma CRP levels in the cholesterol-fed rabbits were significantly higher than those of the control rabbits (Table 1).
3.2. Effects of Cholesterol Exposure on Gross Atherosclerosis
We calculated the AUC, which revealed 2.5-fold (P < 0.001) and 6-fold (P < 0.0001) increases in cholesterol exposure at 16 and 28 weeks, respectively, when compared to 6 weeks (Figure 2(a)). The AUC at 12 weeks also indicated a 1.5-fold increase, but the data were not statistically significant. The sudanophilic en face staining area in the aortic trees of the HCD-fed rabbits gradually increased with prolonged cholesterol exposure at 12 and 16 weeks. At 28 weeks, the lesion area in the aortic arch was 2.5-fold greater (P < 0.0001) than that at 6 weeks (Figures 2(b) and 2(c)). To evaluate the effects of cholesterol exposure on aortic atherosclerosis, we compared cholesterol exposure (AUC of plasma total cholesterol) to the size of gross atherosclerotic lesions. We found that cholesterol exposure was strongly correlated with the progress of gross lesion in the aortic arch (P < 0.001; Figure 2(c)). Moreover, the gross lesions of the thoracic and abdominal aorta were correlated with cholesterol exposure (P < 0.001 and P < 0.05, resp.; data not shown).
Figure 2.
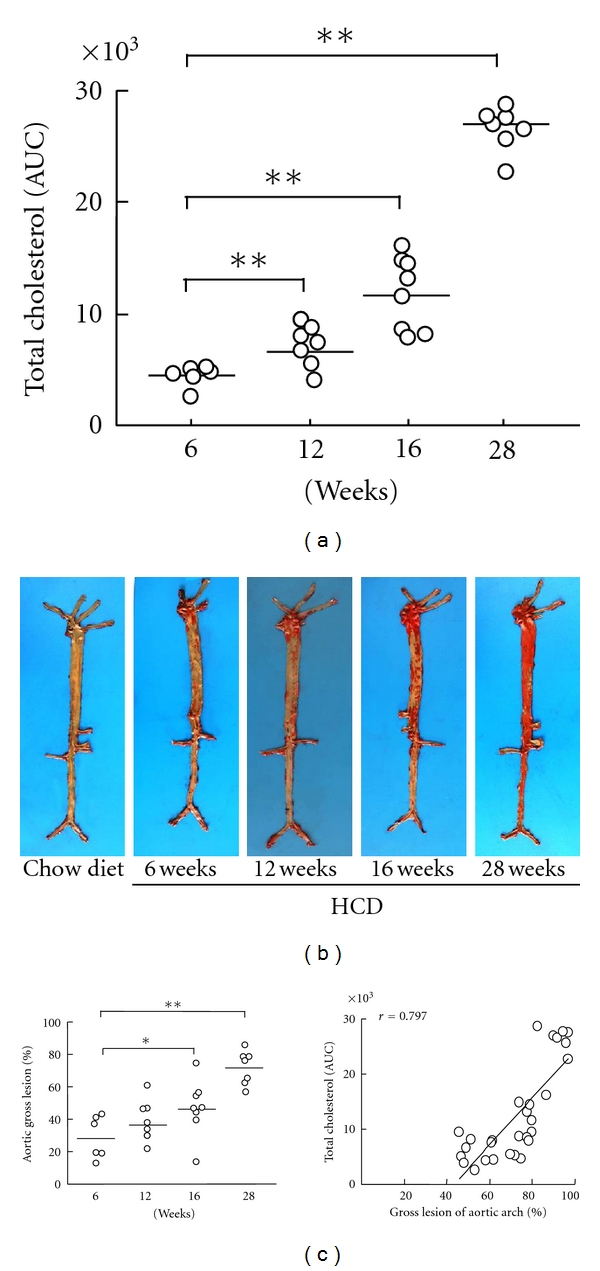
cholesterol exposure and gross atherosclerotic lesions. Cholesterol exposure was evaluated by the area under the curve (AUC) at different experimental time points (a). Representative images of aortic atherosclerosis as stained by Sudan IV (b). Gross lesion areas (left) that were calculated in the aortas of chow- and HCD-fed rabbits at 6, 12, 16 and 28 weeks and the correlations (right) between cholesterol exposure and gross lesion area (c). The data are expressed as the means ± SEM. n = 6–8 for each group. *P < 0.05 and **P < 0.01.
3.3. Effects of Cholesterol Exposure on Microscopic Atherosclerosis
We further calculated the microscopic lesional area of the sections of aortic arch. As expected, the HCD-fed rabbits displayed an increase in this variable that was directly proportional to increased cholesterol exposure (Figure 3(a)). The microscopic lesion sizes were 3.8- (P < 0.05) and 5.9-fold (P ≤ 0.0001) greater at 16 and 28 weeks, respectively, compared to the size at 6 weeks (Figure 3(b)). The lesion size increased at 12 weeks versus 6 weeks (2.0-fold), but this increase was not statistically significant. Microscopic lesions, which are characteristics of gross lesions, were also strongly correlated with cholesterol exposure (P < 0.001) (Figure 3(b)).
Figure 3.
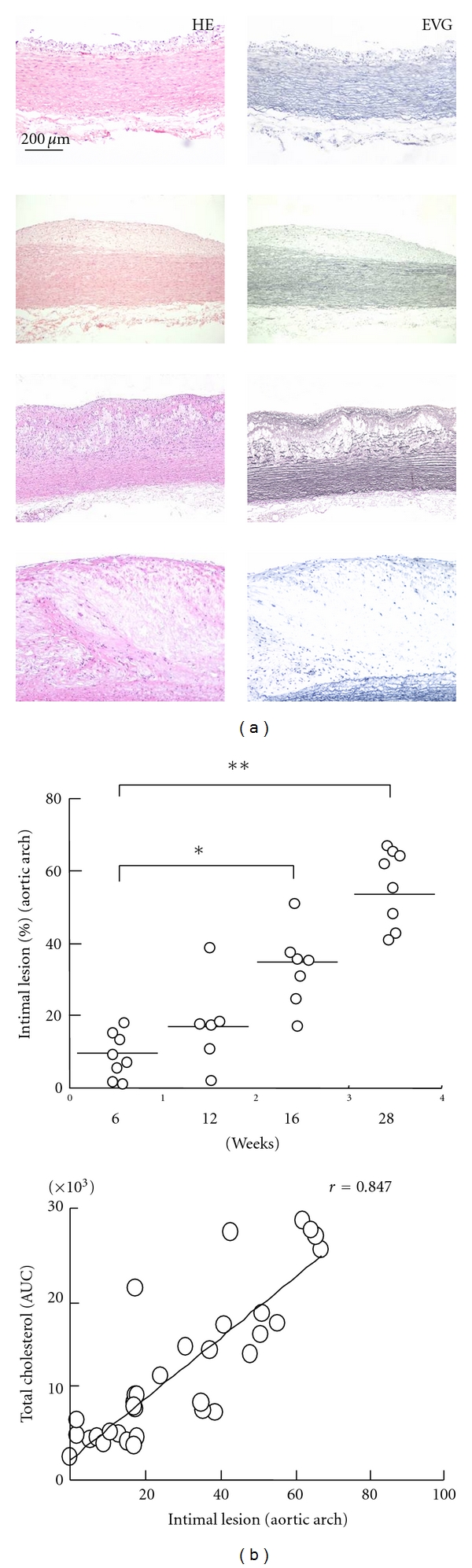
Microscopically atherosclerotic lesions. Representative images of aortic atherosclerosis as stained with H&E and EVG (a). The intima lesional areas (upper) in the sections were measured using the EVG sections and the correlations (beneath) between cholesterol exposure and intima lesions area (b). The data are expressed as the means ± SEM. n = 6–8 for each group. *P < 0.05 and **P < 0.01.
3.4. Aortic Atherosclerosis Analysis
We stained the serial sections with monoclonal antibodies (mAb) against Mφs and found that the lesions at 6 weeks were Mφs-rich and correspond to fatty streaks in humans (Figure 4(a)). The Mφ number increased 2.3-fold (P < 0.05) when cholesterol exposure increased at 16 weeks. Surprisingly, at 28 weeks, the Mφ number decreased 4.6-fold (P < 0.001) when compared to the number at 6 weeks, and the majority of the Mφs were localized at the lesion surface (Figures 3(a) and 3(b)). At 12 weeks, the Mφ level also increased (1.9 fold) when compared to that at 6 weeks, but this increase was not statistically significant. Conversely, the SMC mAb staining revealed that the number of SMCs increased significantly at 12 (P < 0.05), 16 (P < 0.01) and 28 weeks (P < 0.01) when compared to that at 6 weeks (Figure 4(b)). As shown in the representative images (Figure 4(a)), SMCs were present in the intimal and near the surface of the lesions at all of the time points. We next compared the numbers of Mφs and SMCs in the aortic arch with the AUCs and intima lesions. The results of this comparison indicated that the numbers of SMCs were strongly correlated with cholesterol exposure (P < 0.0001), aortic arch microscopic lesions (P < 0.0001) (Figure 4(c)), and the gross lesion area (data not shown). Interestingly, we identified a significant correlation between the Mφs and atherosclerotic aortic lesions (both area and thickness) at 16 weeks; however, when the analysis included the entire 28 weeks, the Mφs did not correlate with the aforementioned parameters (Figure 4(c)).
Figure 4.
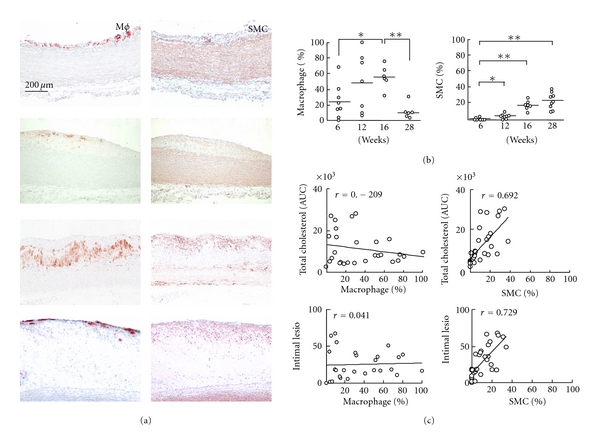
Immunohistochemical investigations of the lesions. Representative images are shown for macrophages (Mφ) and SMC α-actin (a). Mφ- and SMC-positive areas were determined using an image analysis system (b). The Mφs and SMCs were compared with cholesterol exposure (AUC) and intima lesion area, respectively (c). The data are expressed as the means ± SEM. n = 6–8 for each group. *P < 0.05 and **P < 0.001.
As shown in Figure 5, TEM revealed an obvious loss in the integrity of the endothelial layer at 16 weeks, and the increased lipid accumulation led to the compression and deformation of the nuclei of the remaining Mφs at 28 weeks (Figure 5(a)).
Figure 5.
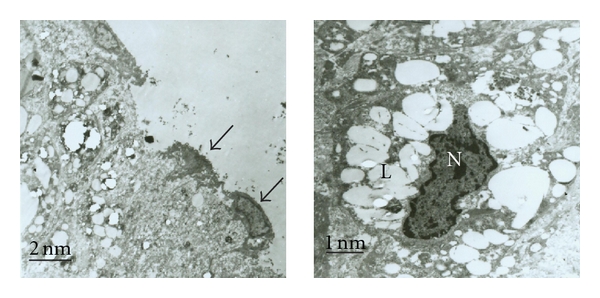
Transmission electron microscopic analysis and the evaluation of lesional inflammation. Damaged endothelial layer (left) and remaining endothelial cells (arrows) at 16 weeks and lipids overladen macrophages (right; L = lipid drop, N = nucleus) at 28 weeks.
4. Discussion
Although hypercholesterolemia is strongly believed to contribute to atherosclerosis development in humans and in animal models, the temporal relationships between cholesterol exposure, atherosclerotic progression, and the changes in lesional cellular composition are not fully understood [3, 16, 17]. When they were fed a high-cholesterol diet, our rabbits developed severe hypercholesterolemia that resulted in aortic atherosclerotic development. The size of the observed gross lesions gradually decreased from the arch to the abdominal aorta, and the aorta demonstrated a susceptibility to diet-induced atherosclerosis that was distinct from that of humans [18]. However, when hypercholesterolemia was induced and sustained for a longer time period, the degree of exposure to cholesterol determined the atherosclerotic lesion area and thickness and led to high inflammatory levels. It is well known that CRP is an excellent marker of inflammation in atherosclerosis [19]. In the current study, a time-dependent increase in plasma CRP suggested that the liver had switched to a predominantly inflammatory state responding to IL-6 or high plasma cholesterol as previously described [19, 20].
Under both long-term cholesterol exposure and proinflammatory conditions, the Mφs in the HCD rabbits accumulated in the early- and middle-stage of atherosclerotic lesions as previously reported [3, 21, 22]. Because the features of early and middle stage of atherosclerotic lesions (6–16 weeks) in rabbits were similar with type I–III of lesion of human, Mφ accumulation in this period might indicate that enhanced monocytes entry and efficient efferocytosis [3, 21, 23]. However, through 28 weeks of cholesterol fed, the advanced atherosclerotic lesions in rabbits may also share some characteristics of type IV-V of lesions in human, the present study provided the novel evidence that the numbers of Mφs were relatively decreased in advanced lesions (they were actually only present on the surface of advanced lesions), and this decrease was independently correlated with cholesterol exposure, gross lesion, and hyperplastic intima of aortic arch [24]. The mechanism of the numbers of Mφs reduction in advanced lesions in rabbits is unclear, but we know that excess accumulation of lipoproteins in advanced lesions causes Mφs death. Mφs death presenting in advanced atherosclerotic lesions of human is deemed as a result of “free cholesterol” and “endoplasmic reticulum stress”, which is usually associated to the secondary necrosis and aggravating inflammation [25]. Conversely, the number of SMCs increased steadily from 6 weeks to 28 weeks, and this increase was strongly correlated with cholesterol exposure, gross lesion development, and microscopic lesion size. In the early lesions, the SMCs infiltrated to the intima from media, but the SMCs were located mainly on the surface when lesions developed to an advanced stage. Their findings suggest that SMCs proliferation may play a protective role in rabbit's advanced lesions. Precious studies also suggested that the SMCs upregulation caused the matrix proliferation, a relatively thick fibrous cap in the lesions, and SMCs apoptosis that directly resulted in the plaque destabilization [1, 5, 22, 26–28]. Moreover, the phenomena of the reduction of Mφs and the proliferation of SMCs in advanced lesions are still unclear and should be studied further to explore the potential mechanism.
In conclusion, these results indicated that long-term cholesterol exposure via high-cholesterol diet intake could induce atherosclerosis development and the reduction in Mφs and increase in SMCs in advanced lesions may affect plaque stability in rabbits, which will help us to understand human atherosclerosis.
Acknowledgment
This work was partly supported by a National Natural Science Foundation of China (81070250), Public Service Platform Grant of Shaanxi Province (2010FWPT-15).
Abbreviations
- AUC:
Area under the curve
- CRP:
C-reactive protein
- EVG:
Elastica van Gieson
- H&E:
Hematoxylin and eosin
- HCD:
High-cholesterol diet
- HDL-C:
High density lipoprotein-cholesterol
- Mφs:
Macrophages
- SMCs:
Smooth muscle cells
- TC:
Total cholesterol
- TEM:
Transmission electron microscopy.
References
- 1.Libby P, Aikawa M. Stabilization of atherosclerotic plaques: new mechanisms and clinical targets. Nature Medicine. 2002;8(11):1257–1262. doi: 10.1038/nm1102-1257. [DOI] [PubMed] [Google Scholar]
- 2.Ross R. Atherosclerosis—an inflammatory disease. New England Journal of Medicine. 1999;340(2):115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 3.Steinberg D. Hypercholesterolemia and inflammation in atherogenesis: two sides of the same coin. Molecular Nutrition and Food Research. 2005;49(11):995–998. doi: 10.1002/mnfr.200500081. [DOI] [PubMed] [Google Scholar]
- 4.Libby P. Changing concepts of atherogenesis. Journal of Internal Medicine. 2000;247(3):349–358. doi: 10.1046/j.1365-2796.2000.00654.x. [DOI] [PubMed] [Google Scholar]
- 5.Bennett MR. Apoptosis of vascular smooth muscle cells in vascular remodelling and atherosclerotic plaque rupture. Cardiovascular Research. 1999;41(2):361–368. doi: 10.1016/s0008-6363(98)00212-0. [DOI] [PubMed] [Google Scholar]
- 6.Boyle JJ, Bowyer DE, Weissberg PL, Bennett MR. Human blood-derived macrophages induce apoptosis in human plaque-derived vascular smooth muscle cells by Fas-ligand/Fas interactions. Arteriosclerosis, Thrombosis, and Vascular Biology. 2001;21(9):1402–1407. doi: 10.1161/hq0901.094279. [DOI] [PubMed] [Google Scholar]
- 7.Fernandez-Ortiz A, Badimon JJ, Falk E, et al. Characterization of the relative thrombogenicity of atherosclerotic plaque components: implications for consequences of plaque rupture. Journal of the American College of Cardiology. 1994;23(7):1562–1569. doi: 10.1016/0735-1097(94)90657-2. [DOI] [PubMed] [Google Scholar]
- 8.Libby P, Geng YJ, Aikawa M, et al. Macrophages and atherosclerotic plaque stability. Current Opinion in Lipidology. 1996;7(5):330–335. doi: 10.1097/00041433-199610000-00012. [DOI] [PubMed] [Google Scholar]
- 9.Lutgens E, De Muinck ED, Kitslaar PJEHM, Tordoir JHM, Wellens HJJ, Daemen MJAP. Biphasic pattern of cell turnover characterizes the progression from fatty streaks to ruptured human atherosclerotic plaques. Cardiovascular Research. 1999;41(2):473–479. doi: 10.1016/s0008-6363(98)00311-3. [DOI] [PubMed] [Google Scholar]
- 10.Waqar AB, Koike T, Yu Y, et al. High-fat diet without excess calories induces metabolic disorders and enhances atherosclerosis in rabbits. Atherosclerosis. 2010;213(1):148–155. doi: 10.1016/j.atherosclerosis.2010.07.051. [DOI] [PubMed] [Google Scholar]
- 11.Liu E, Kitajima S, Higaki Y, et al. High lipoprotein lipase activity increases insulin sensitivity in transgenic rabbits. Metabolism. 2005;54(1):132–138. doi: 10.1016/j.metabol.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 12.Koike T, Kitajima S, Yu Y, et al. Human C-reactive protein does not promote atherosclerosis in transgenic rabbits. Circulation. 2009;120(21):2088–2094. doi: 10.1161/CIRCULATIONAHA.109.872796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang C, Zheng H, Yu Q, et al. A practical method for quantifying atherosclerotic lesions in rabbits. Journal of Comparative Pathology. 2010;142(2-3):122–128. doi: 10.1016/j.jcpa.2009.08.159. [DOI] [PubMed] [Google Scholar]
- 14.Zhao S, Zhang C, Lin Y, et al. The effects of rosiglitazone on aortic atherosclerosis of cholesterol-fed rabbits. Thrombosis Research. 2008;123(2):281–287. doi: 10.1016/j.thromres.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 15.Tokunaga O, Fan J, Watanabe T. Atherosclerosis- and age-related multinucleated variant endothelial cells in primary culture from human aorta. American Journal of Pathology. 1989;135(6):967–976. [PMC free article] [PubMed] [Google Scholar]
- 16.Cullen P, Baetta R, Bellosta S, et al. Rupture of the atherosclerotic plaque: does a good animal model exist? Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23(4):535–542. doi: 10.1161/01.ATV.0000060200.73623.F8. [DOI] [PubMed] [Google Scholar]
- 17.Groot PHE, Van Vlijmen BJM, Benson GM, et al. Quantitative assessment of aortic atherosclerosis in APOE*3 leiden transgenic mice and its relationship to serum cholesterol exposure. Arteriosclerosis, Thrombosis, and Vascular Biology. 1996;16(8):926–933. doi: 10.1161/01.atv.16.8.926. [DOI] [PubMed] [Google Scholar]
- 18.Stary HC, Blankenhorn DH, Chandler AB, et al. A definition of the intima of human arteries and of its atherosclerosis-prone regions: a report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1992;85(1):391–405. doi: 10.1161/01.cir.85.1.391. [DOI] [PubMed] [Google Scholar]
- 19.Libby P, Ridker PM. Inflammation and atherosclerosis: role of C-reactive protein in risk assessment. American Journal of Medicine. 2004;116(6):9–16. doi: 10.1016/j.amjmed.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 20.Kleemann R, Verschuren L, Van Erk MJ, et al. Atherosclerosis and liver inflammation induced by increased dietary cholesterol intake: a combined transcriptomics and metabolomics analysis. Genome Biology. 2007;8(9, article R200) doi: 10.1186/gb-2007-8-9-r200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bobryshev YV. Monocyte recruitment and foam cell formation in atherosclerosis. Micron. 2006;37(3):208–222. doi: 10.1016/j.micron.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 22.Schwartz SM, DeBlois D, O’Brien ERM. The intima: soil for atherosclerosis and restenosis. Circulation Research. 1995;77(3):445–465. doi: 10.1161/01.res.77.3.445. [DOI] [PubMed] [Google Scholar]
- 23.Stary HC, Chandler AB, Glagov S, et al. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis: a report from the committee on vascular lesions of the council on arteriosclerosis, American Heart Association. Circulation. 1994;89(5):2462–2478. doi: 10.1161/01.cir.89.5.2462. [DOI] [PubMed] [Google Scholar]
- 24.Stary HC, Chandler AB, Dinsmore RE, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis: a report from the Committee on Vascular Lesions of the council on arteriosclerosis, American heart association. Circulation. 1995;92(5):1355–1374. doi: 10.1161/01.cir.92.5.1355. [DOI] [PubMed] [Google Scholar]
- 25.DeVries-Seimon T, Li Y, Pin MY, et al. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. Journal of Cell Biology. 2005;171(1):61–73. doi: 10.1083/jcb.200502078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lafont A, Libby P. The smooth muscle cell: sinner or saint in restenosis and the acute coronary syndromes? Journal of the American College of Cardiology. 1998;32(1):283–285. doi: 10.1016/s0735-1097(98)00216-2. [DOI] [PubMed] [Google Scholar]
- 27.Shiomi M, Ito T, Hirouchi Y, Enomoto M. Fibromuscular cap composition is important for the stability of established atherosclerotic plaques in mature WHHL rabbits treated with statins. Atherosclerosis. 2001;157(1):75–84. doi: 10.1016/s0021-9150(00)00708-5. [DOI] [PubMed] [Google Scholar]
- 28.Lusis AJ. Atherosclerosis. Nature. 2000;407(6801):233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]