

Abstract
Some oncolytic viruses, such as myxoma virus (MYXV), can selectively target malignant hematopoietic cells, while sparing normal hematopoietic cells. This capacity for discrimination creates an opportunity to use oncolytic viruses as ex vivo purging agents of autologous hematopoietic cell grafts in patients with hematologic malignancies. However, the mechanisms by which oncolytic viruses select malignant hematopoietic cells are poorly understood. In this study, we investigated how MYXV specifically targets human AML cells. MYXV prevented chloroma formation and bone marrow engraftment of two human AML cell lines, KG-1 and THP-1. The reduction in human leukemia engraftment after ex vivo MYXV treatment was dose-dependent and required a minimum MOI of 3. Both AML cell lines demonstrated MYXV binding to leukemia cell membranes following co-incubation: however, evidence of productive MYXV infection was observed only in THP-1 cells. This observation, that KG-1 can be targeted in vivo even in the absence of in vitro permissive viral infection, contrasts with the current understanding of oncolytic virotherapy, which assumes that virus infection and productive replication is a requirement. Preventing MYXV binding to AML cells with heparin abrogated the purging capacity of MYXV, indicating that binding of infectious virus particles is a necessary step for effective viral oncolysis. Our results challenge the current dogma of oncolytic virotherapy and show that in vitro permissiveness to an oncolytic virus is not necessarily an accurate predictor of oncolytic potency in vivo.
Keywords: leukemia, oncolytic virotherapy, bone marrow, hematopoietic stem cell, animal models
Introduction
Oncolytic viruses are defined as viruses that selectively kill cancer cells.[1] A multitude of oncolytic virus candidates have been identified including: adenovirus, Herpes simplex virus, reovirus, measles virus, Newcastle disease virus and the poxviruses vaccinia and myxoma virus (MYXV) [2]. Significant progress has been made in developing many of these viruses as antineoplastic agents and several are currently in clinical trials [3, 4], including a genetically modified adenovirus, H101, which was recently approved by China’s State Food and Drug Administration for the treatment of head and neck cancer.[5]
Oncolytic viruses have also shown potential in the treatment of hematologic malignancies [6]. Evidence that viruses can selectively cull malignant hematopoietic cells yet spare normal hematopoietic stem and progenitor cells (HSPCs), has led to the proposition of using oncolytic viruses as purging agents for autologous hematopoietic cell transplant (HCT) grafts [2, 7].
We recently demonstrated the ability of MYXV to selectively target primary human acute myeloid leukemia (AML) cells while sparing normal HSPC [8]. However, the mechanisms by which MYXV prevents the engraftment of leukemia cells remain poorly understood. In this study we examined the fundamental requirements of MYXV to specifically target human AML cells and present unexpected, dogma-challenging results that question the reliance of using in vitro infectivity assays to predict oncolytic potency in vivo.
Materials and Methods
Human Leukemia Cell Lines
THP-1 (TIB-202) and KG-1 (CCL-246) cells were purchased from ATCC (Manassas, VA). Cells were cultured at a cell density below 2×106 cells/mL in RPMI 1640 media supplemented with 15% FCS, and 1x pen-strep.
Myxoma Virus and Viral Infections
All viral infections were carried out by incubating cells with vMyx-GFP, a MYXV construct which expresses eGFP at an intergenic location in the viral genome from a synthetic viral early/late promoter.[9] This construct allows viral replication to be detected based on GFP expression within test cells. Human leukemia cells were exposed to vMyx-GFP at a multiplicity of infection (MOI) of 10 for 3 hours in PBS + 10% FBS in a humidified chamber at 37°C and 5% CO2. Mock treated leukemia cells were incubated in PBS plus 10% FBS containing no virus under the same incubation conditions. Treatment with inactivated virus was performed in the same incubation conditions but with inactivated vMyx-GFP prepared by exposing virus to either UV light for 2 hours (UV inactivated) or incubating virus at 55°C for 2 hours (heat inactivated).
Leukemia Xenografts
For systemic leukemia engraftment studies, NOD/Scid/IL2Rγ−/− (NSG) mice were sublethally irradiated using 175 cGy total body irradiation from a Cs137 source. Within twenty-four hours after irradiation, mice were injected through the tail vein with 10×106 THP-1 or 1×106 KG-1 cells that had been either mock-treated or treated with vMyx-GFP. Prophylactic antibiotics were administered in the drinking water for two weeks after transplantation to prevent opportunistic bacterial infection. Six weeks after transplantation, mice were euthanized and bone marrow was harvested. Human leukemia engraftment into bone marrow was quantified using flow cytometry (BD FACSCaliber) for human CD45+ and HLA-A,B,C+ cells. Mice were scored as engrafted if flow cytometry confirmed populations of cells present in bone marrow that were human CD45+/HLA-A,B,C+ double positive. The number of CD45+/HLA-A,B,C+ cells in each bone marrow sample is presented as percent (%) engraftment.
For leukemia chloroma studies, non-irradiated NSG mice were injected subcutaneously with 10×106 KG-1 cells that had been pre-incubated for 3 hrs with mock-, live-, UV-, or heat-treated vMyx-GFP. Chloroma size was measured in two directions using calipers every 3 days after initial chloroma observation. Chloroma volume was calculated using the established formula: 0.5×W×L2.[10] Mice were sacrificed when their chloroma reached 15 mm in any direction in accordance with an approved University of Florida IACUC protocol.
Leukemia Cell In Vitro Functional Assays
For in vitro studies, leukemia cells were mock- or vMyx-GFP treated as above. For viability studies, 1×105 treated leukemia cells were plated in triplicate into 96-well plates. Twenty-four hours after treatment, cell viability was measured using the MTT assay (Pierce) as per the manufacturers recommended procedure. For cell proliferation studies, 1×104 leukemia cells were mock- or vMyx-GFP treated and plated in triplicate into 6 well dishes. Cell number was quantified every 24 hours by manually counting trypan blue excluding cells using a hemocytometer. For colony formation studies, 1×104 leukemia cells were mock- or vMyx-GFP treated and plated into RPMI media containing 1% soft agar and GM-CSF. After ten days of culture, the number of colonies containing greater than 50 cells was determined using light microscopy. For cell adherence studies, 1×106 leukemia cells were mock- or vMyx-GFP treated and plated into 6 well dishes. Twenty-four hours later, cells in suspension were removed and adherent cells were washed gently three times with PBS. Adherent cells were then released from the plate using trypsin and the number of trypan blue excluding cells analysed using a hemocytometer.
Analysis of Virus Infection of Leukemia Cells
To measure initiation of early viral gene expression, leukemia cells were analysed 24 hours after vMyx-GFP exposure for GFP expression using flow cytometry. To measure completion of the viral replication cycle and production of new infectious progeny virus, leukemia cells were harvested at the indicated time points, pelleted and frozen. After harvesting, infectious virus was released by sequential freeze-thaw and the amount of virus in each sample was determined as previously described.[11] Maturation of cells was accomplished by treating with 1ng/mL PMA for 24 hours prior to virus exposure.
MYXV Binding to Leukemia Cells
To measure the binding of vMyx-GFP virions to the cell surface, leukemia cells were exposed to vMyx-GFP at MOI of 10 for 1 hour at 37°C. Cells were then washed 4x with PBS + 10% FBS. The contents of the resulting pellet (cells) as well as the last wash supernatant (wash) were then acid precipitated using trichloroacetic acid (final concentration 30%). Samples were then resuspended in Laemmli buffer, separated on a 15% acrylamide gel, and transferred to PVDF membrane. The presence of viral protein derived from vMyx-GFP virions was then analysed by standard immunoblot analysis using an anti-MYXV rabbit polyclonal serum derived from rabbits that had recovered from infection with an attenuated MYXV construct deleted for the Serp-1 gene [12].
Statistical Analyses
Statistical differences between different experimental groups were determined by one-way analysis of variance and Student’s t-test. The reported values represent the mean plus or minus the standard error of the mean. A P value of less than 0.05 was considered statistically significant.
Results
Inhibition of KG-1 Chloroma Formation by Treatment with Myxoma Virus
To examine the oncolytic effects of live versus inactivated MYXV, immunocompromised NSG mice were inoculated subcutaneously with human KG-1 leukemia cells pre-treated for 3 hours with live MYXV, heat-inactivated MYXV, UV-inactivated MYXV or mock treatment. Leukemia cells treated with live virus showed significantly delayed chloroma formation and reduced tumor volume when compared to the inactivated virus and mock control cohorts (Figure 1A). Live MYXV treatment of KG-1 leukemia cells, but none of the other cohorts, also resulted in prolonged mouse survival (median survival 33 days vs. 71 days, P < 0.005) in this chloroma model (Figure 1B, C).
Figure 1. Inhibition of KG-1 Chloroma Formation after Treatment with MYXV.

Human KG-1 leukemia cells were mock-treated or treated with live, UV-, or heat-inactivated vMyx-GFP, and then injected subcutaneously in immunocompromised NSG mice. (A) Overall chloroma formation was delayed in the cohort receiving live MYXV-treated KG-1 cells. Moreover, the rate of chloroma formation was slower in individual mice receiving live virus-treated cells. (B) Cumulative survival of animals receiving treated KG-1 leukemia cells. (C) Mean survival was significantly longer in animals receiving live MYXV treated KG-1 leukemia compared to those receiving inactivated virus or mock control treatment.
MYXV Prevents Engraftment of KG-1 Leukemia Cells in Bone Marrow of NSG Mice
Because leukemia rarely presents as chloromas, a systemic engraftment model was next used to evaluate the oncolytic potency of ex vivo MYXV treatment against leukemia cells. To do so, immunocompromised NSG mice were sublethally irradiated and transplanted intravenously with KG-1 or THP-1 cells, as two independent models of human AML. Three hours prior to transplant, leukemia cells were treated with either live MYXV or mock treated. As expected, mock treatment resulted in typical leukemia engraftment in bone marrow (Figure 2A). The ex vivo pre-treatment with MYXV, however, significantly inhibited engraftment of both THP-1 and KG-1 cells in the bone marrow of recipient NSG mice (Figure 2A, B, and C). This inhibition could be observed both in significantly reduced levels of engraftment when the leukemia cells were pre-treated with MYXV (2.4% vs. 10.4%, P < 0.05 for KG-1 cells and 0.01% vs. 1.4%, P < 0.04 for THP-1 cells) (Figure 2B) as well as the proportion of mice demonstrating leukemia engraftment (40% vs. 90%, P < 0.05 for KG1 cells and 0% vs. 60% for THP-1 cells) (Figure 2B and C). Importantly, no mouse from any cohort showed any signs of poxvirus related dermal lesions or mucosal ulcerations after transplant of virus-treated cells, indicating that the systemic infusion of cells treated with live MYXV had no observable adverse effects on normal tissues in immunocompromised NSG mice.
Figure 2. MYXV Inhibits Engraftment of Human Leukemia Cells in NSG mice.
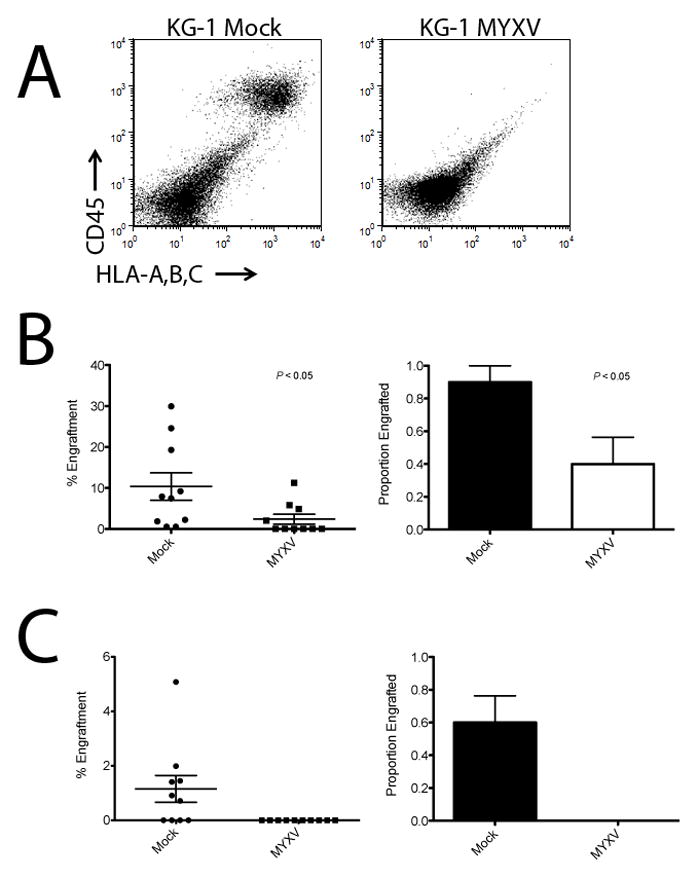
Human KG-1 or THP-1 leukemia cells were treated with live MYXV or mock control, and then transplanted intravenously into immunocompromised NSG mice. (A) Human leukemia engraftment was evaluated six weeks after injection by immunostaining mouse bone marrow for human CD45+ and HLA-ABC+ cells and analyzing by flow cytometry. The presence of double-positive human cells (CD45+/HLA-A,B,C+ in the upper right quadrant) indicated human leukemia engraftment in the mock-treated control. The absence of double-positive cells indicated no engraftment in the MYXV-treated sample. (B) Engraftment of KG-1 leukemia cells was effectively inhibited by treatment with MYXV. Percent KG-1 leukemia cell engraftment and the proportion of mice engrafted were both significantly reduced compared to mock treated control animals (P < 0.05). (C) THP-1 leukemia cells were completely purged by the MYXV treatment (P < 0.05).
Dose Dependent Reduction of Leukemia Engraftment by MYXV
To quantify the effect of MYXV in reducing human AML engraftment, we conducted a limiting dilution assay of MYXV dose. KG-1 cells ex vivo treated with decreasing doses of MYXV (MOI 10, 3, 1, 0.3, 0.1) were transplanted in sublethally irradiated NSG mice and then bone marrow specimens from these mice were evaluated for human AML engraftment six weeks after transplant. Reductions in KG-1 engraftment were clearly evidence in cohorts receiving MYXV MOIs of 10 and 3 (Table 1). However, in animals receiving AML pre-treatment with MYXV MOIs of 1 and 0.3, there was only a trend towards reduced human leukemia engraftment. In animals receiving only MOI 0.1, there was no difference in human AML engraftment.
Table 1.
Human AML (KG1) cells (106) were treated ex vivo with MYXV at the indicated MOIs for 1 hour and then injected IV into sublethally irradiated NSG mice. Six weeks after transplant, bone marrow was harvested from mice and the quantity of human AML cell engraftment compared to mice transplanted with mock treated KG1 cells. A significant decrease in engraftment was observed when mice were transplanted with cells treated with MYXV at either MOI of 10 or 3, while mice transplanted with cells treated with MYXV at either MOI less than 3 showed no statistically significant reduction in human AML engraftment.
| MYXV MOI | P value | Reduction in leukemia engraftment |
|---|---|---|
| 10 | 0.00005 | Significant |
| 3 | 0.04 | Significant |
| 1 | 0.06 | Trend |
| 0.3 | 0.06 | Trend |
| 0.1 | 0.85 | Not Significant |
MYXV Neither Kills Nor Impairs Function of KG-1 Cells In Vitro
Classically, the potency of oncolytic virus candidates for the treatment of target human cancers is first identified using in vitro viral replication and cancer cell killing assays. Thus, to critically examine MYXV as a potential viral oncolytic candidate for different leukemias, we measured the growth of two distinct leukemia cell lines, THP-1 and KG-1, following treatment with MYXV. Since our in vivo results indicated that engraftment of both leukemia cell lines was effectively inhibited by ex vivo MYXV treatment, we anticipated that this treatment would also result in both virus replication and cellular growth defects in vitro. As expected, THP-1 cells treated with MYXV showed significantly reduced cell proliferation compared to mock treatment (P < 0.001 at 7 days, Figure 3A). Surprisingly, KG-1 cells showed no decrease in cell proliferation (P = 0.12 at 7 days, Figure 3A). To further analyse this apparent in vitro discrepancy, viability assays were conducted on both cell lines following viral infection. Similar to the cell proliferation assay, THP-1 cells demonstrated marked reduction of viability after MYXV treatment (64% vs. 100%, P < 0.001) as assessed by MTT assay; whereas, KG-1 cells showed no observable decrease in cell viability in vitro (93% vs. 100%, P = 0.12) (Figure 3B). Furthermore, treatment with MYXV impaired the colony forming potential of THP-1 leukemia cells (47 vs. 67, P = 0.02); however, it did not affect the colony forming potential of KG-1 leukemia cells (42 vs. 42, P = 1.0) (Figure 3C). Finally, leukemia cell adherence to tissue culture plates was assayed as a measure of cell differentiation. THP-1 leukemia cells treated with MYXV showed increased adherence (3.5% vs. 1.0%, P < 0.003); while KG-1 leukemia cells showed no change in adherence after exposure to the live virus (1.0% vs. 0.63%, P = 0.36) (Figure 3D). Thus, by all standard in vitro criteria, cultured THP-1 cells were deemed susceptible to MYXV oncolysis, whereas KG-1 cells were unaffected by MYXV treatment in terms of cell viability, growth characteristics and colony forming ability in vitro.
Figure 3. KG-1 Leukemia Cell Function In Vitro is Not Affected by Treatment with MYXV.
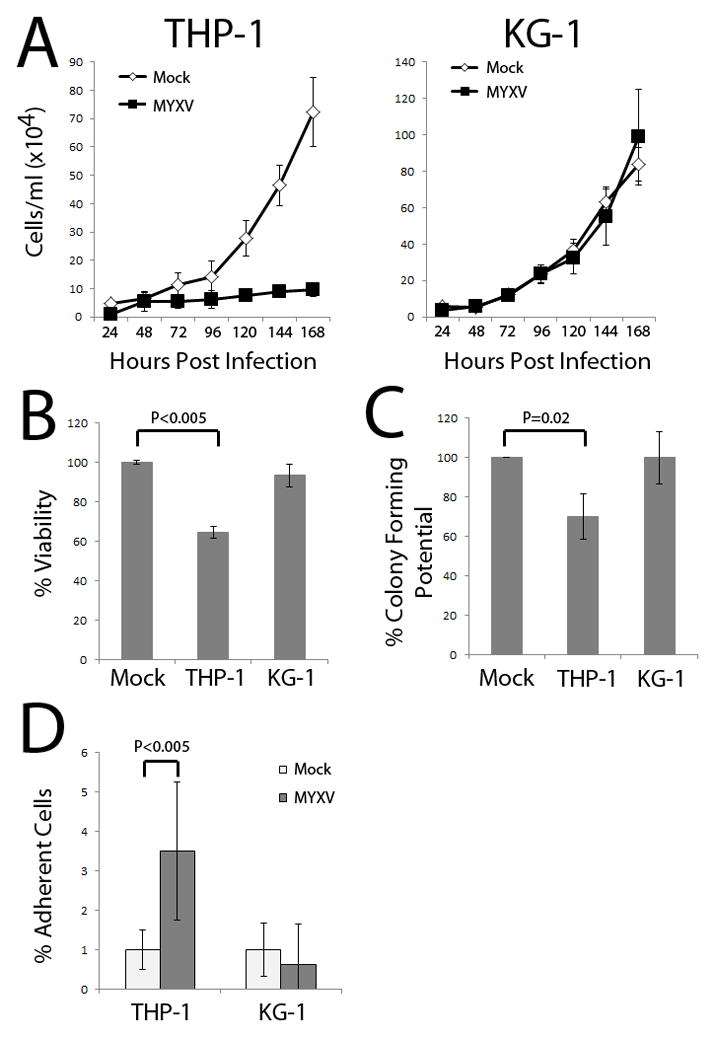
Human KG-1 or THP-1 cells were treated with live MYXV or mock control. (A) MYXV treatment reduced THP-1 leukemia cell proliferation in vitro; however, had no effect on KG-1 cell proliferation. (B) MYXV treatment reduced THP-1 viability as measured by the MTT assay, but did not reduce KG-1 viability. (C) THP-1 leukemia colony forming potential was impaired after MYXV treatment, while KG-1 leukemia colony forming potential was unchanged. (D) The percentage of THP-1 leukemia cells differentiating and adhering to the culture dish was significantly increased after MYXV treatment; whereas, no increase in adherent cells was observed in MYXV-treated KG-1 cells.
KG-1 Leukemia Cells Are Not Permissive for MYXV Infection and Replication
In order to understand the apparent discrepancy between MYXV in vivo purging of KG-1 leukemia cells and the lack of any detectable ability to induce KG-1 oncolysis in vitro, MYXV replication and infection were evaluated in KG-1 and THP-1 cells. Flow cytometry was used to compare the ability of MYXV tagged with eGFP (under a synthetic poxviral early/late promoter) to initiate viral infection within KG-1 and THP-1 leukemia cells. The majority of THP-1 cells expressed eGFP 24 hours after vMyx-GFP exposure (Figure 4A), while the addition of the differentiating agent, phorbol-12-myristate-13-acetate (PMA), enhanced vMyx-GFP infection in THP-1 cells by a relatively small degree. In contrast, KG-1 leukemia cells exposed to vMyxv-GFP showed virtually no evidence of MYXV early or late gene expression as late as 24 hours after virus exposure. Treatment with PMA did not alter this failure to initiate MYXV early gene expression in KG1 cells (Figure 4A).
Figure 4. KG-1 Leukemia Cells are Non-Permissive for MYXV Infection and Replication In Vitro but are Competent for Virus Binding.
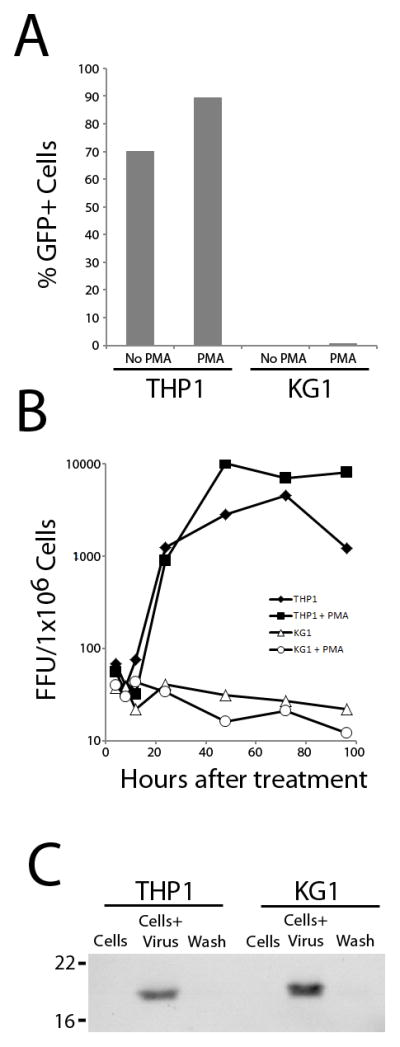
(A) At 24 hours after MYXV treatment, THP-1 or KG-1 leukemia cells were evaluated for evidence of the early stages of MYXV infection (eGFP expression) by flow cytometry. The differentiating agent, PMA, was used in an attempt to augment permissiveness. (B) THP-1 or KG-1 leukemia cells were treated with MYXV and new infectious viral progeny production measured over time. Titers are expressed as log FFU/106 leukemia cells. The differentiating agent, PMA, was used in an attempt to augment viral permissiveness. (C) THP-1 or KG-1 cells were exposed to MYXV for 1 hour, washed extensively and the remaining cell-associated viral protein assessed by Western blotting.
To further determine if THP-1 and KG-1 cells were permissive for productive MYXV replication, we measured production of newly synthesized viral progeny using a single step viral growth curve analysis. MYXV was able to replicate and produce new infectious progeny virus in THP-1 cells indicating a fully permissive phenotype (Figure 4B). In contrast, KG-1 cells treated with MYXV failed to produce any new infectious progeny indicating that these cells were non-permissive for viral replication. The addition of PMA did not alter MYXV progeny levels in either THP-1 (permissive) or KG-1 (nonpermissive) cells. Together, these results indicate that KG-1 cells are completely non-permissive for MYXV replication in vitro and that the block in viral replication occurs prior to early gene expression.
MYXV Binds to the Cell Surface of Both KG-1 and THP-1 Leukemia Cells
Since KG-1 cells demonstrated essentially no viral early gene expression after treatment with MYXV confirmation was sought to verify the physical interaction between MYXV and these leukemia cells. As shown in Figure 4C, MYXV virions readily bound to the surface of both THP-1 and KG-1 leukemia cells. Thus, MYXV binds with comparable efficacy to the cell surface of both KG-1 and THP-1 leukemia cells, but initiates a productive viral infection only in THP-1.
Blocking Virus-Leukemia Cell Binding Abrogates MYXV Oncolysis
Since the in vitro condition that resulted in greatest in vivo purging included virus-bound leukemia cells, we next aimed to test the necessity of this binding for puging leukemia cells. MYXV is thought to bind and enter into target cells (e.g., rabbit cells and human cancer cells) via a non-receptor mediated process. Instead the virus binds to cell surface glycosaminoglycan side chains of cell surface proteoglycans, such as heparan sulfate. Addition of soluble heparin during MYXV infection therefore reduces available binding determinants on the MYXV virion effectively preventing virus-cell binding (Figure 5A).
Figure 5. Virus Binding is Necessary for MYXV Purging of Leukemia Cells.
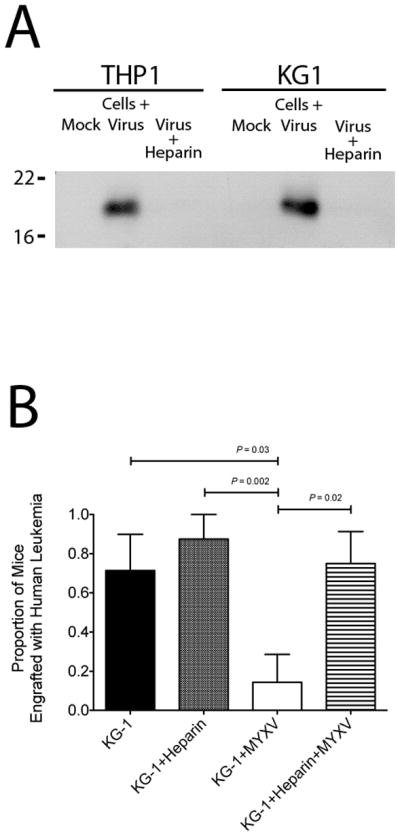
(A) THP-1 or KG-1 cells were exposed to either: mock treatment, MYXV treatment, or MYXV + heparin treatment for 1 hour. The cells were washed extensively and the remaining cell-associated viral protein was assessed by Western blotting. (B) NSG mice were xenotransplanted with KG-1 cells treated ex vivo with KG-1+MYXV (ie virus-bound) or KG-1+Heparin+MYXV (ie virus-unbound) and then evaluated for human leukemia engraftment in the bone marrow after 8 weeks. A significantly decreased proportion of animals receiving MYXV treated KG-1 cells (KG-1+MYXV) showed leukemia engraftment compared to controls. Blocking virus binding with heparin abrogated the oncolytic effect of MYXV and restored KG-1 cell engraftment.
To test the in vivo requirement of MYXV binding to leukemia cells, we transplanted KG-1 cells treated with one of four following conditions: mock treated (control), heparin treated (control), MYXV treated (representing virus bound cells), and MYXV+heparin treated (blocked virus binding). The treated leukemia cells were transplanted into immunocompromised NSG mice and leukemia engraftment was assessed 6 weeks later. As expected, MYXV treatment of the transplanted leukemia graft greatly reduced the proportion of mice engrafted with KG-1 human leukemia cells (14% vs. 71%, P = 0.03) (Figure 5B). However, in the cohort receiving KG-1 cells treated with MYXV and heparin, human leukemia engraftment was equivalent to that seen in control cohorts and significantly increased in comparison to those receiving MYXV treated grafts (75% vs. 14%, P = 0.02) (Figure 5B). These results indicate that blocking MYXV binding to leukemia cells abrogates its oncolytic effects and supports the notion that virion binding, but not subsequent viral replication, is necessary for leukemia cell purging.
Discussion
Several oncolytic viruses are undergoing development in malignant hematology, including coxsackievirus A21 for multiple myeoma [13, 14], reovirus for lymphoma [15, 16], and the Edmonston-B vaccine for multiple myeloma [17, 18]. Previously we demonstrated that ex vivo treatment of transplanted grafts with MYXV prevents primary human AML cells from engrafting in immunocompromised hosts [8]. With mounting interest in harnessing the power of oncolytic viruses, we sought to better understand the interactions between MYXV and target, malignant hematopoietic cells. Based on classical dogma of oncolytic virotherapy, we expected to find that in vivo purging activity was directly associated with in vitro leukemia cell permissiveness to virus infection [3, 19, 20]. For over a decade, demonstrating productive virus replication within the target cancer cell has been a prerequisite for developing any oncolytic virotherapy. However, to our surprise, MYXV efficiently prevented the AML cell line KG-1 from engrafting in immunocompromised hosts despite the failure of these cells to display any evidence of productive viral infection, or any perturbation of cellular growth characteristics in culture. According to the traditional understanding of oncolytic virotherapy, KG-1 leukemia cells were fully “nonpermissive” for MYXV in vitro and should not have succumbed to viral targeting in vivo. In search of a fundamental requirement for purging leukemia cells, we subsequently hypothesized that, at minimum, the virus must physically interact with the target leukemia cell to induce a response incompatible with subsequent engraftment. Indeed, MYXV virion adsorption to KG-1 leukemia cells was readily detectable by direct binding assays, and when we blocked virus binding to cells with heparin, we abrogated its elimination potential. Through these series of experiments, we therefore provide for the first time new insights into oncolytic virotherapy such that binding of MYXV virions, but not direct infection and subsequent viral replication, is necessary and sufficient for leukemia cell targeting.
Results from our study further support the proposed strategy of using MYXV as a purging agent for autologous HCT in patients with hematologic malignancies [2, 7]. Development of MYXV for ex vivo cancer cell purging of autologous grafts in patients with leukemia and multiple myeloma are ongoing [6]. The discovery that virus binding, rather than subsequent virus replication, is necessary informs the translation of oncolytic virotherapy into the clinic by suggesting that, at minimum, binding of infectious virus to patient cancer cells rather than the permissiveness of cancer cells for productive viral replication, may be a more accurate biomarker for treatment response. In the translation of this new purging technology, we anticipate that patients who have malignant hematopoietic cells that bind to live MYXV virions in vitro will benefit from a greater level of purging of contaminating cells in their autologous grafts and achieve improved clinical outcomes (e.g., longer relapse free survival). Our data is also informative for translation to clinical application in that it shows a dose-dependent effect of MYXV in targeting human AML engraftment. In future phase I clinical studies, increasing doses of MYXV may be used to pre-treat autologous hematopoietic cells in patients with haematological malignancies. Our results support a dose escalation trial and suggest that a minimum MOI of 3 will be needed to prevent disease relapse.
Several possibilities could explain why MYXV binding to leukemia cells ex vivo impairs in vivo engraftment. First, virus binding to the leukemia cell surface could evoke an early anti-viral response that triggers intracellular pathways that are incompatible with engraftment into bone marrow. A second possibility to explain how MYXV binding inhibits leukemia engraftment is that binding triggers dysregulation of immunoreactive cell surface antigens, such as HLA or NK receptor expression levels, thus provoking innate immune responses against the transplanted malignant hematopoietic cells. This potential response seems plausible as MYXV generated a greater consequence for both chloroma formation and bone marrow engraftment in vivo than for cell viability in vitro. Prior reports show that class I MHC antigens are downregulated from the surface of highly permissible rabbit cells after MYXV infection [21]. A third possibility is that MYXV is selectively permissive in vitro to a very small population of leukemia stem cells that are exceptionally potent at bone marrow engraftment and leukemia initiation. In this regard, there is known heterogeneity of function within culture-adapted leukemia cell lines such as KG-1 cells. Thus, following this argument, it could be reasoned that whereas the majority of KG-1 leukemia cells are non-permissive of in vitro MYXV infection, a very small subset of leukemia stem cells could be permissive for MYXV infection and rendered incapable of long-term engraftment in xenotransplanted animals. Our discovery of the virus-targetable KG-1 cancer cell line that binds MYXV but is non-permissive for any of the subsequent steps in viral replication highlights opportunities for further investigation of these potential viral oncolytic mechanisms.
In summary, our findings demonstrate that the binding of live infectious MYXV particles, but not the subsequent progression of productive viral replication, is necessary and sufficient to prevent leukemia cells from engrafting in transplanted hosts. Our results challenge the current dogma of oncolytic virotherapy that virus replication in vitro, at least as measured by virus infectivity in cultured cells, is a prerequisite for efficient oncolysis in vivo [22] and suggest that in vitro screening for productive virus replication alone is insufficient for the identification of potentially susceptible cancer cells that could be eliminated from hematopoietic stem cell grafts by virotherapy.
Acknowledgments
We thank Sherin Smallwood for preparing the IACUC protocols, and Dorothy Smith for preparing virus stocks for the study.
Role of the Funding Source
This study was supported by start-up funding to GM from the University of Florida College of Medicine, and NIH grants R01 CA138541 and R21 CA149869 from NCI. CRC, GM and EWS were supported by the Florida Department of Health Bankhead Coley Research Program Team Science grant 1BT02. EB was supported by a grant from STOP! Children’s Cancer, Inc. None of the funding sources were involved in study design, data collection, analysis, interpretation or decision to submit this manuscript for publication.
Footnotes
Authors’ Contributions
GJM, EB, MK, MMR, AM and MCL contributed to acquisition of data, analysis of data, interpretation of data, drafting of manuscript and final approval of the of the version to be submitted. CRC, GM, HVB, and EWS contributed to study design, analysis of data, interpretation of data, drafting of manuscript and final approval of the of the version to be submitted. GJM, EB and MK contributed equally in this study. GM and CRC also contributed equally.
Conflict of Interest
The authors declare no financial conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007;15:651–9. doi: 10.1038/sj.mt.6300108. [DOI] [PubMed] [Google Scholar]
- 2.Rahman MM, Madlambayan GJ, Cogle CR, McFadden G. Oncolytic viral purging of leukemic hematopoietic stem and progenitor cells with Myxoma virus. Cytokine & growth factor reviews. 2010;21:169–75. doi: 10.1016/j.cytogfr.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9:64–71. doi: 10.1038/nrc2545. [DOI] [PubMed] [Google Scholar]
- 4.Hammill AM, Conner J, Cripe TP. Oncolytic virotherapy reaches adolescence. Pediatr Blood Cancer. 2010;55:1253–63. doi: 10.1002/pbc.22724. [DOI] [PubMed] [Google Scholar]
- 5.Yu W, Fang H. Clinical trials with oncolytic adenovirus in China. Curr Cancer Drug Targets. 2007;7:141–8. doi: 10.2174/156800907780058817. [DOI] [PubMed] [Google Scholar]
- 6.Bais S, Rahman M, McFadden G, Cogle C. Oncolytic Virotherapy for Hematologic Malignancies. Advances in Virology. 2012 doi: 10.1155/2012/186512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thirukkumaran CM, Russell JA, Stewart DA, Morris DG. Viral purging of haematological autografts: should we sneeze on the graft? Bone Marrow Transplant. 2007;40:1–12. doi: 10.1038/sj.bmt.1705668. [DOI] [PubMed] [Google Scholar]
- 8.Kim M, Madlambayan GJ, Rahman MM, Smallwood SE, Meacham AM, Hosaka K, et al. Myxoma virus targets primary human leukemic stem and progenitor cells while sparing normal hematopoietic stem and progenitor cells. Leukemia. 2009;23:2313–7. doi: 10.1038/leu.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnston JB, Barrett JW, Chang W, Chung CS, Zeng W, Masters J, et al. Role of the serine-threonine kinase PAK-1 in myxoma virus replication. J Virol. 2003;77:5877–88. doi: 10.1128/JVI.77.10.5877-5888.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989;24:148–54. doi: 10.1007/BF00300234. [DOI] [PubMed] [Google Scholar]
- 11.Smallwood SE, Rahman MM, Smith DW, McFadden G. Myxoma virus: propagation, purification, quantification, and storage. Curr Protoc Microbiol. 2010;Chapter 14(Unit 14A):1. doi: 10.1002/9780471729259.mc14a01s17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Macen JL, Upton C, Nation N, McFadden G. SERP1, a serine proteinase inhibitor encoded by myxoma virus, is a secreted glycoprotein that interferes with inflammation. Virology. 1993;195:348–63. doi: 10.1006/viro.1993.1385. [DOI] [PubMed] [Google Scholar]
- 13.Au GG, Lincz LF, Enno A, Shafren DR. Oncolytic Coxsackievirus A21 as a novel therapy for multiple myeloma. Br J Haematol. 2007;137:133–41. doi: 10.1111/j.1365-2141.2007.06550.x. [DOI] [PubMed] [Google Scholar]
- 14.Shafren DR, Dorahy DJ, Greive SJ, Burns GF, Barry RD. Mouse cells expressing human intercellular adhesion molecule-1 are susceptible to infection by coxsackievirus A21. J Virol. 1997;71:785–9. doi: 10.1128/jvi.71.1.785-789.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thirukkumaran CM, Luider JM, Stewart DA, Cheng T, Lupichuk SM, Nodwell MJ, et al. Reovirus oncolysis as a novel purging strategy for autologous stem cell transplantation. Blood. 2003;102:377–87. doi: 10.1182/blood-2002-08-2508. [DOI] [PubMed] [Google Scholar]
- 16.Alain T, Hirasawa K, Pon KJ, Nishikawa SG, Urbanski SJ, Auer Y, et al. Reovirus therapy of lymphoid malignancies. Blood. 2002;100:4146–53. doi: 10.1182/blood-2002-02-0503. [DOI] [PubMed] [Google Scholar]
- 17.Peng KW, Ahmann GJ, Pham L, Greipp PR, Cattaneo R, Russell SJ. Systemic therapy of myeloma xenografts by an attenuated measles virus. Blood. 2001;98:2002–7. doi: 10.1182/blood.v98.7.2002. [DOI] [PubMed] [Google Scholar]
- 18.Myers RM, Greiner SM, Harvey ME, Griesmann G, Kuffel MJ, Buhrow SA, et al. Preclinical pharmacology and toxicology of intravenous MV-NIS, an oncolytic measles virus administered with or without cyclophosphamide. Clin Pharmacol Ther. 2007;82:700–10. doi: 10.1038/sj.clpt.6100409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parato KA, Senger D, Forsyth PA, Bell JC. Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer. 2005;5:965–76. doi: 10.1038/nrc1750. [DOI] [PubMed] [Google Scholar]
- 20.Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6:821–5. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 21.Boshkov LK, Macen JL, McFadden G. Virus-induced loss of class I MHC antigens from the surface of cells infected with myxoma virus and malignant rabbit fibroma virus. J Immunol. 1992;148:881–7. [PubMed] [Google Scholar]
- 22.Cattaneo R, Miest T, Shashkova EV, Barry MA. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat Rev Microbiol. 2008;6:529–40. doi: 10.1038/nrmicro1927. [DOI] [PMC free article] [PubMed] [Google Scholar]