

Abstract
Background
Neuronal loss in multiple sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (EAE), correlates with permanent neurological dysfunction. Current MS therapies have limited ability to prevent neuronal damage.
Methods
We examined whether oral therapy with SRT501, a pharmaceutical-grade formulation of resveratrol, reduces neuronal loss during relapsing/remitting EAE. Resveratrol activates SIRT1, an NAD+-dependent deacetylase that promotes mitochondrial function.
Results
Oral SRT501 prevented neuronal loss during optic neuritis, an inflammatory optic nerve lesion in MS and EAE. SRT501 also suppressed neurological dysfunction during EAE remission, and spinal cords from SRT501-treated mice had significantly higher axonal density than vehicle-treated mice. Similar neuroprotection was mediated by SRT1720, another SIRT1-activating compound; and sirtinol, a SIRT1 inhibitor, attenuated SRT501 neuroprotective effects. SIRT1 activators did not prevent inflammation.
Conclusions
These studies demonstrate SRT501 attenuates neuronal damage and neurological dysfunction in EAE by a mechanism involving SIRT1 activation. SIRT1 activators are a potential oral therapy in MS.
Keywords: Multiple sclerosis, EAE, resveratrol, SIRT1, neuroprotection
Introduction
MS is a central nervous system demyelinating disease (1) characterized by relapsing/remitting neurological dysfunction associated with acute episodes of inflammation. Significant axonal damage and loss of neurons also occurs in MS, and correlates with permanent neurological disability (2–5). Current MS therapies reduce acute inflammatory episodes by immunomodulation, but have limited effects in preventing permanent neurological deficits, with some patients exhibiting progressive disability despite treatment with first line therapies (6–9). Novel therapies that better prevent neuronal damage are needed to prevent long-term disability in MS patients.
Optic neuritis, an inflammatory demyelinating disease of the optic nerve, often occurs as an acute episode of MS. Similar to other MS lesions, significant neuronal damage, with loss of retinal ganglion cell (RGC) axons, occurs following optic neuritis and correlates with decreased vision (5,10–13). Therapies that prevent permanent neuronal damage during optic neuritis therefore have potential to prevent long-term visual loss, and may have significant neuroprotective effects for other MS lesions.
EAE is an animal model sharing some clinical, immunological, and histopathological features of MS (14), including demyelinating lesions in spinal cord as well as optic nerve (15,16). SJL/J mice immunized with proteolipid protein develop relapsing/remitting EAE, with significant RGC loss beginning several days after acute optic neuritis develops (17). In previous studies, a pharmaceutical-grade formulation of resveratrol (SRT501) attenuated RGC loss during experimental optic neuritis when given by intravitreal injection (18). Resveratrol activates SIRT1, a member of a conserved gene family (sirtuins) encoding NAD+-dependent deacetylases (19–21). Intravitreal SRT501 treatment is limited by the frequency with which injections can be given without inducing significant trauma and intraocular inflammation. In addition, intravitreal SRT501 had no effects on tail or limb paralysis (18), measures of neurological dysfunction from EAE spinal cord lesions. Thus, systemic treatment with SIRT1 activators needs to be examined to determine their potential role as a neuroprotective therapy in MS.
SIRT1 activators in mice are well tolerated systemically at high oral doses, including 1000 mg/kg SRT501 and 100 mg/kg of a more potent SIRT1 activator, SRT1720 (22). SRT501 also has a favorable safety profile in humans and is currently in Phase 2 clinical trials for diabetes. In the current studies, potential neuroprotective effects of SRT501 and SRT1720 are examined in EAE mice. Because SIRT1 activators are well-tolerated with a favorable safety profile in humans, effects seen in EAE may rapidly translate into in clinical trials for optic neuritis and MS.
Methods
Mice
Six week old female SJL/J mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Treatment of animals conformed to Institutional Animal Care and Use Committee guidelines and review.
RGC labeling
Retrograde labeling of RGCs was performed as reported (17). Mice were anesthetized with 0.2 ml solution containing 10 mg/ml ketamine (Sigma, St. Louis, MO) and 1 mg/ml xylazine (Sigma) i.p. Heads were shaved, a mediosagittal incision made, and holes drilled through the skull above each superior colliculus. 2.5 μl of 1.25% hydroxystilbamidine (Fluorogold, Invitrogen, Carlsbad, CA) in PBS was injected stereotactically into each superior colliculus. Fluorogold taken up by axon terminals is retrogradely transported to RGC bodies in the retina.
Induction and evaluation of EAE
Mice were anesthetized with ketamine/xylazine and injected subcutaneously at two sites on the back with 0.1 ml solution containing 0.5 mg/ml proteolipid protein peptide 139–151 (PLP; Protein Chemistry Laboratory, University of Pennsylvania, Philadelphia, PA) emulsified in complete Freund's adjuvant (CFA; Difco, Detroit, MI) containing 2.5 mg/ml mycobacterium tuberculosis (Difco). Control mice were injected with an equal volume of phosphate buffered saline (PBS) and CFA. EAE and control mice were injected i.p. with 200 ng pertussis toxin (List Biological, Campbell, CA) in 0.1 ml PBS on day 0 and day 2. Clinical EAE was scored daily using a 5 point scale (23): no disease=0; partial tail paralysis=0.5; tail paralysis or waddling gait=1.0; partial tail paralysis and waddling gait=1.5; tail paralysis and waddling gait=2.0; partial limb paralysis=2.5; paralysis of one limb=3.0; paralysis of one limb and partial paralysis of another=3.5; paralysis of two limbs=4.0; moribund state=4.5; death=5.0.
SIRT1 activator treatment and bioavailability
SRT501 (Sirtris Pharmaceuticals, Cambridge, MA) was diluted in 2% hydroxypropyl methylcellulose (Shin-Etsu Chemicals, Japan) and 0.2% dioctyl sodium sulfosuccinate (Wilson Laboratories, Mumbai, India); SRT1720 was dissolved in 40% polyethanol glycol 400 and 0.5% tween80 (AG Scientific, San Diego, CA). Mice were treated by oral gavage once daily at indicated doses and time points. To determine the intravitreal bioavailability of orally administered SRT501, mice were anesthetized 1 hr after the final dose. A 30-gauge needle was used to enter the vitreal cavity. Using a Hamilton syringe, a 1 μl sample of vitreous was removed and diluted 1:50 in PBS. Samples were sent for mass spectrometry analysis (Charles River Laboratories, Worcester, MA) to measure SRT501 concentration. For SIRT1 inhibitor studies, sirtinol (Sigma) was injected into the vitreous as described previously (18).
Quantification of RGC numbers
Following sacrifice, eyes were removed, fixed in 4% paraformaldehyde in PBS, and isolated retinas were viewed by fluorescent microscopy. Photographs were taken at 20× magnification in 12 standard fields: 1/6, 3/6, and 5/6 of the retinal radius from the center of the retina in each retinal quadrant, and the number of RGCs was counted using Image-Pro Plus 6.0 (Media Cybernetics, Silver Spring, MD) software as in prior studies (17).
Histopathological evaluation of optic nerves
After sacrifice, optic nerves were removed and fixed in 4% paraformaldehyde in PBS. Processed nerves were embedded in paraffin, cut into 5 μm longitudinal sections, and stained with H&E. Presence of inflammatory cell infiltration was assessed by a grading scale used previously (18,24): No infiltration=0; mild cellular infiltration of optic nerve or optic nerve sheath=1; moderate infiltration=2; severe infiltration=3; massive infiltration=4. Nerves with any detectable inflammation (score 1–4) were considered to have optic neuritis. To assess axonal damage, optic nerve sections were stained by Bielschowsky silver impregnation and axonal density was quantified as in prior studies (25). Briefly, optic nerves were photographed at 40× magnification (one photograph each of the proximal, central, and distal nerve) and the area of silver staining was calculated using ImagePro Plus 6.0 software. Total area examined in three fields/nerve was 38,500 μm2, and data represent the cumulative area of positive staining/nerve.
Spinal cord preparation and histopathology
Mice were transcardially perfused with PBS followed by 4% paraformaldehyde. Spinal cords were removed, post-fixed in 4% paraformaldehyde, embedded in paraffin and cut in 5 μm thick sections. Analysis of inflammatory cell infiltration and axonal area were performed similar to previously described methods (23,26). Sections were stained with H&E, luxol fast blue (LFB), or Bielschowsky silver impregnation and examined by light microscopy. To quantify axonal density, photographs were taken at 40× magnification in 4 standardized fields per section, representing a total area of 50,000 μm2. Axonal density is calculated as the cumulative area of positive silver staining/spinal cord using Image-Pro Plus 6.0.
Inflammatory cell isolation, culture, and flow cytometric analysis
Cells were isolated from EAE spinal cords by methods described previously (27). Briefly, following transcardial perfusion with PBS, spinal cords were removed, pooled, mechanically dissociated through a 100-μm strainer and washed with PBS. The resultant pellet was fractionated on a 60/30% Percoll gradient by centrifugation at 300 × g for 20 min. Microglia and infiltrating mononuclear cells were harvested from the interface, washed, counted, and stained with antibodies for flow cytometry, or alternatively for studies of cytokine expression cells were cultured for 3 hr in RPMI 1640 containing 10% FCS, penicillin/streptomycin, nonessential amino acids, L-glutamine, vitamins and 2-ME and stimulated with phorbol 12-myristate 13-acetate (50 ng/ml), ionomycin (500 ng/ml; Sigma) and treated with GolgiPlug (1 μg per 1 106 cells; BD Pharmingen). Cells were stained for 20 min in the dark at 4 °C with fluorescence-labeled antibodies specific for cell surface markers. For intracellular markers, cells were washed, fixed and permeabilized with Fix & Perm reagents (Caltag Laboratories) then stained with fluorescence-labeled antibodies. All antibodies were used at a concentration of 0.5 ug/ml. All were purchased from BD Bioscience and the details are as follows: (CD45; clone 104), (CD4; clone RM4-5), (CD8; clone 53–6.7), (CD11b; clone m1/70), (IL-17; clone TC11-18H10), (IFN-gamma; clone XMG1.2). Data were acquired on a FACSAria (BD Biosciences) and analyzed with FlowJo software (Treestar). Mononuclear cells (infiltrating immune cells and resident microglia) were gated based on physical parameters (size and granularity).
Statistics
Comparisons of RGC numbers and axonal density were analyzed by one way ANOVA followed by Tukey's Multiple Comparison test using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA). Data represent mean ± SEM number of RGCs or cumulative area of positive axonal staining. Clinical EAE scores were compared between treatment groups by ANOVA for repeated measures using GraphPad Prism 5.0, and the probability of full recovery versus partial recovery during EAE remission between treatment groups was compared by Fisher's exact test. The probability of developing optic neuritis (any positive optic nerve inflammation score, 1–4) versus no optic nerve inflammation was compared between SRT501- and vehicle-treated mice by Fisher's exact test. Similar comparison of the relative degree of optic nerve inflammation was performed between nerves with mild inflammation (score 1) and those with moderate – severe inflammation (scores 2–4). Overall rank distribution of optic nerve inflammation scores were compared by Wilcoxon rank-sum test.
Results
Oral SRT501 penetrates the eye
Intravitreal SRT501 prevents RGC loss from optic neuritis (18), but it is not known whether systemic SRT501 crosses the blood-retina barrier. To examine this, EAE and control mice were treated by oral gavage with SRT501 or placebo (vehicle) beginning on day 8 post-immunization and repeated daily until sacrifice on day 14. At sacrifice, 1 μl vitreal samples were taken (one hour after the final dose), diluted 1:50 in PBS, and SRT501 concentration was measured. No SRT501 was detected in EAE eyes treated with vehicle only (n=3 eyes). SRT501 (2.5±1.3 ng/ml) was detected in EAE eyes treated with 500 mg/kg SRT501 daily (n=5), and intravitreal SRT501 concentration increased with a higher oral dose of 1000 mg/kg SRT501 in both EAE (9.7±5.0 ng/ml; n=6) and control (10.3±5.1 ng/ml; n=3) eyes.
Oral SRT501 treatment prior to onset of optic neuritis attenuates neuronal damage without reducing inflammation
To determine whether oral administration of SRT501 can prevent RGC damage, EAE and control mice were treated with 500 or 1000 mg/kg SRT501, or vehicle, beginning on day 8 post-immunization (one day prior to onset of optic neuritis) and repeated daily until sacrifice on day 14. Optic neuritis was detected in eyes from EAE mice treated with either dose of SRT501, with a similar incidence to vehicle-treated EAE mice (Fig. 1A–C,G). The probability of developing optic neuritis did not differ between eyes of vehicle-treated mice (15 of 26) and 500 (30 of 49; p = 0.8079) or 1000 (11 of 19; p = 1.000) mg/kg SRT501-treated mice. The degree of inflammation, scored on a 4-point scale, also did not differ between 500 mg/kg SRT501-treated (mean±SD score 1.43±0.57, n=30), 1000 mg/kg SRT501-treated (1.54±0.82, n=11) and vehicle-treated (1.33±0.49, n=15) EAE eyes. 10 of the 15 eyes from vehicle-treated mice that had optic neuritis had only mild inflammation (score 1). This was no different from the proportion of eyes from 500 (18 of 30; p = 0.7521) or 1000 (4 of 11; p = 1.000) mg/kg SRT501-treated mice that had mild inflammation. Comparison of the distribution of relative inflammation scores by the Wilcoxon rank sum-test also showed no significant difference between vehicle-treated eyes and those treated with 500 (p = 0.5637) or 1000 (p = 0.4173) mg/kg SRT501.
Fig. 1.
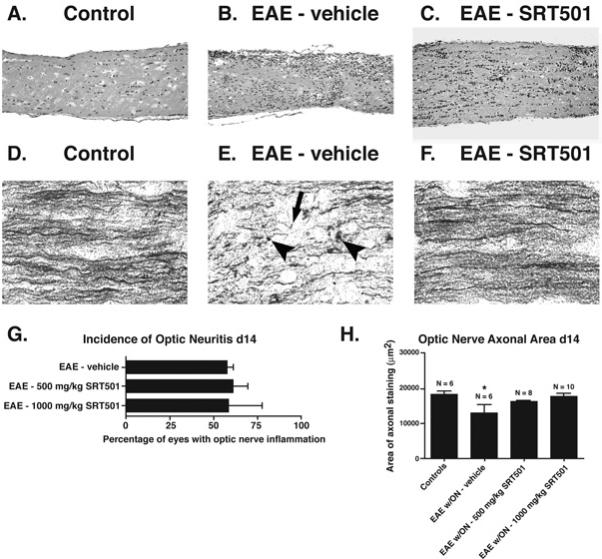
Oral SRT501 reduces axonal damage during optic neuritis without suppressing inflammation. A. A longitudinal section from a control optic nerve stained by H&E shows normal histology. B. A representative optic nerve from an EAE mouse 14 days post-immunization stained by H&E demonstrates increased cellularity from infiltrating inflammatory cells observed during optic neuritis. C. H&E staining of a day 14 optic nerve from an EAE mouse treated with 1000 mg/kg SRT501 daily from d8–14 shows inflammatory infiltrates are still present. D. Normal axonal staining by Bielschowsky silver impregnation in a control optic nerve. E. Axonal damage marked by terminal axonal ovoids (arrowheads) and truncated axons (arrow) found in a nerve with optic neuritis 14 days after induction of EAE. F. Normal optic nerve axonal staining in an eye with optic neuritis from an EAE mouse treated with 1000 mg/kg SRT501 d8–14. G. The percentage of eyes that developed optic neuritis in EAE mice treated with 500 or 1000 mg/kg SRT501 daily from d8–14 was similar to the incidence of optic neuritis in EAE mice treated with vehicle (mean ± SEM of 3 experiments). H. Axonal density, measured as the area of positive silver staining (mean ± SEM), was significantly decreased in eyes with optic neuritis compared to either control eyes or optic neuritis eyes from EAE mice treated with 1000 mg/kg SRT501 daily from d8–14 (*p < 0.05). One experiment of three is shown. Original magnification 20× (AC) and 40× (D–F).
Similar to previous studies (25), optic neuritis was associated with RGC axonal damage, detected by decreased axonal silver staining with truncated axons and terminal axonal ovoids (Fig. 1E). In contrast, optic neuritis nerves from SRT501-treated EAE mice had normal RGC axonal staining (Fig. 1F), comparable to controls (Fig. 1D). Eyes with optic neuritis from EAE mice treated with 500 mg/kg SRT501 showed a trend toward increased axonal density compared to vehicle-treated mice, and at higher doses of 1000 mg/kg, axonal density was significantly higher (Fig. 1H).
RGC numbers in optic neuritis eyes (445.7±87.0 RGCs) were significantly lower than in eyes from control mice treated with 500 mg/kg SRT501 (788.2±83.9) or 1000 mg/kg SRT501 (703.0±42.6; *p<0.05; Fig. 2). Optic neuritis eyes from 500 mg/kg SRT501-treated EAE mice showed a trend toward increased RGC numbers (595.9±54.5), while optic neuritis eyes from 1000 mg/kg SRT501-treated EAE mice had significantly more surviving RGCs (695.0±38.3) than vehicle-treated optic neuritis eyes (**p<0.05). Together, results suggest oral SRT501 can attenuate RGC axonal damage and cell death during acute optic neuritis, and these effects are not mediated by suppressing inflammation.
Fig. 2.
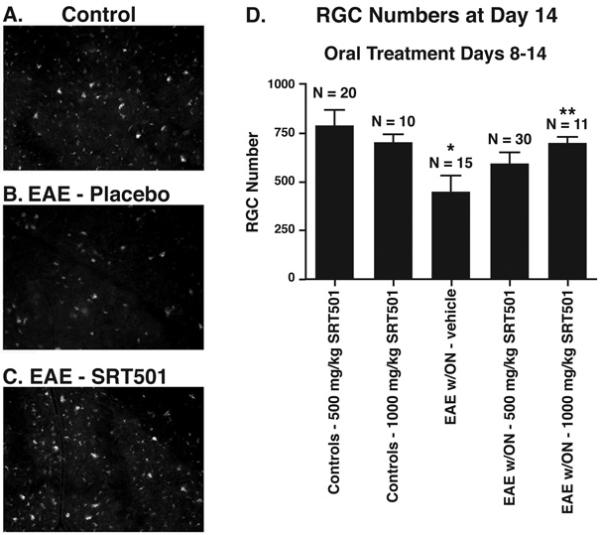
Oral SRT501 treatment from d8–14 post-immunization prevents RGC loss in EAE optic neuritis eyes. A. Numerous fluorescently-labeled RGCs from a representative field in a control retina. B. Fewer RGCs are seen in a corresponding area of retina in an EAE eye with optic neuritis. C. Retina from an optic neuritis eye in an EAE mouse treated with 1000 mg/kg SRT501 contains numerous RGCs similar to controls. D. Optic neuritis induced significant loss of RGCs compared to control eyes (*p < 0.05). 500 mg/kg SRT501 lead to a non-significant trend of increased RGC survival, and 1000 mg/kg SRT501 lead to significantly higher numbers of surviving RGCs in optic neuritis eyes compared to vehicle (**p < 0.05). N = number of eyes. Data represent the mean ± SEM number of RGCs counted in 12 standardized fields/eye.
Oral SRT501 treatment after onset of optic neuritis attenuates neuronal damage
To determine whether SIRT1 activators prevent RGC loss after onset of optic nerve inflammation, correlating to when patients may present clinically, mice were treated with vehicle or 1000 mg/kg SRT501 beginning on day 10 post-immunization and repeated daily until day 14. Prior studies have shown that inflammation begins by day 9, but RGC axonal loss is not detected until day 13 (25). On day 14, RGC numbers in optic neuritis eyes (477.5±66.6) were significantly lower than in eyes from control mice treated with vehicle (802.5±130.0) or SRT501 (739.8±110.5; *p<0.01; Fig. 3A). Optic neuritis eyes from SRT501-treated EAE mice had significantly more RGCs (821.0±136.4) than vehicle-treated optic neuritis eyes (**p<0.01). To examine whether SRT501 prevents RGC loss longer-term, as opposed to merely delaying RGC loss, RGC survival was examined one month after EAE induction. Mice were treated with vehicle or 1000 mg/kg SRT501 from day 10–14 post-immunization (during the first acute optic neuritis episode), then sacrificed at day 30. RGC numbers in vehicle-treated EAE eyes (394.8±52.3) were significantly lower than in eyes from control mice treated with vehicle (608.2±68.5) or SRT501 (624.1±63.5; *p<0.01; Fig. 3B). Eyes from SRT501-treated EAE mice had significantly more RGCs (748.0±44.3) than vehicle-treated EAE eyes (**p<0.001). Correlating with RGC numbers, axonal density in optic nerves at day 14 (Fig. 3C) and day 30 (Fig. 3D) post-immunization demonstrated that SRT501 treatment prevented the loss of axons found in EAE optic neuritis eyes. Thus, SRT501 administered at a clinically-relevant time provides acute neuroprotection with lasting effects after inflammation resolves.
Fig. 3.
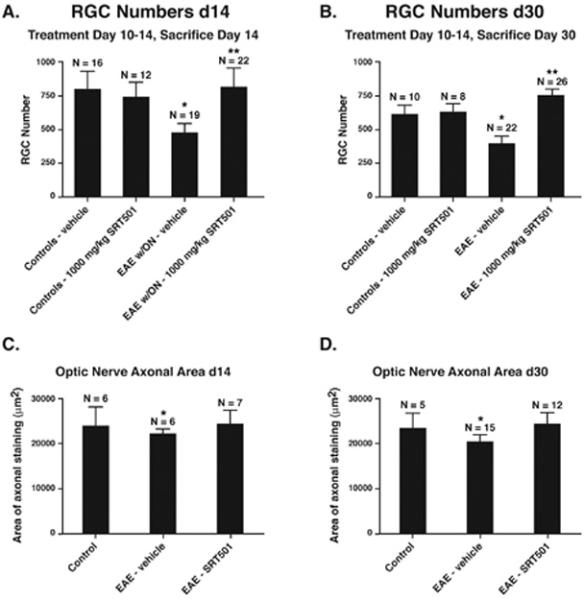
Oral SRT501 treatment beginning after onset of optic neuritis, from d10–14 post-immunization, prevents acute axon and RGC loss at day 14 and maintains these neuroprotective effects at day 30. A. The significant loss of RGCs detected in EAE optic neuritis eyes (*p < 0.01 vs. controls) at day 14 is prevented by 1000 mg/kg SRT501 (**p < 0.01 vs. vehicle-treated EAE optic neuritis eyes). B. Similar RGC loss is detected in eyes from vehicle-treated EAE mice at day 30 (*p < 0.01 vs. controls), with increased RGC survival in SRT501-treated EAE eyes (**p < 0.001 vs. vehicle-treated EAE eyes). C. Decreased area of axonal silver staining in EAE optic neuritis eyes compared to controls at day 14 (*p < 0.05) was prevented by 1000 mg/kg SRT501. D. 1000 mg/kg SRT501 also attenuated axonal loss detected in eyes from EAE mice at day 30 (*p < 0.01). One representative experiment of three is shown.
SRT501 reduces residual neurological dysfunction during EAE remission
Effects of SRT501 treatment on clinical EAE were evaluated. No difference in onset of acute EAE through day 14 post-immunization was observed between vehicle-treated EAE mice and EAE mice treated with 500 or 1000 mg/kg SRT501 (Fig. 4A) from days 8–14. In EAE mice treated from days 10–14 with vehicle or 1000 mg/kg SRT501, there also was no difference in clinical EAE score between vehicle- or SRT501-treated mice through the peak of symptoms during the first clinical episode (Fig. 4B). Following the peak at day 14, both vehicle- and SRT501-treated mice showed remission of EAE as seen previously in this relapsing/remitting disease. SRT501-treated mice had improved recovery, with a significant (p<0.05) decrease in EAE score compared with vehicle-treated mice. In three experiments, the combined probability of complete remission was significantly higher (21 of 24 mice) in SRT501-treated EAE mice than in vehicle-treated (6 of 19) mice (p=0.0003).
Fig. 4.
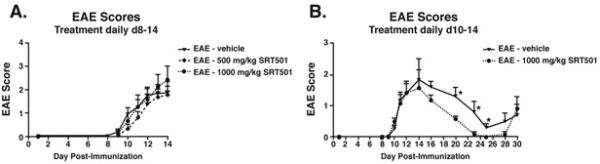
SRT501 suppresses EAE remission but not acute exacerbations. A. No difference was found between the degree of EAE in mice treated with vehicle (n = 19), 500 mg/kg SRT501 (n = 30), or 1000 mg/kg SRT501 (n = 13) given by oral gavage daily from d8–14 up to the peak of disease at sacrifice on day 14. B. In an extended treatment study, no difference in development of acute EAE was detected between vehicle-treated (n = 5) and 1000 mg/kg SRT501-treated (n = 6) EAE mice treated d10–14. During remission at days 20–25, EAE was significantly suppressed in SRT501-treated vs. vehicle-treated mice (*p < 0.05). One of three experiments is shown.
SRT501 attenuates axonal loss in EAE spinal cords without suppressing inflammation or demyelination
Development of EAE in SRT501-treated mice suggests acute spinal cord inflammation may not be suppressed, whereas improved recovery during remission indicates SRT501 likely prevents permanent neuronal damage following acute inflammation. To examine this further, control and EAE mice treated with vehicle or SRT501 were perfused at day 14 or day 30, and spinal cord cross-sections were evaluated for inflammation and axonal loss. 14 days post-immunization, focal areas of inflammatory cell infiltration were observed at multiple spinal cord levels in EAE, but not control, mice (Fig. 5A–E). The incidence of inflammation (Fig. 5F) and demyelination (Fig. 5G) were similar between EAE mice treated with vehicle, 500 mg/kg SRT501 or 1000 mg/kg SRT501. Silver staining showed no difference in axonal density between vehicle- or SRT501-treated mice at day 14 (data not shown). However, 30 days post-immunization, focal areas of axonal loss were observed in vehicle-treated spinal cords (Fig. 6B,E) but not in controls (Fig. 6A,D) or SRT501-treated mice (Fig. 6C,F). Significantly decreased axonal density at day 30 was measured in vehicle-treated, but not SRT501-treated, EAE spinal cords (Fig. 6G).
Fig. 5.
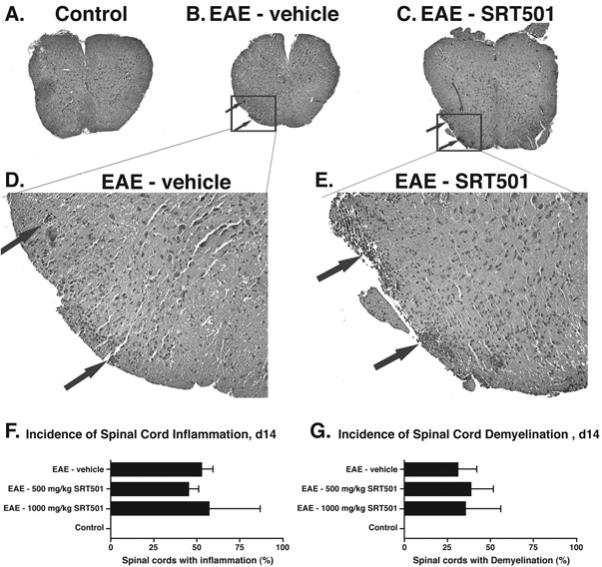
SRT501 does not reduce spinal cord inflammation or demyelination at day 14 post-immunization. A. An H&E stained cross-section through the spinal cord of a control mouse demonstrates normal histology at 4× original magnification. B. Focal areas containing inflammatory cell infiltrates (arrows) are shown in a spinal cord from an EAE mouse. C. Similar foci of inflammation (arrows) are present in the spinal cord of an EAE mouse treated with 1000 mg/kg SRT501 daily from d8–14. D. Higher magnification (40×) photograph of areas of inflammation shown in B. E. Higher magnification (40×) photograph of areas of inflammation shown in C. F. The percentage of spinal cord sections that contained inflammation in EAE mice treated with 500 or 1000 mg/kg SRT501 daily from d8–14 was similar to the incidence in EAE mice treated with vehicle (mean ± SEM of 3 experiments), with no inflamed spinal cords found in control mice. G. The percentage of spinal cord sections that contained demyelination, assessed by LFB staining, in EAE mice treated with 500 or 1000 mg/kg SRT501 daily from d8–14 was similar to the incidence in EAE mice treated with vehicle (mean ± SEM of 3 experiments), with no demyelination found in control mice.
Fig. 6.
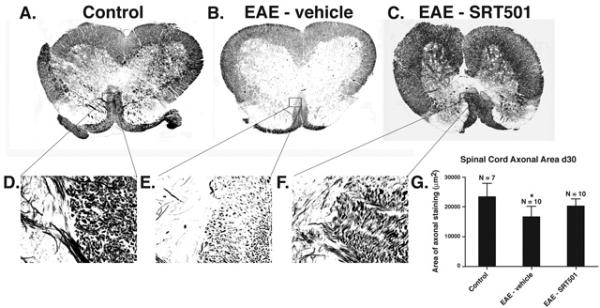
SRT501 treatment during acute inflammation on d10–14 prevents loss of axons in EAE spinal cord 30 days post-immunization. A. Bielschowsky silver stained section of control spinal cord shows normal axonal staining. Higher magnification of boxed area is shown in D, with normal gray matter (left half) and medial corticospinal tract (right half) demonstrating high density of silver stained axons. B. Spinal cord from an EAE mouse contains focal areas of axonal loss (arrowheads) and mild diffuse decrease in silver staining. Higher magnification of boxed area shown in E demonstrates a focal area of medial corticospinal tract with notable decrease in axons compared to control spinal cord. C. Normal axonal staining observed in the spinal cord of an EAE mouse treated with 1000 mg/kg SRT501. Higher magnification of boxed area shown in F demonstrates higher axonal concentration than vehicle-treated mouse spinal cord. G. Quantification of axonal staining demonstrates that significant axonal loss compared to controls occurs in spinal cords from vehicle-treated (*p < 0.001), but not SRT501-treated, EAE mice. One of three experiments is shown. Photographs A–C shown at 4× original magnification, D–F at 40× original magnification.
SRT501 treatment during acute EAE does not modulate the immune response in spinal cord
While gross levels of inflammation were not reduced by SRT501 treatment during active EAE, it is possible that SRT501 may alter the phenotype of the inflammatory cells present in the CNS. To examine whether SRT501 affects the type of cells found in spinal cord infiltrates, EAE mice were treated daily from day 10 through day 14 post-immunization with vehicle or 1000 mg/kg SRT501. Mice were perfused on day 14, at the peak of EAE, and microglia and infiltrating monocytes were isolated from spinal cords pooled from 6 mice/group. Freshly isolated cells were labeled with antibodies to cell surface markers and analyzed by flow cytometry to determine the composition of inflammatory cells present. Additional cells isolated from SRT501- and vehicle-treated mice were cultured for three hours, then viable cells were permeabilized and labeled with antibodies to cytokines IL-17 and interferon-gamma (IFNg). T cells comprised less than one quarter of all isolated cells, with no significant difference in the percentage of CD4+ or CD8+ T cells between vehicle-treated and SRT501-treated EAE mice. The majority (two-thirds) of CNS cells were CD11b+ macrophages/microglia, with no significant difference between vehicle-treated and SRT501-treated EAE mice. Similarly, following three hours in culture, no significant differences in expression of the Th1 cytokine IFNg or the Th17 cytokine IL-17 were found between cells from vehicle-treated and SRT501-treated EAE mice. Interestingly, there was a non-significant trend toward increased numbers of inflammatory cells that were isolated from each SRT501-treated mouse spinal cord as compared to vehicle-treated EAE mice, with no significant difference in the average number of CD4+, CD8+, CD11b+, CD45hi+, IFNg+, or IL-17+ cells per mouse with or without SRT501 treatment.
Mechanism of SRT501 neuroprotection in EAE is mediated by SIRT1 activation
Resveratrol exerts effects by several mechanisms (28), including activation of SIRT1. To examine whether the mechanism of SRT501-mediated neuroprotection in EAE involves SIRT1 activation, EAE mice were treated with SRT1720, another SIRT1 activator that activates SIRT1 by a distinct mechanism and with higher potency than SRT501 (22). Similar to SRT501, SRT1720 did not prevent spinal cord or optic nerve inflammation, but did attenuate neuronal damage. Mice were treated daily from day 10–14 post-immunization with vehicle or 100 mg/kg SRT1720, then sacrificed on days 14 or 30. No difference in EAE score occurred during acute disease, but SRT1720-treated mice had significantly better recovery during remission than vehicle-treated mice (Fig. 7A), with increased probability of complete remission (p=0.024) and preserved spinal cord axons (Fig. 7E). While optic neuritis incidence did not differ between vehicle- and SRT1720-treated EAE mice (Fig. 7B), SRT1720 did reduce optic nerve axonal loss and RGC loss at day 14 (data not shown) and day 30 (Fig. 7C,D).
Fig. 7.
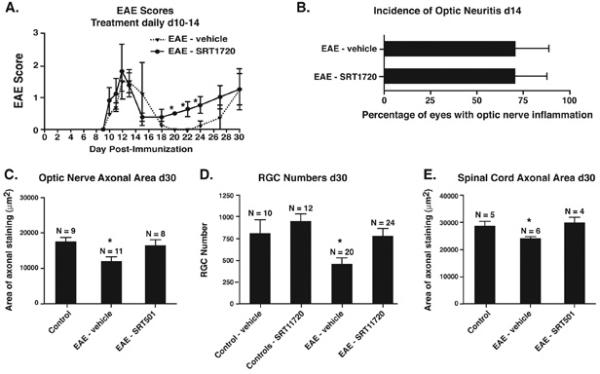
SRT1720 treatment exhibits similar neuroprotective effects in EAE as SRT501, without reduction of inflammation. A. No difference in development of acute EAE was detected between vehicle-treated (n = 5) and 100 mg/kg SRT1720-treated (n = 5) EAE mice treated d10–14. During remission at days 20–24, EAE was significantly suppressed in SRT1720-treated vs. vehicle-treated mice (*p < 0.05). One of two experiments is shown. B. The incidence of optic neuritis (mean ± SEM of 3 experiments) did not differ between vehicle-treated and SRT1720-treated EAE mice 14 days post-immunization. C. SRT1720 prevents the significant loss of axons that occurs in EAE optic nerves at day 30 (*p < 0.05). Data represent the mean ± SEM area of positive silver staining measured in one of two experiments. D. SRT1720 also prevents the significant loss of RGCs that occurs in EAE eyes at day 30 (*p < 0.01). Data represent the mean ± SEM number of RGCs measured in each eye. E. SRT1720 has similar neuroprotective effects for spinal cord axons, preventing the significant loss that occurs in EAE spinal cords at day 30 (*p < 0.05). Data represent the mean ± SEM area of positive silver staining measured in one of two experiments.
The ability of two distinct SIRT1 activators to prevent neurological dysfunction and neuronal damage in EAE suggests the mechanism of these neuroprotective effects involves SIRT1 activation. To further confirm this, EAE mice treated with 1000 mg/kg SRT501 on days 10–14 were given an intravitreal injection of sirtinol (100 μM), a SIRT1 inhibitor (18). Sirtinol administration significantly reduced the ability of SRT501 to prevent RGC loss 14 days post-immunization (Fig. 8).
Fig. 8.
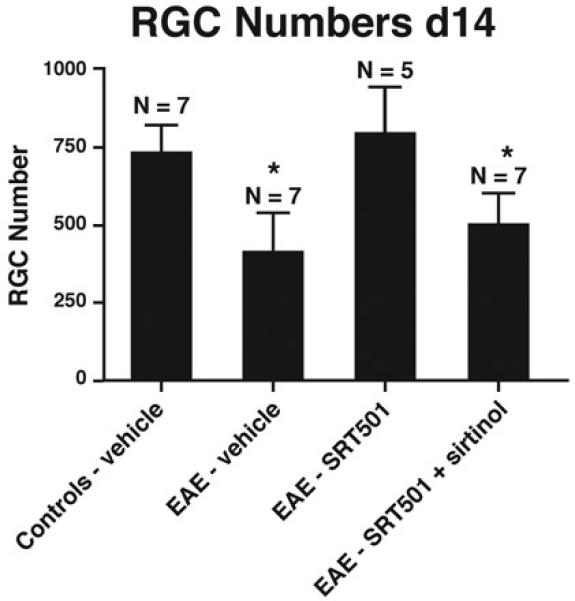
The SIRT1 inhibitor sirtinol reduces neuroprotective effects of SRT501 on RGCs during optic neuritis. 1000 mg/kg SRT501 administered daily from d10–14 in EAE mice results in significantly higher RGC numbers compared to optic neuritis eyes from vehicle-treated EAE mice, but intravitreal injection of sirtinol (100 μM) in SRT501-treated mice on d11 results in decreased RGC numbers (*p < 0.05) following sacrifice on day 14.
Discussion
The current studies show that orally administered SRT501 attenuates neuronal damage in EAE optic nerves and spinal cords. Observed neuroprotective effects on RGCs are similar to effects seen previously with intravitreal administration (18), but in those studies no effect on EAE or spinal cord pathology occurred. SRT501 is well tolerated in the eye (18), as well as systemically (22). Based on the current results and its favorable safety profile, SRT501 is an important potential oral therapy to prevent permanent neurological disability in MS.
MS involves a complex autoimmune inflammatory response to components of CNS myelin (1), and neuronal damage, including RGC loss during optic neuritis in experimental models, can occur secondary to inflammation (17). The mechanism of neuroprotection by SRT501 in EAE, however, is not mediated by reducing inflammation, as incidence of optic nerve and spinal cord inflammation was not reduced by SRT501 treatment. SRT501 also did not alter the phenotype of inflammatory cell infiltrates in the spinal cord at the peak of the acute EAE attack. In addition, EAE scores during acute disease episodes, signs that reflect active spinal cord inflammation, were not reduced by SRT501 treatment. EAE was suppressed during disease remission, with preserved axonal density, suggesting SRT501 may be able to prevent permanent neurological dysfunction and neuronal damage in MS after acute spinal cord inflammation resolves. While treatment during the first clinical EAE episode, day 10–14 post-immunization, led to improved functional recovery during remission, this treatment course did not prevent the onset of EAE relapse at day 30. Further studies with extended treatment will be important to examine longer-term effects of SRT501.
Results suggest the mechanism of SRT501 neuroprotection in EAE involves SIRT1 activation. SRT501 activates SIRT1 in vitro (22). Administration of the SIRT1 inhibitor sirtinol blocks SRT501-mediated RGC neuroprotection, and the fact that a second distinct SIRT1 activator, SRT1720, induced similar neuroprotective effects further suggests SIRT1 activation is a requisite mechanism for the observed neuroprotection. Precise molecular pathways driven by SIRT1 activity leading to neuroprotection require further examination.
Interestingly, while the current studies showed no suppression of EAE inflammation by SRT501, resveratrol has been shown to partially suppress EAE and modulate inflammation in two other studies (29,30), although there are important differences between those studies and the current studies. Singh and colleagues (29) used a different EAE model with a chronic disease course, and in their studies resveratrol treatment was initiated right after immunization, long before clinical EAE develops, suggesting that it may affect development of the disease model as opposed to treating active disease. In addition, mechanisms of neuronal loss in chronic EAE can differ from relapsing/remitting EAE, as neuronal loss occurs several days after immunization and prior to onset of inflammation (31), as opposed to neuronal loss only beginning after inflammation develops in the relapsing/remitting model (17). Thus, both the mechanism of neuronal loss and the timing of treatment are likely important factors for resveratrol neuroprotective effects. In the studies by Imler and Petro (30), resveratrol treatment was also initiated prior to disease onset, at the time of immunization, and while continuous daily treatment did result in long-term EAE suppression through multiple relapses, even in their studies resveratrol effects were limited during the first episode of EAE. Together, these studies suggest resveratrol may modulate immune responses in EAE during the induction phase of disease, and it is possible that SRT501 may alter immune responses at other time points along the EAE disease course, but our results show that once the immune response has been induced and clinical disease developed, SRT501 no longer suppresses inflammation but does retain significant ability to prevent neuronal damage. As the timing of SRT501 treatment may play a role in its ability to prevent neuronal injury, it is important to note that neuroprotective effects were seen when treatment was initiated after onset of inflammation, but prior to optic nerve axonal loss (25). If a similar window of time can be indentified in optic neuritis patients, then there would be a potential opportunity to initiate neuroprotective therapies.
Overall, our results demonstrate a significant neuroprotective effect of SIRT1 activators in EAE mice, reducing neurological dysfunction during EAE remission and preventing axonal damage and neuronal loss. SIRT1 activators have a potential therapeutic role in preventing permanent visual loss following optic neuritis and permanent neurological dysfunction in MS patients. SRT501 itself has shown a favorable safety profile without retinal toxicity in EAE and is also safe in humans. Therefore, our findings likely can be translated into clinical trials to assess potential neuroprotection in optic neuritis and MS patients. Because SIRT1 activation does not prevent inflammation when treatment is initiated after disease onset, SIRT1 activators have potential to work synergistically with current immunomodulatory therapies.
Acknowledgments
The authors thank Elsa Aglow for expert technical assistance.
Acknowledgment of funding sources
This work was supported by National Institutes of Health grants EY015098 and EY019014 (KSS), grant RG 4214-A-1 from the National Multiple Sclerosis Society (KSS), a Career Development Award from Research to Prevent Blindness (KSS), grants from Sirtris Pharmaceuticals (AMR), and the F. M. Kirby Foundation (KSS).
Footnotes
Conflict of interest statement: The SRT501 and SRT1720 compounds used in these studies are made by Sirtris Pharmaceuticals, a GSK company, and were provided without charge. Peter Elliot was employed full time by Sirtris during these studies, and a portion of the funding for experiments was provided by Sirtris.
References
- 1.Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Eng J Med. 2002;343:938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 2.Davie CA, Barker GJ, Webb S, Tofts PS, Thompson AJ, Harding AE, McDonald WI, Miller DH. Persistent functional deficit in multiple sclerosis and autosomal dominant ataxia associated with axon loss. Brain. 1995;118:1583–1592. doi: 10.1093/brain/118.6.1583. [DOI] [PubMed] [Google Scholar]
- 3.Losseff NA, Webb SL, O'Riordan JI, Page R, Wang L, Barker GJ, Tofts PS, McDonald WI, Miller DH, Thompson AJ. Spinal cord atrophy and disability in multiple sclerosis: a new reproducible and sensitive MRI method with potential to monitor disease progression. Brain. 1996;119:701–708. doi: 10.1093/brain/119.3.701. [DOI] [PubMed] [Google Scholar]
- 4.Losseff NA, Wang L, Lai HM, Yoo DS, Gawne-Cain ML, McDonald WI, Miller DH, Thompson AJ. Progressive cerebral atrophy in multiple sclerosis: a serial MRI study. Brain. 1996;119:2009–2019. doi: 10.1093/brain/119.6.2009. [DOI] [PubMed] [Google Scholar]
- 5.Fisher JB, Jacobs DA, Markowitz CE, Galetta SL, Volpe NJ, Nano-Schiavi ML, Baier ML, Frohman EM, Winslow H, Frohman TC, Calabresi PA, Maguire MG, Cutter GR, Balcer LJ. Relation of visual function to retinal nerve fiber layer thickness in multiple sclerosis. Ophthalmol. 2006;113:324–332. doi: 10.1016/j.ophtha.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 6.Parry A, Corkill R, Blamire AM, Palace J, Narayanan S, Arnold D, Styles P, Matthews PM. Beta-Interferon treatment does not always slow the progression of axonal injury in multiple sclerosis. J Neurol. 2003;250:171–178. doi: 10.1007/s00415-003-0965-8. [DOI] [PubMed] [Google Scholar]
- 7.Hickman SJ, Kapoor R, Jones SJ, Altmann DR, Plant GT, Miller DH. Corticosteroids do not prevent optic nerve atrophy following optic neuritis. J Neurol Neurosurg Psychiatry. 2003;74:1139–1141. doi: 10.1136/jnnp.74.8.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobs LD, Cookfair DL, Rudick RA, Herndon RM, Richert JR, Salazar AM, Fischer JS, Goodkin DE, Granger CV, Simon JH, Alam JJ, Bartoszak DM, Bourdette DM, Braiman J, Brownscheidle CM, Coats ME, Cohan SL, Dougherty DS, Kinkel RP, Mass MK, Munschauer FE, III, Priore RL, Pullicino PM, Scherokman BJ, Weinstock-Guttman B, Whitham RH, The Multiple Sclerosis Collaborative Research Group Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. Ann Neurol. 1996;39:285–294. doi: 10.1002/ana.410390304. [DOI] [PubMed] [Google Scholar]
- 9.Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, Myers LW, Panitch HS, Rose JW, Schiffer RB, Vollmer T, Weiner LP, Wolinsky JS, the Copolymer 1 Multiple Sclerosis Study Group Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind, placebo-controlled trial. Neurology. 1995;45:1268–1276. doi: 10.1212/wnl.45.7.1268. [DOI] [PubMed] [Google Scholar]
- 10.Steel DHW, Waldock A. Measurement of the retinal nerve fibre layer with scanning laser polarimetry in patients with previous demyelinating optic neuritis. J Neurol Neurosurg Psychiatry. 1998;64:505–509. doi: 10.1136/jnnp.64.4.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parisi V, Manni G, Spadaro M, Colacino G, Restuccia R, Marchi S, Bucci MG, Pierelli F. Correlation between morphological and functional retinal impairment in multiple sclerosis patients. Invest Ophthalmol Vis Sci. 1999;40:2520–2527. [PubMed] [Google Scholar]
- 12.Trip SA, Schlottmann PG, Jones SJ, Altmann DR, Garway-Heath DF, Thompson AJ, Plant GT, Miller DH. Retinal nerve fiber layer axonal loss and visual dysfunction in optic neuritis. Ann Neurol. 2005;58:383–391. doi: 10.1002/ana.20575. [DOI] [PubMed] [Google Scholar]
- 13.Costello F, Coupland S, Hodge W, Lorello GR, Koroluk J, Pan YI, Freedman MS, Zackon DH, Kardon RH. Quantifying axonal loss after optic neuritis with optical coherence tomography. Ann Neurol. 2006;59:963–969. doi: 10.1002/ana.20851. [DOI] [PubMed] [Google Scholar]
- 14.Constantinescu CS, Hilliard B, Fujioka T, Bhopale MK, Calida D, Rostami AM. Pathogenesis of neuroimmunologic diseases. Immunol Res. 1998;17:217–227. doi: 10.1007/BF02786446. [DOI] [PubMed] [Google Scholar]
- 15.Potter NT, Bigazzi PE. Acute optic neuritis associated with immunization with the CNS myelin proteolipid protein. Invest Ophthalmol Vis Sci. 1992;33:1717–1722. [PubMed] [Google Scholar]
- 16.Guy J, Qi X, Hauswirth WW. Adeno-associated viral-mediated catalase expression suppresses optic neuritis in experimental allergic encephalomyelitis. Proc Natl Acad Sci. 1998;95:13847–13852. doi: 10.1073/pnas.95.23.13847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shindler KS, Guan Y, Ventura E, Bennett J, Rostami A. Retinal ganglion cell loss induced by acute optic neuritis in a relapsing model of multiple sclerosis. Mult Scler. 2006;12:526–532. doi: 10.1177/1352458506070629. [DOI] [PubMed] [Google Scholar]
- 18.Shindler KS, Ventura E, Rex TS, Elliott P, Rostami A. SIRT1 activation confers neuroprotection in experimental optic neuritis. Invest Ophthalmol Vis Sci. 2007;48:3602–3609. doi: 10.1167/iovs.07-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 20.Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R. The silencing protein SIR2 and its homologs are NAD dependent protein deacetylases. Proc Natl Acad Sci USA. 2000;97:5807–5811. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith JS, Brachmann CB, Celic I, Kenna MA, Muhammad S, Starai VJ, Avalos JL, Escalante-Semerena JC, Grubmeyer C, Wolberger C, Boeke JD. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci USA. 2000;97:6658–6663. doi: 10.1073/pnas.97.12.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gran B, Zhang G-X, Yu S, Li J, Chen XH, Ventura ES, Kamoun M, Rostami A. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J Immunol. 2002;169:7104–7110. doi: 10.4049/jimmunol.169.12.7104. [DOI] [PubMed] [Google Scholar]
- 24.Shao H, Huang Z, Sun SL, Kaplan HJ, Sun D. Myelin/oligodendrocyte glycoprotein-specific T-cells induce severe optic neuritis in the C57BL/6 mouse. Invest Ophthalmol Vis Sci. 2004;45:4060–4065. doi: 10.1167/iovs.04-0554. [DOI] [PubMed] [Google Scholar]
- 25.Shindler KS, Ventura E, Dutt M, Rostami AM. Inflammatory demyelination induces axonal injury and retinal ganglion cell apoptosis in experimental optic neuritis. Exp Eye Res. 2008;87:208–213. doi: 10.1016/j.exer.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paintlia AS, Paintlia MK, Singh I, Singh AK. Immunomodulatory effect of combination therapy with lovastatin and 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside alleviates neurodegeneration in experimental autoimmune encephalomyelitis. Am J Pathol. 2006;169:1012–1025. doi: 10.2353/ajpath.2006.051309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fitzgerald DC, Ciric B, Touil T, Harle H, Grammatikopolou J, Das Sarma J, Gran B, Zhang GX, Rostami A. Suppressive effect of IL-27 on encephalitogenic Th17 cells and the effector phase of experimental autoimmune encephalomyelitis. J Immunol. 2007;179:3268–3275. doi: 10.4049/jimmunol.179.5.3268. [DOI] [PubMed] [Google Scholar]
- 28.de la Lastra CA, Villegas I. Resveratrol as an anti-inflammatory and anti-aging agent: mechanisms and clinical implications. Mol Nutr Food Res. 2005;49:405–430. doi: 10.1002/mnfr.200500022. [DOI] [PubMed] [Google Scholar]
- 29.Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh NP, Hegde VL, Hofseth LJ, Nagarkatti M, Nagarkatti P. Resveratrol (trans-3,5,4'-trihydroxystilbene) ameliorates experimental allergic encephalomyelitis, primarily via induction of apoptosis in T cells involving activation of aryl hydrocarbon receptor and estrogen receptor. Mol Pharmacol. 2007;72:1508–1521. doi: 10.1124/mol.107.038984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imler TJ, Jr, Petro TM. Decreased severity of experimental autoimmune encephalomyelitis during resveratrol administration is associated with increased IL-17+IL-10+ T cells, CD4(−) IFN-gamma+ cells, and decreased macrophage IL-6 expression. Int Immunopharmacol. 2009;9:134–143. doi: 10.1016/j.intimp.2008.10.015. 2009. [DOI] [PubMed] [Google Scholar]
- 32.Hobom M, Storch MK, Weissert R, Maier K, Radhakrishnan A, Kramer B, Bähr M, Diem R. Mechanisms and time course of neuronal degeneration in experimental autoimmune encephalomyelitis. Brain Pathol. 2004;14:148–157. doi: 10.1111/j.1750-3639.2004.tb00047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]