

Abstract
Currently, about 20 crystal structures per day are released and deposited in the Protein Data Bank. A significant fraction of these structures is produced by research groups associated with the structural genomics consortium. The biological function of many of these proteins is generally unknown or not validated by experiment. Therefore, a growing need for functional prediction of protein structures has emerged. Here we present an integrated bioinformatics method that combines sequence-based relationships and three-dimensional (3D) structural similarity of transcriptional regulators with computer prediction of their cognate DNA binding sequences. We applied this method to the AraC/XylS family of transcription factors, which is a large family of transcriptional regulators found in many bacteria controlling the expression of genes involved in diverse biological functions. Three putative new members of this family with known 3D structure but unknown function were identified for which a probable functional classification is provided. Our bioinformatics analyses suggest that they could be involved in plant cell wall degradation (Lin2118 protein from Listeria innocua, PDB code 3oou), symbiotic nitrogen fixation (protein from Chromobacterium violaceum, PDB code 3oio), and either metabolism of plant-derived biomass or nitrogen fixation (protein from Rhodopseudomonas palustris, PDB code 3mn2).
1. Introduction
Due to recent advances in high-throughput structure determination, structural genomics initiatives are proceeding fast. The Protein Structure Initiative (PSI) reports the determination of over 5,500 proteins structures in its Structural Genomics Knowledgebase as of November 2011 [1]. A total of 3,020 proteins (>50%) are of unknown function or only minimally characterized [1]. Hence, there is an urgent need for computational annotation methods of these structures of unknown function. We present here an integrated bioinformatics method for the functional annotation of transcription factors that combines sequence-based relationships and three-dimensional (3D) structural similarity of transcriptional regulators with computer prediction of their cognate DNA binding sequences. We applied this method to the AraC/XylS family of transcription factors.
AraC/XylS is a large family of transcriptional regulators found in many bacteria controlling the expression of genes with diverse biological functions involved in metabolism, stress response, and virulence [2–5]. Most members of this family are comprised of 250 to 300 amino acids and contain two domains: a conserved DNA binding domain (DBD) of ca. 100 residues found at the C-terminus of most regulators and a variable domain thought to be responsible for effector binding or multimerization [2]. A few exceptions include regulators which are considerably smaller (ca. 150 residues) containing only the DBD domain (e.g., MarA and SoxS) [6], substantially larger (e.g., HrpB, 477 residues), or that contain the DBD at the N-terminus (e.g., Rob) [6] or in the central domain (e.g., Ada) [7].
The DBD assumes a conserved tertiary structure of two helix-turn-helix (HTH) domains, each made up of three α-helices, which are connected by a longer central α-helix [8]. The two HTH domains bind to the major groove of two adjacent turns on the same side of the DNA helix. Residues stabilizing the hydrophobic core are highly conserved, while the overall sequence identity is low (approximately 24%). Key residues of the N-terminal HTH domain are more variable and this domain is thought to define the individual DNA binding specificities of the family members, while the C-terminal HTH is more conserved and provides improved binding affinity [2]. The multifunctional protein Ada is an exception to this general topology, as its N-terminal domain (N-Ada) contains a DNA repair domain with a distinct fold joined to a single AraC-like HTH domain by a flexible linker [7, 9]. In contrast to the conserved DBD, the effector and multimerization domain is variable among family members and its molecular function is not always well understood [2].
Historically, members of the AraC/XylS family were assigned to one of the three general categories “metabolism”, “stress response”, and “virulence” based on the genes that they regulate [2, 3, 10]. The metabolism group includes regulators involved in carbohydrate metabolism (e.g., ChbR and AraC from E. coli), metabolism of benzene derivatives (XylS from Pseudomonas putida), alkane metabolism (AlkR from Acinetobacter), and amine metabolism (FeaR from E. coli). Regulators of the stress response group are involved in the direct or indirect response to stress factors such as Ada (alkylating agents in E. coli), AdiY (acid resistance in E. coli), MarA (multiple antibiotic resistance in E. coli), SoxS (oxidative stress in E. coli), and RipA from Corynebacterium (iron limitation stress). The third group of pathogenesis or virulence contains family members, such as VirF which regulates the expression of proteins of the type III secretion system (TTSS) in Shigella flexneri, Rns from E. coli which regulates cell adhesion proteins, and Caf1R from Yersinia pestis which is involved in capsule formation [11]. Here, we have included a fourth category, dubbed bacteria-plant interaction, which includes RhrA from Rhizobium meliloti that regulates iron concentration required for nitrogen fixation by production of a siderophore and Y4fK from Rhizobium sp. (strain NGR234), which induces formation of nodules in plant roots where nitrogen fixation takes place.
In a recent update on the AraC/XylS family of transcriptional regulators, Ibarra et al. identified 58 well-characterized members and found a total of 1,974 known and putative members in 149 bacterial genomes [3]. They generated dendrograms from the multiple sequence alignment of the DBD to functionally classify putative family members.
The aim of the present study was the development of a bioinformatics method for the functional annotation of putative transcriptional regulators with solved tertiary structure but unknown function. We applied this method to members of the AraC/XylS family of transcriptional regulators. Similar to Ibarra et al., we analyzed the DBD to perform a functional classification. However, our method is not restricted to sequence information only but rather incorporates primary and tertiary structure information from the transcriptional regulator protein sequence, structure, and DNA binding site. We performed three-dimensional (3D) structure similarity searches against the whole Protein Data Bank (PDB) and identified three putative new members of the AraC/XylS family with uncharacterized function. Detailed structural analyses and sequence comparison between these three proteins and 62 well-characterized AraC/XylS family members suggest that they could be involved in plant cell wall degradation (Lin2118 protein, PDB code: 3oou), symbiotic nitrogen fixation (PDB code: 3oio), and either metabolism of plant-derived biomass or nitrogen fixation (PDB code: 3mn2). Structure-based DNA binding site prediction for these transcription factors is concordant with these functional assignments.
2. Materials and Methods
2.1. Structural Similarity Search
Scanning of the complete PDB for target proteins sharing a similar tertiary structure with a given query protein was performed with the web server COPS–TopSearch of the Center of Applied Molecular Engineering (University of Salzburg) available at http://www.came.sbg.ac.at/ [12]. TopSearch is a fast three-dimensional (3D) search method that relies on the TopMatch structure alignment software to rank protein structures available in the PDB (target structures t) according to their absolute similarity S(q,t) to a provided query structure q [13, 14]. The absolute similarity is defined as the number of structurally equivalent residue pairs or the length of the structural alignment. Starting with MarA from E. coli (PDB code: 1bl0), the top 100 most similar proteins were manually inspected based on their structural alignment with the query and functional annotations available from the PDB and the UniProt database (http://www.uniprot.org/) [15]. The search was repeated for each known and putative AraC/XylS family member identified. Structures were manually modified to contain the DNA binding domain only, where necessary. More than 70,000 protein structures (PDB release: 6 June 2011) were searched for similarity and a total of thirteen (10 known and 3 putative) structures of AraC/XylS-family transcription factors were identified.
2.2. Multiple-Sequence Alignment
Protein sequences that belong to the HTH-AraC family (PFAM family PF00165) were retrieved from the UniProt database [15, 18]. Only those entries marked as reviewed by UniProt and with experimental demonstration of their functional role were selected. A total of 62 sequences fulfilled the criteria (Table S1 see in Supplementary Material available online at doi:10.1155/2012/103132.). Sequences from four PDB structures (PDB codes: 3lsg, 3mn2, 3oio, and 3oou) identified from the structural similarity search were also incorporated into this dataset.
A multiple-sequence alignment was constructed with the MAFFT 6 web server available at http://mafft.cbrc.jp/alignment/server/ [19]. The gap penalty was set to a value of 3.0. Based on the multiple sequence alignment and available structural information, we defined a region of 100 amino acids as the DBD (corresponding to Ile-13 to Thr-112 in the sequence of MarA from E. coli, PDB code: 1bl0). Assignment of biological roles (regulated biological processes) to the sequences was transferred from evidence described in the literature where possible, following a similar nomenclature to that already proposed [2, 3].
2.3. Dendrogram Generation
The multiple-sequence alignment obtained as described previously was used to build a dendrogram of HTH-AraC family members with experimental evidence of their biological role. The tree was constructed employing the maximum likelihood optimization criteria with the software MEGA5 [20]. The WAG amino acid substitution model was selected using the ProtTest server and applied as previously described [21, 22]. Bootstrap values were calculated with one hundred replicates. Phylogenetic tree images were produced with the iTOL web server tool [23] available at http://itol.embl.de/.
2.4. Multiple-Structure Alignment
PDB files were manually modified to include only amino acids of the defined 100-residue region of the DBD. Then, a multiple-structure alignment of the DBD was constructed with the SALIGN module from the MODELLER version 9.9 software package [16, 25]. The SALIGN module reports a table with the number of equivalent Cα positions (the alignment length; 3.5 Å cut-off), the root mean squared (RMS) distance of equivalent positions, and the sequence identity of equivalent residues for all pairs of proteins, as well as the multiple-sequence alignment (MSA) derived from the multiple optimal superposition of protein structures. A dendrogram of the structure-derived MSA was generated as described previously.
2.5. Comparative Modeling
MODELLER version 9.9 was employed to generate full-atom 3D comparative models of protein-DNA complexes [25]. The complex of MarA with the 22-mer DNA binding site mar (PDB code: 1bl0) was defined as template. DNA was treated as rigid body (nucleotides were defined as “block residues”) and ten models were initially generated. For each target, a single model with reasonable orientation of key residues of helices three and six was finally selected by visual inspection and energy minimized with the Molecular Operating Environment (MOE) version 2010.10 [17]. Hydrogen atoms were added, nucleotide atoms were fixed, protein backbone atoms were restrained by a quadratic force term, and the protein part was energy minimized with the AMBER99 [26] force field and a Born solvation term until a gradient of 0.05 kcal/(mol Å) was reached.
2.6. DNA Binding Site Prediction
The prepared comparative models were further processed to construct static protein-DNA models with varied DNA sequence. Briefly, the protein part and the DNA backbone were fixed and DNA bases were permitted to vary, thereby preserving the double-helix structural parameters of the initial model. The software 3DNA [27] was employed to build a new DNA double helix with varied sequence while at the same time retaining the helical parameters of the original DNA structure. For each initial model, 10,000 models with randomized DNA sequences were built. Next, from the Protein-DNA Interface database [28], a nonredundant set of 208 protein-DNA complexes was obtained. This set was used to derive statistical distance-dependent pairwise potentials at the protein-DNA interface. The statistical potential parameters were the same as those previously described [29, 30]. These potentials were then utilized to score the observed protein-DNA interactions in the comparative models. From each ensemble of random models, sets of low-energy structures were selected according to the 0.5% lower tail of each distribution of energy scores. Position-weight matrices (PWMs) were finally derived from the low energy score sets, and sequence logos were generated with the software WebLogo [31]. PWMs were converted into linear vectors, the all-against-all Euclidean distances calculated for all possible pairs of them and recorded as a distance table, which was finally used to perform a hierarchical clustering with Ward's method.
2.7. Genomic Context of Predicted DNA Binding Sites
Genomes and annotations were downloaded from the EMBL suite of databases and web servers (http://www.ebi.ac.uk/genomes/). High-scoring binding sequences were mapped with the software bowtie version 0.12.7 [33], allowing up to three mismatches. SeqMonk version 0.16.0 (http://www.bioinformatics.bbsrc.ac.uk/projects/seqmonk/) and the EMBL genome browser, Genome Reviews [34], were used to determine the neighboring genes of the putative binding sites. The six genes closest to the binding site, including those overlapping with the binding sites, were selected for further analysis. To establish functional relations for each gene, we employed the databases UniProt (http://www.uniprot.org/) [15], InterPro (http://www.ebi.ac.uk/interpro/) [35], and KEGG (http://www.genome.jp/kegg/) [36].
3. Results and Discussion
3.1. PDB Search for AraC/XylS Family Members
Employing the fast web server COPS-TopSearch of the Center of Applied Molecular Engineering (University of Salzburg) [12], we identified ten structures of proteins known to be members of the AraC/XylS family and three structures of uncharacterized proteins (Table 1). The ten known family members include MarA (PDB codes: 1bl0 [8], 1xs9 [37]), Rob (PDB code: 1d5y [38]), and Ada (PDB codes: 1u8b and 1zgw [7], 1wpk [9]), all involved in stress response, AraC (PDB code: 2k9s [39]) and YesN (PDB code: 3lsg) related to carbohydrate metabolism (though the function of YesN is not well characterized), GadX (PDB code: 3mkl) involved in acid resistance, and TcpN, also called ToxT (PDB code: 3gbg [40]), which is involved in virulence. The three uncharacterized, putative members include the Lin2118 protein from Listeria innocua (PDB code: 3oou), a protein from Chromobacterium violaceum with the gene name argR (PDB code: 3oio), and a protein from Rhodopseudomonas palustris (PDB code: 3mn2).
Table 1.
Known and putative TFs of the AraC/XylS family identified by a structural similarity search in the PDB.
| Name (PDB code) | UniProt ID | DNA complex(a) | Technique (Res.)(b) | Species | Biological process |
|---|---|---|---|---|---|
| MarA (1bl0) | P0ACH5 | Yes | X-ray (2.30 Å) | Escherichia coli | Multiple antibiotic resistance |
| MarA (1xs9) | P0ACH5 | Yes | NMR | Escherichia coli | Multiple antibiotic resistance |
| Rob (1d5y) | P0ACI0 | Yes | X-ray (2.70 Å) | Escherichia coli | Antibiotic resistance, organic solvent tolerance and heavy-metal resistance |
| Ada (1u8b) | P06134 | Yes | X-ray (2.10 Å) | Escherichia coli | Repair of and response to alkylated DNA |
| Ada (1zgw) | P06134 | Yes | NMR | Escherichia coli | Repair of and response to alkylated DNA |
| Ada (1wpk) | P06134 | No | NMR | Escherichia coli | Repair of and response to alkylated DNA |
| GadX (3mkl) | B1X7X1 | No | X-ray (2.15 Å) | Escherichia coli | Acid resistance |
| AraC (2k9s) | P0A9E0 | No | NMR | Escherichia coli | Transport and catabolism of L-arabinose |
| TcpN or ToxT (3gbg) | A5F384 | No | X-ray (1.90 Å) | Vibrio cholerae | Biosynthesis and assembly of the toxin-coregulated pilus |
| YesN (3lsg) | Q8RGT8 | No | X-ray (2.05 Å) | Fusobacterium nucleatum | Possibly involved in plant cell wall degradation |
| Lin2118 protein (3oou) | Q92A04 | No | X-ray (1.57 Å) | Listeria innocua | Unknown |
| 3oio | Q7NTG7 | No | X-ray (1.65 Å) | Chromobacterium violaceum | Unknown |
| 3mn2 | Q6NCA5 | No | X-ray (1.80 Å) | Rhodopseudomonas palustris | Unknown |
(a)Indicates whether the 3D protein structure was cocrystallized with DNA or not.
(b)Numbers in parenthesis refer to the resolution of crystal structures.
Structural similarity, as quantified by the number of equivalent residue pairs (length of the structural alignment, cf. Materials and methods), of ten of the thirteen structures is high (Table 2 and Figure 1). The average number of equivalent Cα positions of ten structures without Ada is 89.5 (of a maximum of 100.1), the average root mean squared (RMS) distance is 1.9 Å, and the mean sequence identity of equivalent residue pairs is 23%, which indicates high structural similarity and moderate levels of sequence identity. We removed the three structures of Ada from this analysis, since this protein consists of only a single HTH domain joined to an unrelated DNA repair domain and thus has a structural coverage of roughly 50% with the other structures. We scanned the PDB with any of the ten non-Ada structures as query structure and retrieved the remaining nine structures ranked as top hits in all cases. Two of the uncharacterized proteins (PDB codes: 3oou, 3oio) are currently classified as members of the AraC family in the Pfam database [18] and our structural analysis confirms membership to this family. Structure 3mn2 with unknown function was titled as “probable AraC family” by the authors of the crystal structure, and through our structural analysis, we confirm this membership, as well. It should be noted that the gene of the 3oio protein was named argR. The gene product of argR is the transcriptional regulator arginine repressor (ArgR), which controls the expression of operons involved in arginine biosynthesis. However, structural alignment of 3oio with the DNA binding domain of ArgR (e.g., PDB code: 3fhz) reveals only moderate similarity (equivalent residue pairs: 40). ArgR binds DNA as a hexamer and its DBD consists of a single three-helix domain with low sequence identity (15%) to 3oio. Based on this structural analysis, we conclude that 3oio is more likely a member of the AraC/XylS family rather than of the ArgR family of transcriptional regulators.
Table 2.
Matrix of equivalent residue pairs of available AraC/XylS 3D structures(a).
| 1bl0 | 1xs9 | 1d5y | 3oou | 2k9s | 3gbg | 3mkl | 3mn2 | 3oio | 3lsg | 1u8b | 1wpk | 1zgw | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1bl0 | 100 | 100 | 100 | 96 | 84 | 81 | 88 | 82 | 94 | 97 | 47 | 44 | 42 | 1bl0 |
| 0.0 | 0.7 | 0.9 | 1.8 | 1.9 | 2.2 | 1.9 | 2.2 | 1.9 | 1.3 | 1.6 | 2.2 | 2.3 | ||
| 100% | 100% | 50% | 18% | 16% | 15% | 16% | 14% | 19% | 25% | 15% | 13% | 15% | ||
| 1xs9 | 100 | 100 | 93 | 82 | 78 | 88 | 84 | 93 | 97 | 48 | 45 | 45 | 1xs9 | |
| 0.0 | 1.1 | 1.7 | 1.8 | 2.0 | 1.8 | 2.0 | 1.7 | 1.4 | 1.7 | 2.3 | 2.3 | |||
| 100% | 50% | 18% | 16% | 15% | 16% | 14% | 19% | 25% | 15% | 13% | 15% | |||
| 1d5y | 100 | 95 | 80 | 81 | 88 | 88 | 95 | 97 | 45 | 44 | 41 | 1d5y | ||
| 0.0 | 1.5 | 1.7 | 2.1 | 1.8 | 2.0 | 1.8 | 1.2 | 1.7 | 2.3 | 2.3 | ||||
| 100% | 20% | 21% | 16% | 11% | 16% | 21% | 25% | 25% | 18% | 17% | ||||
| 3oou | 100 | 72 | 84 | 87 | 88 | 87 | 97 | 47 | 42 | 37 | 3oou | |||
| 0.0 | 2.0 | 2.3 | 1.8 | 1.8 | 1.9 | 1.4 | 1.7 | 2.3 | 2.3 | |||||
| 100% | 23% | 16% | 17% | 20% | 20% | 31% | 15% | 13% | 11% | |||||
| 2k9s | 101 | 89 | 91 | 84 | 95 | 86 | 45 | 41 | 33 | 2k9s | ||||
| 0.0 | 2.0 | 2.0 | 2.3 | 1.8 | 1.9 | 2.1 | 2.3 | 2.6 | ||||||
| 100% | 21% | 24% | 22% | 25% | 26% | 23% | 10% | 9% | ||||||
| 3gbg | 99 | 90 | 84 | 95 | 81 | 35 | 36 | 23 | 3gbg | |||||
| 0.0 | 1.5 | 2.2 | 1.6 | 2.0 | 2.3 | 2.4 | 2.6 | |||||||
| 100% | 28% | 15% | 20% | 23% | 8% | 3% | 6% | |||||||
| 3mkl | 99 | 96 | 95 | 88 | 45 | 32 | 35 | 3mkl | ||||||
| 0.0 | 1.7 | 1.3 | 1.7 | 1.9 | 2.4 | 2.5 | ||||||||
| 100% | 21% | 26% | 23% | 19% | 10% | 8% | ||||||||
| 3mn2 | 102 | 100 | 85 | 44 | 40 | 28 | 3mn2 | |||||||
| 0.0 | 1.6 | 2.0 | 2.1 | 2.2 | 2.8 | |||||||||
| 100% | 28% | 15% | 21% | 12% | 13% | |||||||||
| 3oio | 100 | 93 | 51 | 45 | 35 | 3oio | ||||||||
| 0.0 | 1.8 | 2.0 | 2.0 | 2.7 | ||||||||||
| 100% | 18% | 33% | 22% | 21% | ||||||||||
| 3lsg | 100 | 45 | 48 | 41 | 3lsg | |||||||||
| 0.0 | 1.5 | 2.4 | 2.2 | |||||||||||
| 100% | 15% | 13% | 19% | |||||||||||
| 1u8b | 52 | 41 | 36 | 1u8b | ||||||||||
| 0.0 | 2.5 | 2.2 | ||||||||||||
| 100% | 62% | 44% | ||||||||||||
| 1wpk | 60 | 28 | 1wpk | |||||||||||
| 0.0 | 2.5 | |||||||||||||
| 100% | 28% | |||||||||||||
| 1zgw | 53 | 1zgw | ||||||||||||
| 0.0 | ||||||||||||||
| 100% |
(a)The top number indicates the number of equivalent Cα positions of the aligned structures (length of the alignment). The middle number denotes the root mean square (RMS) deviation of equivalent Cα atoms (3.5 Å distance cutoff) and the lower number denotes the sequence identity of equivalent residues. Structures were aligned with SALIGN [16]. Structures of unknown biological role are indicated in bold.
Figure 1.
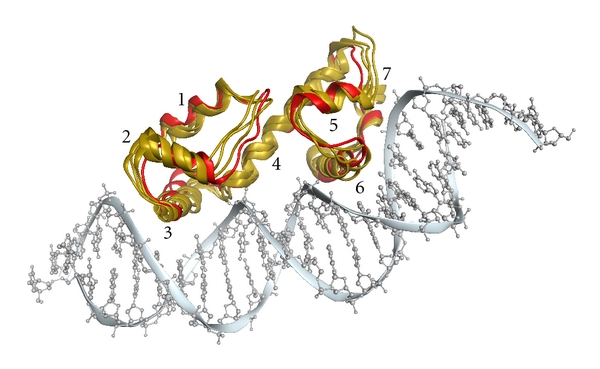
Structural alignment of three putative AraC/XylS family members (PDB codes: 3mn2, 3oio, and 3oou; gold ribbons) with MarA (PDB code: 1bl0; red ribbons). The DNA molecule of the MarA structure is shown for illustration (gray); however 3mn2, 3oio, and 3oou were crystallized without DNA. Numbers of α-helices are given next to the respective α-helix. The alignment was generated with SALIGN [16] and the figure was created with the Molecular Operating Environment (MOE) [17].
3.2. Functional Annotation
Despite their similar structures, the transcriptional regulators of the AraC/XylS family are known to act on various genes involved in distinct biological processes. To attempt a possible functional classification of the new putative family members on sequence level, we followed an approach similar to that of Ibarra et al. [3]. We compiled a list of 62 well-characterized AraC/XylS family members with known biological roles (Supplementary Table S1) and generated a maximum likelihood dendrogram (Figure 3) from the multiple-sequence alignment (MSA) of the DNA binding domains (DBDs) of the family members (Figure 2). The sequences of the three putative family members (PDB codes: 3mn2, 3oio, and 3oou) and the sequence of the poorly characterized protein YesN from F. nucleatum (PDB code: 3lsg) were added to the MSA and dendrogram in order to permit annotation of their biological roles based on their neighborhood in the tree.
Figure 3.
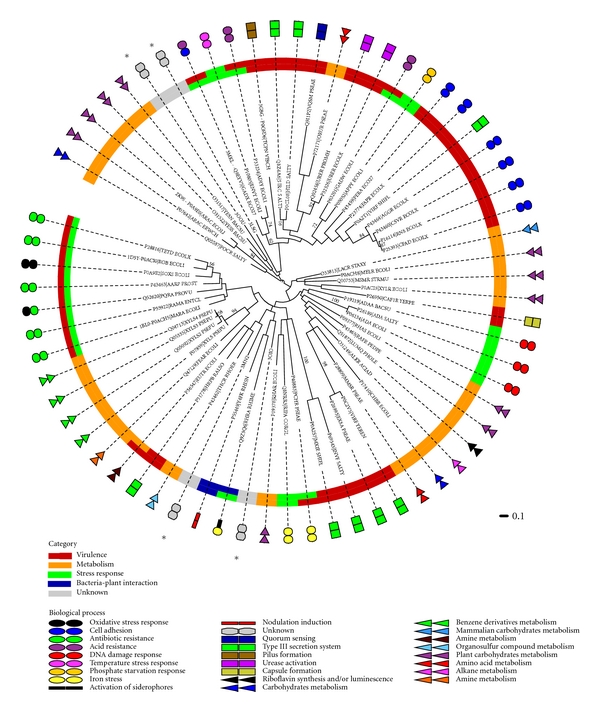
Dendrogram of AraC/XylS family members annotated with functional categories. A dendrogram of 62 AraC/XylS family members and four proteins found by structural similarity search and whose biological role is unknown or poorly characterized is presented (marked with an asterisk). The tree was constructed using the maximum likelihood optimization criteria and a bootstrap test was conducted with 100 replicates. Bootstrap values greater than 50 are shown as number in the corresponding nodes of the tree. Leaves contain the UniProt or PDB accession code and the protein name. Two classification levels are included in this dendrogram. The inner color strip represents a primary classification (functional category) that contains four general classes as in Figure 2. The outer circle contains several colored shapes that represent a secondary and more specific classification scheme, which provides more detail to the biological process associated with each protein. One or two functional categories were assigned.
Figure 2.
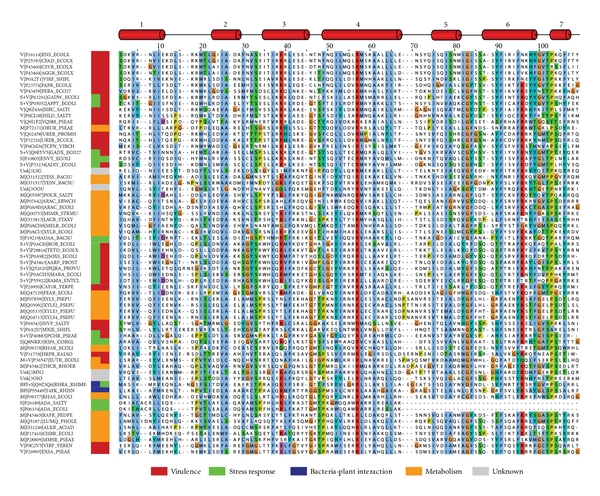
Multiple-sequence alignment of the DNA binding domain of 62 AraC/XylS family members with experimental evidence of their biological role. Four proteins found by the structural similarity search and whose function is unknown or poorly characterized were included in this multiple sequence alignment. Each sequence label contains the general functional category, the UniProt accession code, and the protein name. Functional categories are BPI: bacteria-plant interaction; M: metabolism; S: stress response; V: virulence and Unk: unknown. One or two functional categories were assigned. The sequences are sorted according to the tree order (see Figure 3). Secondary structure elements are given at the top (according to MarA (PDB code: 1bl0)). The alignment was plotted with JalView version 2.6.1 [24]. Color legend: light blue: hydrophobic; green: polar and aliphatic; turquois: polar and aromatic; red: basic; purple: acidic; orange: glycine; yellow: proline.
The putative AraC/XylS family member Lin2118 protein from Listeria innocua (PDB code: 3oou) is found in a branch together with YesN and YesS from Bacillus subtilis. In the next neighboring branch, we find the sequence of YesN from Fusobacterium nucleatum subsp. Nucleatum (PDB code: 3lsg). YesS is a probable transcription factor regulating a pathway responsible for rhamnogalacturonan depolymerization, which is a carbohydrate product of plant cell wall degradation [41]. YesN is a member of the two-component regulatory system yesM/yesN. Its biological role is not well understood. Genes upregulated by YesN, which include yesS, are thought to be involved in rhamnogalacturonan degradation in Bacillus subtilis, and an ortholog of YesN from Paenibacillus sp. is thought to be involved in hemicellulose degradation [42–44]. A similar role of the Lin2118 protein (PDB code: 3oou) in plant cell wall degradation seems, therefore, likely. The three proteins, YesN from B. subtilis, YesN from F. nucleatum (PDB code: 3lsg), and Lin2188 from L. innocua (PDB code: 3oou), share a common additional response regulator receiver domain of the CheY-like superfamily at their N-terminus. This domain contains a phosphoacceptor site that is phosphorylated by histidine kinase homologs, for instance, YesM [43]. The common regulatory domain further supports a possible involvement in the biological process of plant cell wall degradation.
The sequence of structure 3oio from Chromobacterium violaceum is found in a common branch together with Y4fK, encoded on the pNGR234a plasmid of Rhizobium sp. (strain NGR234), and RhrA, encoded on the plasmid pSymA of Rhizobium meliloti. Y4fk activates the transcription of nod genes that play a role in the formation of plant root nodules, where symbiotic nitrogen fixation takes place [45]. RhrA activates the expression of a siderophore necessary to maintain low levels of iron, required for efficient nitrogen fixation. Both plasmids are essential for the symbiosis between plant and bacteria in the rhizosphere [46, 47]. Based on these similarities, and the fact that the habitat of Chromobacterium violaceum is primarly soil and water, a tentative role in symbiotic nitrogen fixation seems possible.
The last putative AraC/XylS member (PDB code: 3mn2) from Rhodopseudomonas palustris is located in between the previously described nitrogen fixation branch of the dendrogram and a branch that contains the regulators ThcR, EutR, and HrpB. ThcR from Rhodococcus erythropolis is responsible for degradation of a thiocarbamate herbicide [48], and EutR from Escherichia coli is involved in metabolism of ethanolamine derived from phosphatidylethanolamine contained in biological membranes. EutR was also linked to virulence in both animals and plants [49]. Finally, HrpB, which is from the phytopathogen Ralstonia solanacearum, activates the expression of a type III secretion system required for plant host cell infection [50, 51]. It is known that symbiotic rhizobia and phytopathogenic bacteria share a common mechanism for plant host recognition [52], which could explain the observed proximity of the two branches associated to symbiotic and pathogenic behavior.
A pathogenic role of R. palustris has not been reported. R. palustris is a free living bacterium and is capable of acquiring carbon from many types of green plant-derived compounds. In addition, the bacterium is able to assimilate atmospheric nitrogen, albeit not in symbiosis with plants, as in the neighboring nitrogen fixation branch of the dendrogram [53]. R. palustris is one of the most metabolically versatile bacteria known [53] and one of a few prokaryotes described so far that possess three types of nitrogenases [54]. Prediction of a possible biological role of the putative AraC/XylS transcription factor with PDB code 3mn2 is not straightforward, due to the heterogeneity of the available data. A tentative role in either metabolism of plant-derived biomass or nitrogen fixation seems possible.
3.3. Structural Comparison
Taking advantage of the protein structures available, we extended the sequence-based functional annotation with structural data. All thirteen identified AraC/XylS structures were aligned with the software SALIGN [16] and an MSA was derived from the resulting structural alignment (Figure 4(a)). The structures aligned nicely with a typical fold: two HTH domains of each three α-helices are connected by a longer, central α-helix. The two HTH domains themselves are superimposable. One α-helix of each HTH domain (α-helix three and six) inserts into the major groove of DNA establishing base-specific contacts (Figure 1). The orientation of the C-terminal HTH domain (HTH2) is conserved while the N-terminal domain (HTH1) is more variable. Domain rotation of up to approximately 30 degrees with respect to MarA (PDB code: 1bl0) is observable for HTH1. The position of α-helix two is shifted by up to 3.5 Å in some structures and the position of the connecting loops to helices one and three is variable. On sequence level, the average pairwise sequence identity of HTH1 is 17%, while it is 27% for HTH2. These observations are in agreement with the current hypothesis that DNA binding affinity and specific recognition by different regulators are governed by HTH1, while HTH2 might additionally enhance affinity and binding site discrimination through a more conserved mechanism common to regulators of the AraC/XylS family [2, 38, 55]. The reported variability of HTH1 could further indicate a possible induced fit mechanism upon DNA binding [39].
Figure 4.
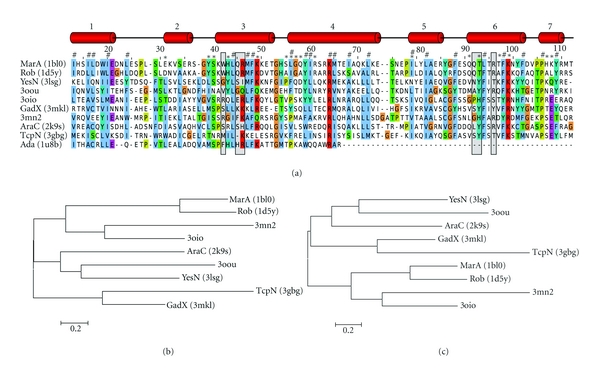
Structure-derived multiple-sequence alignment and dendrogram of known and putative AraC/XylS-family transcription factors. (a) The structures were aligned with SALIGN [16] and a multiple sequence alignment was derived from equivalent residue positions. Secondary structure elements are given at the top. Residues marked with a gray-shaded box are engaged in DNA base interactions, those marked with an asterisk contact the DNA sugar-phosphate backbone, and a hash sign denotes residues of the hydrophobic core. Numbering scheme and annotations are according to MarA protein (PDB code: 1bl0). Colors are as in Figure 2. (b) Maximum likelihood dendrogram derived from the structure-based MSA in (a) generated with the software MEGA5 [20]. Distances in the dendrogram refer to the number of amino acid substitutions per site. Only structures with two HTH domains (omitting Ada) were included. (c) For comparison, a more concise view of the dendrogram in Figure 3 is given, showing only relevant proteins.
Conserved residues in the multiple sequence alignment in Figure 4 are mostly related to hydrophobic amino acids stabilizing the hydrophobic core of the two HTH domains or small amino acids at the beginning and end of α-helices. A prominent exception are the three conserved residues Leu-28, Leu-30, and Val-33 (1bl0 numbering, Figure 4(a)). Due to the structural flexibility of α-helix two, these residues are shifted by one or two positions in the MSA in Figure 4 for MarA (PDB code: 1bl0) and Rob (PDB code: 1d5y), in contrast to all other structures. In the case of MarA and Rob, these residues are pointing toward the solvent, whereby they point inside, toward the hydrophobic core in all other structures. It is conceivable that this conformational change is part of the already mentioned induced fit mechanism. MarA and Rob structures were crystallized in complex with DNA, in contrast to the other bipartite HTH structures. However, in structures of Ada (PDB codes: 1u8b, 1zgw), which were solved in complex with DNA, these residues point inward as well. Another possible explanation is that this conformational change relates to the biological role of the respective transcriptional regulator. MarA and Rob are closely related regulators involved in stress response. However, movement of α-helix two seems to be unrelated to both conditions (biological role; complexed with DNA). The observed flexibility could thus also be an artifact of the structure determination experiment.
We further calculated the electrostatic potential surfaces of the AraC/XylS protein structures with the software DelPhi version 4 (Supplementary Figure S1). We observed a general dipolar character of the protein structures, with a positive potential on the side facing the DNA double helix and a negative potential on the opposite side. However, the detailed distribution of potential charges was generally diverse, and any relation to the biological role of the transcription factors was not obvious.
To perform a more systematic structure-based analysis of the relationship of bipartite HTH AraC/XylS family members, we generated a dendrogram from the structure-derived MSA (Figure 4(b)). We compared this structure-based dendrogram with the sequence-based dendrogram in Figure 3. Figure 4(c) shows an extract of the latter dendrogram containing the same AraC/XylS members as in Figure 4(b), for easier comparison. The two trees are highly similar with only a minor topology change involving branch GadX/TcpN. Consistency of the structure and sequence-based MSAs and dendrograms provides further support for the provided functional annotation.
3.4. DNA Binding Site Prediction
All analyses up to this point focused on the transcription factor proteins (sequence and structure). We will now turn to their cognate DNA binding sequences. Since the DNA binding sites for most identified AraC/XylS members are unknown, we devised a computational method to predict these sequences. The modeling of transcription factor-DNA complexes puts a strong focus on the DNA binding interface of the proteins and thus highlights key residues of helices three and six involved in specific base recognition.
In this structure-based approach, we generated initial comparative models based on MarA (PDB code: 1bl0) as a template structure and replaced the DNA sequence of the crystallized DNA molecule, while retaining the helical parameters of the original DNA structure. A total of 10,000 models with randomized full duplex DNA sequences were generated for all structures except Ada, which lacks the second HTH domain. A statistical potential was employed to score these protein-DNA models and position weight matrices (PWMs), along with sequence logos, were generated from top-scoring complexes (Figure 5(a)). A dendrogram was produced from the PWMs based on their Euclidean distance (Figure 5(b)).
Figure 5.
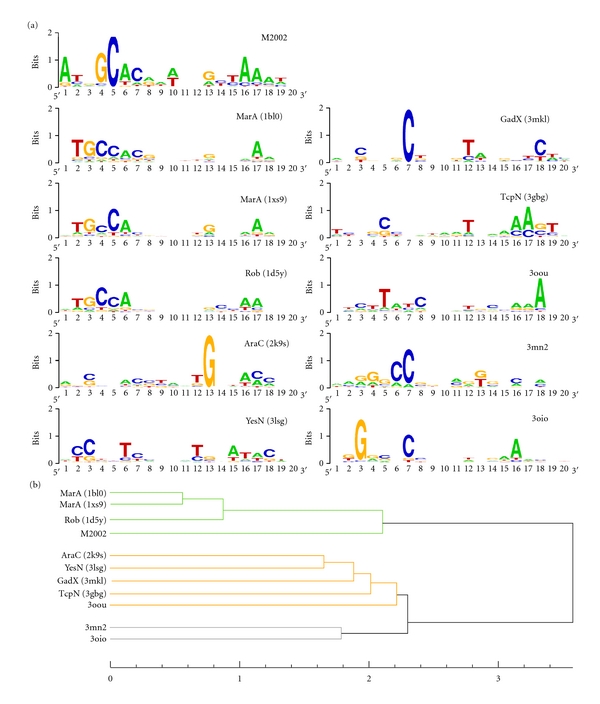
Sequence logos of predicted DNA binding sites and hierarchical clustering of the corresponding PWMs. (a) Sequence logos generated from an ensemble of top-scored random DNA sequences of protein-DNA complexes modeled after MarA (PDB code: 1bl0). M2002 denotes a set of 24 known binding sequences of the marA/rob/soxS regulon reported by Martin and Rosner in 2002 [32]. (b) Hierarchical clustering generated by calculating the Euclidean distance of the PWMs and applying the minimum variance clustering method (Ward's method). Three clusters are highlighted.
We included the PWM generated from 24 known DNA binding sequences of the marA/rob/soxS regulon as a positive control [32]. The results show that our structure-based modeling/scoring approach is capable of reproducing the experimentally determined DNA binding sequence of MarA and Rob reasonably well. As expected, the PWMs of MarA and Rob form a distinct cluster together with the known MarA/Rob/SoxS binding sequences in the dendrogram (Figure 5(b)). Another cluster is formed by the two putative AraC/XylS family members with PDB codes 3mn2 and 3oio. This result is in agreement with the relative proximity of the two proteins observed in our DBD sequence-based dendrogram (Figure 3). A third cluster contains the remaining structures of YesN (PDB code: 3lsg), Lin2118 (PDB code: 3oou), GadX (PDB code: 3mkl), AraC (PDB code: 2k9s), and TcpN (PDB code: 3gbg). Despite distinct biological roles, these proteins were organized in the same major branch of the sequence-based dendrogram, as well.
A functional classification based only on the binding site prediction was not feasible, mainly due to the small amount of available structures (ten in total) and the small amount structures complexed with DNA (three in total). In addition, a more stringent calibration of our structure-based method on available binding site data is required in the future. However, the similar grouping of the transcriptional regulators in the sequence-based, structure-based, and DNA binding site-based dendrograms is promising and increases the confidence of the functional annotation of the three putative AraC/XylS members. It should also be noted that the comparison of DNA binding specificity treats function on a molecular level, whereas the analysis of the sequence-based dendrogram treats function on a level of biological processes. The two functional levels are connected in the next section by matching the predicted binding sites against the genomes of the source organism and analyzing the genetic context of the matched binding sites.
An interesting observation is the preference of thymine in position five of the predicted binding sequences of the Lin2118 protein (PDB code: 3oou). This position corresponds to a cytosine in the crystal structures of MarA (C-32) and Rob (C-7). In these two structures, we observe a specific hydrophobic interaction of a tryptophan residue (Trp-42 in MarA and Trp-36 in Rob) with the base of this cytosine nucleotide in the major groove. This tryptophan residue is conserved in family members related to stress response and in fact seems to be a unique characteristic of this group (Figure 2). This is remarkable as tryptophan has a low statistical propensity to interact with DNA, but its propensity to interact with either cytosine or thymine is similar (Refs: [56] and A. Schüller, unpublished data). In the Lin2118 protein (PDB code: 3oou) this tryptophan residue is substituted by valine (Val-36). Valine has an over threefold increased propensity to interact with thymine in comparison with any of the other bases, and this preference is reflected in the corresponding predicted binding sequences of the Lin2118 protein.
3.5. Genomic Context of Predicted DNA Binding Sites
We further analyzed the predicted DNA binding sites by mapping the predicted nucleotide sequences against the genomes of the respective source organism. We included the three putative AraC/XylS members (PDB codes: 3oou, 3mn2, and 3oio) and the poorly characterized transcriptional regulator YesN (PDB code: 3lsg) in this analysis. A total of 195 top-scoring sequences were retrieved for the four structures and, of these, only 6 sequences could be mapped to their respective genomes, allowing up to three mismatches. A detailed analysis of the genomic context of the binding sites (three genes upstream and three genes downstream) is provided in the Supplementary Material (Figure S2 and Table S2).
The transcriptional regulator YesN (PDB code: 3lsg; F. nucleatum) is thought to be involved in plant cell wall degradation [43, 44]. Mapping 39 high-scoring, predicted DNA binding sites of YesN against the genome of F. nucleatum retrieved a single match. 1,438 base pairs (bp) upstream of the binding site we found the xylose repressor gene xylR. XylR is a transcriptional regulator of the xylose operon that contains genes required for degradation of xylose, the most abundant sugar monomer of hemicelluloses [57]. The vicinity of a gene related to hemicellulose depolymerization is consistent with the proposed biological process of plant cell wall degradation.
64 high-scoring binding sites were generated for the Lin2118 protein from L. innocua (PDB code: 3oou). Based on its vicinity to YesN and YesS in the dendrogram of AraC/XylS family members, we proposed an involvement in plant cell wall degradation for this protein. Of the 64 binding sites, only two matched against the genome of L. innocua. 1,404 and 1,026 bp downstream of the first matched site we found the two genes crcB1 and crcB2, respectively (Supplementary Figure S2). It has been shown in E. coli that overexpression of these crcB homologs (along with crcA and cspE) protected cells from the DNA decondensing agent camphor [58]. Camphor is a terpenoid found in Lauraceae and Lamiaceae families of angiosperms. Although unrelated to plant carbohydrate metabolism, the two genes are involved in the response to an antimicrobial substance produced by certain plants.
The second binding site is located in a cluster of three genes with similarity to proteins of the phosphoenolpyruvate-dependent sugar phosphotransferase system (PTS), which is involved in the uptake and phosphorylation of specific carbohydrates from the extracellular environment. The predicted binding site matched inside the lin2833 gene that encodes a protein similar to domain IIA of enzyme II. Enzyme II is a carbohydrate-specific permease responsible for sugar uptake and a component of PTS [59]. The other two genes have similarity with domains IIB (lin2831 gene, 1,610 bp upstream) and IIC (lin2832 gene, 166 bp upstream) of enzyme II (Supplementary Figure S2). The identified enzyme II domains are of the cellobiose-specific subfamily. Cellobiose is a major component of cellulose found in plant cell walls. These data agree well with a possible role in plant cell wall degradation.
Based on our AraC/XylS-family sequence analysis we predicted a role in symbiotic nitrogen fixation for the uncharacterized protein with PDB code 3oio from C. violaceum. 72 sequences were predicted as high-scoring DNA binding sites, of which only two sequences mapped against the genome of C. violaceum. In the vicinity of the first binding site, we found the ibeB gene (726 bp upstream), which by homology encodes an outer membrane efflux protein (OEP) (Supplementary Figure S2). IbeB is a member of the NodT subfamily of the resistance-nodulation-cell division (RND) type efflux systems involved in Nod factor secretion, which are important for nodulation in species of Rhizobium [60]. Root nodules are specialized plant organs where symbiotic nitrogen fixation takes place. This finding is well in agreement with a proposed biological role in nitrogen fixation.
In the genetic context of the second binding site, we identified the gene locus CV_3010 (1261 bp upstream) (Supplementary Figure S2). By sequence similarity, this gene contains a molybdopterin cofactor binding domain found in a variety of oxidoreductases. Main members of this family are nitrate reductase and sulfite oxidase. The presence of an assimilatory nitrate reductase domain is consistent with a proposed biological role in nitrogen fixation [61].
Twenty high-scoring binding site sequences were predicted for the uncharacterized protein with PDB code 3mn2 from R. palustris, which produced a single match in the genome. Based on our analysis of DBD sequences, we proposed a tentative biological role in either metabolism of plant-derived biomass or nitrogen fixation. The locus Rpal_1214 is found 1,510 bp upstream of the binding site and encodes a DSBA oxidoreductase (Supplementary Figure S2) that is involved in cellular respiration [62]. Nitrogen fixation is an energetically expensive process. It has been shown, in another strain of R. palustris, that genes involved in the electron transfer of cellular respiration increase their expression under conditions of high nitrogen fixation [63]. Electron transfer has been proposed as rate-limiting for nitrogenase activity [64]. Moreover, between 95 and 295 genes of R. palustris that are not directly associated with nitrogenase synthesis and assembly were induced under nitrogen-fixing conditions [54]. We found a second gene 2,688 bp upstream of the predicted DNA binding site (locus Rpal_1216, Supplementary Figure S2), which encodes a flavoenzyme ferric reductase. This protein is involved in the electron transfer system and might be related to obtaining energy for nitrogen assimilation. However, proteins associated with cellular respiration may be induced by a multitude of pathways and a false positive binding site prediction cannot be ruled out.
In summary, by matching high-scoring predicted binding sites of uncharacterized transcriptional regulators against their host genomes, in five of six cases we were able to identify related genes in the genetic context of the binding sites. It should be noted that the majority of the analyzed genes encode hypothetical proteins identified by sequence similarity/homology lacking experimental validation. However, the fact that database annotations of these genes are consistent with the biological roles that we proposed based on the analysis of AraC/XylS-family DBD sequences is promising. Yet, the presented analysis is, by no means, complete. Of the 422 (≈2 × 1013) possible binding site sequences we generated a subset of 10,000 sequences, which is a mere 6 × 10−8 percent of the possible sequence space. This number was slightly increased by allowing up to three mismatches; however, we expect a large number of false negatives, that is, undetected binding sites. Increasing the sampling rate and switching to an optimized sampling algorithm will improve this limitation in the future.
4. Conclusions
We presented an integrated bioinformatics method that combined sequence-based relationships, structural similarity, and prediction of DNA binding sites of transcriptional regulators. The advantage of the proposed method is the utilization of information derived from different structural levels (primary and tertiary) and different biological entities (proteins and DNA binding sites). The prediction of DNA binding sequences is a promising approach to complement available information, where experimental data are scarce. Mapping of the predicted binding sites to the genomes of the source organisms and analysis of the genetic context was in good agreement with sequence-derived functional annotations. Although the presented method needs further refinement and validation in the future, the consistency of the presented results is promising. We applied the method to the AraC/XylS family of transcriptional regulators and predicted the biological roles of the three putative new family members with PDB codes 3mn2, 3oio, and 3oou, which originated from structural genomics initiatives. Our bioinformatics analyses suggest that they could be involved in plant cell wall degradation (PDB code 3oou), symbiotic nitrogen fixation (PDB code 3oio), and either metabolism of plant-derived biomass or nitrogen fixation (PDB code 3mn2).
The utilization of combined information derived from structure-based and sequence-based analysis of transcription factor proteins and binding sites is proposed as a convenient way to assign a biological role to structures of unknown function and may guide biologists in conducting proper experimental characterization.
Supplementary Material
Figure S1: Electrostatic potential surface representations of AraC/XylS family members calculated with the software DelPhi; Figure S2: A description of the genetic context of predicted binding sites of the uncharacterized AraC/XylS transcription factors; Table S1: The list of 62 well–]characterized AraC/XylS- family transcriptional regulators with known biological roles from which the dendrogram was calculated; Table S2: Details of the analysis of the genetic context of predicted binding sites of the uncharacterized AraC/XylS transcription factors.
Acknowledgments
A. Schüller is grateful for a FONDECYT (Fondo Nacional de Desarrollo Científico y Tecnológico, Chile) postdoctoral research grant (no. 3110009), and A. W. Slater is grateful for a CONICYT (Comisión Nacional de Investigación Científica y Tecnológica, Chile) graduate scholarship. This paper was funded by grants from FONDECYT (no. 1110400) and ICM (Iniciativa Científica Milenio, Chile; no. P09-016-F).
References
- 1.Berman HM, Westbrook JD, Gabanyi MJ, et al. The protein structure initiative structural genomics knowledgebase. Nucleic Acids Research. 2009;37(1):D365–D368. doi: 10.1093/nar/gkn790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gallegos MT, Schleif R, Bairoch A, Hofmann K, Ramos JL. AraC/Xyls family of transcriptional regulators. Microbiology and Molecular Biology Reviews. 1997;61(4):393–410. doi: 10.1128/mmbr.61.4.393-410.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ibarra JA, Pérez-Rueda E, Segovia L, Puente JL. The DNA-binding domain as a functional indicator: the case of the AraC/XylS family of transcription factors. Genetica. 2008;133(1):65–76. doi: 10.1007/s10709-007-9185-y. [DOI] [PubMed] [Google Scholar]
- 4.Martin RG, Rosner JL. The AraC transcriptional activators. Current Opinion in Microbiology. 2001;4(2):132–137. doi: 10.1016/s1369-5274(00)00178-8. [DOI] [PubMed] [Google Scholar]
- 5.Egan SM. Growing repertoire of AraC/XylS activators. Journal of Bacteriology. 2002;184(20):5529–5532. doi: 10.1128/JB.184.20.5529-5532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alekshun MN, Levy SB. Regulation of chromosomally mediated multiple antibiotic resistance: the mar regulon. Antimicrobial Agents and Chemotherapy. 1997;41(10):2067–2075. doi: 10.1128/aac.41.10.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He C, Hus JC, Li JS, et al. A methylation-dependent electrostatic switch controls DNA repair and transcriptional activation by E. coli Ada. Molecular Cell. 2005;20(1):117–129. doi: 10.1016/j.molcel.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 8.Rhee S, Martin RG, Rosner JL, Davies DR. A novel DNA-binding motif in MarA: the first structure for an AraC family transcriptional activator. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(18):10413–10418. doi: 10.1073/pnas.95.18.10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takinowaki H, Matsuda Y, Yoshida T, Kobayashi Y, Ohkubo T. The solution structure of the methylated form of the N-terminal 16-kDa domain of Escherichia coli Ada protein. Protein Science. 2006;15(3):487–497. doi: 10.1110/ps.051786306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tobes R, Ramos JL. AraC-XylS database: a family of positive transcriptional regulators in bacteria. Nucleic Acids Research. 2002;30(1):318–321. doi: 10.1093/nar/30.1.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang J, Tauschek M, Robins-Browne RM. Control of bacterial virulence by AraC-like regulators that respond to chemical signals. Trends in Microbiology. 2011;19(3):128–135. doi: 10.1016/j.tim.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Suhrer SJ, Wiederstein M, Gruber M, Sippl MJ. COPS—a novel workbench for explorations in fold space. Nucleic Acids Research. 2009;37(2):W539–W544. doi: 10.1093/nar/gkp411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sippl MJ. On distance and similarity in fold space. Bioinformatics. 2008;24(6):872–873. doi: 10.1093/bioinformatics/btn040. [DOI] [PubMed] [Google Scholar]
- 14.Sippl MJ, Wiederstein M. A note on difficult structure alignment problems. Bioinformatics. 2008;24(3):426–427. doi: 10.1093/bioinformatics/btm622. [DOI] [PubMed] [Google Scholar]
- 15.Apweiler R, Martin MJ, O'Donovan C, et al. Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Research. 2011;39(supplement 1):D214–D219. doi: 10.1093/nar/gkq1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marti-Renom MA, Madhusudhan MS, Sali A. Alignment of protein sequences by their profiles. Protein Science. 2004;13(4):1071–1087. doi: 10.1110/ps.03379804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Molecular Operating Environment (MOE), Chemical Computing Group, Montreal, Canada.
- 18.Finn RD, Mistry J, Tate J, et al. The Pfam protein families database. Nucleic Acids Research. 2010;38(1):D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katoh K, Misawa K, Kuma KI, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research. 2002;30(14):3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang HC, Li K, Susko E, Roger AJ. A class frequency mixture model that adjusts for site-specific amino acid frequencies and improves inference of protein phylogeny. BMC Evolutionary Biology. 2008;8(1, article 331) doi: 10.1186/1471-2148-8-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21(9):2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 23.Letunic I, Bork P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23(1):127–128. doi: 10.1093/bioinformatics/btl529. [DOI] [PubMed] [Google Scholar]
- 24.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25(9):1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. Journal of Molecular Biology. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 26.Wang J, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? Journal of Computational Chemistry. 2000;21(12):1049–1074. [Google Scholar]
- 27.Lu XJ, Olson WK. 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nature Protocols. 2008;3(7):1213–1227. doi: 10.1038/nprot.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Norambuena T, Melo F. The Protein-DNA Interface database. BMC Bioinformatics. 2010;11, article 262 doi: 10.1186/1471-2105-11-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Melo F, Feytmans E. Novel knowledge-based mean force potential at atomic. Journal of Molecular Biology. 1997;267(1):207–222. doi: 10.1006/jmbi.1996.0868. [DOI] [PubMed] [Google Scholar]
- 30.Capriotti E, Norambuena T, Marti-Renom MA, Melo F. All-atom knowledge-based potential for RNA structure prediction and assessment. Bioinformatics. 2011;27(8):1086–1093. doi: 10.1093/bioinformatics/btr093. [DOI] [PubMed] [Google Scholar]
- 31.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Research. 2004;14(6):1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin RG, Rosner JL. Genomics of the marA/soxS/rob regulon of Escherichia coli: identification of directly activated promoters by application of molecular genetics and informatics to microarray data. Molecular Microbiology. 2002;44(6):1611–1624. doi: 10.1046/j.1365-2958.2002.02985.x. [DOI] [PubMed] [Google Scholar]
- 33.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10(3, article R25) doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kersey P, Bower L, Morris L, et al. Integr8 and Genome Reviews: integrated views of complete genomes and proteomes. Nucleic Acids Research. 2005;33:D297–D302. doi: 10.1093/nar/gki039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hunter S, Apweiler R, Attwood TK, et al. InterPro: the integrative protein signature database. Nucleic Acids Research. 2009;37(1):D211–D215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Research. 2011;40(1):D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dangi B, Gronenborn AM, Rosner JL, Martin RG. Versatility of the carboxy-terminal domain of the α subunit of RNA polymerase in transcriptional activation: use of the DNA contact site as a protein contact site for MarA. Molecular Microbiology. 2004;54(1):45–59. doi: 10.1111/j.1365-2958.2004.04250.x. [DOI] [PubMed] [Google Scholar]
- 38.Kwon HJ, Bennik MHJ, Demple B, Ellenberger T. Crystal structure of the Escherichia coli Rob transcription factor in complex with DNA. Nature Structural Biology. 2000;7(5):424–430. doi: 10.1038/75213. [DOI] [PubMed] [Google Scholar]
- 39.Rodgers ME, Schleif R. Solution structure of the DNA binding domain of AraC protein. Proteins. 2009;77(1):202–208. doi: 10.1002/prot.22431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lowden MJ, Skorupski K, Pellegrini M, Chiorazzo MG, Taylor RK, Kull FJ. Structure of Vibrio cholerae ToxT reveals a mechanism for fatty acid regulation of virulence genes. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(7):2860–2865. doi: 10.1073/pnas.0915021107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ochiai A, Itoh T, Kawamata A, Hashimoto W, Murata K. Plant cell wall degradation by saprophytic Bacillus subtilis strains: gene clusters responsible for rhamnogalacturonan depolymerization. Applied and Environmental Microbiology. 2007;73(12):3803–3813. doi: 10.1128/AEM.00147-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kobayashi K, Ogura M, Yamaguchi H, et al. Comprehensive DNA microarray analysis of Bacillus subtilis two-component regulatory systems. Journal of Bacteriology. 2001;183(24):7365–7370. doi: 10.1128/JB.183.24.7365-7370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chow V, Nong G, Preston JF. Structure, function, and regulation of the aldouronate utilization gene cluster from Paenibacillus sp. strain JDR-2. Journal of Bacteriology. 2007;189(24):8863–8870. doi: 10.1128/JB.01141-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poncet S, Soret M, Mervelet P, Deutscher J, Noirot P. Transcriptional activator YesS is stimulated by histidine-phosphorylated HPr of the Bacillus subtilis phosphotransferase system. Journal of Biological Chemistry. 2009;284(41):28188–28197. doi: 10.1074/jbc.M109.046334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vlassak KM, Snoeck C, Luyten E, de Wilde P, van Rhijn P, Vanderleyden J. The Rhizobium sp. BR816 NodD3 gene is regulated by a transcriptional regulator of the AraC/XylS family. In: Elmerich C, Kondorosi A, Newton WE, editors. Biological Nitrogen Fixation for the 21st Century. Springer; 1998. (Proceedings of the 11th International Congress on Nitrogen Fixation, Institut Pasteur, Paris, France, July 20-25, 1997). [Google Scholar]
- 46.Blanca-Ordóñez H, Oliva-García JJ, Pérez-Mendoza D, et al. pSymA-dependent mobilization of the Sinorhizobium meliloti pSymB megaplasmid. Journal of Bacteriology. 2010;192(23):6309–6312. doi: 10.1128/JB.00549-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmeisser C, Liesegang H, Krysciak D, et al. Rhizobium sp. strain NGR234 possesses a remarkable number of secretion systems. Applied and Environmental Microbiology. 2009;75(12):4035–4045. doi: 10.1128/AEM.00515-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagy I, Schoofs G, Compernolle F, Proost P, Vanderleyden J, De Mot R. Degradation of the thiocarbamate herbicide EPTC (S-ethyl dipropylcarbamothioate) and biosafening by Rhodococcus sp. Strain NI86/21 involve an inducible cytochrome P-450 system and aldehyde dehydrogenase. Journal of Bacteriology. 1995;177(3):676–687. doi: 10.1128/jb.177.3.676-687.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garsin DA. Ethanolamine utilization in bacterial pathogens: roles and regulation. Nature Reviews Microbiology. 2010;8(4):290–295. doi: 10.1038/nrmicro2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Büttner D, Bonas U. Getting across—bacterial type III effector proteins on their way to the plant cell. EMBO Journal. 2002;21(20):5313–5322. doi: 10.1093/emboj/cdf536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cunnac S, Boucher C, Genin S. Characterization of the cis-acting regulatory element controlling HrpB-mediated activation of the type III secretion system and effector genes in Ralstonia solanacearum. Journal of Bacteriology. 2004;186(8):2309–2318. doi: 10.1128/JB.186.8.2309-2318.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soto MJ, Sanjuán J, Olivares J. Rhizobia and plant-pathogenic bacteria: common infection weapons. Microbiology. 2006;152(11):3167–3174. doi: 10.1099/mic.0.29112-0. [DOI] [PubMed] [Google Scholar]
- 53.Larimer FW, Chain P, Hauser L, et al. Complete genome sequence of the metabolically versatile photosynthetic bacterium Rhodopseudomonas palustris. Nature Biotechnology. 2004;22(1):55–61. doi: 10.1038/nbt923. [DOI] [PubMed] [Google Scholar]
- 54.Oda Y, Samanta SK, Rey FE, et al. Functional genomic analysis of three nitrogenase isozymes in the photosynthetic bacterium Rhodopseudomonas palustris. Journal of Bacteriology. 2005;187(22):7784–7794. doi: 10.1128/JB.187.22.7784-7794.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Griffith KL, Wolf RE. A comprehensive alanine scanning mutagenesis of the Escherichia coli transcriptional activator SoxS: identifying amino acids important for DNA binding and transcription activation. Journal of Molecular Biology. 2002;322(2):237–257. doi: 10.1016/s0022-2836(02)00782-9. [DOI] [PubMed] [Google Scholar]
- 56.Luscombe NM, Laskowski RA, Thornton JM. Amino acid-base interactions: a three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Research. 2001;29(13):2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heo GY, Kim WC, Joo GJ, et al. Deletion of xylR gene enhances expression of xylose isomerase in Streptomyces lividans TK24. Journal of Microbiology and Biotechnology. 2008;18(5):837–844. [PubMed] [Google Scholar]
- 58.Sand O, Gingras M, Beck N, Hall C, Trun N. Phenotypic characterization of overexpression or deletion of the Escherichia coli crcA, cspE and crcB genes. Microbiology. 2003;149(8):2107–2117. doi: 10.1099/mic.0.26363-0. [DOI] [PubMed] [Google Scholar]
- 59.Sliz P, Engelmann R, Hengstenberg W, Pai EF. The structure of enzyme IIA(lactose) from Lactococcus lactis reveals a new fold and points to possible interactions of a multicomponent system. Structure. 1997;5(6):775–788. doi: 10.1016/s0969-2126(97)00232-3. [DOI] [PubMed] [Google Scholar]
- 60.Rivilla R, Sutton JM, Downie JA. Rhizobium leguminosarum NodT is related to a family of outer-membrane transport proteins that includes TolC, PrtF, CyaE and AprF. Gene. 1995;161(1):27–31. doi: 10.1016/0378-1119(95)00235-x. [DOI] [PubMed] [Google Scholar]
- 61.Solomonson LP, Barber MJ. Assimilatory nitrate reductase: functional properties and regulation. Annual Review of Plant Physiology and Plant Molecular Biology. 1990;41(1):225–253. [Google Scholar]
- 62.Deshmukh M, Turkarslan S, Astor D, Valkova-Valchanova M, Daldal F. The dithiol:disulfide oxidoreductases DsbA and DsbB of Rhodobacter capsulatus are not directly involved in cytochrome c biogenesis, but their inactivation restores the cytochrome c biogenesis defect of CcdA-null mutants. Journal of Bacteriology. 2003;185(11):3361–3372. doi: 10.1128/JB.185.11.3361-3372.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rey FE, Heiniger EK, Harwood CS. Redirection of metabolism for biological hydrogen production. Applied and Environmental Microbiology. 2007;73(5):1665–1671. doi: 10.1128/AEM.02565-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jeong HS, Jouanneau Y. Enhanced nitrogenase activity in strains of Rhodobacter capsulatus that overexpress the rnf genes. Journal of Bacteriology. 2000;182(5):1208–1214. doi: 10.1128/jb.182.5.1208-1214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Electrostatic potential surface representations of AraC/XylS family members calculated with the software DelPhi; Figure S2: A description of the genetic context of predicted binding sites of the uncharacterized AraC/XylS transcription factors; Table S1: The list of 62 well–]characterized AraC/XylS- family transcriptional regulators with known biological roles from which the dendrogram was calculated; Table S2: Details of the analysis of the genetic context of predicted binding sites of the uncharacterized AraC/XylS transcription factors.