

It is believed that the adaptation level of codons to the relative concentrations of the corresponding tRNA species affects the translation efficiency of genes. Indeed, in unicellular organisms, codon bias correlates with the genomic copy number of the matching tRNA genes. In this study, the authors extend this view by analyzing experimental tRNA measurements in humans. They show that some technologies of tRNA measurements show a strong correlation with codon usage. Furthermore, they show that the relative composition of tRNA isoacceptor groups in healthy, cancerous, or transformed cells remains essentially constant, suggesting a global regulation of tRNA genes across a broad range of conditions and tissues.
Keywords: codon usage, ENCODE, RNA polymerase III, noncoding RNA, translation elongation
Abstract
Elongation in protein translation is strongly dependent on the availability of mature transfer RNAs (tRNAs). The relative concentrations of the tRNA isoacceptors determine the translation efficiency in unicellular organisms. However, the degree of correspondence of codons and the relevant tRNA isoacceptors serves as an estimator for translation efficiency in all organisms. In this study, we focus on the translational capacity of the human proteome. We show that the correspondence between the codon usage and tRNAs can be improved by combining experimental measurements with the genomic copy number of isoacceptor groups. We show that there are technologies of tRNA measurements that are useful for our analysis. However, fragments of tRNAs do not agree with translational capacity. It was shown that there is a significant increase in the absolute levels of tRNA genes in cancerous cells in comparison to healthy cells. However, we find that the relative composition of tRNA isoacceptors in healthy, cancerous, or transformed cells remains almost identical. This result may indicate that maintaining the relative tRNA composition in cancerous cells is advantageous via its stabilizing of the effectiveness of translation.
INTRODUCTION
Translation elongation efficiency
Translation must be tightly controlled for coping with the cell demands and its limited resources. Energetically, it is the most expensive process in dividing cells (Arava et al. 2003; Ingolia et al. 2009; Plotkin and Kudla 2010; Tuller et al. 2010a; Gingold and Pilpel 2011). Thus, an appropriate regulation of the rate of translation reduces the ribosomal drop-off and the translation errors and improves overall ribosomal allocation (Zhang et al. 2010; Gingold and Pilpel 2011). The relative genomic abundance of the synonymous codons varies in all organisms from bacteria to mammals (Sharp and Matassi 1994; Stenico et al. 1994). Furthermore, codon usage in different genes tends to be related to their expression levels (Marais and Duret 2001; dos Reis et al. 2004; Plotkin and Kudla 2010; Tuller et al. 2010b). Specifically, highly expressed genes (e.g., ribosomal proteins) usually include codons that are recognized by more abundant tRNA molecules, suggesting that the control of the translation process is under selective pressure (Anderson 1969; Bulmer 1987).
In all organisms, less than 61 tRNA types carry out the decoding of all codons. For example, there are only 40 tRNA types (called “tRNA isoacceptors”) in Escherichia coli K12, 44 tRNA types in Drosophila melanogaster, 48 tRNA types in Caenorhabditis elegans, and 51 tRNA types in humans. The decoding of mRNA molecules to proteins in most organisms is therefore based on the presence of some tRNAs that use the same anticodon for recognizing more than one codon (according to the wobble restricted rules) (Percudani 2001; Duret 2002).
In unicellular organisms such as bacteria and fungi, the genomic tRNA copy number correlates with the intracellular tRNA levels (Ikemura 1981; Sorensen and Pedersen 1991; Dong et al. 1996; Percudani et al. 1997; Kanaya et al. 1999; Dittmar et al. 2004). Thus, in general, translation efficiency can be analyzed at the level of amino acids, codon usage, tRNA isoacceptors, and genomic tRNA copy number.
Technologies for large-scale quantifying protein levels have lagged behind the methodologies for mRNA level quantification such as microarray or deep sequencing. Thus, a common assumption in the field is that the transcriptome signature of a cell is an appropriate reflection of its proteome. However, in mouse and humans, it has been shown that the mRNA levels explain only 27%–40% of the protein level variation (Ghazalpour et al. 2011; Schwanhausser et al. 2011). In addition, the level of tRNA molecules is the best-known approximation for the translational rate and the efficiency of codon usage. Unfortunately, measuring the intracellular levels of tRNA molecules remains technologically challenging (Dittmar et al. 2006). Specifically, conventional technologies such as DNA microarray, tilling platform, and PCR-based sequencing methods fail to determine the expression of tRNA molecules. Because tRNAs are short and extensively modified molecules, the routine molecular manipulations (e.g., preparing cRNA) may not be straightforward (Juhling et al. 2009). Thus, currently, the genomic tRNA dosage has been used as a proxy for the actual tRNA cellular abundance (dos Reis et al. 2004; Man and Pilpel 2007; Tuller et al. 2011).
Measurements of short RNAs from deep sequencing platforms are archived in deepBase (Yang et al. 2009). Among these sequences, tRNAs occupied a substantial amount. These data provide an opportunity to study the relations between the actual tRNA measurements and translational-related properties such as the transcribed amino acids, the codon usage, and the tRNA genomic copy number.
The tRNA adaptation index (tAI) (dos Reis et al. 2004; Man and Pilpel 2007; Tuller et al. 2010b) is a measure of the adaptation of a gene (or a codon) to the cellular pool of tRNAs. In practice, to calculate this measure, the genomic tRNA copy number is combined with thermodynamic considerations of the codon–anticodon interaction (Man and Pilpel 2007; Tuller et al. 2010b). The tAI is based on the assumption that the concentrations of the tRNA molecules that recognize a codon have a strong effect on the efficiency and speed of translation.
In this study, we analyzed large-scale genomic and transcriptomic data that were generated by technologies that provide accurate measurements of the relative quantities of tRNA molecules. Based on these data, we found that the relative concentrations of tRNA molecules in different cell types and pathological states remain remarkably stable. We conclude that, for a wide variety of healthy, transformed, and cancerous cells, the tRNA molecules act as stabilizers by providing balanced tRNA pools that resemble the pools of healthy cells.
RESULTS
In the following sections, we report various analyses that we performed with the tRNA measurements. We have focused on human cells for which there are measurements of the intracellular tRNA levels and gene expression, under identical conditions.
Figure 1A illustrates the levels of resolution that were addressed in this study. There are 21 amino acids (including selanocysteine, which is encoded by the codon UAG). Each amino acid is decoded using the tRNA codon–anticodon hybridization and according to the restricted wobble rules (Percudani 2001). In human, there are 51 different isoacceptor types. When considering transcription of genes, each of the codons (64, including stop codons) is represented. In human, the number of tRNA genes for an individual tRNA isoacceptor is between one gene and 32 genes. The total number of tRNA genes in the genome is referred to as the “genomic tRNA copy number.” In human (according to hg19 version), there are potentially 512 functional tRNA genes that cover the 21 amino acids. An additional 100 pseudogenes are also found, but they will not be further discussed. Each tRNA is identified by its anticodon and a numeric value (e.g., tRNA13–AlaCGC). There are tRNA genes that share the same sequences throughout their length. According to this definition, the total number of unique tRNA genes is reduced to 434.
FIGURE 1.

The levels of resolution in tRNAs analyses. (A) An illustration for tRNA quantitative analyses at varying levels of resolution is shown. (i) There are 21 amino acids that are decoded by tRNAs including selanocysteine. (ii) tRNA isoacceptor groups specified by the number of different tRNA carrying different anticodons. There are 51 such tRNA types that are grouped to match the 21 amino acids. Each amino acid has a different number of isoacceptor groups. In this example, alanine (Ala) is decoded by three isoacceptor groups. (iii) Codons that encode each amino acid. There are 62 codons in total. (iv) Each tRNA isoacceptor group has a different number of tRNA genes, referred to as genomic tRNA copy number (CN). For example, the 43 tRNAs for Ala are grouped into 29, 5, and 9 groups. The 512 tRNAs are grouped into the 51 tRNA isoacceptor groups, some with only a single tRNA (Tyr for the ATA codon) and others with as high as 32 tRNAs (Asn for the GTT codon). Among the tRNAs, some of the genes share the same sequences (gray), resulting in only 434 sequence-unique tRNA genes. (B) A flow diagram of the analyses performed in this study is shown. We considered experimental data from deep sequencing (referred to as “Reads”), tRNA probe array (referred to as “Hybridization”), and transcriptomic gene expression (referred to as “Codon Usage”). Experimentally, data were compared among themselves (blue arrows) and for the various human cell lines and tissues (brown). Additional quantitative data are derived from the genomic data of the genomic tRNA copy numbers (referred to as “CN”). We analyzed the correlations between the experimental data of tRNA, the cell transcriptome, and the isoacceptor groups by the genomic CN (purple arrows). See details in the text.
Our research flow is shown in Figure 1B. We start with the comparison of the experimental tRNA measurements that are based on several deep-sequencing resources and tRNA hybridization arrays. The experimental codon usage is calculated from the gene expression transcriptomic data of the analyzed cells. These data are collected from different human cells and tissues. Then, a comparison to the genomic tRNA copy number is performed. A refined estimation of the effect of the tRNA abundance on the efficiency of the translation rate of codons is achieved from the tRNA adaptation index (tAI; see Materials and Methods) (dos Reis et al. 2004). We further compare the tRNA genomic copy number and the codons that are used according to the transcriptomic data and according to various accepted measures of codon preference (Sharp and Li 1987; Duret 2000) (Materials and Methods). We investigate the correlation of the tRNA level measurements based on hybridization arrays and the deep-sequencing reads with the tRNA copy numbers (CN). All analyses were performed for healthy and transformed breast cell lines and for healthy and cancerous tissues (Fig. 1B).
tRNA abundance from RNA-seq correlates with the amino acid frequency
We have focused on cells for which we have different experimental measurements including the direct measurement of tRNA levels and the gene expression profiles that were collected under identical conditions. Figure 2 includes the correlation between the amino acid frequencies (as determined from the transcriptome data) and the number of tRNA genes that recognize the codons of each amino acid (e.g., 43 tRNA genes for Ala) (Fig. 1A). The data were collected from the MCF-10A epithelial breast cell line. A high correlation (r = 0.765, P-value = 3.3 × 10−5) indicates that the number of tRNA genes that decode the codons of an amino acid is in accordance with their genomic abundance (Fig. 2).
FIGURE 2.

Correlation between the genomic copy number and the amino acid usage from epithelial normal cell line MCF-10A. Each codon from the cell transcriptome (10,132 identified expressed genes) was multiplied by the relative gene expression signal. The number of tRNA genes grouped by tRNA isoacceptor groups specifies each amino acid. The 21 amino acids are abbreviated according to standard convention, with SeCys denoting selanocysteine. The maximal value in the x-axis is for alanine (A) with a total of 43 tRNA genes.
tRNA abundance from RNA-seq correlates with the genomic tRNA copy number
We further tested whether such a trend occurs between the experimental measurements of the tRNA molecules and their genomic copy number. Data sets that include measurements of tRNA genes were generated by the main sequencing platforms. For such analyses, we include data from deepBase (Yang et al. 2009) that includes about 60 tRNA experiments (only 20 of them are of a substantial coverage). The entire collection of reads apparently covers all known tRNA genes (510 of the 512 genomic tRNAs). The correlation of tRNA reads and their genomic copy number was moderate but significant (r = 0.34, P-value = 0.012) (Supplemental Fig. S1). This unbiased analysis emphasizes the need for high-quality and high-coverage deep-sequencing data for accurate tRNA abundance measurements.
At the next stage, we focused on individual high-coverage experiments. Such an unbiased data set was extracted from the ENCODE short RNA-seq project (Washietl et al. 2007). Millions of reads for short RNA molecules (e.g., tRNA and miRNA) are reported. The data include short RNA sequences (20–200 nt) without poly(A) that were extracted from several human cell types.
We analyzed the 52,893 and 28,959 reads that were associated with the erythrocytic leukemia cells (K562) and the B-lymphoblastoid cells (GM12878), respectively. The two cell lines differ in their origins as well as in their chromatic state (Ernst et al. 2011). For each tRNA gene that was uniquely defined, we compared the number of reads that were recorded and its genomic copy number. The reads for the GM12878 cells according to their partitioning to tRNA isoacceptor groups (a total of 51) (Fig. 1A) are shown in Figure 3A. As can be seen in the figure, some tRNA isoacceptor groups are assigned to only a few reads, while others are assigned to >10% of the reads.
FIGURE 3.

Expression levels of tRNA types for GM12878 cells by RNA-seq sequencing technology. The total reads that match tRNA genes from GM12878 samples are indicated. (A–C) The ENCODE data set is based on short (20–200 nt) non-poly(A) RNA. (D–F) The data set was extracted from the RNA polymerase III (Pol III) immunoprecipitation experiment. The reads were normalized and presented as a sorted list according to the relative read values (A,D). (B,E) The list includes the relative log abundance above and below the expectation value according to the 51 available tRNA types (note that tRNAs that had no reads are not shown). The correlation of the absolute number of reads and the genomics tRNA copy numbers is plotted. The correlation coefficient (r) value and the P-value are indicated (C,F). Note that the correlations and the P-values were calculated from the original dot plots. The data are shown in a log scale for data compression graphical reasons.
Comparison of the relative abundance of the 51 groups of tRNA reads to their genomic copy number is shown in Figure 3B. The two distributions are far from being identical. Specifically, the tRNA levels of the anticodons CAG and TTT are over-represented in the GM12878 data set, while many tRNAs are under-represented in these cells. Among the under-represented anticodons are GAA and GCA but also the anticodons for Ser (T/C)GA, Arg (T/C)CT, and more. Nevertheless, the correlation between experimentally measured tRNA levels (according to the unique reads) and their genomic copy number for the 51 isoacceptor groups was r = 0.3813 (P-value = 0.0058) (Fig. 3C).
A similar analysis was performed for the K562 cells. However, for the K562 cells, a poor and not significant correlation was measured. In these cells, the number of the tRNA isoacceptor types represented is low (34/49) (Supplemental Fig. S2). We suggest that the tRNAs from deep sequencing are a useful source for tRNA sampling and quantization. However, the experiments differ in term of their reproducibility, coverage, and quality.
In the next step, we seek an independent source for tRNA sequences. The immunoprecipitation experiment using polymerase III (Pol III) fulfills this criterion (Raha et al. 2010). The isolated sequences represent an enriched fraction of genes that were directly attached to Pol III (Figs. 3D–F). The experiments included the K562 and GM12878 cell lines. In both cell lines, a significant correlation of the tRNA measurements and the 51 groups of the tRNA types clustered from the genomic copy number was obtained (Fig. 3F; Supplemental Fig. S2). Figure 3E shows the relative abundance of tRNA reads in comparison to their genomic copy number. The reported correlation for the GM12878 data set was high (r = 0.548, P-value = 3.13 × 10−5). A similar analysis was performed for the K562 cell line (Supplemental Fig. S2).
A direct correlation between the tRNA assigned reads obtained from the two experimental settings (ENCODE and Pol III) for the GM12878 cells is r = 0.475 (P-value = 4.28 × 10−4). This relatively high correlation suggests a sufficient consistency in the tRNA reads from the deep-sequencing methodology, despite substantial variations among the experiments (Supplemental Fig. S3).
Fragmented tRNAs do not correlate significantly with the tRNA genomic copy number
The RNA-seq methodology using deep sequencing can estimate the abundance of sequences that are related to the several steps in the life cycle of tRNA genes. For example, the 3′-tRNA trailers are subsequences from the 3′ end of the pre-tRNA. These sequences are cleaved by a specific endonuclease (RNaseZ) during the process of tRNA maturation (Nashimoto 1997). In Liao et al. (2010), the presence of the 3′-tRNA trailers was quantified in the cytoplasm and the nucleus of the human nasopharyngeal carcinoma (NPC) 5-8F cell line. Interestingly, the estimated levels of the 3′-tRNA trailers showed no correlation with the genomic copy number of tRNA isoacceptors (r = −0.1169, P-value = 0.4140). Importantly, the measurements of the 3′-tRNA trailers are of high quality, because the correlation between different samples (nuclear or cytosolic fractions) is extremely high (r = 0.9461, P-value = 3.715 × 10−24). Furthermore, the number of uniquely matched reads reaches nearly half a million, suggesting a high coverage of the experiment.
Another set of RNA-seq experiments was conducted on HeLa cells with the aim of tracing extremely short stable RNA species (<30 nt) (Cole et al. 2009). In this study, a surprisingly high number of tRNA fragments were detected. Despite the sequencing depth, only a few tRNAs dominate (e.g., tRNA isoacceptors for Lys, Val, Glu, and Arg). In this case, we did not get a significant correlation at the amino acid resolution (r = 0.295, P-value = 0.18) (Supplemental Fig. S4).
tRNAs detected by microarray hybridization strongly correlate with tRNA genomic copy number
In a study published by Pavon-Eternod et al. (2009), a specific microarray for identifying tRNAs by the hybridization signal was designed. Each probe was designed to complement either a single tRNA type or a combination of some of the isoacceptor tRNAs. Consequently, all amino acids (except proline) were covered. Whereas in the study of Pavon-Eternod et al. the relative changes in the expression levels of tRNA (each gene compared to itself) were emphasized, in the present study we focused on the ranking between the expression levels of individual tRNAs. We have used the raw data (Pavon-Eternod et al. 2009) from the normal MCF-10A breast cell line to generalize our findings across technologies.
As mentioned above, some of the probes cross-react with several isoacceptor tRNAs. Consequently, the contribution of each specific tRNA cannot be deduced. Indeed, when the probe hybridization intensity was compared with its isoacceptor group according to the tRNA copy number, no correlation was found (MCF-10A cell line).
We therefore refined the data by applying a strict definition for the tRNAs and the probe sequences. We eliminated the data derived from the mitochondrial tRNAs and focused on the nuclear set. Furthermore, we filtered out the results by analyzing only the subset of probes that perfectly match only a single tRNA gene. Under such a criterion, we reduce the discussion to only 22 valid tRNA isoacceptors (see Materials and Methods). Following such probe–tRNA match selection, the correlation increases but remains insignificant (r = 0.323, P-value = 0.1424).
Many tRNA genes share a remarkably high sequence similarity. Thus, at the next step, we considered only the 183 tRNA genes having an unequivocal hybridization potential by the 22 selected probes. The correlation of the hybridization intensity with these validated tRNA genes was considerably improved (r = 0.4275, P-value = 0.047).
The strong dependency of the analysis on the selected probes encouraged us to increase the number of probes in the analyses. We thus used the definitions that were provided by Pavon-Eternod et al. (2009) for a uniquely matched probe. Thirty tRNA probes were considered based on this definition (some probes matching tRNAs within the same isoacceptor group).
The rest of the analysis is based on the uniquely defined 30 probes (associated with 25 different tRNA isoacceptor groups). Using the experimental hybridization intensities, we found that the correlation between these measured expression levels of the selected probes and the genomic copy number of the relevant tRNA isoacceptor groups was quite high (r = 0.639, P-value = 1.4 × 10−4) (Fig. 4).
FIGURE 4.

Correlation of the hybridization intensity and tRNA genomics copy number of MCF-10A cells. The correlation is according to the tRNA copy number and the unique 30 probes from the tRNA microarray experiments described in Pavon-Eternod et al. (2009). The 30 tRNA probes cover 25 tRNA isoacceptor groups. Some of the outliers are indicated by their codons.
Correspondence of experimental data with various measures of codon usage
In this section, we evaluate the comparison of the hybridization intensity to common scores of codon usage.
Specifically, we tested classical measures including the RGF (the relative tRNA gene frequency from the isoacceptors of a specific amino acid) and the partition between the synonymous codons as measured by RSCU (i.e., the relative usage of a codon among all codons for an amino acid). We included a measure of the dKL (Kullback–Leibler divergence) that provides an overall estimation for the similarities of the various measures (Prat et al. 2009). (For formal definitions, see Materials and Methods.)
Figure 5 shows that for most isoacceptor groups, the codons that had the highest RSCU within each amino acid were those that are decoded by the tRNA isoacceptor with the highest RGF values (the genomic tRNA copy numbers used are listed in Supplemental Table S1). The wobble-restricted rules do not explicitly include the affinity or specificity for the cognate codon–anticodon and the wobble codon. A naive view suggests an equal decoding capacity by a tRNA for the perfectly matched codon and the wobble-codon. While it is clearly an oversimplification, the results show that, for the 16/19 tRNA isoacceptor groups, a full correspondence of the top RGF and the top RSCU is achieved (note that the 6-based decoding of leucine, arginine, and serine was separated according to the genetic code table; see Materials and Methods). Some tRNA isoacceptors cannot be ranked either because the number of tRNA genes within a codon group is identical (Fig. 5B, green background) or because there is only one anticodon that corresponds to the amino acid (Fig. 5B, blue background).
FIGURE 5.

Schematic view of the relation between the tRNA isoacceptor groups and codon usage. (A) Restricted wobble rules are indicated. (Red) The cognate-matched corresponding codon; (orange) the match with the wobble codon. (Bottom table) The values for human threonine (Thr). There are four potential anticodons; however, in human, one of them is missing (GGT). When sorted by the relative copy number (RGF), this anticodon has the highest number of tRNA genes in the Thr isoacceptor group. However, it is ranked only third by the RSCU (red). The RSCU reflects the codon usage within the relevant isoacceptor group. The codon ACC that is decoded by the wobble rule is sorted on the top of the RSCU (orange). (B) Codons are grouped by their detailed tRNA isoacceptor groups. The six-codon amino acids (Arg, Leu, Ser) were fractionated to their groups according to their identity in positions 2 and 3 of their anticodons [(i) four-codon and (ii) two-codon groups]. The perfectly matched codon–anticodon (red); the wobble codon (orange). The matrix is colored by the rank of the codons sorted according to the RGF (as in A). In the case in which the copy number for the tRNAs within an isoacceptor group is identical, it is colored green. Amino acids that are decoded by a single tRNA type (blue). (Dark yellow) Potentially a wobble codon; but a tRNA exists that perfectly matches the indicated codon. In 16/19 isoacceptor groups, the perfectly matched anticodon or the wobble codon is also the codon that is used the most (based on RSCU of the MCF-10A transcriptome). The source data are provided in Supplemental Table S2. (C) Distributions of mRNA codon usage and copy number. The mRNA codon usage (red bars) was calculated based on the gene expression array from the MCF-10A cell line and was compared with the genomic tRNA copy number (blue bars). The data are used for calculating the dKL between the two distributions.
The dKL calculates the difference in the distributions of the experimental tRNA measurements and the expressed codon as compiled from the gene expression transcriptomic data. The minimal dKL value for the distribution of genomic copy numbers and the codon usage is low (0.1717), supporting the relatedness between these distributions (Fig. 5C).
Estimating the tRNA abundance using tAI-based measurements
In this subsection, we report on the correlation of codon bias (based on gene expression microarray; see Materials and Methods) and tRNA levels or copy numbers. In the case of the MCF-10A cell line, the correlation between mRNA level codon bias and tRNA copy number was 0.38 (P-value = 0.0016). This correlation is based on the 51 tRNA type copy numbers that have at least one gene and an additional 12 codons that have no perfectly matched tRNA (for definitions, see Fig. 1A; Supplemental Table S2). The “missing” codons are decoded according to the restricted wobble rules (Fig. 5A).
We also tested the quality of the correlation of the mRNA codon usage (based on the transcriptomic data for MSF-10A cells) and the tAI values of the codons (based on tRNA copy number). The correlation coefficient of the tAI computed by genomic tRNA copy number and the codon usage according to the gene expression array was r = 0.57, P-value = 8.6 × 10−7.
We repeated the calculation of the correlation between the partial set of experimental tRNA levels based on probe data (Pavon-Eternod et al. 2009) and the genomic copy number. However, in this stage, when values for the probe hybridization were missing (based on uniquely assigned 30 probes) (Fig. 4), we estimated them based on the relevant genomic tRNA copy number (see Materials and Methods). The combined tAI obtained a higher and more significant correlation of r = 0.70 (P-value = 1.4 × 10−10; see Materials and Methods) with mRNA codon usage (cf. Fig. 6A,B).
FIGURE 6.

Correlation of the tRNA measurements and the codon usage of MCF-10A cells. (A) The tAI value of each codon was computed using the genomic tRNA copy number of the tRNA genes. Recall that the calculation by the tAI covers all 62 codons. (B) The tAI values of each codon were computed using the normalized values from the hybridization intensity levels. The missing values are inferred based on the genomic tRNA copy number (triangles). Note that the overall correlation was significantly increased when the tAI was calculated based on a combination of the genomic tRNA copy number and the actual experimental data (based on the hybridization intensity from the 30 unique probes).
The correlation between the two measures for the tAI is exceptionally high, supporting the validity of the approach used for the missing values (r = 0.814, P-value = 1.4 × 10−16).
The tRNAs and the codon usage vary in a coordinated way in different cell types
Evidently, the human genomic tRNA copy number is identical in all cells (considering the normal karyotype). We have shown that the codon usage correlates with the hybridization intensity directly (Figs. 4, 5B,C) or through the refined tAI measure (Fig. 6A,B). We therefore analyzed the impact of the alteration in the expression levels in MCF-10A (mammary epithelial cells) and ZR-075 (a breast transformed cell line), which represent normal and cancerous breast cell lines, respectively. These cells were also used for measuring the tRNA abundance (see Materials and Methods). We have compared the mRNA codon bias with their genomic tRNA copy number and with the tRNA expression levels obtained based on hybridization (for the subset of 183 validated tRNA genes) (Fig. 7, indicated as tRNA probe 25).
FIGURE 7.

Correlation between codon usage and tRNA approximations. The correlation according to the tRNA copy number (CN) and the codon usage based on the MCF-10A transcriptome (A) and the same data analysis from the cancer ZR-75-1 cell line (B). The CN is based on those that were selected for the 25 tRNA isoacceptor groups. Note that these are identical to the 30 elected probes from the tRNA microarray experiments. The tRNA probes applied the actual measurements from the 30 probes that are associated with 25 tRNA isoacceptor groups. Note that few of the tRNA probes hybridize to the same isoacceptor tRNA type. The correlation with all genes (blue bar) concerns all expressed genes in the transcriptome (10,132 genes). (Red bar) The 200 most highly expressed genes in the array; (dark green bar) the 200 lowly expressed genes. (C) Correlation of tRNA probes hybridization intensity. (D) The tAI computed by the combination of the tRNA copy number with the hybridization intensity. The correlations performed for normal (MCF-10A) and cancerous (ZR-75-1) cell lines. The raw data were from Pavon-Eternod et al. (2009). The correlation and the P-values are reported.
Overexpressing genes are primarily composed of housekeeping genes that are optimally expressed. This optimization is monitored by the codon adaptation index (CAI), which is maximal for highly abundant genes (e.g., ribosomal proteins). We tested whether such support is also valid for the calculated tAI. We performed the analysis for three complementary data sets: (i) all genes in the gene expression array; (ii) the 200 most highly expressed genes; and (iii) the 200 most lowly expressed genes. The tests were performed for the normal breast cell line MCF-10A and the cancerous breast cell line ZR-075.
Figure 7 demonstrates that there is a strong correlation for the tRNA probes when we consider all expressed genes (more than 10,000 genes) with r = 0.69 (P-value = 1.3 × 10−4) for the normal cells, and r = 0.71 for the cancerous cell lines (P-value = 7.5 × 10−5).
When the 200 most highly expressed genes were analyzed separately, a slightly higher correlation between the mRNA codon bias and the genomic tRNA copy number (r = 0.54, P-value = 3.8 × 10−5 for the normal cell line; r = 0.53, P-value = 0.05 for the cancerous cell line) was obtained. As shown (Fig. 6), when experimental tRNA levels and tRNA copy numbers are combined, the correlation becomes stronger (Fig. 7, labeled as tRNA probe −25).
Importantly, both cells show a similar correlation trend for multiple selections of tRNA measurements (cf. Fig. 7A and 7B). We tested the direct correlation between the tRNA levels in these apparently different cell lines based on the experimental hybridization intensity (25 unique isoacceptors) (Fig. 7C). In a similar way, we applied the refined measure based on tAI that is based on tRNA hybridization experiments and tRNA copy numbers (when the data are missing) (Fig. 7D). In both instances, when the normal cells were compared with the cancerous cell line, an almost perfect correlation was revealed (r = 0.98, P-value = 1.23 × 10−45).
Human cell lines and tissues maintain a stable composition of tRNAs in pathological conditions
We studied whether the strong and consistent correlation found between tRNA levels and tRNA copy numbers in cell lines is comparable to these correlations in healthy and cancerous breast tissues (Table 1).
TABLE 1.
Correlation (and the P-values) between the genomic tRNA copy number and the codon usage (see Supplemental Table S4)

The analyzed cancerous tissues included three main subtypes of breast cancer (a total of nine samples): luminal (ER+, HER2−), basal (ER−, HER2+), and ER−/HER2−. When we computed the correlation of the tRNA probe hybridization intensity for the normal (average of three healthy tissues) and the cancerous tissues (average of nine breast cancer tissues), we found it to be significant (r = 0.38, P-value = 0.037).
At the next step, we analyzed the internal correlations between the 12 different tRNA measurements of the raw data in human tissue (Pavon-Eternod et al. 2009). Specifically, we tested the internal correlations, the mutual dKL, and the correspondence of the tRNA levels (measured by the hybridization intensity for the 25 uniquely defined tRNA isoacceptors) to the genomic tRNA copy number. The results according to the dKL calculations are shown in Figure 8. There is high similarity of the tRNA distributions among all tested tissues (Fig. 8; Supplemental Table S3). Unsupervised clustering supports two main clusters of the healthy (three samples) and the cancerous (nine samples) tissues. Interestingly, among the cancerous tissues, clustering of the results failed to indicate their cancer typing (in terms of the expression of ER and HER2).
FIGURE 8.

Calculation of the dKL of the tRNA expression levels in healthy and diseased tissues. The values of the dKL measures are shown by a color gradient (black to red). The calculations are based on the hybridization signals from the 30 unique tRNA probes for healthy (three samples) and cancerous tissue samples (nine samples). The symmetric matrix indicates the clustering of the 13 columns in the matrix. The diagonal is indicated as dKL = 0. (The left column and the bottom row of the matrix) The dKL for the tRNA hybridization intensity and the genomic tRNA copy number (CN). (Red) A weaker correspondence (higher dKL value). The columns are sorted based on the clustering. The correlations, the minimum dKL, and the P-values are listed in Supplemental Table S3. The samples are colored by their labels as ER−/HER2−: 59826, 60046, 62706, 62944 (blue); ER−/HER2+: 46258, 58955 (red); ER+/HER2−: 41299, 57731, 45163 (orange); and healthy breast tissues: A-01, A-06, and S-23 (green). Note that there is no clear separation between ER−/HER2+ and ER+/HER2− by this measure.
The results (Fig. 8; Table 1; Supplemental Table S3) are consistent with the notion that while the absolute level of tRNAs had changed drastically (Pavon-Eternod et al. 2009), the relative abundance of each tRNA type is quite robust. A stable tRNA composition is valid among cell lines and healthy and diseased tissues.
DISCUSSION
Advances in tRNA measurements
In this study, we analyzed tRNA measurements from RNA-seq that originated from all leading technological platforms (454, SoliD, and Illumina). Our analyses covered several cellular settings including healthy, transformed cell lines, and cancerous tissues. We focused on most of the reported experiments that measured tRNAs by different methodologies. These methodologies include the Pol III immunoprecipitation and a survey for short non-poly(A) RNA conducted by the ENCODE project. We had validated the feasibility of the deep-sequencing data to provide a reproducible source for tRNA concentrations. However, the high level of modifications in human tRNAs may lead to a failure in the required reverse transcriptase reaction. Consequently, all further steps in the sequencing protocol will be affected. Indeed, in the ENCODE data (RNA of 20–200 nt), the tRNAs occupy only 5% and 10% of the entire sample for K562 and GM12878 cells, respectively. In the Pol III data (Fig. 3; Raha et al. 2010), the tRNA fraction occupies only 5% of all of the reads. We attribute this low recovery of tRNAs to an actual methodology limitation. The challenges and biases in identifying the small noncoding RNAs, including tRNA molecules by deep-sequencing technologies, were recently discussed (Beck et al. 2011).
On average, tRNAs occupy 30% of all RNA molecules in a cell. Thus, no amplification step was needed for the hybridization experiments using the tRNA probe microarray (Pavon-Eternod et al. 2009). We assume that by avoiding a reverse transcription step on the RNA sample, potential biases were removed. An additional obstacle in estimating the tRNA abundance stems from misalignment of the RNA-seq reads on the chromosomal segments that are rich in repeats in the vicinity of tRNA gene clusters.
tRNA gene regulation
The expression of a tRNA gene that is not subjected to a regulation is expected to be similar to its respective copy number. Genes with the highest skew from this rule may be candidates for some tissue-specific regulation. Figure 3, B and E, highlights such instances. For example, the tRNAs that recognize the anticodons CGA and TTT (Fig. 3B) are suspected to be the most up-regulated in the ENCODE project data of GM12878 cells, while the tRNA molecules that recognize the codons GAA and GCA are down-regulated in this tissue. Additional tRNA genes appear as outliers in the plot of tRNA levels versus tRNA copy number (for example, Fig. 6A,B). The results reported in this study suggest that individual tRNA genes are regulated more significantly than it was initially anticipated. The involvement of epigenetic signature and chromatin state is a plausible explanation for the observed difference in specific tRNA expression (Ernst et al. 2011).
tRNA fragments do not exhibit a significant correlation with tRNA abundance
We found that there is a poor correlation between tRNA copy numbers and tRNA fragments (Cole et al. 2009; Liao et al. 2010). Thus, in all of the reported studies, the relationship between measured fragments of tRNAs and translation elongation efficiency is not supported.
Recently, another study used deep sequencing (based on 454 sequencing) for estimating the expression levels of small RNAs from prostate cancer cell lines (Lee et al. 2009). Many of the reads were assigned to the processed tRNA fragments that were derived from the regions that overlap with the 3′-tRNA-trailers (Liao et al. 2010). Our results reject the hypothesis that the frequencies of these fragments reflect the expression levels of tRNA genes. Thus, these tRNA fragments may have cellular functions not related to translation. For example, it was suggested that tRNA fragments have a regulatory role in the apoptotic pathway (Mei et al. 2010) and in regulating cell proliferation (Lee et al. 2009). Furthermore, it was also suggested that the tRNA fragments act as regulators of miRNA by competing on the miRNA processing (Lee et al. 2009; Pederson 2010). Our analysis is in accord with the notion that tRNA fragments have some yet-unknown functionality (Okamura and Lai 2008).
A robust expression of tRNAs in various cell types
We have rigorously analyzed several types of cells including healthy, transformed, and cancerous tissues. The absolute tRNA level in the transformed cells is about 20-fold higher than the levels in normal cells (Pavon-Eternod et al. 2009). As we showed, the measured level of tRNA expression is in a strong accordance with the mRNA codon bias extracted from a global gene expression analysis.
We argue that the tRNAs that change their overall expression roughly maintain their relative concentrations upon a wide range of conditions. Moreover, the change in codon usage among cell lines of different identity is negligible (Fig. 7). The relatively constant ranking of the concentrations of tRNAs under a broad range of cells and conditions may indicate that fine-tuned tissue-specific changes in the gene translation rate are probably mostly a result of an additional layer of regulation such as epigenetic, transcriptional, and miRNAs, and not a result of a programmed change in the tRNA levels.
The first systematic analysis that was based only on a careful measure of the relative hybridization intensities (Pavon-Eternod et al. 2009) indicated that tRNAs carrying specific amino acids (such as S, T, and Y) are mostly overexpressed in breast cancer cell lines and breast tumors. In our study, we show that the ranked order of tRNAs (and not necessarily the total amount) is similar whether it is tested by direct experimental data (e.g., RNA-seq) or under a rich model that includes thermodynamic codon–anticodon parameters (e.g., tAI) (dos Reis et al. 2004). The same trends hold when we compare the RGF to the RSCU (Fig. 5). Thus, expression of a balanced, stable ranking of the isoacceptor tRNAs dominates our study.
A detailed study of tRNA relative expression in tissues and cell lines showed that specific tRNA isoacceptors have higher-than-expected variation in some tissues (Dittmar et al. 2006). Our results suggest that the variations in specific isoacceptors are insignificant relative to the overall trend showing a wide variation in the amount of the entire set of tRNAs for a number of tissues and cell lines (as measured in Fig. 8). In agreement with the results from this study, it was noted that the relative expression of tRNAs in HeLa and HEK293 cell lines is similar among the isoacceptors (Dittmar et al. 2006), even though they are derived from different tissues (cervix and embryonic kidney, respectively).
The high correlation between tRNA (or tAI) levels in cells with different transcriptomic profiles (Fig. 7C,D) supports fundamental robustness in the process of protein translation across a range of conditions and tissues. According to our findings, the global rate of translation is probably altered under pathological conditions, while the difference in the relative translation rates of specific genes is less likely to occur.
The correlation of the tRNA abundance and the codon usage is extremely robust in the healthy and transformed cells. Thus, we propose a model in which the tRNA levels change either by an excess of RNA Pol III, by instability of the karyotype, or other indirect cellular scenarios. However, the changes in tRNA gene expression are general and occur across the entire tRNA gene sets, maintaining the relative expression levels of tRNA genes. In support of this intuitively unexpected phenomena, we noted that also the correlation between the calculated tAIs of the normal cells and the transformed cells is extremely high (r = 0.9784, P-value = 3.879 × 10−44). Thus, as a first approximation, one can order genes according to their translation efficiency in the same conditions by considering the genomic tRNA copy numbers and pre-calculated factorization of the codons (as in Fig. 6). However, because we expect global changes (and changes that do not affect the ranking of the expression levels of individual tRNAs) in tRNA levels across tissues or conditions, the tRNA copy number should not be used for a comparison of translation efficiency of a specific gene across different conditions/tissues.
Breast cancerous tissues of different origin display similar tRNA composition
Probably, cell lines were adapted for a stable, constant growth. This may lead to a loss of regulatory mechanisms of the tRNA genes while maximizing the expression levels of each tRNA isoacceptor to meet the constant need for cell divisions. However, we showed that the strong and significant resemblance of the tRNA expression levels also occurs in human healthy and cancerous tissues. All of the 12 samples (three healthy and nine cancerous) show a strong correlation. However, the correlations (and the dKL) of the tRNA levels with the tRNA copy number are considerably weaker for each of the tested samples (Fig. 8; Supplemental Table S3). The average value (Table 1) demonstrates that, despite a strong coherence in the results among all tissues, the genomic tRNA copy number is not a perfect approximation of the tRNA abundance. Several reasons may have reduced the correlations (Fig. 8; Table 1): (1) Data from the 30 tRNA probes (covering 25 tRNA isoacceptors) may not be an optimal sampling for the entire 64 codons and may include different sources of noise and bias. (2) The tRNA copy number may be inaccurate. For example, the identification of functional tRNAs is based on algorithmic arbitrary thresholds, and it is known that the functionality of some tRNAs remains uncertain (Lowe and Eddy 1997). In addition, some pseudogenes that have been excluded from the copy number calculation are expressed (based on the deepBase data) (Yang et al. 2009). (3) Different tRNA genes have different levels of regulations that reduce the correlation between copy number and tRNA levels.
Based on the results reported in this study, we conclude that, in human, the genomic tRNA copy number is a reliable and valid approximation for their expression levels. Thus, when performing a large-scale transcriptomic study, the tRNA copy number can be safely used for estimating global translation efficiency. However, we showed that data from deep sequencing or tRNA microarrays are useful because they potentially improve the estimation of the tRNA levels. At present, accurate measurements of processed, functional tRNAs are still fragmented and mostly missing. Thus, high-quality data are urgently needed.
Our comprehensive study that is based on collecting most available experimental data led to new insights on translation efficiency in a wide range of cellular settings. Ample research studies showed that due to genomic instability and changes in chromatin structure, the expression of hundreds of genes is altered in cancer relative to healthy cells. In sharp contrast, at the level of translation, such alterations seem to be tamed and attenuated. Our findings argue that a regulation of tRNA expression is not at the gene level or the chromosomal level, but instead it is performed globally on the entire collection of tRNA genes. We raised the notion that translation, being the most energetically expensive process in dividing cells, acts as a stabilizer that maintains a balanced translation potential even under unstable cellular conditions.
MATERIALS AND METHODS
Genomic copy number
The data of genomic tRNA copy number, chromosomal locations, and the sequence identity tRNA genes were taken from the Genomic tRNA Database using human genome hg19 (NCBI Build 37.1, Feb 2009) (Lowe and Eddy 1997). For each tRNA gene, the number of copies was counted, ignoring pseudogenes but including selanocysteine (for a detailed list, see Supplemental Table S1).
tRNA gene counting
The convention is based on the algorithms described in Lowe and Eddy (1997). The tRNA probe reanalysis is based on a replacement of the degenerate base. The degenerative probes are indicated by one-letter codes (http://www.bioinformatics.org/sms/iupac.html). Each tRNA is designated by the anticodon that is depicted by three bases. For consistency, all codons and anticodons are described with the base thymidine (T) instead of uridine (U). For example, the tRNA Met (CAT) decodes the codon ATG. We kept the notation of the tissues used by Pavon-Eternod et al. (2009).
RNA-seq sequencing data of tRNAs
tRNA 3′-trailers
The data for the tRNAs 3′-trailers were based on a cytoplasmic and a nuclear extraction from human nasopharyngeal carcinoma (NPC) 5-8F (Liao et al. 2010).
ENCODE
The data were downloaded from the small RNA [non-poly(A)] ENCODE project at http://genome.ucsc.edu/. The tRNA measurements were collected from the K562 and GM12878 cell lines. Each validated read was assigned to the appropriate genomic location (contigs), and the number of reads that were detected for each genomic sequence was recorded. All sequences that overlap (even partially) a tRNA gene were considered. The chromosomal location of each tRNA gene was based on the data of the UCSC Genome Browser (Raney et al. 2010).
Pol III immunoprecipitation
Reads of tRNA from the K562 and GM12878 cells were collected using an antibody against Pol III (Raha et al. 2010). The number of reads for each tRNA gene was used as a measure for the tRNA abundance.
deepBase
deepBase compiles 59 individual experiments; among them about 20 are of high coverage (more than 1 million reads). A total of 625 tRNAs are reported as expressed genes by deep sequencing. The list includes 99 genes that were assigned as pseudogenes. Out of the complete list of tRNAs, only 12 were unidentified. It is important to note that for sequence-identical tRNAs, no unique identification is possible, and therefore some of the reads should be considered as a sum of the multiple tRNAs that are identical in sequence.
Analyzing tRNA probe hybridization intensity data
The data of tRNA probe hybridization intensity were taken from Pavon-Eternod et al. (2009). The set of probes was used to measure the differences in tRNA levels between the three samples of MCF-10A (used as reference for the epithelial normal cell line) and nine additional samples from different stages and types of breast cancer tissues (Pavon-Eternod et al. 2009).
For each biological sample, two probe arrays with a mixture of Cy3 and Cy5 dyes were used to produce the hybridization data. To reduce the staining bias, the cell line that was used as a reference (a non-cancer-derived breast epithelial MCF-10A) was dyed with each of the two dyes, separately. The variation between the measured hybridization intensities was used to estimate the bias. While the experiment was not designed for estimating absolute measurements of tRNA genes, we considered the average of the hybridization intensities measured from the dye swapped arrays as a quantity of the tRNA hybridization. The complete data set (Pavon-Eternod et al. 2009) covers the 50 different nuclear tRNA probes. However, among these probes, only 30 were designed to match a single tRNA type. The rest were designed to match several tRNA isoacceptors. The 30 unique probes were used throughout our study. This set covers 25 of the isoacceptor groups.
Additional filtration was applied to ensure a perfect match for the hybridization reaction. To this end, the sequence of each probe was aligned using the tRNA genomic BLAST tool. The aligned sequences were filtered to include only the tRNA genes that had a perfect matched alignment of ≥20 sequential nucleotides. Only 22 legitimate probes passed this filter. We refined the assignment of a tRNA to its probe by defining higher constraints on the probe–tRNA hybridization.
Gene expression and codon usage analysis
Data from gene expression arrays of a normal cell line (MCF-10A) and a cancerous cell line (ZR-075) were used in this study. Genes that were reported as expressed were retrieved from Ensembl (Flicek et al. 2010). The Ensembl annotation of each probe was based on the HG-U133A Affymetrix annotation file. Genes with no matched sequence or without an ATG initiator codon were discarded. Out of 22,215 potential human genes, 10,132 genes were further analyzed. The mRNA codon usage was computed by accumulating over all of the genes the product of the codon occurrences in each gene multiplied by the actual expression intensity of the gene. The intensity was retrieved from the relevant HG-U133A microarray. A naive view on gene expression is based on the Ensembl gene list (22,215 potential human genes). This analysis was applied for instances in which detailed transcriptomic data were missing.
tRNA gene frequency (RGF) and relative synonymous codon usage (RSCU)
For each tRNA isoacceptor that encodes a specific amino acid, the relative gene frequency was computed in the following way: In each amino acid isoacceptor group, let CNi be the copy number of the i-th isoacceptor. Let Mcnj be the average of the copy number of all isoacceptor within the j-th group. The i-th isoacceptor is part of the j-th group (Fig. 1A). The RGF of isoacceptor i, RGFi, is defined as:
We partitioned the six-based codons of Leu, Ser, and Arg to their subgroups according to the genetic code table. The partition to six-based codons to four- and two-based codons. A total of 24 isoacceptor groups were considered; among them there are 21 isoacceptor groups with two or more codons.
Similarly, the relative synonymous copy number was computed: Let CUi be the codon usage of codon i. Let Mcuj be the average of the codon usage within a group of the j-th amino acid synonymous codons. The i-th isoacceptor is part of the j-th group.
The RSCU of each codon, RSCUi, is defined as:
Kullback–Leibler divergence (dKL)
The difference of the probability distribution between two data sets was computed using the Kullback–Leibler divergence definition (Prat et al. 2009). Let P and Q be the probability distribution of each data set. We applied this measure for normalized distribution of the copy numbers and the codon usage.
The dKL is defined as:
Computing the tRNA adaptation index (tAI)
tAI was computed according to dos Reis et al. (2004). This measure gauges the availability of tRNAs for each codon along an mRNA. Because codon–anticodon coupling is not unique due to wobble interactions, practically, several anticodons can recognize the same codon, with somewhat different efficiency.
Let ni be the number of tRNA isoacceptors recognizing codon i. Let tCGNij be the copy number of the j-th tRNA that recognizes the i-th codon, and let Sij be the selective constraint on the efficiency of the codon–anticodon coupling. We define the absolute adaptiveness Wi for each codon i as:
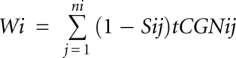 |
From Wi we obtain wi, which is the relative adaptiveness value of codon i, by normalizing the Wi values (dividing them by the maximal of all the 61 Wi).
Computing tAI for missing data
Reliable experimental data are limited to the 30 tRNA genes for which 30 unique probes are used to estimate the tRNA levels. The rest of the tRNA genes are estimated based on the genomic copy number. In this case, for a tRNA that has no matching probe, the value of the tRNA abundance was normalized with the total genomic tRNA copy number in the following manner: Let tpi be the hybridization intensity value of probe i. Let Mp be the mean of all of the probe hybridization intensities. Let Mcn be the mean of all genomic tRNA copy numbers. The tRNA abundance estimator tEi for tRNAi is defined as:
 |
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Mariana Pavon-Eternod for her assistance and personal help throughout this study. We thank Nathan Linial for his advice and useful suggestions. The work is partially supported by grants (to M.L.) Prospects (EU FrVII), the ISF 592/07, and the BSF 2007219 (to M.L.). S.M. is a fellow of the SCCB, the Sudarsky Center for Computational Biology.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.030775.111.
REFERENCES
- Anderson WF 1969. The effect of tRNA concentration on the rate of protein synthesis. Proc Natl Acad Sci 62: 566–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arava Y, Wang Y, Storey JD, Liu CL, Brown PO, Herschlag D 2003. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc Natl Acad Sci 100: 3889–3894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck D, Ayers S, Wen J, Brandl MB, Pham TD, Webb P, Chang CC, Zhou X 2011. Integrative analysis of next generation sequencing for small non-coding RNAs and transcriptional regulation in myelodysplastic syndromes. BMC Med Genomics 4: 19 doi: 10.1186/1755-8794-4-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulmer M 1987. Coevolution of codon usage and transfer RNA abundance. Nature 325: 728–730 [DOI] [PubMed] [Google Scholar]
- Cole C, Sobala A, Lu C, Thatcher SR, Bowman A, Brown JW, Green PJ, Barton GJ, Hutvagner G 2009. Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA 15: 2147–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmar KA, Mobley EM, Radek AJ, Pan T 2004. Exploring the regulation of tRNA distribution on the genomic scale. J Mol Biol 337: 31–47 [DOI] [PubMed] [Google Scholar]
- Dittmar KA, Goodenbour JM, Pan T 2006. Tissue-specific differences in human transfer RNA expression. PLoS Genet 2: e221 doi: 10.1371/journal.pgen.0020221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Nilsson L, Kurland CG 1996. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J Mol Biol 260: 649–663 [DOI] [PubMed] [Google Scholar]
- dos Reis M, Savva R, Wernisch L 2004. Solving the riddle of codon usage preferences: A test for translational selection. Nucleic Acids Res 32: 5036–5044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L 2000. tRNA gene number and codon usage in the C. elegans genome are co-adapted for optimal translation of highly expressed genes. Trends Genet 16: 287–289 [DOI] [PubMed] [Google Scholar]
- Duret L 2002. Evolution of synonymous codon usage in metazoans. Curr Opin Genet Dev 12: 640–649 [DOI] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Brent S, Chen Y, Clapham P, Coates G, Fairley S, Fitzgerald S, et al. 2010. Ensembl 2011. Nucleic Acids Res 39: D800–D806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazalpour A, Bennett B, Petyuk VA, Orozco L, Hagopian R, Mungrue IN, Farber CR, Sinsheimer J, Kang HM, Furlotte N, et al. 2011. Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet 7: e1001393 doi: 10.1371/journal.pgen.1001393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingold H, Pilpel Y 2011. Determinants of translation efficiency and accuracy. Mol Syst Biol 7: 481 doi: 10.1038/msb.2011.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemura T 1981. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J Mol Biol 146: 1–21 [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhling F, Morl M, Hartmann RK, Sprinzl M, Stadler PF, Putz J 2009. tRNAdb 2009: Compilation of tRNA sequences and tRNA genes. Nucleic Acids Res 37: D159–D162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaya S, Yamada Y, Kudo Y, Ikemura T 1999. Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: Gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene 238: 143–155 [DOI] [PubMed] [Google Scholar]
- Lee YS, Shibata Y, Malhotra A, Dutta A 2009. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev 23: 2639–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao JY, Ma LM, Guo YH, Zhang YC, Zhou H, Shao P, Chen YQ, Qu LH 2010. Deep sequencing of human nuclear and cytoplasmic small RNAs reveals an unexpectedly complex subcellular distribution of miRNAs and tRNA 3′ trailers. PLoS ONE 5: e10563 doi: 10.1371/journal.pone.0010563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR 1997. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25: 955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man O, Pilpel Y 2007. Differential translation efficiency of orthologous genes is involved in phenotypic divergence of yeast species. Nat Genet 39: 415–421 [DOI] [PubMed] [Google Scholar]
- Marais G, Duret L 2001. Synonymous codon usage, accuracy of translation, and gene length in Caenorhabditis elegans. J Mol Evol 52: 275–280 [DOI] [PubMed] [Google Scholar]
- Mei Y, Yong J, Liu H, Shi Y, Meinkoth J, Dreyfuss G, Yang X 2010. tRNA binds to cytochrome c and inhibits caspase activation. Mol Cell 37: 668–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashimoto M 1997. Distribution of both lengths and 5′ terminal nucleotides of mammalian pre-tRNA 3′ trailers reflects properties of 3′ processing endoribonuclease. Nucleic Acids Res 25: 1148–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Lai EC 2008. Endogenous small interfering RNAs in animals. Nat Rev Mol Cell Biol 9: 673–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavon-Eternod M, Gomes S, Geslain R, Dai Q, Rosner MR, Pan T 2009. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res 37: 7268–7280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson T 2010. Regulatory RNAs derived from transfer RNA? RNA 16: 1865–1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percudani R 2001. Restricted wobble rules for eukaryotic genomes. Trends Genet 17: 133–135 [DOI] [PubMed] [Google Scholar]
- Percudani R, Pavesi A, Ottonello S 1997. Transfer RNA gene redundancy and translational selection in Saccharomyces cerevisiae. J Mol Biol 268: 322–330 [DOI] [PubMed] [Google Scholar]
- Plotkin JB, Kudla G 2010. Synonymous but not the same: The causes and consequences of codon bias. Nat Rev Genet 12: 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat Y, Fromer M, Linial N, Linial M 2009. Codon usage is associated with the evolutionary age of genes in metazoan genomes. BMC Evol Biol 9: 285 doi: 10.1186/1471-2148-9-285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raha D, Wang Z, Moqtaderi Z, Wu L, Zhong G, Gerstein M, Struhl K, Snyder M 2010. Close association of RNA polymerase II and many transcription factors with Pol III genes. Proc Natl Acad Sci 107: 3639–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raney BJ, Cline MS, Rosenbloom KR, Dreszer TR, Learned K, Barber GP, Meyer LR, Sloan CA, Malladi VS, Roskin KM, et al. 2010. ENCODE whole-genome data in the UCSC genome browser (2011 update). Nucleic Acids Res 39: D871–D875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M 2011. Global quantification of mammalian gene expression control. Nature 473: 337–342 [DOI] [PubMed] [Google Scholar]
- Sharp PM, Li WH 1987. The codon Adaptation Index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res 15: 1281–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Matassi G 1994. Codon usage and genome evolution. Curr Opin Genet Dev 4: 851–860 [DOI] [PubMed] [Google Scholar]
- Sorensen MA, Pedersen S 1991. Absolute in vivo translation rates of individual codons in Escherichia coli. The two glutamic acid codons GAA and GAG are translated with a threefold difference in rate. J Mol Biol 222: 265–280 [DOI] [PubMed] [Google Scholar]
- Stenico M, Lloyd AT, Sharp PM 1994. Codon usage in Caenorhabditis elegans: Delineation of translational selection and mutational biases. Nucleic Acids Res 22: 2437–2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, Pan T, Dahan O, Furman I, Pilpel Y 2010a. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141: 344–354 [DOI] [PubMed] [Google Scholar]
- Tuller T, Waldman YY, Kupiec M, Ruppin E 2010b. Translation efficiency is determined by both codon bias and folding energy. Proc Natl Acad Sci 107: 3645–3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuller T, Girshovich Y, Sella Y, Kreimer A, Freilich S, Kupiec M, Gophna U, Ruppin E 2011. Association between translation efficiency and horizontal gene transfer within microbial communities. Nucleic Acids Res 39: 4743–4755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S, Pedersen JS, Korbel JO, Stocsits C, Gruber AR, Hackermuller J, Hertel J, Lindemeyer M, Reiche K, Tanzer A, et al. 2007. Structured RNAs in the ENCODE selected regions of the human genome. Genome Res 17: 852–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JH, Shao P, Zhou H, Chen YQ, Qu LH 2009. deepBase: A database for deeply annotating and mining deep sequencing data. Nucleic Acids Res 38: D123–D130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Fedyunin I, Miekley O, Valleriani A, Moura A, Ignatova Z 2010. Global and local depletion of ternary complex limits translational elongation. Nucleic Acids Res 38: 4778–4787 [DOI] [PMC free article] [PubMed] [Google Scholar]