

Abstract
Gut inflammation occurring in patients with inflammatory bowel diseases (IBD) is associated with an excessive immune response that is directed against constituents of the normal bacterial flora and results in the production of large amounts of inflammatory cytokines. Anti-cytokine compounds, such as the neutralizing TNF antibodies, have been employed with clinical success in patients with IBD. However, nearly half of IBD patients are refractory to such treatments, response can wane with time, and anti-TNF treatment can associate with severe side effects and/or development/exacerbation of extra-intestinal immune-mediated pathologies. These observations, and the demonstration that, in IBD, the pathological process is also characterized by defects in the production and/or activity of counter-regulatory cytokines, have boosted further studies aimed at delineating novel strategies to combat the IBD-associated tissue-damaging immune response.
Keywords: IBD, Crohn's disease, ulcerative colitis, anti-TNF, anti-IL-12, anti-IL-6R, cytokines
Introduction
Inflammatory bowel diseases (IBD) is the general term indicating Crohn's disease (CD) and ulcerative colitis (UC), two chronic inflammatory disorders of the intestine that have different morphological, immunological and clinical characteristics. The aetiology of IBD is unknown, but there is evidence that the liability to develop CD or UC is influenced by a wide range of genetic and environmental factors that promote an exaggerated mucosal immune response that is direct against components of the gut microflora (Kaser et al., 2010). The novel therapeutic strategies for IBD patients are based on inhibiting or modulating the mucosal immune system. In this context, a major effort for researchers is to clarify all the inflammatory pathways of tissue damage in order to optimize the pharmacological interventions. In both CD and UC, the lesion occurs in mucosal areas heavily infiltrated with leucocytes, which through the release of cytokines interact with other immune and non-immune cells thus contributing to the IBD-associated tissue-damaging inflammatory response (MacDonald et al., 1999). In this article, we review the available data justifying the use of compounds that modulate cytokine function in the therapy of IBD.
Anti-TNF therapy
TNF is a cytokine produced by multiple immune and non-immune cells, including macrophages, lymphocytes, mast cells, endothelial cells, fibroblasts and adipocytes, and involved in the regulation of various inflammatory processes (Bazzoni and Beutler, 1996). TNF is primarily produced as a type II transmembrane protein (tmTNF), which can be cleaved by the metalloprotease TNF converting enzyme (TACE) in a soluble form (sTNF) (Black et al., 1997). The biological activity of both tmTNF and sTNF is mediated by binding to specific receptors, namely TNF-receptor type 1 (TNF-R1) and TNF-R2. While TNF-R1 is expressed in most tissues and by several cell types, TNF-R2 is expressed by immune lineage cells only (MacEwan, 2002). Following interaction with the ligand, TNF-R1 and 2 initiate a cascade of intracellular events culminating in the activation of either mitogen-activated-protein-kinases and NF-κB or death-inducing signals (MacEwan, 2002). TNF is highly produced in the intestine of mice with experimental inflammation (Neurath et al., 1997) and it is supposed to play a major role in driving the pathological process (Kontoyiannis et al., 1999). Indeed, administration of TNF blockers to mice suppresses colitis (Powrie et al., 1994; Neurath et al., 1997). Excessive production of TNF is also seen in the inflamed mucosa of IBD patients (MacDonald et al., 1990), where it targets several immune and non-immune cells and triggers multiple inflammatory pathways (e.g. production of chemokines and cytokines, induction of adhesion molecules on endothelial cells, synthesis of non-specific mediators of inflammation and tissue-degrading proteases) (Efimov et al., 2009). Therefore, neutralization of TNF has been considered as a possible strategy for the treatment of active IBD patients. The first TNF blocker used in IBD has been infliximab, a monoclonal chimeric IgG1 anti-TNF neutralizing antibody that is effective in adult and pediatric patients with active, luminal and perianal CD (Targan et al., 1997; Present et al., 1999), and adult patients with active UC (Rutgeerts et al., 2005). Importantly, clinical benefit seen in infliximab-treated patients associates with healing of the inflamed intestinal mucosa (D'Haens et al., 1999). It has also been shown that scheduled therapy (administration every 8 weeks) can maintain clinical remission up to 1 year (Hanauer et al., 2002). Unfortunately, however, response wanes over time probably due to the development of antibodies to infliximab (ATI), which are reported in high percentages in patients undergoing long-term infliximab therapy, and which neutralize and reduce the circulating levels of the drug. A strong inverse correlation exists between serum trough levels of infliximab and levels of ATI. High trough levels of infliximab have been associated with a more durable maintenance of clinical response, and low trough concentrations with potential loss of response (Baert et al., 2003).
Shortening the infusion intervals (i.e. administration every 4 weeks) and/or increasing the dose of the drug (from 5 mg·kg–1 to 10 mg·kg–1 body weight) are useful strategies for increasing trough levels of infliximab and restoring the response to the drug (D'Haens et al., 2011). At the same time, the concomitant administration of immunosuppressive drugs (i.e. thiopurines or methotrexate) could help prevent ATI development (Baert et al., 2003). The spectrum of TNF-neutralizing drugs has recently been enriched by the commercialization of adalimumab, a fully human monoclonal IgG1 anti-TNF antibody, and certolizumab pegol, a monovalent Fab anti-TNF antibody fragment covalently linked to polyethylene glycol (Figure 1). Indications for both adalimumab and certolizumab pegol include induction and maintenance of remission in adult patients with luminal and perianal CD (Schreiber et al., 2005; Colombel et al., 2007). Administration of adalimumab or certolizumab is accompanied by a reduced production of anti-drug antibodies as compared with infliximab (Anderson et al., 2005), confirming that chimeric antibodies are generally more immunogenic than humanized or human antibodies. No study has been yet carried to compare clinical efficacy of the three commercially available TNF antagonists in patients with IBD, but data from clinical trials suggest that they have similar efficacy and adverse-event profiles. The exact mechanism by which anti-TNF drugs reduce intestinal inflammation is not fully understood. The three compounds seem to have high binding affinities for both soluble (s) and tmTNF, but they differ in their ability to activate specific cellular pathways. For example, infliximab and adalimumab are able to bind FcγR, a natural receptor for antibodies, fix complement and induce antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity (Arora et al., 2009) Infliximab and adalimumab enhance also the rate of caspase-dependent apoptosis in lamina propria lymphocytes, a mechanism mediated by tmTNF-activated reverse signalling (ten Hove et al., 2002). Certolizumab lacks these effector functions because it has no Fc region (Weir et al., 2006), and does not induce apoptosis (Fossati and Nesbitt, 2005) probably because it is not able to crosslink tmTNF.
Figure 1.
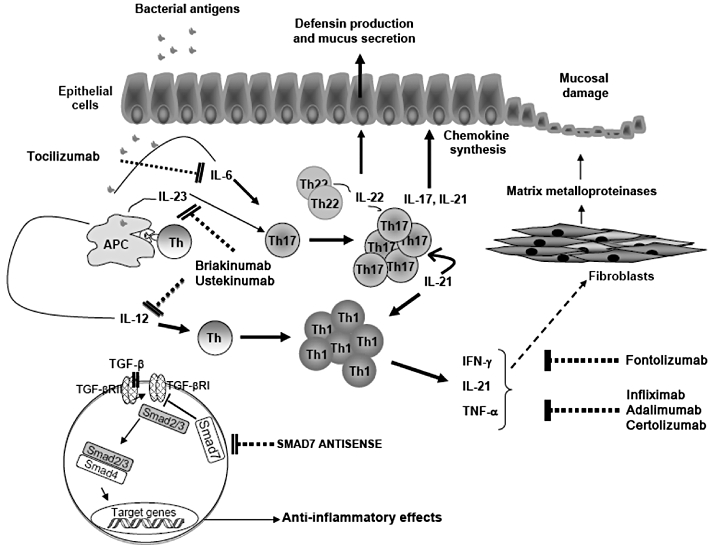
Schematic view of potential targets and site of action of some biologics in IBD. Following stimulation with bacterial products/components, antigen presenting cells (APCs) produce IL-12, which facilitates the differentiation of Th1 cells. Moreover, APCs make IL-23 and IL-6, which contribute to the polarization and expansion of Th17 cells. Briakinumab and ustekinumab, two monoclonal antibodies directed against p40, a subunit shared by IL-12 and IL-23, could interfere with the activation of both Th1 and Th17 cells. Commitment of naive T cells along the Th17 cell pathway and survival of such cells could also be inhibited by tocilizumab, an antibody blocking IL-6 signalling. The biological activity of IFN-γ, a typical Th1 cytokine, can be suppressed by fontolizumab, while anti-TNF antibodies include infliximab, adalimumab and certolizumab. Th17-derived cytokines, such as IL-17A and IL-21, contribute to amplify the ongoing mucosal inflammation by stimulating epithelial cells to make chemokines, thus enhancing the recruitment of inflammatory cells to the intestinal lamina propria. TNF and IL-21 can also stimulate fibroblasts to synthesize MMPs, a family of enzymes that play a major role in the mucosal degradation and tissue damage occurring in IBD. Moreover, IL-21 enhances the production of IFN-γ by Th1 cells, thereby expanding Th1 cell responses. Attenuation of the mucosal inflammation could be obtained using IL-22, a cytokine made by both Th17 cells and Th22 cells, which targets epithelial cells and stimulates the production of defensins and mucins. In IBD, there is a defective activity of TGF-β1, a powerful anti-inflammatory cytokine, due to high Smad7. Smad7 is an inhibitor Smad that interacts with type I TGF-β receptor and prevents Smad3/3 phosphorylation, a phenomenon that is induced by activated type I TGF-β receptor following binding of TGF-β1 to type II TGF-β receptor. Inhibition of TGF-β-induced Smad2/3 phosphorylation by Smad7 prevents the interaction of Smad2/3 with Smad4 and subsequent migration of the complex to the nucleus, where Smad2/3+Smad4 inhibit the expression of many inflammatory genes. Therefore, inhibition of Smad7 with a specific antisense oligonucleotide could restore TGF-β1 activity and facilitate the resolution of the inflammatory process.
Because TNF is involved in the host defence against pathogens, particularly intracellular bacteria, it is not surprising that some patients can develop serious infections (Bongartz et al., 2006) and have reactivation of opportunistic infections and latent tuberculosis following anti-TNF therapy (Keane et al., 2001). Long-term therapy with infliximab can induce serum sickness and allergic-like infusion reactions, which have been linked to circulating ATI (Vermeire et al., 2003). Moreover, infliximab and adalimumab therapy has been associated with de novo emergence of autoimmune conditions, such as drug-induced lupus or psoriasis (Wollina et al., 2008). The reason by which anti-TNF therapy causes/exacerbates these immune-mediated pathologies is not known, even though studies in experimental models of psoriasis have shown that blockade of TNF can enhance T helper (Th)17 cell responses (Ma et al., 2010), which are supposed to be pathogenic in the skin. By contrast, no increased risk of developing tumours has been documented in IBD patients receiving TNF blockers (Biancone et al., 2006), even if a recent meta-analysis has demonstrated that these drugs can dose dependently increase the risk of malignancies in patients with rheumatoid arthritis (Bongartz et al., 2006).
Abrogation of IL-6 signalling with a neutralizing IL-6 receptor antibody
IL-6 can be produced by many cell types, but major sources of this cytokine are monocytes and macrophages during acute inflammation and T cells in chronic inflammation (Nishimoto and Kishimoto, 2006). IL-6 biological activity is mediated by binding of IL-6 with a non-signalling membrane-bound receptor, termed IL-6 receptor (IL-6R)α. This interaction facilitates the recruitment of homodimers of the signal-transducing receptor gp130, with the downstream effect of activating the protein kinase JAK1 and Stat3, a transcription factor involved in the growth and resistance of cells to apoptotic stimuli as well as in the differentiation of Th17 lymphocytes (Atreya et al., 2006). An alternative IL-6 signalling pathway, known as IL-6 trans-signalling, is triggered by interaction of IL-6 to the soluble form of IL-6Rα (sIL-6Rα) and subsequent binding of this complex to membrane-bound gp130 (Rose-John et al., 2006). IL-6 trans-signalling is responsible for the majority of IL-6 biological effects during chronic inflammatory processes and blockade of this signalling with an IL6R neutralizing antibody enhances mucosal T cell apoptosis and attenuates experimental colitis in mice (Atreya et al., 2000). Expression of IL-6, sIL-6Rα and gp130 is up-regulated in IBD (Hyams et al., 1993; Gustot et al., 2005), and there is preliminary evidence indicating that administration of tocilizumab, formerly known as MRA, a humanized monoclonal antibody against IL-6R, to CD patients associates with higher clinical response and remission and induction of intestinal T cell apoptosis as compared with placebo (Ito et al., 2004) (Figure 1). Nonetheless, tocilizumab does not seem to attenuate the endoscopic and histologic lesion (Ito et al., 2004). It is thus conceivable that blockade of IL-6 signalling is not sufficient to fully inhibit the ongoing inflammation perhaps due to the existence of IL-6-independent inflammatory pathways that sustain the mucosal damage in patients treated with tocilizumab (Figure 1).
What do we need to effectively block inflammatory Th cell response in IBD?
As previously pointed out, CD and UC are immunologically distinct diseases. In particular, it has been long recognized that CD bears the stigmata of a T helper type 1 (Th1)-related disease, characterized by excessive production of IL-12, the major inducing factor of Th1 cell response in humans, and IFN-γ (Fuss et al., 1996; Monteleone et al., 1997). Consistently, CD4+ T cells isolated from the inflamed gut of CD patients over-express the IL-12Rβ2 chain necessary for IL-12 signalling, and contain high levels of active Stat4 and T-bet (Parrello et al., 2000; Neurath et al., 2002), two transcription factors necessary for driving and sustaining Th1 cell responses. IL-12 is a heterodimeric cytokine produced by macrophages/dendritic cells mostly in response to bacterial stimulation and shares the p40 subunit with IL-23, another dendritic cell-derived cytokine involved in the regulation of both Th1 and Th17 cell responses (Trinchieri, 2003) (Figure 1). The nature of antigen(s) eliciting IL-12 production in CD remains unknown, but interactions between genes and components of the enteric flora may favour the shift of T cell responses towards the Th1 subtype. Studies in mice with lack or mutations of NOD2 (CARD15), a gene linked to CD and implicated in the response to bacteria, have shown that NOD2 activation is necessary to negatively regulate the production of IL-12 following to Toll-like receptor (TLR)-2 stimuli (Watanabe et al., 2004). These studies raise the intriguing possibility that high IL-12 production and Th1 cell activation in CD may be secondary to a failure of NOD2 to inhibit TLR2 signalling, and that blockade of Th1-associated cytokines/transcription factors or inhibitors of TLR2 could be useful for attenuating the CD-associated Th1-driven inflammatory response. This hypothesis is supported by the demonstration that, in vivo in mice, administration of a neutralizing IL-12/p40 antibody reduces the ongoing Th1 cell response and ameliorates CD-like colitis induced by intrarectal administration of 2,4,6-trinitrobenzene sulfonic acid (Neurath et al., 1995). Unfortunately, however, results of clinical studies testing the efficacy of two distinct neutralizing monoclonal anti-IL-12p40 antibodies (briakinumab, formerly known as ABT-874 and ustekinumab) in patients with moderate-to-severe active luminal CD were quite disappointing, as these antibodies were only slightly superior to placebo in inducing clinical remission (Mannon et al., 2004; Sandborn et al., 2008) (Figure 1). However, ustekinumab appeared more effective than placebo in inducing clinical response in patients who had previously received anti-TNF therapy, suggesting the possibility that anti-IL-12/p40 can be used in patients who failed or are resistant to TNF blockers (Sandborn et al., 2008).
Similarly, no significant clinical response has been documented in active CD patients treated with fontolizumab, an anti-IFN-γ antibody (Hommes et al., 2006; Reinisch et al., 2006, 2010), as well as in patients receiving apilimod mesylate, an oral inhibitor of IL-12/IL-23 synthesis (Sands et al., 2010) (Figure 1). The reason why anti-Th1 cytokine therapy has failed in CD remains unknown. A possible explanation is that these therapeutic approaches block the ongoing Th1 cell-related inflammation but leave unchanged or enhance further pathways of tissue damage. Indeed, it is now well known that the gut of CD patients contains high numbers of Th17 cells, another subset of Th cells that secrete IL-17A, IL-17F, IL-22 and IL-26, whose production/activity is not inhibitable by anti-IL-12/p40 antibodies or fontolizumab (Brand, 2009). If so, one could speculate that compounds targeting simultaneously both Th1 and Th17 cells rather than a targeted approach aimed exclusively at one or the other are more useful for combating CD-related inflammation. A strategy to reach this goal is to inhibit the expression/activity of IL-21, because this cytokine is produced in excess in the inflamed gut of CD patients (Monteleone et al., 2005) and controls positively the production of both Th1 and Th17 cytokines (Monteleone et al., 2005; Fina et al., 2008). Moreover, preclinical studies have shown that IL-21-deficient mice are resistant against Th1/Th17-cell-driven colitis and administration of an antagonist IL-21 receptor fusion protein to mice suppresses experimental colitis (Fina et al., 2008). The inflammatory role of IL-21 in the gut is also supported by the ability of this cytokine to enhance the synthesis of chemokines by epithelial cells (Caruso et al., 2007) and secretion of MMPs by stromal cells (Monteleone et al., 2006) and to make effector CD4+ T cells resistant to regulatory T cell-mediated immunesuppression (Peluso et al., 2007) (Figure 1).
In UC, the T cell response is less polarized, even though UC T-LPL produces more IL-5 and IL-13, two Th2-related cytokines, as compared with normal controls (Fuss et al., 2004). Consistently, studies in the oxazolone model of colitis, which shows striking similarities with UC, indicate that an excessive Th2-cell response contributes to the pathogenesis of the intestinal inflammation. Oxazolone-induced colitis is mediated by CD1-reactive natural-killer T (NKT) cells that make high levels of IL-13 (Heller et al., 2002). Indeed, elimination of NKT cells or neutralization of IL-13 prevents the development of colitis. Overall, these data suggest that IL-13 is a potential therapeutic target for controlling mucosal inflammation in UC.
Like CD, UC is characterized by enhanced production of IL-17A and IL-21 (Fujino et al., 2003; Monteleone et al., 2005). Therefore, it is conceivable that simultaneous blockade of Th2 and Th17 cytokines could be useful for inhibiting UC-associated immune-inflammatory response.
Enhancing the activity of counter-regulatory cytokines as a novel approach to treat IBD
In recent years, a considerable amount of work has been produced to show that IBD-associated immune response is marked by a defective production and/or activity of counter-regulatory cytokines. Furthermore, preclinical studies have shown that restoring/enhancing the activity of such molecules is useful to suppress gut inflammation. The first example of such defects involves the intracellular pathway triggered by TGF-β1, a powerful immune-regulatory cytokine, which is able to target multiple immune and non-immune cell types and suppress inflammatory pathways (Gorelik and Flavell, 2002). The anti-inflammatory properties of TGF-β1 are largely dependent on activation of an intracellular signal program, which is triggered by binding of the cytokine to a heterodimeric membrane-bound receptor and promotes phosphorylation (activation) of Smad2/3, two intracellular proteins that interact with and are activated by TGF-β receptor (Heldin et al., 1997). Although TGF-β1 is highly produced in IBD tissue (Babyatsky et al., 1996), activation of Smad2/3 is defective due to the abundance of Smad7. Smad7 is an inhibitor Smad that binds the TGF-β receptor and prevents TGF-β driven Smad2/3 activation (Monteleone et al., 2001). Consistently, knockdown of Smad7 with a specific antisense oligonucleotide restores TGF-β1-induced Smad2/3 activation(Monteleone et al., 2001), thereby suppressing inflammatory cytokine production and experimental colitis in mice (Boirivant et al., 2006) (Figure 1). Studies are now ongoing to assess the efficacy of Smad7 antisense oligonucleotide in patients with active CD.
IBD patients exhibit defects in the colonic production of IL-25 and thymic stromal lymphopoietin, two cytokines synthesized constitutively by epithelial cells and able to suppress the production of inflammatory cytokines (Caruso et al., 2009; Iliev et al., 2009). Importantly, administration of IL-25 prevents and cures experimental models of colitis, raising the possibility that IL-25-based therapy can enter into the therapeutic armamentarium of IBD patients in the next future (Caruso et al., 2009).
Another cytokine with regulatory function is IL-22, a T cell-derived cytokine which is essential for the integrity of the intestinal epithelial barrier (Ouyang, 2010) (Figure 1). We have recently shown that intestinal production of IL-22 can be enhanced by activation of aryl hydrocarbon receptor (Monteleone et al., 2011), a finding that well fits with the demonstration that the therapeutic effect of aryl hydrocarbon receptor ligands in murine models of colitis is preventable by neutralization of IL-22.
Another approach to potentate anti-inflammatory signals in the gut is to use IL-10, a cytokine, which inhibits the development of Th1-type responses, suppresses Th2 cell-mediated allergic reactions and macrophage/dendritic cell activation, and enhances the differentiation of regulatory T cells (Moore et al., 2001). Additionally, IL-10 can target stromal cells and inhibit the production of tissue-degrading proteases (Pender et al., 1998). IL-10-null mice spontaneously develop bacteria-induced IL-12-driven enterocolitis, thus confirming the key role of this cytokine in preventing detrimental inflammatory responses in the gut (Sellon et al., 1998). Germline mutations in IL-10R genes have been documented in some patients with early-onset and severe IBD, raising the possibility that defects in IL-10 activity may have a key role in sustaining the IBD-related pathogenic response (Glocker et al., 2009). Unfortunately, however, subcutaneous administration of recombinant human IL-10 to patients with active and steroid-dependent CD was not effective in inducing clinical response/remission (Schreiber et al., 2000). It is conceivable that the failure of these clinical studies relies on the fact that IL-10 did not reach therapeutic concentrations in the gut following systemic administration (Braat et al., 2006). To overcome this issue, IL-10-encoding probiotics have been developed. Despite an encouraging phase I trial in CD patients (Braat et al., 2006), studies testing the therapeutic effect of IL-10-secreting lactobacilli in IBD patients have not yet published.
Conclusions
In the last decades, studies in various experimental models of IBD have advanced our understanding of the contribution of cytokines in the pathogenesis of gut inflammation. It has also become evident that distinct subsets of inflammatory cytokines can be produced at the same time in the same patient. Therefore, targeting simultaneously two or more of these signals could be more advantageous than inhibiting selectively single cytokine pathways. Various approaches for inhibiting cytokines that govern Th cell polarization and/or activity have already been developed and tested in IBD patients. Other compounds are now ready to move into the clinic. However, in designing clinical interventions around inflammatory cytokines, we should take into consideration that many of these molecules are involved in the host response against infective agents and neoplasias. Therefore, the use of cytokine blockers could associate with severe side effects (Table 1). In this context, a more advantageous approach could consist in restoring/enhancing the production/activity of counter-regulatory cytokines, which are defective in IBD. As it is highly plausible that no drug will work in all IBD patients, further experimentation will be however needed to ascertain whether and which patient could benefit from specific cytokine-based therapies.
Table 1.
Side-effects associated with the use of anti-cytokine antibodies in patients with inflammatory bowel diseases
| Compound | Route of administration | Patients | % of patients with side effects | Severity of side effects | Side effects | References |
|---|---|---|---|---|---|---|
| Infliximab | Intravenous | CD/UC | 0.1–20 | Potentially severe | Allergic-like infusion reactions, serum sickness, infections including TB reactivation, worsening of cardiac decompensation, demyelinating disease, hepatosplenic T cell lymphoma (rare), psoriasiform eruptions | Targan et al., 1997 |
| Hanauer et al., 2002 | ||||||
| Peyrin-Biroulet et al., 2008 | ||||||
| Adalimumab | Subcutaneous | CD/UC | 0.1–10 | Potentially severe | Injection site reactions, headache, infections including TB reactivation, worsening of cardiac decompensation, demyelinating disease, hepatosplenic T cell lymphoma (rare), psoriasiform eruptions | Hanauer et al., 2006 |
| Colombel et al., 2007 | ||||||
| Peyrin-Biroulet et al., 2008 | ||||||
| Certolizumab pegol | Subcutaneous | CD | 0.1–5 | Potentially severe | Injection site reactions, allergic reactions, headache, perianal abscess, bleeding, infections including TB reactivation | Schreiber et al., 2005 |
| Schoepfer et al. 2010 | ||||||
| Tocilizumab (anti-IL-6R) | Intravenous | CD | 9 | Mild to moderate | Gastrointestinal bleeding | Ito et al., 2004 |
| Fontolizumab (anti-IFN-gamma) | Intravenous | CD | 5–8 | Mild | Dry skin, rash, sinusitis | Reinisch et al., 2006 |
| Reinisch et al. 2010 | ||||||
| Hommes et al., 2006 | ||||||
| Ustekinumab (anti IL-12/p40) | Intravenous, subcutaneous | CD | 16 | Potentially severe | Pruritus, anxiety, infusion reactions (pyrexia, flushing), viral gastroenteritis (1 case), disseminated histoplasmosis (1 case) | Sandborn et al., 2008 |
| ABT-874/J695 (anti IL-12/p40) | Subcutaneous | CD | 10–88 | Mild | Injection site reactions, headache, bronchitis, urinary tract infections | Mannon et al., 2004 |
Acknowledgments
The authors received support for their work from ‘Fondazione Umberto Di Mario’ (Rome, Italy) and Giuliani Spa (Milan, Italy). GM is inventor of a patent entitled ‘Antisense oligonucleotides (odn) against Smad7 and uses thereof in medical field’ Application No. 12264058.
Glossary
- ATI
antibodies to infliximab
- CD
Crohn's disease
- IBD
inflammatory bowel diseases
- JAK
Janus-activated kinase
- Stat
signal transducer and activator of transcription
- Th
cell, T helper cell
- T-LPL
T-lamina propria lymphocytes
- tmTNF
trans-membrane TNF
- UC
ulcerative colitis
Conflict of interest
GM has filed a patent entitled ‘A treatment for inflammatory diseases’ (patent No. 08154101.3), while the remaining authors have no conflict of interests to disclose.
References
- Anderson P, Louie J, Lau A, Broder M. Mechanisms of differential immunogenicity of tumor necrosis factor inhibitors. Curr Rheumatol Rep. 2005;7:3–9. doi: 10.1007/s11926-005-0002-2. [DOI] [PubMed] [Google Scholar]
- Arora T, Padaki R, Liu L, Hamburger AE, Ellison AR, Stevens SR, et al. Differences in binding and effector functions between classes of TNF antagonists. Cytokine. 2009;45:124–131. doi: 10.1016/j.cyto.2008.11.008. [DOI] [PubMed] [Google Scholar]
- Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in Crohn disease and experimental colitis in vivo. Nat Med. 2000;6:583–588. doi: 10.1038/75068. [DOI] [PubMed] [Google Scholar]
- Atreya R, Atreya I, Neurath MF. Novel signal transduction pathways: analysis of STAT-3 and Rac-1 signaling in inflammatory bowel disease. Ann N Y Acad Sci. 2006;1072:98–113. doi: 10.1196/annals.1326.001. [DOI] [PubMed] [Google Scholar]
- Babyatsky MW, Rossiter G, Podolsky DK. Expression of transforming growth factors alpha and beta in colonic mucosa in inflammatory bowel disease. Gastroenterology. 1996;110:975–984. doi: 10.1053/gast.1996.v110.pm8613031. [DOI] [PubMed] [Google Scholar]
- Baert F, Noman M, Vermeire S, Van Assche G, D'Haens G, Carbonez A, et al. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn's disease. N Engl J Med. 2003;348:601–608. doi: 10.1056/NEJMoa020888. [DOI] [PubMed] [Google Scholar]
- Bazzoni F, Beutler B. The tumor necrosis factor ligand and receptor families. N Engl J Med. 1996;334:1717–1725. doi: 10.1056/NEJM199606273342607. [DOI] [PubMed] [Google Scholar]
- Biancone L, Orlando A, Kohn A, Colombo E, Sostegni R, Angelucci E, et al. Infliximab and newly diagnosed neoplasia in Crohn's disease: a multicentre matched pair study. Gut. 2006;55:228–233. doi: 10.1136/gut.2005.075937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- Boirivant M, Pallone F, Di Giacinto C, Fina D, Monteleone I, Marinaro M, et al. Inhibition of Smad7 with a specific antisense oligonucleotide facilitates TGF-beta1-mediated suppression of colitis. Gastroenterology. 2006;131:1786–1798. doi: 10.1053/j.gastro.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA. 2006;295:2275–2285. doi: 10.1001/jama.295.19.2275. [DOI] [PubMed] [Google Scholar]
- Braat H, Rottiers P, Hommes DW, Huyghebaert N, Remaut E, Remon JP, et al. A phase I trial with transgenic bacteria expressing interleukin-10 in Crohn's disease. Clin Gastroenterol Hepatol. 2006;4:754–759. doi: 10.1016/j.cgh.2006.03.028. [DOI] [PubMed] [Google Scholar]
- Brand S. Crohn's disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn's disease. Gut. 2009;58:1152–1167. doi: 10.1136/gut.2008.163667. [DOI] [PubMed] [Google Scholar]
- Caruso R, Fina D, Peluso I, Stolfi C, Fantini MC, Gioia V, et al. A functional role for interleukin-21 in promoting the synthesis of the T-cell chemoattractant, MIP-3alpha, by gut epithelial cells. Gastroenterology. 2007;132:166–175. doi: 10.1053/j.gastro.2006.09.053. [DOI] [PubMed] [Google Scholar]
- Caruso R, Sarra M, Stolfi C, Rizzo A, Fina D, Fantini MC, et al. Interleukin-25 inhibits interleukin-12 production and Th1 cell-driven inflammation in the gut. Gastroenterology. 2009;136:2270–2279. doi: 10.1053/j.gastro.2009.02.049. [DOI] [PubMed] [Google Scholar]
- Colombel JF, Sandborn WJ, Rutgeerts P, Enns R, Hanauer SB, Panaccione R, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn's disease: the CHARM trial. Gastroenterology. 2007;132:52–65. doi: 10.1053/j.gastro.2006.11.041. [DOI] [PubMed] [Google Scholar]
- D'Haens G, Van Deventer S, Van Hogezand R, Chalmers D, Kothe C, Baert F, et al. Endoscopic and histological healing with infliximab anti-tumor necrosis factor antibodies in Crohn's disease: a European multicenter trial. Gastroenterology. 1999;116:1029–1034. doi: 10.1016/s0016-5085(99)70005-3. [DOI] [PubMed] [Google Scholar]
- D'Haens GR, Panaccione R, Higgins PD, Vermeire S, Gassull M, Chowers Y, et al. The London Position Statement of the World Congress of Gastroenterology on Biological Therapy for IBD with the European Crohn's and Colitis Organization: when to start, when to stop, which drug to choose, and how to predict response? Am J Gastroenterol. 2011;106:199–212. doi: 10.1038/ajg.2010.392. [DOI] [PubMed] [Google Scholar]
- Efimov GA, Kruglov AA, Tillib SV, Kuprash DV, Nedospasov SA. Tumor necrosis factor and the consequences of its ablation in vivo. Mol Immunol. 2009;47:19–27. doi: 10.1016/j.molimm.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Fina D, Sarra M, Fantini MC, Rizzo A, Caruso R, Caprioli F, et al. Regulation of gut inflammation and th17 cell response by interleukin-21. Gastroenterology. 2008;134:1038–1048. doi: 10.1053/j.gastro.2008.01.041. [DOI] [PubMed] [Google Scholar]
- Fossati G, Nesbitt AM. Effect of the anti-TNF agents, adalimumab, etanercept, infliximab, and certolizumab pegol (CDP870) on the induction of apoptosis in activated peripheral blood lymphocytes and monocytes. Am J Gastroenterol. 2005;100(Suppl. S):S298–S299. [Google Scholar]
- Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuss IJ, Neurath M, Boirivant M, Klein JS, Motte C, Strong SA, et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–1270. [PubMed] [Google Scholar]
- Fuss IJ, Heller F, Boirivant M, Leon F, Yoshida M, Fichtner-Feigl S, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113:1490–1497. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik L, Flavell RA. Transforming growth factor-beta in T-cell biology. Nat Rev Immunol. 2002;2:46–53. doi: 10.1038/nri704. [DOI] [PubMed] [Google Scholar]
- Gustot T, Lemmers A, Louis E, Nicaise C, Quertinmont E, Belaiche J, et al. Profile of soluble cytokine receptors in Crohn's disease. Gut. 2005;54:488–495. doi: 10.1136/gut.2004.043554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanauer SB, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, et al. Maintenance infliximab for Crohn's disease: the ACCENT I randomised trial. Lancet. 2002;359:1541–1549. doi: 10.1016/S0140-6736(02)08512-4. [DOI] [PubMed] [Google Scholar]
- Hanauer SB, Sandborn WJ, Rutgeerts P, Fedorak RN, Lukas M, MacIntosh D, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn's disease: the CLASSIC-I trial. Gastroenterology. 2006;130:323–333. doi: 10.1053/j.gastro.2005.11.030. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- Heller F, Fuss IJ, Nieuwenhuis EE, Blumberg RS, Strober W. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity. 2002;17:629–638. doi: 10.1016/s1074-7613(02)00453-3. [DOI] [PubMed] [Google Scholar]
- Hommes DW, Mikhajlova TL, Stoinov S, Stimac D, Vucelic B, Lonovics J, et al. Fontolizumab, a humanised anti-interferon gamma antibody, demonstrates safety and clinical activity in patients with moderate to severe Crohn's disease. Gut. 2006;55:1131–1137. doi: 10.1136/gut.2005.079392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Hove T, van Montfrans C, Peppelenbosch MP, van Deventer SJ. Infliximab treatment induces apoptosis of lamina propria T lymphocytes in Crohn's disease. Gut. 2002;50:206–211. doi: 10.1136/gut.50.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyams JS, Fitzgerald JE, Treem WR, Wyzga N, Kreutzer DL. Relationship of functional and antigenic interleukin 6 to disease activity in inflammatory bowel disease. Gastroenterology. 1993;104:1285–1292. doi: 10.1016/0016-5085(93)90336-b. [DOI] [PubMed] [Google Scholar]
- Iliev ID, Spadoni I, Mileti E, Matteoli G, Sonzogni A, Sampietro GM, et al. Human intestinal epithelial cells promote the differentiation of tolerogenic dendritic cells. Gut. 2009;58:1481–1489. doi: 10.1136/gut.2008.175166. [DOI] [PubMed] [Google Scholar]
- Ito H, Takazoe M, Fukuda Y, Hibi T, Kusugami K, Andoh A, et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn's disease. Gastroenterology. 2004;126:989–996. doi: 10.1053/j.gastro.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. 2001;345:1098–1104. doi: 10.1056/NEJMoa011110. [DOI] [PubMed] [Google Scholar]
- Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–398. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- Ma HL, Napierata L, Stedman N, Benoit S, Collins M, Nickerson-Nutter C, et al. Tumor necrosis factor alpha blockade exacerbates murine psoriasis-like disease by enhancing Th17 function and decreasing expansion of Treg cells. Arthritis Rheum. 2010;62:430–440. doi: 10.1002/art.27203. [DOI] [PubMed] [Google Scholar]
- MacDonald TT, Hutchings P, Choy MY, Murch S, Cooke A. Tumour necrosis factor-alpha and interferon-gamma production measured at the single cell level in normal and inflamed human intestine. Clin Exp Immunol. 1990;81:301–305. doi: 10.1111/j.1365-2249.1990.tb03334.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald TT, Bajaj-Elliott M, Pender SL. T cells orchestrate intestinal mucosal shape and integrity. Immunol Today. 1999;20:505–510. doi: 10.1016/s0167-5699(99)01536-4. [DOI] [PubMed] [Google Scholar]
- MacEwan DJ. TNF ligands and receptors – a matter of life and death. Br J Pharmacol. 2002;135:855–875. doi: 10.1038/sj.bjp.0704549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, et al. Anti-interleukin-12 antibody for active Crohn's disease. N Engl J Med. 2004;351:2069–2079. doi: 10.1056/NEJMoa033402. [DOI] [PubMed] [Google Scholar]
- Monteleone G, Biancone L, Marasco R, Morrone G, Marasco O, Luzza F, et al. Interleukin 12 is expressed and actively released by Crohn's disease intestinal lamina propria mononuclear cells. Gastroenterology. 1997;112:1169–1178. doi: 10.1016/s0016-5085(97)70128-8. [DOI] [PubMed] [Google Scholar]
- Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. Blocking Smad7 restores TGF-beta1 signaling in chronic inflammatory bowel disease. J Clin Invest. 2001;108:601–609. doi: 10.1172/JCI12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteleone G, Monteleone I, Fina D, Vavassori P, Del Vecchio Blanco G, Caruso R, et al. Interleukin-21 enhances T-helper cell type I signaling and interferon-gamma production in Crohn's disease. Gastroenterology. 2005;128:687–694. doi: 10.1053/j.gastro.2004.12.042. [DOI] [PubMed] [Google Scholar]
- Monteleone G, Caruso R, Fina D, Peluso I, Gioia V, Stolfi C, et al. Control of matrix metalloproteinase production in human intestinal fibroblasts by interleukin 21. Gut. 2006;55:1774–1780. doi: 10.1136/gut.2006.093187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L, et al. Aryl hydrocarbon receptor-induced signals upregulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology. 2011;141:237–248.e1. doi: 10.1053/j.gastro.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Moore KW, Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- Neurath MF, Fuss I, Kelsall BL, Stuber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1281–1290. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurath MF, Fuss I, Pasparakis M, Alexopoulou L, Haralambous S, Meyer zum Buschenfelde KH, et al. Predominant pathogenic role of tumor necrosis factor in experimental colitis in mice. Eur J Immunol. 1997;27:1743–1750. doi: 10.1002/eji.1830270722. [DOI] [PubMed] [Google Scholar]
- Neurath MF, Weigmann B, Finotto S, Glickman J, Nieuwenhuis E, Iijima H, et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn's disease. J Exp Med. 2002;195:1129–1143. doi: 10.1084/jem.20011956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto N, Kishimoto T. Interleukin 6: from bench to bedside. Nat Clin Pract Rheumatol. 2006;2:619–626. doi: 10.1038/ncprheum0338. [DOI] [PubMed] [Google Scholar]
- Ouyang W. Distinct roles of IL-22 in human psoriasis and inflammatory bowel disease. Cytokine Growth Factor Rev. 2010;21:435–441. doi: 10.1016/j.cytogfr.2010.10.007. [DOI] [PubMed] [Google Scholar]
- Parrello T, Monteleone G, Cucchiara S, Monteleone I, Sebkova L, Doldo P, et al. Up-regulation of the IL-12 receptor beta 2 chain in Crohn's disease. J Immunol. 2000;165:7234–7239. doi: 10.4049/jimmunol.165.12.7234. [DOI] [PubMed] [Google Scholar]
- Peluso I, Fantini MC, Fina D, Caruso R, Boirivant M, MacDonald TT, et al. IL-21 counteracts the regulatory T cell-mediated suppression of human CD4+ T lymphocytes. J Immunol. 2007;178:732–739. doi: 10.4049/jimmunol.178.2.732. [DOI] [PubMed] [Google Scholar]
- Pender SL, Breese EJ, Gunther U, Howie D, Wathen NC, Schuppan D, et al. Suppression of T cell-mediated injury in human gut by interleukin 10: role of matrix metalloproteinases. Gastroenterology. 1998;115:573–583. doi: 10.1016/s0016-5085(98)70136-2. [DOI] [PubMed] [Google Scholar]
- Peyrin-Biroulet L, Deltenre P, de Suray N, Branche J, Sandborn WJ, Colombel JF. Efficacy and safety of tumor necrosis factor antagonists in Crohn's disease: meta-analysis of placebo-controlled trials. Clin Gastroenterol Hepatol. 2008;6:644–653. doi: 10.1016/j.cgh.2008.03.014. [DOI] [PubMed] [Google Scholar]
- Powrie F, Leach MW, Mauze S, Menon S, Caddle LB, Coffman RL. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1994;1:553–562. doi: 10.1016/1074-7613(94)90045-0. [DOI] [PubMed] [Google Scholar]
- Present DH, Rutgeerts P, Targan S, Hanauer SB, Mayer L, van Hogezand RA, et al. Infliximab for the treatment of fistulas in patients with Crohn's disease. N Engl J Med. 1999;340:1398–1405. doi: 10.1056/NEJM199905063401804. [DOI] [PubMed] [Google Scholar]
- Reinisch W, Hommes DW, Van Assche G, Colombel JF, Gendre JP, Oldenburg B, et al. A dose escalating, placebo controlled, double blind, single dose and multidose, safety and tolerability study of fontolizumab, a humanised anti-interferon gamma antibody, in patients with moderate to severe Crohn's disease. Gut. 2006;55:1138–1144. doi: 10.1136/gut.2005.079434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinisch W, de Villiers W, Bene L, Simon L, Racz I, Katz S, et al. Fontolizumab in moderate to severe Crohn's disease: a phase 2, randomized, double-blind, placebo-controlled, multiple-dose study. Inflamm Bowel Dis. 2010;16:233–242. doi: 10.1002/ibd.21038. [DOI] [PubMed] [Google Scholar]
- Rose-John S, Scheller J, Elson G, Jones SA. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: role in inflammation and cancer. J Leukoc Biol. 2006;80:227–236. doi: 10.1189/jlb.1105674. [DOI] [PubMed] [Google Scholar]
- Rutgeerts P, Sandborn WJ, Feagan BG, Reinisch W, Olson A, Johanns J, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353:2462–2476. doi: 10.1056/NEJMoa050516. [DOI] [PubMed] [Google Scholar]
- Sandborn WJ, Feagan BG, Fedorak RN, Scherl E, Fleisher MR, Katz S, et al. A randomized trial of Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn's disease. Gastroenterology. 2008;135:1130–1141. doi: 10.1053/j.gastro.2008.07.014. [DOI] [PubMed] [Google Scholar]
- Sands BE, Jacobson EW, Sylwestrowicz T, Younes Z, Dryden G, Fedorak R, et al. Randomized, double-blind, placebo-controlled trial of the oral interleukin-12/23 inhibitor apilimod mesylate for treatment of active Crohn's disease. Inflamm Bowel Dis. 2010;16:1209–1218. doi: 10.1002/ibd.21159. [DOI] [PubMed] [Google Scholar]
- Schoepfer AM, Vavricka SR, Binek J, Felley C, Geyer M, Manz M, et al. Efficacy and safety of certolizumab pegol induction therapy in an unselected Crohn's disease population: results of the FACTS survey. Inflamm Bowel Dis. 2010;16:933–938. doi: 10.1002/ibd.21127. [DOI] [PubMed] [Google Scholar]
- Schreiber S, Fedorak RN, Nielsen OH, Wild G, Williams CN, Nikolaus S, et al. Safety and efficacy of recombinant human interleukin 10 in chronic active Crohn's disease. Crohn's Disease IL-10 Cooperative Study Group. Gastroenterology. 2000;119:1461–1472. doi: 10.1053/gast.2000.20196. [DOI] [PubMed] [Google Scholar]
- Schreiber S, Rutgeerts P, Fedorak RN, Khaliq-Kareemi M, Kamm MA, Boivin M, et al. A randomized, placebo-controlled trial of certolizumab pegol (CDP870) for treatment of Crohn's disease. Gastroenterology. 2005;129:807–818. doi: 10.1053/j.gastro.2005.06.064. [DOI] [PubMed] [Google Scholar]
- Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–5231. doi: 10.1128/iai.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Targan SR, Hanauer SB, van Deventer SJ, Mayer L, Present DH, Braakman T, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn's disease. Crohn's Disease cA2 Study Group. N Engl J Med. 1997;337:1029–1035. doi: 10.1056/NEJM199710093371502. [DOI] [PubMed] [Google Scholar]
- Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- Vermeire S, Noman M, Van Assche G, Baert F, Van Steen K, Esters N, et al. Autoimmunity associated with anti-tumor necrosis factor alpha treatment in Crohn's disease: a prospective cohort study. Gastroenterology. 2003;125:32–39. doi: 10.1016/s0016-5085(03)00701-7. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004;5:800–808. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- Weir N, Athwal D, Brown D, Foulkes R, Kollias GI, Nesbitt A, et al. A new generation of high-affinity humanized PEGylated Fab' fragment anti-tumor necrosis factor-(alpha) monoclonal antibodies. Therapy. 2006;3:535–545. [Google Scholar]
- Wollina U, Hansel G, Koch A, Schonlebe J, Kostler E, Haroske G. Tumor necrosis factor-alpha inhibitor-induced psoriasis or psoriasiform exanthemata: first 120 cases from the literature including a series of six new patients. Am J Clin Dermatol. 2008;9:1–14. doi: 10.2165/00128071-200809010-00001. [DOI] [PubMed] [Google Scholar]