

Abstract
Purpose
Evaluation of genomic tests is often challenging because of the lack of direct evidence of clinical benefit compared to usual care and unclear evidence requirements. To address these issues, this study presents a risk-benefit framework for assessing the health-related utility of genomic tests.
Methods
We incorporated approaches from a variety of established fields, including decision science, outcomes research, and health technology assessment to develop the framework. Additionally, we considered genomic test stakeholder perspectives and case studies.
Results
We developed a 3-tiered framework: first, we use decision-analytic modeling techniques to synthesize data, project incidence of clinical events, and assess uncertainty. Second, we define the health-related utility of genomic tests as improvement in health outcomes as measured by clinical event rates, life expectancy, and quality-adjusted life-years (QALYs). Finally, we display results using a risk-benefit policy matrix to facilitate the interpretation and implementation of findings from these analyses.
Conclusion
A formal risk-benefit framework may accelerate the utilization and practice-based evidence development of genomic tests that pose low risk and offer plausible clinical benefit, while discouraging premature use of tests that provide little benefit or pose significant health risks compared to usual care
INTRODUCTION
Few genomic testing technologies have reached routine clinical practice or been incorporated into clinical guidelines to date.(1-4) Nonetheless, a multitude of genomic tests are marketed to consumers and physicians, and genome-wide assays are available to consumers for several hundred dollars.(5) These assays, coupled with the rapid growth of somatic gene expression profiling in oncology, present a significant challenge to clinicians and policy makers seeking to establish clinical practices that maximize benefit for patients while minimizing harm.
The efficient and appropriate translation of genomic discoveries into clinical practice is particularly challenging due to an interrelated combination of factors.(6) First, there is a notable lack of comparative effectiveness data for genomic applications due to regulatory and reimbursement policies that neither require nor incentivize investment in such studies.(7-9) Consequently, while randomized trials have been initiated for select genomic applications such as CYP2C9/VKORC1 testing with warfarin therapy,(10, 11) CYP2D6 testing with antidepressant use,(12) and gene expression profiling in breast cancer treatment,(13) there are generally few prospective comparative genomic tests evaluations planned or underway.(14)
Second, the ease of market access for genomic tests makes the aforementioned lack of evidence more problematic.(15, 16) For example, when investigators from the National Institute of Mental Health reported an association between two genetic variants and suicidal ideation in patients taking citalopram, (17) within a week a genomic testing company announced plans to offer testing to “help to reduce a recently announced spike in suicide rates among US youth”.(18) This situation is partly related to regulatory policy, but is also related to the fact that providing information about genomic susceptibilities does not require specialized medical facilities or training, and involves very little direct risk of immediate harm to patients.
Lastly, there is a lack of consensus on evidence requirements or thresholds for genomic test evaluation [19]. Some stakeholders accept the findings of retrospective analyses and clinical plausibility, while others expect controlled clinical trial data.(20, 21) For example, in the case of the anticoagulant warfarin, variants of the genes CYP2C9 and VKORC1 are clearly associated with lower dose requirements, but no study to date has definitively demonstrated that using this information improves patient outcomes.(22) Alternatively, warfarin patients concomitantly taking amiodarone also require lower warfarin dosing (due to inhibition of CYP2C9), and doing so is considered standard of care.(23) This lack of consistency in evidence requirements, in addition to the other factors outlined above, creates a roadblock on the translational pathway for genomic tests.
The Secretary’s Advisory Committee on Genetics, Health, and Society (SACGHS) recently issued a report (15) emphasizing the importance of assessing and weighing potential harm against potential benefit, so that patients do not inadvertently forgo real benefit because of small or hypothetical harms. Additionally, regulatory authorities have shown heightened interest in the use of quantitative approaches to assess risk-benefit tradeoffs for pharmaceuticals.(24-31) A recent Institute of Medicine (IOM) study advised that FDA “develop and continually improve a systematic approach to risk-benefit analysis.”(32) FDA is currently evaluating various approaches to incorporate risk-benefit analyses into their assessment processes. Although approaches have been developed to incorporate indirect evidence (e.g., non-comparative data) in a semi-quantitative fashion, and decision-analytic techniques are beginning to be applied in the assessment of genomic tests,(4, 33) quantitative assessment of risk-benefit tradeoffs, and the uncertainty surrounding them, have not been explicitly included in genomic test evidence recommendations to date.
We believe there is a significant opportunity to use existing decision modeling methods to synthesize genomic, clinical, epidemiological, and patient outcome data to explicitly evaluate risk and benefit trade-offs of genomic tests, and the uncertainty surrounding their utility. The objective of this study was to develop a systematic and comprehensive approach to help clinicians and policy-makers estimate health outcomes of genomic testing in the absence of definitive data. The novel aspect of the risk-benefit framework described in this manuscript is the synthesis of approaches from a variety of fields in order to systematically and quantitatively evaluate the risk-benefit profile of genomic tests – the use of decision modeling, the projection of multiple clinical outcomes (including quality-adjusted life-years, QALYs, as a summary measure of clinical utility), and a recommendation framework that enables utilization of the information generated. These estimates are intended to help guide decisions about clinical test use and coverage, and provide a framework for encouraging practice-based evidence development for tests with plausible net health benefit.
Methods
The risk-benefit framework presented herein is based on work from the fields of decision science, outcomes research, and health technology assessment. Traditional evidence-based processes have generally relied on direct evidence of clinical utility (e.g. data from RCTs). Recently, however, advisory bodies have recognized that direct evidence will not always be available to answer questions of interest. For example, the U.S. Preventive Services Task Force (USPSTF) developed an approach for evaluating indirect evidence with a focus on evaluating net health benefit, and the uncertainty around estimates.(34, 35) The Task Force constructs a “chain of evidence” within an analytic framework, and assesses the level of certainty based on specific questions. If the certainty of net benefit is moderate or high, the magnitude of benefit is assessed, and modeled event rates are provided in an outcomes table. For example, Nelson and colleagues utilized this approach to evaluate BRCA mutation testing for breast and ovarian cancer susceptibility, although a summary measure of the net health benefit was not determined.(36)
More recently, the U.S. Centers for Disease Control and Prevention (CDC) has sponsored the Evaluation of Genomic Applications in Practice and Prevention (EGAPP) initiative.(37) EGAPP’s methods are analogous to that of the USPSTF, and involve use of an analytic framework to assess indirect evidence. Three of the seven EGAPP evidence reports commissioned to date have explicitly conducted decision-analytic modeling. (38) In two cases, evidence supporting a valid association between variants and clinical outcomes was lacking, and the models were used in an exploratory capacity.(39, 40) In the other case, the model was used to assess efficiency of case detection, but not patient outcomes.(41) A summary measure of net health benefit was not calculated in any of these cases.
The private sector has also pursued analogous, evidence-based approaches. Notably, the BlueCross BlueShield Association’s Technology Evaluation Center (TEC) has conducted extensive evidence-based evaluations of genomic tests.(42) The TEC uses five criteria to evaluate health technologies such as genomic tests: 1) it must have regulatory approval, 2) the evidence must permit conclusions regarding its effect on health outcomes, 3) it must result in an improvement in net health outcomes, 4) it must be at least as good as current alternatives, and 5) it’s benefits must be attainable outside of the investigational setting. Quantitative evaluation of indirect evidence has not been utilized for TEC assessments to date.
In summary, although approaches to date have incorporated various aspects of a quantitative risk-benefit framework, they have not included a formal and explicit approach to assessing indirect evidence, a summary measure of risk-benefit, and a decision-making framework that synthesizes this information. Below, we propose a quantitative risk-benefit approach that incorporates these aspects within a single framework. We utilized stakeholder feedback and previous experience with case studies and regulatory science to inform development of the framework. (19, 43-45)
I. Decision-analytic framework
Decision-analytic modeling provides an explicit framework for evaluating technologies by incorporating data from various sources in a quantitative and transparent fashion, and comparing the likely results of technology use versus the next best alternative. By assessing the incremental outcomes compared to the next best alternative (e.g., no genomic testing), the ‘opportunity cost’ of genomic testing can be captured. Weinstein and Fineberg characterize the decision-analytic approach as: 1) identifying and bounding the decision problem, 2) structuring the decision problem over time, 3) characterizing the information needed to inform the structure, 4) choosing a preferred course of action.(46) This approach is advantageous in that there is an explicit framework for evaluating risks and benefits, decision makers must identify quantitative estimates of risks and benefits, the approach can be applied to a wide variety of technologies, and complexity and timing of analyses can be suited to the decision-making task.(24)
To illustrate the decision-modeling process, we consider a hypothetical cohort of patients initiating long-term warfarin therapy for the prevention of thromboembolic events. During the warfarin initiation period, determination of the dose required to achieve an optimal level of anticoagulation can be challenging. Clinicians monitor the international normalized ratio (INR), a measure of anticoagulation status that can serve as a surrogate marker for adverse events. INR values between 2 and 3 are considered within therapeutic range for most patients - INR values above 3 are associated with higher risk of serious bleeding events, while INR values below 2 are associated with increased risk of thromboembolic events. Most patients are initiated on 5mg warfarin per day, and clinical and demographic variables that indicate warfarin sensitivity such as older age, drug interactions, or co-morbidities are used to adjust doses downward. Information about the patients’ CYP2C9 and VKORC1 gene status (hereafter referred to as “genotype-guided” dosing) also could be incorporated in the initial dose selection. Below, we demonstrate how decision modeling can be used to quantitatively evaluate the risks and benefits of each approach based on an analysis conducted as part of this risk-benefit framework project, as well as the results of a previously published warfarin decision analytic model.(43, 47)
Decision structure, data sources, and outcomes
At the core of the risk-benefit framework is what could be described as a clinical disease-based model. The goal of this approach is to incorporate relevant clinical effects attributable to a genomic test and subsequent actions in order to estimate impact on patient outcomes. A schematic of the process is depicted in Figure 1.
Figure 1.
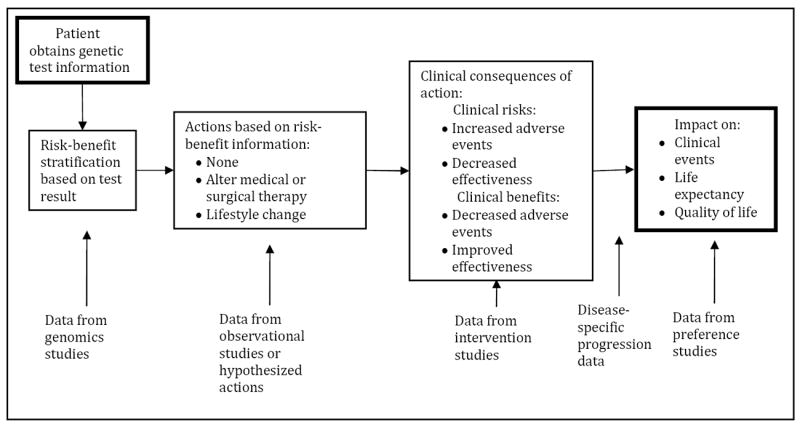
Schematic diagram of disease-based model
Consider this approach in the context of genomic testing to guide warfarin therapy described earlier. First, clinicians receive genomic test results reporting patients’ CYP2C9 and VKORC1 genomic status. Next, informed by the test results, an estimated initial dose is calculated and warfarin therapy is administered – assuming the clinician and patient agree with the suggested dosing. During the subsequent weeks, clinicians will monitor INR and adjust warfarin dose in response. The goal is to achieve stable INR values in therapeutic range, and a standard measure of anticoagulation management success is the time in therapeutic range (TTR) over the first month (or months) of warfarin therapy. (48)
We developed a risk-benefit analysis for warfarin pharmacogenomic testing based on extensive interaction with various stakeholders, particularly practicing anticoagulation clinicians.(47) Because of their familiarity with INR as an outcome, clinicians indicated that a model that projected bleed and clot events based on INR over the first 1-3 months of warfarin therapy would be most useful to assess the potential net benefit of testing (Figure 2). The probabilities of achieving different levels of INR control ideally would be informed by the results of comparative, randomized clinical trials. In this instance we utilized results from the highest quality RCT available to date conducted by Anderson and colleagues (N=200).(49) Additionally, the relationship between time in INR range and the risk of clinical events can be derived from longitudinal cohort studies, as was done with data from van Walraven and colleagues in the warfarin model under consideration.(50) These probabilities are then multiplied to compare the overall likelihood of having specific events within each dosing strategy.
Figure 2.
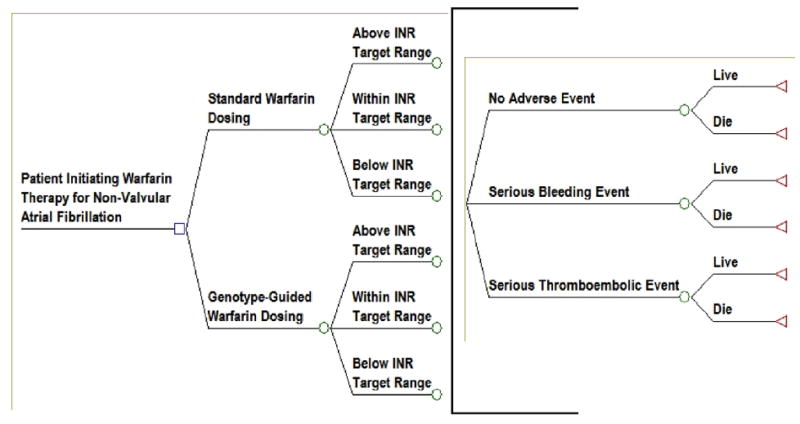
Warfarin Pharmacogenomics Decision Tree
We estimated in a cohort of 10,000 patients observed for the first month of warfarin therapy approximately 44 and 45 patients would experience serious bleeding events and approximately 27 and 28 would experience serious thromboembolic events in the genotype-guided and standard dosing strategies, respectively.(47).
Uncertainty: Scenario and Sensitivity Analysis
The decision structure described above does not allow traditional statistical analyses and hypothesis testing because not all data are derived from the same study, nor typically obtained at the patient level. However, it is possible to explicitly evaluate uncertainty – particularly related to lack of data. To accomplish this, scenario analyses can be conducted in which model inputs are varied over plausible ranges, and the impact on results assessed – for instance in ‘most likely’, ‘best case’, and ‘worst case’ scenarios. Each model input can be varied individually to identify inputs that drive the analysis and are associated with the greatest uncertainty in the results.
For example, in the decision tree depicted in Figure 2, uncertainty about the proportion of time patients spend within the target INR range in the first month of warfarin therapy could be explored by examining the modeled outcomes of a plausible range of values. Perhaps in the “most likely”, “best”, “worst” scenarios patients are within the INR target range for 66%, 82%, and 50% of the time, respectively. Downstream outcomes of these times in target INR range can be modeled to see how use of genotype-guided dosing compares to standard dosing under each assumption. In the model, the “most likely”, “best”, and “worst” genotype-guided dosing scenarios are estimated to result in approximately 44, 40, and 52 serious bleeding events and 27, 24, 28, serious thromboembolic events, respectively.(47)
Additionally, overall uncertainty related to data inputs can be evaluated using probabilistic sensitivity analysis (PSA), in which distributions are assigned to the model inputs, and Monte Carlo simulation is used to repeatedly draw sets of model inputs.(51, 52) Although the use of PSA is considered best practice for health outcomes modeling, use of individual parameter sensitivity analyses and multiple-parameter scenario analyses may be more intuitive for stakeholders.(53, 19)
II. Summary Measure of Health-Related Utility
Analogous to the USPSTF approach to presenting the results of indirect evidence assessments, we suggest presenting both benefits and risks in an outcomes table, as well as reporting ranges of results obtained from evaluations of uncertainty and assumptions, as described above.(35) However, assessing the overall balance of risks and benefits can be more challenging. Clinical events differ in their severity and frequency, and projecting their impact without an explicit framework or summary outcome measure is difficult. For example, considering the warfarin therapy cohort, should serious bleeding events experienced or thromboembolic events avoided receive a greater weight? Projected life expectancy is an important summary measure of mortality and should be assessed in all risk-benefit analyses for which there is uncertainty about clinical utility. However, life expectancy does not account for patient morbidity and quality of life impacts.
Quality-Adjusted Life-Years
The challenge of comparing different types of outcomes across different diseases and interventions has been addressed in health outcomes research using the metric of the quality-adjusted life-year (QALY).(54) The use of QALYs as the preferred measure in health outcomes research has been established in the US and a variety of other countries.(54, 55) In addition, the recent IOM Committee to Evaluate Measures of Health Benefits for Environmental Health and Safety Regulation in the United States stated that analyses that “integrate morbidity and mortality impacts in a single effectiveness measure should use the quality-adjusted life year to represent net health effects.”(56)
The QALY represents an adjustment to length of life for the estimated quality of life. Quality of life is measured with a preference scale or index, where 0 represents the value or “utility score” for death and 1 represents normal “full” health. Thus, ten years of life expectancy at a utility of 0.5 is equivalent to five years with full health. There are several approaches to measuring preferences including time trade-off, standard gamble, and population-weighted surveys.(54) These measures evaluate physical, mental, emotional, and social functioning domains to varying extent, and can be general or condition specific.
Grosse and Khoury suggested using the term ‘utility’ to include both ‘clinical utility’ (health-related outcomes) and ‘social utility’ (primarily psychological effects).(57) We propose defining the utility of genomic testing from a health policy perspective as an improvement in life expectancy or quality of life for patients and their families, and term this measure ‘health-related utility’ (HRU). The psychological impacts of testing, whether benefits or harms, would be included if they have a measurable impact on patient’s health-related quality of life, defined in general as mental, emotional, or social functioning related to their knowledge of genomic test results. In this construct, clinical events can be assessed through their impact on patient life expectancy (i.e., attributable mortality) and morbidity (i.e., patient quality of life). This definition combines attributes of ‘clinical utility’ and ‘social utility’, but does not include effects, such as impact on diagnostic thinking, if there is no associated influence on clinical outcomes or quality of life.
Returning to the warfarin case study, assessment of the potential impact of clinical events on life expectancy and QALYs requires the tracking of events, mortality, and quality of life over the lifetime of a patient cohort, which is commonly achieved in decision modeling through the use of Markov models.(58) We previously developed a warfarin pharmacogenomics health policy model employing such techniques, and estimated in the base-case analysis that testing could lead to an improvement in QALYs of 0.003 (1 day).(43) Notably, uncertainty analyses indicated that the difference in QALYs could range from -0.005 to +0.010. These findings are relatively consistent with the results of similar analyses recently conducted by Eckman and colleagues and Patrick and colleagues. (59,60)
Limitations of QALYs
There are several limitations to the use of QALYs as a summary measure of HRU for genomic tests. First, there are limited data on the impact of testing on patient and family quality of life or preferences.(61) Second, measuring the psychological impacts of testing using a preference approach is challenging, as most instruments likely are not sensitive, and disease and test specific instruments will need to be developed.(61) Third, there is significant uncertainty associated with most preference estimates, further complicating interpretation of the results of risk-benefit analyses. Fourth, different individuals will vary in their utility ratings of the same health state, so clinical guidelines should allow clinicians flexibility to address individual preferences, though population-level clinical policies will generally aim to consider average or typical preferences.
Many of these concerns have been noted by genomic test stakeholders in the literature.(19) Specifically, we found that while stakeholders are receptive to the concept of using decision-analytic methods to evaluate genomic tests, many have concerns about lack of consistency in the methods used to elicit preferences, the ability of QALYs to capture the psychological value of test results, and the use of QALYs as a summary measure of HRU.(19) Perhaps most importantly, stakeholders noted that use of QALYs as a summary measure of HRU is likely to lead to arguments about preference elicitation methods, and could ultimately limit the use of decision-analysis to evaluate genomic tests.(19)
These limitations highlight the need for ongoing stakeholder dialogue in relation to the development and use of decision-analytic methods to evaluate genomic tests, consideration of the impact of patient treatment preferences on health outcomes, and the importance of outcome measures in addition to QALYs. To help address these issues, we suggest that analyses report a multitude of health outcomes, including: 1) proportion of patients with a reclassified risk status, 2) proportion of patients indicated to receive an alternative treatment strategy, 3) proportion of patients likely to choose the alternative treatment 4) clinical events (benefits and harms), 5) life expectancy, and 6) quality-adjusted life-expectancy (Table 1).
Table 1.
Risk-Benefit outcomes table: Examples of potential outcomes for different types of genomic tests
| Incremental results for testing vs. comparator strategy | Type of Genomic Test | ||
|---|---|---|---|
| Pharmacogenomics | Disease Risk | Newborn Screening | |
| Proportion of patients with a reclassified risk status | % patients with reclassified risk status | % individuals with reclassified risk status | % children with reclassified risk status |
| Proportion of patients indicated to receive an alternative treatment strategy | % patients with dose or drug change | % individuals initiating lifestyle change or preventative interventions | % children receiving medical or dietary intervention |
| Proportion of patients likely to choose the alternative treatment | % patients choosing indicated treatment strategy | % individuals choosing indicated lifestyle change or preventative intervention | % children receiving indicated medical or dietary intervention |
| Incidence of clinical events: Benefits and Harms (including NNT, NNS, NNB, NNH) | serious adverse drug reaction, myocardial infarction, stroke, cancer recurrence | myocardial infarction, stroke, cancer recurrence | growth, mental functioning |
| Life expectancy | life years | life years | life years |
| Quality of life (and patient preferences) | impact of clinical events on quality of life | impact of increased fear vs. empowerment | impact of ‘right to know’ vs. harm from uncertainty in diagnosis or treatment |
| Quality-Adjusted Life-Years | QALYs | QALYs | QALYs |
NNT=Number Needed to Treat, NNS=Number Needed to Screen, NNB=Number Needed to Benefit, NNH=Number Needed to Harm, QALY=Quality-Adjusted Life-Year
III. Risk-benefit policy matrix
Health policy evaluations of genomic tests are complex and warrant a variety of clinical, social, and political considerations. The framework established above serves to anchor one of these domains, health-related utility. Given the results of a quantitative risk-benefit analysis, in addition to other factors, decision-makers are faced with three options: 1) recommend the technology, 2) reject it, or 3) wait and collect more data.
In reference to the latter option, there has been increasing regulatory interest in the use of ‘coverage with evidence development’ (CED). CED programs provide patients access to technology while developing evidence to inform future policy decisions.(62) The U.S. Medicare program has recently applied this approach in other areas where there is limited evidence available (e.g. surgical interventions and medical devices). In such programs, healthcare payers agree to cover medical services or technologies under the condition that beneficiaries enroll in studies or registries to collect additional data on the use and outcomes of the therapy. Thus, CED provides a process for moving technologies along the translational pathway. For example, based on recommendation from the Medicare Evidence Development & Coverage Advisory Committee (MEDCAC), CMS recently implemented a CED policy for pharmacogenomic testing with warfarin therapy.(63)
To implement CED in a manner consistent with facilitating the appropriate translation of genomics into healthcare, a ‘technology triage’ mechanism is needed to identify potential candidates. We believe quantitative risk-benefit assessment can serve this important role. Risk-benefit policy matrices can be used to categorize genomic tests based on potential magnitude of HRU, and the uncertainty around these estimates. Our draft matrix (Figure 3) provides 5 recommendation options, aiming to discourage use (or clinical development) of tests that have a reasonable chance of overall ‘negative’ HRU, while encouraging entry into a ‘post-marketing’ development pathway for tests that offer substantial promise but lack evidence of HRU.
Figure 3.
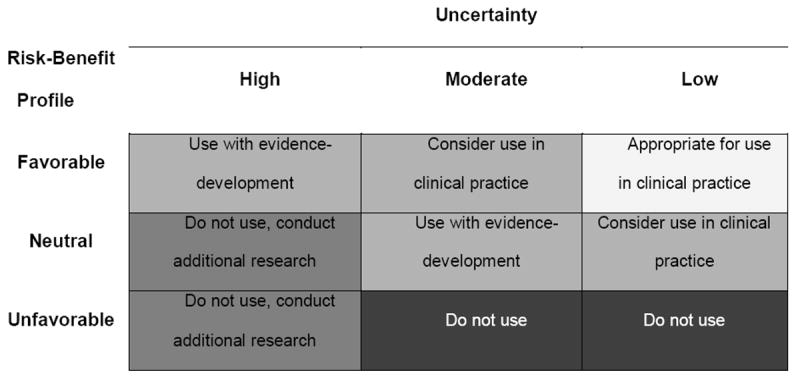
Risk-Benefit Policy Matrix
For example, in the warfarin case study, model estimates indicated that genotype-guided dosing would result in a small increase in QALYs relative to standard dosing, but probabilistic sensitivity analyses estimated that genotype-guided dosing would increase QALYs in 84% of simulations and decrease QALYs in 16% of simulations.(43) Given these findings indicate an approximately ‘neutral’ risk-benefit profile and a ‘moderate’ degree of uncertainty, genotype-guided warfarin dosing could be classified as ‘use with evidence development.’ In fact, this was the conclusion reached by the Centers for Medicare and Medicaid Services (CMS) in August of 2009.(64) Although formal decision modeling did not appear to have a direct role in this decision, CMS “considered the evidence in the hierarchical framework of Fryback and Thornbury where Level 2 addresses diagnostic accuracy, sensitivity, and specificity of the test; Level 3 focuses on whether the information produces change in the physician’s diagnostic thinking; Level 4 concerns the effect on the patient management plan and Level 5 measures the effect of the diagnostic information on patient outcomes.”(65) While the evidence considered was similar, a formal risk-benefit approach may have provided greater transparency in regard to synthesis across levels of evidence, and quantification of the potential net benefit and associated uncertainty. Uncertainty analyses also could highlight the evidence gap in regard to the effectiveness of testing, and the value of conducting an RCT.
The recommendation categories we propose for the risk-benefit framework offer a starting point for stakeholders to develop dialogue about the merits of genomic tests. As with most policy frameworks, we expect this approach to evolve over time, and be modified as needed by individual stakeholder groups. For certain stakeholders, other considerations such as cost and equity will be important and should be evaluated. Indeed, in our previous study of stakeholder perspectives, payers indicated that tests with lower budget impacts might be evaluated using a simpler matrix primarily focused on potential harm, while more expensive tests or ones that have larger downstream cost impacts would require a careful evaluation of risk-benefit as well as cost-effectiveness. (19)
Challenges
In some cases, formal risk-benefit assessment of genomic tests will be limited by lack of sufficient or valid data to make utilization recommendations. In these cases, health outcomes modeling can be used to conduct exploratory evaluations to identify key parameter values that are required to produce health-related utility. For example, this approach was taken in the recent EGAPP evidence reports evaluating CYP-P450 testing for antidepressant therapy and ovarian cancer susceptibility testing.(39, 40) In the CYP-P450 for antidepressant therapy evaluation, a decision analysis was conducted to examine under which conditions genetic testing could lead to a better clinical outcome at six weeks, with the outcome of proportion responders.(40) In the ovarian cancer susceptibility testing evaluation, a Markov model with a lifetime horizon was used to assess what combinations of inputs would be required to achieve a target of 20% reduction in cancer mortality.(39)
Quantitative evaluation of genomic tests is also complicated by their diverse applications (66), and distinct ethical and policy implications, based on predictive value and the availability of treatment for patients who test positive.(67) In this sense, the risks and benefits of genomic testing extend beyond the usual endpoints measured in health technology assessment. Consideration of risks like stigma and discrimination, false reassurance, opportunity costs, and use of unproven therapies must be weighed against potential benefits of genomic diagnosis to family members, and the value placed on risk information by both patients and providers.(57, 67-71) Whether these risks and benefits should drive health care decision-making is an open question, to be determined in part by their relative weight compared to medical outcomes of testing.
Lastly, given the required assumptions and potential complexity of analyses, stakeholder acceptance of a modeling approach is likely to be a major challenge.(72, 73) To address this issue, collaboration with stakeholders to specify optimal approaches, interpretation, and recognition of limitations is critical to the success of a genomic testing risk-benefit framework. Such efforts are underway, but additional work is needed in this area. (19) Issues that need to be addressed include: 1) data to be included in risk-benefit analyses, 2) outcomes generated by the analyses, 3) a decision-making framework and corresponding thresholds, and 4) transparency, acceptance, and communication of the results of the analyses.
Summary
We believe a formal risk-benefit framework is useful for evaluating genomic tests for several reasons. First, while it must be recognized that the gold standard for direct evidence of health-related utility of genomic tests will come from prospective randomized controlled clinical trials, there is an opportunity to utilize quantitative risk-benefit analysis to derive at least preliminary estimates of HRU. This approach could be particularly valuable for genomic tests with a clear course of action that has been well studied.
Second, there will be a significant shortage of direct evidence of HRU for genomic tests in the near future. In some cases, indirect evidence of a favorable risk-benefit profile will suffice to recommend a test for use in clinical practice. Formal risk-benefit analysis offers a pragmatic approach to assessing HRU in a reasonably timely yet systematic manner. Thus, safe and potentially valuable genomic technologies will not be withheld from clinical use due to lack of direct evidence.
Third, risk-benefit analysis provides a tool to quantify the risk of interventions that result from testing in relation to potential benefit. Specifying risks can also aid in communicating such risks to providers and policymakers, thus protecting patients’ and the public’s health.
Finally, there will be significant uncertainty surrounding the HRU of most genomic tests. Scenario analysis and formal sensitivity analysis provide a mechanism for the quantification of uncertainty in health-related utility. Risk-benefit analyses also provide a foundation for assessing the value of additional research to reduce uncertainty and guide prioritization of comparative effectiveness research in genomics.
In summary, quantitative risk-benefit analysis provides a valuable tool for prioritizing genomic tests for development in the translational pathway. Specifically, tests that appear to have a reasonable risk profile, but with significant uncertainty with regard to the magnitude of benefit, can be recommended for use in clinical practice in coverage with evidence development programs. These strategies provide a viable route to generating evidence of health-related utility in the de facto ‘post-marketing’ environment of genomic tests. This approach could serve as a foundation for assessment of population health impacts, regulatory decisions, health economics studies, and for the incorporation of the personal utility of prognostic information.
Acknowledgments
This publication was supported by Cooperative Agreement No. 1U18GD000005 from the Centers for Disease Control and Prevention. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the Centers for Disease Control and Prevention.
References
- 1.Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med. 2009;11(1):35–41. doi: 10.1097/GIM.0b013e31818fa2ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Recommendations from the EGAPP Working Group: can UGT1A1 genotyping reduce morbidity and mortality in patients with metastatic colorectal cancer treated with irinotecan? Genet Med. 2009;11(1):15–20. doi: 10.1097/GIM.0b013e31818efd9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Recommendations from the EGAPP Working Group: can tumor gene expression profiling improve outcomes in patients with breast cancer? Genet Med. 2009;11(1):66–73. doi: 10.1097/GIM.0b013e3181928f56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Recommendations from the EGAPP Working Group: testing for cytochrome P450 polymorphisms in adults with nonpsychotic depression treated with selective serotonin reuptake inhibitors. Genet Med. 2007;9(12):819–25. doi: 10.1097/gim.0b013e31815bf9a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khoury MJ, McBride CM, Schully SD, et al. The Scientific Foundation for personal genomics: recommendations from a National Institutes of Health-Centers for Disease Control and Prevention multidisciplinary workshop. Genet Med. 2009;11(8):559–67. doi: 10.1097/GIM.0b013e3181b13a6c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khoury MJ, Feero WG, Reyes M, et al. The genomic applications in practice and prevention network. Genet Med. 2009;11(7):488–94. doi: 10.1097/GIM.0b013e3181a551cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramsey SD, Veenstra DL, Garrison LP, Jr, et al. Toward evidence-based assessment for coverage and reimbursement of laboratory-based diagnostic and genetic tests. Am J Manag Care. 2006;12(4):197–202. [PubMed] [Google Scholar]
- 8.Garrison LP, Jr, Carlson RJ, Carlson JJ, Kuszler PC, Meckley LM, Veenstra DL. A review of public policy issues in promoting the development and commercialization of pharmacogenomic applications: challenges and implications. Drug Metab Rev. 2008;40(2):377–401. doi: 10.1080/03602530801952500. [DOI] [PubMed] [Google Scholar]
- 9.Garrison LP, Austin MJ. The Economics of Personalized Medicine: A Model of Incentives for Value Creation and Capture. Drug Information Journal. 2007;41(4) [Google Scholar]
- 10.Hillman MA, Wilke RA, Yale SH, et al. A prospective, randomized pilot trial of model-based warfarin dose initiation using CYP2C9 genotype and clinical data. Clin Med Res. 2005;3(3):137–45. doi: 10.3121/cmr.3.3.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voora D, Eby C, Linder MW, et al. Prospective dosing of warfarin based on cytochrome P-450 2C9 genotype. Thromb Haemost. 2005;93(4):700–5. doi: 10.1160/TH04-08-0542. [DOI] [PubMed] [Google Scholar]
- 12.Murphy GM, Jr, Kremer C, Rodrigues HE, Schatzberg AF. Pharmacogenetics of antidepressant medication intolerance. Am J Psychiatry. 2003;160(10):1830–5. doi: 10.1176/appi.ajp.160.10.1830. [DOI] [PubMed] [Google Scholar]
- 13.Genomic Health. Oncotype Dx breast cancer assay. 2005 [Google Scholar]
- 14.Khoury MJ, Rich EC, Randhawa G, Teutsch SM, Niederhuber J. Comparative effectiveness research and genomic medicine: an evolving partnership for 21st century medicine. Genet Med. 2009;11(10):707–11. doi: 10.1097/GIM.0b013e3181b99b90. [DOI] [PubMed] [Google Scholar]
- 15.Secretary’s Advisory Committee on Genetics H, and Society (SACGHS) U.S. System of Oversight of Genetic Testing: A Response to the Charge of the Secretary of HHS. Draft Report of the Secretary’s Advisory Committee on Genetics, Health, and Society. 2007 [Google Scholar]
- 16.Hudson KL, Murphy JA, Kaufman DJ, Javitt GH, Katsanis SH, Scott J. Oversight of US genetic testing laboratories. Nat Biotechnol. 2006;24(9):1083–90. doi: 10.1038/nbt0906-1083. [DOI] [PubMed] [Google Scholar]
- 17.Laje G, Paddock S, Manji H, et al. Genetic markers of suicidal ideation emerging during citalopram treatment of major depression. Am J Psychiatry. 2007;164(10):1530–8. doi: 10.1176/appi.ajp.2007.06122018. [DOI] [PubMed] [Google Scholar]
- 18.Ray T. NeuroMark Launches Suicidal Ideation Dx For Investigational Use Pending Final Study. Pharmacogenomics Reporter. 2007 [Google Scholar]
- 19.Roth JA, Garrison LP, Burke W, Ramsey SD, Carlson R, Veenstra DL. Stakeholder perspectives on a risk-benefit framework for genetic testing. Public Health Genomics. doi: 10.1159/000290452. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lesko LJ. The critical path of warfarin dosing: finding an optimal dosing strategy using pharmacogenetics. Clin Pharmacol Ther. 2008;84(3):301–3. doi: 10.1038/clpt.2008.133. [DOI] [PubMed] [Google Scholar]
- 21.Garcia DA. Warfarin and pharmacogenomic testing: the case for restraint. Clin Pharmacol Ther. 2008;84(3):303–5. doi: 10.1038/clpt.2008.131. [DOI] [PubMed] [Google Scholar]
- 22.Kangelaris KN, Bent S, Nussbaum RL, Garcia DA, Tice JA. Genetic testing before anticoagulation? A systematic review of pharmacogenetic dosing of warfarin. J Gen Intern Med. 2009;24(5):656–64. doi: 10.1007/s11606-009-0949-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. (8) 2008;133(6 Suppl):160S–198S. doi: 10.1378/chest.08-0670. [DOI] [PubMed] [Google Scholar]
- 24.Garrison LP, Jr, Towse A, Bresnahan BW. Assessing a structured, quantitative health outcomes approach to drug risk-benefit analysis. Health Aff (Millwood) 2007;26(3):684–95. doi: 10.1377/hlthaff.26.3.684. [DOI] [PubMed] [Google Scholar]
- 25.Meltzer D. Risks and benefits of risk-management plans. Health Aff (Millwood) 2007;26(3):681–3. doi: 10.1377/hlthaff.26.3.681. [DOI] [PubMed] [Google Scholar]
- 26.Pauly MV. Risks and benefits in health care: the view from economics. Health Aff (Millwood) 2007;26(3):653–62. doi: 10.1377/hlthaff.26.3.653. [DOI] [PubMed] [Google Scholar]
- 27.Peters E, Hibbard J, Slovic P, Dieckmann N. Numeracy skill and the communication, comprehension, and use of risk-benefit information. Health Aff (Millwood) 2007;26(3):741–8. doi: 10.1377/hlthaff.26.3.741. [DOI] [PubMed] [Google Scholar]
- 28.Juhn P, Phillips A, Buto K. Balancing modern medical benefits and risks. Health Aff (Millwood) 2007;26(3):647–52. doi: 10.1377/hlthaff.26.3.647. [DOI] [PubMed] [Google Scholar]
- 29.Cohen JT, Neumann PJ. What’s more dangerous, your aspirin or your car? Thinking rationally about drug risks (and benefits) Health Aff (Millwood) 2007;26(3):636–46. doi: 10.1377/hlthaff.26.3.636. [DOI] [PubMed] [Google Scholar]
- 30.Graham J, Hu J. The risk-benefit balance in the United States: who decides? Health Aff (Millwood) 2007;26(3):625–35. doi: 10.1377/hlthaff.26.3.625. [DOI] [PubMed] [Google Scholar]
- 31.Califf RM. Defining the balance of risk and benefit in the era of genomics and proteomics. Health Aff (Millwood) 2004;23(1):77–87. doi: 10.1377/hlthaff.23.1.77. [DOI] [PubMed] [Google Scholar]
- 32.(IOM) IoM. (Institute of Medicine of the National Academies) The Future of Drug Safety: Promoting and Protecting the Health of the Public. 2006 [Google Scholar]
- 33.Teutsch SM, Bradley LA, Palomaki GE, et al. The Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Initiative: methods of the EGAPP Working Group. Genet Med. 2009;11(1):3–14. doi: 10.1097/GIM.0b013e318184137c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calonge N. New USPSTF guidelines: integrating into clinical practice. US Preventive Services Task Force. Am J Prev Med. 2001;20(3 Suppl):7–9. doi: 10.1016/s0749-3797(01)00264-1. [DOI] [PubMed] [Google Scholar]
- 35.Sawaya GF, Guirguis-Blake J, LeFevre M, Harris R, Petitti D. Update on the methods of the U.S. Preventive Services Task Force: estimating certainty and magnitude of net benefit. Ann Intern Med. 2007;147(12):871–5. doi: 10.7326/0003-4819-147-12-200712180-00007. [DOI] [PubMed] [Google Scholar]
- 36.Nelson HD, Huffman LH, Fu R, Harris EL. Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: systematic evidence review for the U.S. Preventive Services Task Force. Ann Intern Med. 2005;143(5):362–79. doi: 10.7326/0003-4819-143-5-200509060-00012. [DOI] [PubMed] [Google Scholar]
- 37.CDC. Evaluation of Genomic Applications in Practice and Prevention (EGAPP): Implementation and Evaluation of a Model Approach. Vol. 2006. Atlanta: 2006. [Google Scholar]
- 38.Evaluation of Genomic Applications in Practice and Prevention (EGAPP) 2009 doi: 10.1097/GIM.0b013e31818fa2ff. Working Group: Evidence Reports. http://www.egappreviews.org/workingrp/reports.htm. [DOI] [PMC free article] [PubMed]
- 39.Myers ER, Havrilesky LJ, Kulasingam SL, et al. Genomic tests for ovarian cancer detection and management. Evid Rep Technol Assess (Full Rep) 2006;145:1–100. [PMC free article] [PubMed] [Google Scholar]
- 40.Matchar D, Thakur M, Grossman I, et al. Testing for Cytochrome P450 Polymorphisms in Adults With Non-Psychotic Depression Treated With Selective Serotonin Reuptake Inhibitors (SSRIs) Vol. Evidence Report/Technology Assessment No. 146. (Prepared by the Duke Evidence-based Practice Center under Contract No. 290-02-0025.): Agency for Healthcare Research and Quality. 2007 [PMC free article] [PubMed] [Google Scholar]
- 41.Bonis P, Trikalinos T, Chung M, et al. Hereditary Nonpolyposis Colorectal Cancer: Diagnostic Strategies and Their Implications. Evidence Report/Technology Assessment No. 150 (Prepared by Tufts-New England Medical Center Evidence-based Practice Center under Contract No. 290-02-0022)(AHRQ Publication No. 07-E008. 2007 [PMC free article] [PubMed] [Google Scholar]
- 42.Technology Evaluation Center (TEC) BlueCross BlueShield Association. 2008 [Google Scholar]
- 43.Meckley LM, Wittkowsky AK, Rieder MJ, Rettie AE, Veenstra DL. A policy model to evaluate the benefits, risks and costs of warfarin pharmacogenomic testing. Pharmacoeconomics. 2010;28:61–74. doi: 10.2165/11318240-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 44.Veenstra DL, Harris J, Gibson RL, Rosenfeld M, Burke W, Watts C. Pharmacogenomic testing to prevent aminoglycoside-induced hearing loss in cystic fibrosis patients: potential impact on clinical, patient, and economic outcomes. Genetics in Medicine. 2007;9(10):695–704. doi: 10.1097/gim.0b013e318156dd07. [DOI] [PubMed] [Google Scholar]
- 45.Garrison LP, Jr, Carlson RJ, Carlson JJ, Kuszler PC, Meckley LM, Veenstra DL. A review of public policy issues in promoting the development and commercialization of pharmacogenomic applications: challenges and implications. Drug Metab Rev. 2008;40(2):377–401. doi: 10.1080/03602530801952500. [DOI] [PubMed] [Google Scholar]
- 46.Weinstein M, Fineberg H. Clinical Decision Analysis. Philadelphia: Saunders; 1980. [Google Scholar]
- 47.University of Washington Center for Genomics and Public Health. Risk-Benefit Framework for Genetic Tests. 2010 http://depts.washington.edu/cgph/riskbenefit.htm.
- 48.Garcia DA, Witt DM, Hylek E, Wittkowsky AK, Nutescu EA, Jacobson A, Moll S, Merli GJ, Crowther M, Earl L, Becker RC, Oertel L, Jaffer A, Ansell JE Anticoagulation Forum. Delivery of optimized anticoagulant therapy: consensus statement from the anticoagulation forum. Ann Pharmacother. 2008;42(7):979–988. doi: 10.1345/aph.1L098. [DOI] [PubMed] [Google Scholar]
- 49.Anderson JL, Horne BD, Stevens SM, Grove AS, Barton S, Nicholas ZP, Kahn SF, May HT, Samuelson KM, Muhlestein JB, Carlquist JF Couma-Gen Investigators. Randomized Trial of Genotype-Guided Versus Standard Warfarin Dosing in Patients Initiating Oral Anticoagulation. Circulation. 2007;116(22):2563–2570. doi: 10.1161/CIRCULATIONAHA.107.737312. [DOI] [PubMed] [Google Scholar]
- 50.Van Walraven C, Oake N, Wells PS, Forster AJ. Burden of Potentially Avoidable Anticoagulant-Associated Hemorrhagic and Thromboembolic Events in the Elderly. Chest. 2007;131(5):1508–1515. doi: 10.1378/chest.06-2628. [DOI] [PubMed] [Google Scholar]
- 51.Doubilet P, Begg CB, Weinstein MC, Braun P, McNeil BJ. Probabilistic sensitivity analysis using Monte Carlo simulation. A practical approach. Med Decis Making. 1985;5:157–77. doi: 10.1177/0272989X8500500205. [DOI] [PubMed] [Google Scholar]
- 52.Briggs AH. Handling uncertainty in cost-effectiveness models. Pharmacoeconomics. 2000;17:479–500. doi: 10.2165/00019053-200017050-00006. [DOI] [PubMed] [Google Scholar]
- 53.Weinstein MC, O’Brien B, Hornberger J, Jackson J, Johannesson M, McCabe C, Luce BR ISPOR Task Force on Good Research Practices--Modeling Studies. Principles of good practice for decision analytic modeling in healthcare evaluation: report of the ISPOR task force on good research practices-modeling studies. Value in Health. 2003;6(1):9–17. doi: 10.1046/j.1524-4733.2003.00234.x. [DOI] [PubMed] [Google Scholar]
- 54.Gold ME, Russell LB, Siegel JE, Weinstein ME, editors. Cost-effectiveness in Health and Medicine. New York: Oxford University Press; 1996. [Google Scholar]
- 55.NICE. (National Institute for Health and Clinical Excellence) Guide to the Methods of Technology Appraisal (Reference N0515) 2004 [PubMed] [Google Scholar]
- 56.(IOM) IoM. (Institute of Medicine of the National Academies) IOM Committee to Evaluate Measures of Health Benefits for Environmental Health, and Safety Regulation in the United States. 2006 [Google Scholar]
- 57.Grosse SD, Khoury MJ. What is the clinical utility of genetic testing? Genet Med. 2006;8(7):448–50. doi: 10.1097/01.gim.0000227935.26763.c6. [DOI] [PubMed] [Google Scholar]
- 58.Briggs AH. New methods for analyzing cost-effectiveness. BMJ. 2007;335:622–623. doi: 10.1136/bmj.39332.587581.BE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eckman MH, Rosand J, Greenberg SM, Gage BF. Cost-effectiveness of using pharmacogenetic information in warfarin dosing for patients with non-valvular atrial fibrillation. Annals of Internal Medicine. 2009;150(2):73–83. doi: 10.7326/0003-4819-150-2-200901200-00005. [DOI] [PubMed] [Google Scholar]
- 60.Patrick AR, Avorn J, Choudhry NK. Cost-Effectiveness of Genotype-Guided Warfarin Dosing for Patients with Atrial Fibrillation. Circulation: Cardiovascular Quality and Outcomes. 2009;2:429–436. doi: 10.1161/CIRCOUTCOMES.108.808592. [DOI] [PubMed] [Google Scholar]
- 61.Grosse SD, Wordsworth S, Payne K. Economic methods for valuing the outcomes of genetic testing: beyond cost-effectiveness analysis. Genet Med. 2008;10(9):648–54. doi: 10.1097/gim.0b013e3181837217. [DOI] [PubMed] [Google Scholar]
- 62.Tunis SR, Pearson SD. Coverage options for promising technologies: Medicare’s coverage with evidence development’. Health Aff (Millwood) 2006;25(5):1218–30. doi: 10.1377/hlthaff.25.5.1218. [DOI] [PubMed] [Google Scholar]
- 63.CMS. Proposed Decision Memo for Pharmacogenomic Testing for Warfarin Response (CAG-00400N) Washington DC: 2009. [Google Scholar]
- 64.Jensen SK, Jacques LB, Ciccanti M, Long K, Eggleston L, Roche J. Decision Memo for Pharmacogenomic Testing for Warfarin Response (CAG-00400N) Centers for Medicare and Medicaid Services. 2009 August 3; Memo. [PubMed] [Google Scholar]
- 65.Fryback DG, Thornbury JR. The efficacy of diagnostic imaging. Medical Decision Making. 1991;11(2):88–94. doi: 10.1177/0272989X9101100203. [DOI] [PubMed] [Google Scholar]
- 66.Burke W, Zimmern RL, Kroese M. Defining purpose: a key step in genetic test evaluation. Genet Med. 2007;9(10):675–81. doi: 10.1097/gim.0b013e318156e45b. [DOI] [PubMed] [Google Scholar]
- 67.Burke W, Pinsky LE, Press NA. Categorizing genetic tests to identify their ethical, legal, and social implications. Am J Med Genet. 2001;106(3):233–40. doi: 10.1002/ajmg.10011. [DOI] [PubMed] [Google Scholar]
- 68.Johnson FR, Ozdemir S, Mansfield C, Hass S, Miller DW, Siegel CA, Sands BE. Crohn’s disease patients’ risk-benefit preferences: Serious adverse event risks versus treatment efficacy. Gastroenterology. 2007 Sep;133(3):769–79. doi: 10.1053/j.gastro.2007.04.075. [DOI] [PubMed] [Google Scholar]
- 69.Burke W, Atkins D, Gwinn M, et al. Genetic test evaluation: information needs of clinicians, policy makers, and the public. Am J Epidemiol. 2002;156(4):311–8. doi: 10.1093/aje/kwf055. [DOI] [PubMed] [Google Scholar]
- 70.Burke W, Psaty BM. Personalized medicine in the era of genomics. Jama. 2007;298(14):1682–4. doi: 10.1001/jama.298.14.1682. [DOI] [PubMed] [Google Scholar]
- 71.Clayton EW. Ethical, legal, and social implications of genomic medicine. N Engl J Med. 2003;349(6):562–9. doi: 10.1056/NEJMra012577. [DOI] [PubMed] [Google Scholar]
- 72.Hughes DA, Bayoumi AM, Pirmohamed M. Current assessment of risk-benefit by regulators: is it time to introduce decision analyses? Clin Pharmacol Ther. 2007;82(2):123–7. doi: 10.1038/sj.clpt.6100240. [DOI] [PubMed] [Google Scholar]
- 73.Temple R. Quantitative decision analysis: a work in progress. Clin Pharmacol Ther. 2007;82(2):127–30. doi: 10.1038/sj.clpt.6100239. [DOI] [PubMed] [Google Scholar]