

Abstract
Peripheral neuropathy develops in human subjects with prediabetes and metabolic syndrome, prior to overt hyperglycemia. The contributions of impaired glucose tolerance and insulin signaling, hypertriglyceridemia and/or increased NEFA, and hypercholesterolemia to this condition remain unknown. Niacin and its derivatives alleviate dyslipidemia with a minor effect on glucose homeostasis. This study evaluated the roles of impaired glucose tolerance versus dyslipidemia in prediabetic neuropathy using Zucker fatty (fa/fa) rats and the niacin derivative acipimox, as well as the interplay of hypertriglyceridemia, increased NEFA, and oxidative-nitrosative stress. 16 wk-old Zucker fatty rats with impaired glucose tolerance, obesity, hyperinsulinemia, hypertriglyceridemia, hypercholesterolemia, and increased NEFA, displayed sensory nerve conduction velocity deficit, thermal and mechanical hypoalgesia, and tactile allodynia. Acipimox (100 mgkg−1d−1, 4 weeks) reduced serum insulin, NEFA, and triglyceride concentrations without affecting glucose tolerance and hypercholesterolemia. It alleviated sensory nerve conduction velocity deficit, changes in behavioral measures of sensory function, and corrected oxidative-nitrosative stress, but not impaired insulin signaling, in peripheral nerve. Elevated NEFA increased total and mitochondrial superoxide production and NAD(P)H oxidase activity in cultured human Schwann cells. In conclusion, hypertriglyceridemia and/or increased NEFA concentrations cause prediabetic neuropathy through oxidative-nitrosative stress. Lipid-lowering agents and antioxidants may find use in management of this condition.
Keywords: Acipimox, dyslipidemia, human Schwann cells, insulin signaling, NAD(P)H oxidase, nitrotyrosine, oxidative-nitrosative stress, prediabetic neuropathy, superoxide, Zucker fatty (fa/fa) rat
Introduction
Evidence for the importance of factors other than overt hyperglycemia, i.e., insulin resistance and impaired insulin signaling in the peripheral nervous system [1–4], hypertension [5], and increased body mass index/obesity [5], in diabetic peripheral neuropathy is emerging. Several studies revealed significant positive associations between the presence and/or severity of diabetic peripheral neuropathy and dyslipidemia, and, in particular, total cholesterol [5], LDL cholesterol [5], and triglycerides [5–7]. A multifactorial etiology of diabetic neuropathy is supported by a higher incidence of neuropathic changes in human subjects with impaired glucose tolerance [8, 9] and metabolic syndrome [10], although the existence of an association between impaired fasting glucose or impaired glucose tolerance and neuropathy is not uniformly accepted [11,12]. The mechanisms of neuropathic changes preceding overt diabetes are unknown, and their exploration is complicated by the lack of animal models that develop prediabetes first and then spontaneously transit to overt diabetes. For this reason, the mechanisms underlying prediabetes per se as well as end-organ damage associated with this condition are studied in high-fat diet fed mice [13–15] and Zucker fatty (fa/fa) rats [16–20] that maintain metabolic abnormalities characteristic for prediabetic condition i.e., hyperinsulinemia, impaired glucose tolerance in the absence of overt hyperglycemia, hypertriglyceridemia and/or increased non-esterified fatty acid abundance, as well as hypercholesterolemia, during their whole life span. Both models exhibit nerve conduction deficit, small sensory nerve fiber dysfunction, and clearly manifested oxidative-nitrosative stress in the peripheral nerve and vasa nervorum [21–25] and are, therefore, suitable for dissection of relative contribution of these phenomena to peripheral neuropathy in prediabetes. Zucker fa/fa rat with genetically predetermined obesity, hyperinsulinemia, and other afore-mentioned metabolic abnormalities is the preferential model for this kind of studies, as many pharmacological interventions, including those alleviating oxidative stress, have been reported to interfere with the prediabetic condition per se in high-fat diet fed rodents [26–28].
Dissecting a relative contribution of impaired glucose tolerance/overt hyperglycemia vs dyslipidemia to complications associated with Type 2 diabetes or prediabetes is quite challenging, as a vast majority of therapeutic agents affects both carbohydrate and lipid metabolism. Niacin reduces triglyceride concentration by inhibiting hepatic synthesis of fatty acids and triglycerides, and hepatic VLDL secretion as well as release of fatty acids from adipose tissue [29, 30]. In a clinical study in 468 participants including 125 with diabetes mellitus [31], niacin improved lipid profile, without affecting blood glucose control, in human subjects with and without diabetes. The niacin derivative acipimox (6-methyl-1-oxidopyrazin-1-ium-2-carboxylic acid) that can be used in lower doses and has less marked adverse side effects was also reported to alleviate dyslipidemia, but not hyperglycemia, when administered to Type 2 diabetic subjects long-term [32]. Furthermore, acute administration of acipimox reduced hyperinsulinemia and non-esterified fatty acid (NEFA) concentrations in subjects with metabolic syndrome [33]. We therefore used acipimox for gaining new insights into the roles of impaired glucose tolerance, insulin sensitivity, and dyslipidemia in the development of prediabetic neuropathy using Zucker fatty (fa/fa) rat model. Identification of a causative role of elevated triglyceride and NEFA, together with the current knowledge on the role for both factors in reactive oxygen species (ROS) generation [34, 35], consequently led us to evaluation of the interplay of hypertriglyceridemia, fatty acidemia, and oxidative-nitrosative stress in the peripheral nerve, using the material from the afore-mentioned in vivo study, as well as in cultured human Schwann cells (HSC).
Methods
A. Reagents
Unless otherwise stated, all chemicals were of reagent-grade quality, and were purchased from Sigma-Aldrich Chemical Co., St. Louis, MO, USA. The Cholesterol Quantitation Kit for assessment of serum total cholesterol concentration was obtained from MBL International, Woburn, MA. The Triglyceride Quantification Kit and HDL and LDL/VLDL Cholesterol Assay Kit for measurements of serum triglyceride and VLDL/LDL-cholesterol concentrations were purchased from Abcam, Cambridge, MA. The HR Series NEFA-HR(2) kit for assessment of serum non-esterified fatty acid (NEFA) concentrations was obtained from Wako Pure Chemical Industries, Ltd., Osaka, Japan. The Ultra Sensitive Rat Insulin ELISA Kit from Crystal Chem Inc., Downers Grove, IL, was used for measurements of serum insulin concentrations. For Western blot analyses of variables of insulin signaling, rabbit monoclonal (4B8) anti-Insulin Receptor β (IRβ) antibody, rabbit monoclonal (59G8) anti-Insulin receptor substrate 1 (IRS-1) antibody, rabbit polyclonal (Ser307) anti-Phospho-Insulin receptor substrate 1 (phospho-IRS-1) antibody, and rabbit polyclonal antibodies for total and phosphorylated Akt were obtained from Cell Signaling Technology, Inc., Danvers, MA, USA. The OxiSelect Nitrotyrosine ELISA kit for nitrotyrosine assay in the sciatic nerve was obtained from Cell Biolabs, San Diego, CA. Dihydroxyethidium and MitoSOX™ for assessment of oxidative stress variables in cell culture experiments were purchased from Invitrogen, Carlsbad, CA.
B. Animals
The experiments were performed in accordance with regulations specified by the National Institutes of Health “Principles of Laboratory Animal Care, 1985 Revised Version” and Pennington Biomedical Research Center Protocol for Animal Studies. 10 wk-old male Zucker fatty (fa/fa) and Zucker lean rats (Charles River, Wilmington, MA) were fed a standard rat chow (PMI Nutrition Int., Brentwood, MO) and had access to water ad libitum. At 16 wks of age, all the rats were weighed. Blood samples for glucose measurements were taken from the tail vein. Zucker fatty and Zucker lean rats were randomly divided into groups maintained with or without acipimox treatment, 100 mg kg−1d−1, for another four weeks. Glucose tolerance test (2 g glucose, i.p., after 12-h fasting), and measurements of serum insulin, NEFA, triglyceride, total cholesterol, VLDL/LDL-cholesterol, motor nerve conduction velocity, sensory nerve conduction velocity, thermal and mechanical algesia, and tactile response thresholds were conducted in 16 wk-old Zucker fatty and Zucker lean rats before acipimox treatment, as well as in 20 wk-old untreated and acipimox-treated Zucker fatty and Zucker lean rats at the end of experiment. After completion of functional studies, the rats were sedated by CO2, and immediately sacrificed by cervical dislocation. Sciatic nerves were rapidly removed, frozen in liquid nitrogen, and stored at −80°C prior to assessment of variables of insulin signaling and oxidative-nitrosative stress.
C. Specific Methods
Measurements of serum insulin, total and VLDL/LDL cholesterol, triglyceride, and NEFA were performed in accordance with the manufacturers' instructions. Sciatic motor and hind-limb digital sensory nerve conduction velocities (MNCV and SNCV), thermal algesia (Hargreaves method), mechanical algesia (Randall-Selitto test), tactile response thresholds (flexible von Frey filament test), and sciatic nerve nitrotyrosine concentrations were evaluated as we described previously [36–39]. Nitrotyrosine, a stable “footprint” of peroxynitrite (ONOO-) action, was chosen over other variables of oxidative stress, for several major reasons. First, evidence for the important role of peroxynitrite in cell injury and pathological conditions associated with oxidative stress is emerging [40, 41]. Second, peroxynitrite is the major contributor to the pathogenesis of diabetes and diabetic complications [40–43]. Third, a shortened lag time to plasma ONOO- production (pholasin test) was identified as an independent risk factor for the severity of diabetic polyneuropathy [6]. Fourth, our recent experimental study demonstrated diabetes-associated nitrotyrosine accumulation in all major cell targets for diabetic peripheral neuropathy, including endothelial and Schwann cells of the peripheral nerve, neurons, astrocytes, and oligodendrocytes of the spinal cord, and neurons and glial cells of the dorsal root ganglia [38]. Furhermore, sciatic nerve nitrotyrosine concentrations inversely correlated with motor and sensory nerve conduction velocities and myelin thickness. Variables of insulin signaling in the sciatic nerve were assessed by Western blot analysis. We employed 7.5% sodium dodecyl sulfate-polyacrylamide gel for IRβ, phospho-IRS-1 and IRS-1, and 10% gel for phospho-Akt and Akt. The electrophoresis was conducted for 2 h. Protein bands were visualized with the Amersham™ ECL™ Western Blotting Detection Reagents (GE Healthcare, Buckinghamshire, UK). Membranes were then stripped and reprobed with β-actin antibody to verify equal protein loading. The data were quantified by densitometry (Quantity One 4.5.0 software, Bio-Rad Laboratories, Richmond, CA). All other details of Western blot analysis were described by us in detail previously.
D. Cell culture experiments
Schwann cells play a key role in the pathology of various inflammatory, metabolic and hereditary polyneuropathies, including diabetic peripheral neuropathy [44]. Previous studies demonstrated that cultured HSC (cell line cat. #1700, ScienCell, Carlsbad, CA) manifest increased superoxide production, accumulation of nitrated and poly(ADP-ribosyl)ated proteins and 4-hydroxynonenal adducts, inducible nitric oxide synthase overexpression, 12/15-lipoxygenase overexpression and activation, downregulation of taurine transporter, as well as impaired insulin signaling early (1–7 d) after exposure to high glucose [45–49]. They therefore represent a good cell culture model for studying mechanisms of diabetes-associated oxidative-nitrosative stress in the peripheral nerve. Our pilot experiments revealed that, like other cell types [50], HSC produce ROS shortly after exposure to elevated concentrations of NEFA, consistent with a recent report for other immortalized Schwann cells [35]. We, therefore, used HSC to compare the effects of NEFA in the concentration range identified in non-diabetic subjects vs those with prediabetes and metabolic syndrome, on total and mitochondrial superoxide production and NAD(P)H oxidase activity. NEFA mixture consisted of 20% linoleic acid, 43% oleic acid, 10% stearic acid, and 27% palmitic acid, and was prepared as described [51]. In addition to NEFA mixture, we also used palmitate recently reported to be the most toxic fatty acid for Schwann cells [35]. HSC (passages 7–10) were cultured in 12-well plates in commercial media containing 5.5 mM D-glucose, at 37° C. At ~80% confluency, the commercial media were replaced with the ones containing 0.2, 0.4, 0.6, and 0.8 mEq NEFA or palmitate, for 48 h. Then the cells were washed twice with Dulbecco's Phosphate-Buffered solution and loaded with dihydroethidium (30 μM in commercial media containing the afore-mentioned NEFA concentrations), for 30 min. Total superoxide fluorescence measurements were performed at λ excitation 520 nm and λ emission 610 nm, using FlexStation 1 scanning fluorometer (Molecular Devices Corporation, Sunnyvale, CA). Then the NAD(P)H oxidase inhibitor apocynin (1 mM) was added for 30 min to the same wells, and superoxide fluorescence measurements were performed again. NAD(P)H oxidase activity was calculated as a difference between total and residual (non-responsive to apocynin) superoxide fluorescence. For assessment of mitochondrial superoxide production, parallel HSC cultures were loaded with MitoSOX™ (1 μM) for 15 min. Fluorescence measurements (λ excitation 510 nm and λ emission 580 nm) were performed using the fluorometer described above. Total and mitochondrial superoxide production and NAD(P)H oxidase activity have been corrected for autofluorescence measured before addition of fluorescent probes. All values were normalized for 10−4 HSC. Cell counts were performed immediately after fluorescence measurements.
E. Statistical analysis
The results are expressed as mean ± standard errors. Individual comparisons between 16 wk-old Zucker fatty and Zucker lean rats and HSC cultured in normal and elevated NEFA or palmitate concentrations were made using the unpaired two-tailed Student's t-test or Mann-Whitney rank sum test where appropriate. Taking into consideration the range of NEFA concentrations in non-diabetic subjects (~0.2–0.4 mEq) as well as in subjects with prediabetes/metabolic syndrome (~0.6/0.8 mEq), we made the comparisons between oxidative stress variables obtained with both lower range (0.2 and 0.6 mEq) and higher range (0.4 and 0.8 mEq) concentrations. Significance was defined at p ≤ 0.05. For multiple group comparisons (20 wk-old untreated and acipimox-treated Zucker fatty and Zucker lean rats), data were subjected to equality of variance F test, and then to log transformation, if necessary, before one-way analysis of variance. When overall significance (p < 0.05) was attained, individual between group comparisons were made using the Student-Newman-Keuls multiple range test. Significance was defined at p ≤ 0.05. When between-group variance differences could not be normalized by log transformation (datasets for final body weights and plasma glucose), the data were analyzed by the nonparametric Kruskal-Wallis one-way analysis of variance, followed by the Bonferroni/Dunn or Fisher's PLSD tests for multiple comparisons.
Results
Body weights and non-fasting blood glucose concentrations were increased by 56% and 41%, respectively, in the 16 wk-old Zucker fatty rats, compared with the age-matched Zucker lean rats (P < 0.01 for both comparisons, Table 1). Similar differences in the two variables were observed between 20 wk-old Zucker fatty and Zucker lean rats. Acipimox treatment did not affect weight gain and non-fasting blood glucose concentrations in either Zucker fatty or Zucker lean rats.
Table 1.
Body weights and non-fasting blood glucose and serum insulin concentrations in Zucker fatty and Zucker lean rats before and after acipimox treatment
| Zucker lean | Zucker lean + A | Zucker fatty | Zucker fatty + A | |
|---|---|---|---|---|
| Before treatment | ||||
| Body weight, g | 430.4±10.9 | 671.8±13.1** | ||
| Blood glucose, mmol/L | 6.2±0.6 | 8.7±1.0** | ||
| Insulin, ng/mL | 0.51±0.07 | 3.7±0.48** | ||
| After treatment | ||||
| Body weight, g | 462.8±10.8 | 467.6±10.9 | 719.9±27.9** | 718.7±16.6** |
| Blood glucose, mmol/L | 6.2±0.1 | 6.5±0.1 | 8.8±1.1** | 9.0±1.2** |
| Insulin, ng/mL | 0.34±0.02 | 0.49±0.06 | 1.81±0.40** | 0.94±0.12*,## |
Mean ± SEM, n = 7–10. A – acipimox.
p < 0.05 and < 0.01 vs age-matched Zucker lean groups;
p < 0.05 and < 0.01 vs age-matched Zucker lean groups;
p < 0.01 vs Zucker fatty rats treated with acipimox
Despite a severe hyperinsulinemia, with serum insulin concentrations 7.3 and 5.3 times greater than in the corresponding Zucker lean groups (P < 0.01 for both comparisons, Table 1), both 16 wk-old and 20 wk-old Zucker fatty rats displayed impaired glucose tolerance (Fig.1). Serum insulin concentrations in the 20 wk-old acipimox-treated Zucker fatty rats were 1.9-fold lower than in the corresponding untreated group (P < 0.01), and 2.8-fold higher than in the age-matched Zucker lean group (P < 0.05). Acipimox did reduce serum insulin concentrations in Zucker lean rats. Glucose tolerance curves were not affected by acipimox treatment in either Zucker fatty or Zucker lean rats.
Fig. 1.

Glucose tolerance curves in Zucker fatty and Zucker lean rats before and after acipimox treatment. Mean ± SEM, n = 8–10 per group. ** p < 0.01 vs age-matched Zucker lean rats. 1- Zucker lean rats; 2 – Zucker lean rats treated with acipimox; 3 – Zucker fatty rats; 4 – Zucker fatty rats treated with acipimox.
Zucker fatty rats displayed clearly manifest dyslipidemia (Table 2). Serum total cholesterol concentrations were 2.9–fold and 3.6–fold greater in 16 wk-old and 20 wk-old Zucker fatty rats compared with the age-matched Zucker lean groups (P < 0.01 for both comparisons). The significant differences between Zucker fatty and Zucker lean groups were also observed for VLDL/LDL cholesterol (2.6-fold and 2.6-fold, P < 0.01 and < 0.05, respectively), triglyceride (4.4-fold and 5.0-fold, P < 0.01 for both comparisons), and NEFA (5-fold and 9.2-fold, P < 0.01 for both comparisons). Triglyceride and NEFA concentrations were 1.6-fold and 1.7-fold lower in acipimox-treated Zucker fatty rats than in the corresponding untreated group (P < 0.01). Total and VLDL/LDL-cholesterol concentrations were not significantly different between untreated and acipimox-treated Zucker fatty rats. Acipimox did not affect lipid profile in Zucker lean rats.
Table 2.
Lipid profiles in Zucker fatty and Zucker lean rats before and after acipimox treatment
| Zucker lean | Zucker lean + A | Zucker fatty | Zucker fatty + A | |
|---|---|---|---|---|
| Before treatment | ||||
| Total cholesterol, mg/dL | 147.0±43.5 | 424.4±33.3** | ||
| VLDL/LDL cholesterol, mg/dL | 27.2±1.3 | 70.0±11.3** | ||
| Triglycerides, mmol/L | 2.1±0.1 | 9.3±0.7** | ||
| NEFA, mEq/L | 0.24 ±0.03 | 1.19±0.21** | ||
| After treatment | ||||
| Total cholesterol, mg/dL | 161.8±19.8 | 165.2±17.3 | 589.5±44.8** | 532.6±39.9** |
| VLDL/LDL cholesterol, mg/dL | 24.2±1.3 | 27.7±4.6 | 62.7±25.9* | 63.6±20.6* |
| Triglycerides, mmol/L | 2.1±0.2 | 2.0±0.2 | 10.6±2.3** | 6.6±0.6**,## |
| NEFA, mEq/L | 0.15±0.03 | 0.15±0.03 | 1.40±0.25** | 0.81±0.19**,## |
Mean ± SEM, n = 7–10. A – acipimox.
p < 0.05 and < 0.01 vs age-matched Zucker lean groups;
p < 0.05 and < 0.01 vs age-matched Zucker lean groups;
p < 0.01 vs Zucker fatty rats treated with acipimox
MNCVs were similar in 16 wk-old and 20 wk-old Zucker fatty and Zucker lean rats, and were not affected by acipimox treatment in either group (Table 3). SNCVs were reduced by 12% and 15% in 16 wk-old and 20 wk-old Zucker fatty rats, compared with the age-matched Zucker lean rats (P < 0.01 for both comparisons). Acipimox treatment reversed SNCV deficit in Zucker fatty rats (P < 0.01 compared with the corresponding untreated group), without affecting SNCV in Zucker lean rats.
Table 3.
Indices of peripheral nerve function in Zucker fatty and Zucker lean rats before and after acipimox treatment
| Zucker lean | Zucker lean + A | Zucker fatty | Zucker fatty + A | |
|---|---|---|---|---|
| Before treatment | ||||
| MNCV, m/s | 53.9±0.8 | 53.6±1.0 | ||
| SNCV, m/s | 41.5±0.4 | 36.7±0.9** | ||
| Thermal response latency, s | 14.6±0.9 | 21.2±1.4** | ||
| Tactile response thresholds, g | 16.0±1.9 | 8.4±0.8** | ||
| Mechanical withdrawal thresholds, g | 11.6±0.6 | 17.8±1.0** | ||
| After treatment | ||||
| MNCV, m/s | 54.9±0.7 | 53.4±0.5 | 54.1±0.7 | 54.9±1.3 |
| SNCV, m/s | 45.1±1.1 | 44.1±0.8 | 38.4±0.9** | 44.1±0.8## |
| Thermal response latency, s | 10.7±0.2 | 11.3±0.4 | 22.8±1.0** | 16.5±0.5**,## |
| Tactile response thresholds, g | 18.7±0.8 | 20.4±1.8 | 7.1±0.5** | 8.9±0.3** |
| Mechanical withdrawal thresholds, g | 10.5±0.6 | 11.2±0.5 | 16.3±0.7** | 12.6±0.4**,** |
Mean ± SEM, n = 7–9. A – acipimox.
p < 0.01 vs age-matched Zucker lean groups;
p < 0.01 vs Zucker fatty rats treated with acipimox
Thermal response latencies were increased by 45% and 113% in 16 wk-old and 20 wk-old Zucker fatty rats, compared with the age-matched Zucker lean rats (P < 0.01 for both comparisons), consistent with the presence of thermal hypoalgesia. This condition was alleviated, although not completely corrected, by acipimox treatment. Thermal response latencies in acipimox-treated Zucker fatty rats were 28% lower than in the corresponding untreated group, and 54% higher than in the age-matched Zucker lean rats (P < 0.01 for both comparisons). Acipimox treatment had no effect on thermal algesia in Zucker lean rats.
In addition to thermal hypoalgesia, Zucker fatty rats displayed reduced sensitivity to mechanical noxious stimuli. Mechanical withdrawal thresholds were 53% and 55% higher in 16 wk-old and 20 wk-old Zucker fatty rats, compared with the age-matched Zucker lean rats (P < 0.01 for both comparisons). Acipimox treatment alleviated mechanical hypoalgesia in Zucker fatty rats, without affecting mechanical withdrawal thresholds in Zucker lean rats.
Tactile response thresholds were reduced by 47% and 62% in 16 wk-old and 20 wk-old Zucker fatty rats, compared with the age-matched Zucker lean rats (P < 0.01 for both comparisons). This is consistent with the presence of tactile allodynia i.e., a condition when light touch is perceived as painful. Acipimox treatment did not affect tactile response thresholds in either Zucker fatty or Zucker lean rats.
Sciatic nerve phospho-Akt/Akt ratio was reduced in 20 wk-old Zucker fatty rats compared with the age-matched Zucker lean rats (P < 0.05, Fig.2, A), indicative of impaired insulin signaling. IRβ expression (Fig.2, B) and phospho-IRS/IRS ratios (Fig.2, C) were similar between the two groups. Acipimox treatment did not affect any variables of insulin signaling in either Zucker fatty or Zucker lean rats.
Fig. 2.

Variables of insulin signaling in the sciatic nerve in Zucker fatty and Zucker lean rats maintained with or without acipimox treatment. Mean ± SEM, n = 6–8 per group. * p < 0.05 vs age-matched Zucker lean rats. 1- Zucker lean rats; 2 – Zucker lean rats treated with acipimox; 3 – Zucker fatty rats; 4 – Zucker fatty rats treated with acipimox. IRβ– insulin receptor β. IRS – insulin receptor substrate.
Sciatic nerve nitrotyrosine concentration was 88% higher in 20 wk-old Zucker fatty rats, than in the age-matched Zucker lean rats (P < 0.05, Fig.3). Acipimox treatment blunted oxidative-nitrosative stress in Zucker fatty rats, without affecting sciatic nerve nitrotyrosine concentration in Zucker lean rats.
Fig. 3.
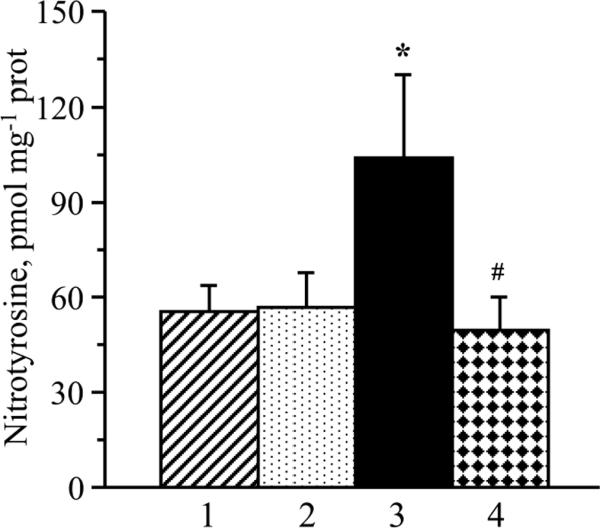
Sciatic nerve nitrotyrosine concentrations in Zucker fatty and Zucker lean rats maintained with or without acipimox treatment. Mean ± SEM, n = 7–9 per group. * p < 0.05 vs age-matched Zucker lean rats. # p < 0.05 vs untreated Zucker fatty rats. 1- Zucker lean rats; 2 – Zucker lean rats treated with acipimox; 3 – Zucker fatty rats; 4 – Zucker fatty rats treated with acipimox.
Despite certain challenges and limitations of ROS measurements with fluorescent probes [52], our data clearly show that elevated NEFA and palmitate concentrations enhanced oxidative stress in HSC. In particular, both elevated NEFA and palmitate concentrations increased total superoxide production in cultured HSC (Fig.4). The elevation in NEFA concentration from 0.4 to 0.8 mEq (higher range in non-diabetic subjects and those with metabolic syndrome, respectively) increased total superoxide production by 25% (P < 0.05). The elevation in palmitate concentration from 0.2 to 0.6 mEq (lower range in non-diabetic subjects and those with metabolic syndrome, respectively) and from 0.4 to 0.8 mEq increased total superoxide production by 64% and by 45% (P < 0.01 for both comparisons). Elevated NEFA, but not palmitate, concentrations increased mitochondrial superoxide production in cultured HSC (Fig.5). The elevation in NEFA concentration from 0.4 to 0.8 mEq increased mitochondrial superoxide production by 34% (P < 0.05). No significant increases in mitochondrial superoxide production was observed with elevations in palmitate concentration from 0.2 to 0.6 mEq and from 0.4 to 0.8 mEq. Both elevated NEFA and palmitate concentrations increased NAD(P)H oxidase activity in cultured HSC (Fig.6). The elevation in NEFA concentration from 0.4 to 0.8 mEq increased NAD(P)H oxidase activity by 140 % (P < 0.05). The elevation in palmitate concentration from 0.2 to 0.6 mEq and from 0.4 to 0.8 mEq increased NAD(P)H oxidase activity by 193% and by 160% (P < 0.01 for both comparisons).
Fig.4.

Total superoxide production in human Schwann cells cultured in increasing concentrations of non-esterified fatty acids (left) and palmitate (right). RFU – relative fluorescence units. Mean ± SEM, n = 9–10 per group. *** p < 0.05 and < 0.01 compared with cells cultured in normal non-esterified fatty acid or palmitate concentrations.
Fig.5.

Mitochondrial superoxide production in human Schwann cells cultured in increasing concentrations of non-esterified fatty acids (left) and palmitate (right). RFU – relative fluorescence units. Mean ± SEM, n = 10 per group. * p < 0.05 compared with cells cultured in normal non-esterified fatty acid concentrations.
Fig.6.

NAD(P)H oxidase activity in human Schwann cells cultured in increasing concentrations of non-esterified fatty acids (left) and palmitate (right). RFU – relative fluorescence units. Mean ± SEM, n = 10 per group. *,** p < 0.05 and < 0.01 compared with cells cultured in normal non-sterified fatty acid or palmitate concentrations.
Discussion
In the present study in Zucker fatty rats, the niacin derivative acipimox, which reduced serum insulin, NEFA, and triglyceride concentrations without affecting impaired glucose tolerance and total and VLDL-LDL cholesterol concentrations, corrected peripheral nerve oxidative-nitrosative stress, SNCV deficit, and alleviated changes in behavioral measures of sensory function associated with a metabolic condition mimicking prediabetes and obesity in humans. These findings have a number of important implications for understanding the contribution of hypertriglyceridemia and/or fatty acidemia and resultant oxidative-nitrosative stress versus other phenomena, such as glucose intolerance, hypercholesterolemia, and impaired insulin signaling in the peripheral nerve, to prediabetic neuropathy.
Our results provide the first experimental evidence for a key role of hypertriglyceridemia and/or increased fatty acid concentrations in peripheral nerve dysfunction associated with prediabetes. The fact that alleviation of hypertriglyceridemia and free fatty acidemia, in the absence of improvement of glucose tolerance and decrease in total and VLDL/LDL cholesterol concentrations, led to a complete normalization of SNCV in acipimox-treated Zucker fatty rats, indicates that prediabetes-associated sensory nerve conduction slowing develops solely due to the detrimental effects of elevated triglyceride/NEFA, whereas glucose intolerance and hypercholesterolemia are not involved. Note, however, that the contribution of the last two factors to prediabetic small sensory nerve fiber neuropathy can not be excluded on the basis of the current study. Neither hypertriglyceridemia/free fatty acidemia, nor manifestations of sensory neuropathy such as thermal or mechanical hypoalgesia were completely reversed by acipimox treatment. It, therefore, remains to be established whether elevated triglyceride/NEFA concentrations is the only factor causing both sensory disorders, or other metabolic disturbances associated with prediabetes also play a role. This can be achieved in studies with more potent, than acipimox, and more specific hypolipidemic agents e.g., recently developed dual inhibitors of fatty acid binding proteins 4 and 5 which reduce plasma triglyceride/fatty acids, but have no effect on insulin sensitivity [53].
Our study in high-fat diet fed mice, another model of a metabolic condition mimicking prediabetes and alimentary obesity in humans, suggests that many biochemical changes characteristic for diabetic peripheral neuropathy develop prior to overt hyperglycemia [22]. Those include augmented sorbitol pathway activity in the peripheral nerve, as well as 4-hydroxynonenal adduct, nitrotyrosine, and poly(ADP-ribose) accumulation and 12/15-lipoxygenase overexpression in the peripheral nerve and dorsal root ganglion neurons [22]. The presence of enhanced oxidative-nitrosative stress, manifest by increased hydroxyoctadecadienoic acid, dityrosine, and nitrotyrosine levels, in the peripheral nerve of high-fat diet fed mice has also been reported by others [23]. Zucker fatty rats display increased production of superoxide and peroxynitrite in vasa nervorum [24], consistent with nitrotyrosine accumulation in the peripheral nerve in the current study. Numerous publications of both our group [55–58] and others [59–67] suggest that ROS and peroxynitrite play a key role in nerve conduction slowing, neurovascular dysfunction, changes in behavioral measures of sensory function, and morphological manifestations of diabetic peripheral neuropathy. Furthermore, oxidative-nitrosative stress leads to formation of advanced glycation end products and proinflammatory response, two other major contributors to neuropathic changes in diabetes [68–72] and, potentially, prediabetes [73]. Interestingly and surprisingly, enhanced peripheral nerve oxidative-nitrosative stress was associated with SNCV, but not MNCV, deficit in 20 wk-old Zucker fa/fa rats. Note, however, that whereas numerous studies in animal models of diabetes and prediabetes revealed inverse correlations between peripheral nerve oxidative stress and MNCV [54–58, 60–63], at least, two groups [47,74,75] demonstrated that prevention of diabetes-induced oxidative stress in peripheral nerve does not necessarily arrest motor nerve conduction slowing and, alternatively, is not essentially required for preservation of normal MNCV. It is possible that development of MNCV deficit requires certain level of oxidative stress in vasa nervorum. Zucker fa/fa rats manifested increased superoxide production in sciatic nerve epineurial arterioles starting from 28 wks of age, and MNCV deficit starting from 32 wks of age [21]. In the same study, 20 wk-old Zucker fa/fa rats did not develop MNCV deficit, which is consistent with our findings. A different susceptibility of large motor and sensory fiber axons to atrophy was identified in streptozotocin-diabetic Swiss-Webster mice [76] which exhibited a greater dropout of large caliber axons in sural (sensory) than in sciatic (motor) nerve.
Evidence for the contribution of factors, other than elevated glucose concentrations, to oxidative-nitrosative stress in tissue-sites for diabetic complications is emerging. Oxidized low-density lipoproteins have been shown to cause oxidative stress in cultured dorsal root ganglion neurons by mechanisms independent of hyperglycemia [23]. The present study suggests that prediabetes-associated oxidative-nitrosative stress is a likely mediator of the negative effects of hypertriglyceridemia and increased NEFA concentrations on peripheral nerve function. Fatty-acid-induced ROS overproduction caused endothelial dysfunction, known to play an important role in diabetic peripheral neuropathy [56,59,61–63,71,75], in Zucker diabetic fatty rats [77]. In our previous study [50], increased palmitate concentrations induced oxidative-nitrosative injury in cultured microvascular cells. In the present experiments, increased superoxide production was manifest after exposure of cultured HSC to either NEFA mixture with the composition mimicking the one in the human blood or to equimolar concentrations of palmitate alone. Thus NEFA, in the concentration range found in subjects with prediabetes/metabolic syndrome, increase both mitochondrial and extramitochondrial [NAD(P)H oxidase-mediated] superoxide production in cultured HSC. Therefore, despite limitations of in vitro studies in cultured cells, our findings raise the possibility that increased NEFA and NEFA-generated mitochondrial and extramitochondrial oxidative-nitrosative stress play a role in neuropathic changes developing in subjects with prediabetes/metabolic syndrome. NEFA can penetrate through the blood-nerve barrier after binding with glycated albumin [78], as well as during ischemia-reperfusion injury [79]. The latter has been shown to lead to enhanced oxidative stress and apoptosis in Schwann cells of diabetic rats [65].
In previous studies, several B vitamins i.e., thiamine (B1), pyridoxine (B6), and methylcobalamin (B12), ameliorated diabetes-induced peripheral nerve dysfunction [80–82]. Furthermore, experimental studies in streptozotocin-diabetic rats [83, 84] and a small open-label clinical trial [85] with another niacin derivative, niceritrol, demonstrated reduction of serum triglyceride concentration and alleviation of peripheral neuropathy, in the absence of amelioration of diabetic hyperglycemia. This apparent similarity of action between niacin (vitamin B3)/its derivatives and other B vitamins is not surprising because niacin [85], its derivative acipimox in the current study, and other afore-mentioned B vitamins [82, 86, 87] counteract oxidative-nitrosative stress. Note, however, that only niacin and its derivatives, but not other B vitamins, display potent hypolipidemic activity. With consideration of 1) the important role of oxidative-nitrosative stress in neuropathy and its presence prior to the development of overt diabetes; 2) generation of oxidative stress by NEFA; and 3) alleviation of both hypertriglyceridemia/fatty acidemia and peripheral nerve oxidative-nitrosative stress by acipimox, it is reasonable to suggest that acipimox alleviates peripheral nerve dysfunction through inhibition of triglyceride/NEFA-generated-oxidative-nitrosative stress. Thus, the mechanisms of neuroprotective effects of acipimox (and, potentially, of niacin and other niacin derivatives) are principally different from other B vitamins, which counteract oxidative-nitrosative stress through inhibition of methylglyoxal and advanced glycation end-product formation, correction of the glutathione redox state, and chelation of transition metals [82, 86, 87].
Findings from several laboratories suggest that impaired insulin signaling in the peripheral nerve [88] and dorsal root ganglion neurons [2–4] is an important and independent of hyperglycemia mechanism in the pathogenesis of diabetic neuropathy. Suboptimal doses of insulin administered systemically or intrathecally have been reported to counteract MNCV and SNCV deficits [2] and to cause intraepidermal nerve fiber regeneration [3] in streptozotocin-diabetic rats. In the present study, impaired insulin signaling manifest in reduced phospho-Akt/Akt ratio, in the absence of any changes in IR expression or phospho-IRS/IRS ratio, has been identified in the peripheral nerve of Zucker fatty rats, thus suggesting the presence of this phenomenon at the prediabetic stage. A complete reversal of SNCV deficit in the absence of any changes in peripheral nerve insulin signaling in acipimox-treated Zucker rats indicates that the role of this factor in prediabetes-associated nerve conduction slowing is minor.
In conclusion, hypertriglyceridemia and/or increased NEFA concentrations play an important role in sensory nerve conduction slowing and changes in behavioral measures of sensory function associated with prediabetes. The underlying mechanisms are likely to involve oxidative-nitrosative stress in the peripheral nerve, and, in particular, in Schwann cells. The findings suggest that lipid-lowering agents and antioxidants may find use in management of peripheral neuropathy in subjects with prediabetes. Further studies are needed to establish whether reduction of hypertriglyceridemia and free fatty acidemia alleviates oxidative-nitrosative stress and neuropathic changes in overt diabetes.
Highlights
-
-
NEFA increase mitochondrial superoxide and NAD(P)H oxidase activity in human Schwann cells.
-
-
Acipimox reduces triglyceride/NEFA and alleviates neuropathy in Zucker fatty rats.
-
-
Hypertriglyceridemia and NEFA, but not glucose intolerance, cause prediabetic neuropathy.
-
-
Lipid-lowering agents and antioxidants may find use in management of this condition.
Acknowledgments
The study was supported in part by the National Institutes of Health Grants RO1DK074517, RO1DK077141, and RO1DK081147 (all to I.G.O.).
Abbreviations
- HSC
human Schwann cells
- IRβ
insulin receptor β
- IRS-1
insulin receptor substrate 1
- MNCV
sciatic motor nerve conduction velocity
- NEFA
non-esterified fatty acids
- ROS
reactive oxygen species
- SNCV
hind-limb digital sensory nerve conduction velocity
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Sahin M, Karatas M, Sahin M, Ertugrul D, Kulaksizoğlu M, Dogruk A, Gokcel A, Tutuncu NB, Guvener ND, Kutlu M. High prevalence of neuropathy in patients with impaired 60-minute oral glucose tolerance test but normal fasting and 120-minute glucose levels. Minerva Endocrinol. 2008;33:289–296. [PubMed] [Google Scholar]
- [2].Huang TJ, Price SA, Chilton L, Calcutt NA, Tomlinson DR, Verkhratsky A, Fernyhough P. Insulin prevents depolarization of the mitochondrial inner membrane in sensory neurons of type 1 diabetic rats in the presence of sustained hyperglycemia. Diabetes. 2003;52:2129–2136. doi: 10.2337/diabetes.52.8.2129. [DOI] [PubMed] [Google Scholar]
- [3].Toth C, Brussee V, Zochodne DW. Remote neurotrophic support of epidermal nerve fibres in experimental diabetes. Diabetologia. 2006;49:1081–1088. doi: 10.1007/s00125-006-0169-8. [DOI] [PubMed] [Google Scholar]
- [4].Francis G, Martinez J, Liu W, Nguyen T, Ayer A, Fine J, Zochodne D, Hanson LR, Frey WH, 2nd, Toth C. Intranasal insulin ameliorates experimental diabetic neuropathy. Diabetes. 2009;58:934–945. doi: 10.2337/db08-1287. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [5].Tesfaye S, Chaturvedi N, Eaton SE, Ward JD, Manes C, Ionescu-Tirgoviste C, Witte DR, Fuller JH, EURODIAB Prospective Complications Study Group Vascular risk factors and diabetic neuropathy. N. Engl. J. Med. 2005;352:341–350. doi: 10.1056/NEJMoa032782. [DOI] [PubMed] [Google Scholar]
- [6].Ziegler D, Sohr CG, Nourooz-Zadeh J. Oxidative stress and antioxidant defense in relation to the severity of diabetic polyneuropathy and cardiovascular autonomic neuropathy. Diabetes Care. 2004;27:2178–2183. doi: 10.2337/diacare.27.9.2178. [DOI] [PubMed] [Google Scholar]
- [7].Wiggin TD, Sullivan KA, Pop-Busui R, Amato A, Sima AA, Feldman EL. Elevated triglycerides correlate with progression of diabetic neuropathy. Diabetes. 2009;58:1634–1640. doi: 10.2337/db08-1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sumner CJ, Sheth S, Griffin JW, Cornblath DR, Polydefkis M. The spectrum of neuropathy in diabetes and impaired glucose tolerance. Neurology. 2003;60:108–111. doi: 10.1212/wnl.60.1.108. [DOI] [PubMed] [Google Scholar]
- [9].Smith AG, Singleton JR. Impaired glucose tolerance and neuropathy. Neurologist. 2008;14:23–29. doi: 10.1097/NRL.0b013e31815a3956. [DOI] [PubMed] [Google Scholar]
- [10].Papanas N, Vinik AI, Ziegler D. Neuropathy in prediabetes: does the clock start ticking early? Nat. Rev. Endocrinol. 2011;7:682–690. doi: 10.1038/nrendo.2011.113. [DOI] [PubMed] [Google Scholar]
- [11].Hughes RA, Umapathi T, Gray IA, Gregson NA, Noori M, Pannala AS, Proteggente A, Swan AV. A controlled investigation of the cause of chronic idiopathic axonal polyneuropathy. Brain. 2004;127:1723–1730. doi: 10.1093/brain/awh192. [DOI] [PubMed] [Google Scholar]
- [12].Dyck PJ, Dyck PJ, Klein CJ, Weigand SD. Does impaired glucose metabolism cause polyneuropathy? Review of previous studies and design of a prospective controlled population-based study. Muscle Nerve. 2007;36:536–541. doi: 10.1002/mus.20846. [DOI] [PubMed] [Google Scholar]
- [13].Sparks LM, Xie H, Koza RA, Mynatt R, Hulver MW, Bray GA, Smith SR. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes. 2005;54:1926–1933. doi: 10.2337/diabetes.54.7.1926. [DOI] [PubMed] [Google Scholar]
- [14].Longo KA, Govek EK, Nolan A, McDonagh T, Charoenthongtrakul S, Giuliana DJ, Morgan K, Hixon J, Zhou C, Kelder B, Kopchick JJ, Saunders JO, Navia MA, Curtis R, DiStefano PS, Geddes BJ. Pharmacologic inhibition of ghrelin receptor signaling is insulin sparing and promotes insulin sensitivity. J. Pharmacol. Exp. Ther. 2011;339:115–124. doi: 10.1124/jpet.111.183764. [DOI] [PubMed] [Google Scholar]
- [15].Shevalye H, Lupachyk S, Watcho P, Stavniichuk R, Khazim K, Abboud HE, Obrosova IG. Prediabetic Nephropathy as an Early Consequence of the High-Calorie/High-Fat Diet: Relation to Oxidative Stress. Endocrinology. 2012 doi: 10.1210/en.2011-1997. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zawalich WS, Zawalich KC, Kelley GG, Shulman GI. Islet phosphoinositide hydrolysis and insulin secretory responses from prediabetic fa/fa ZDF rats. Biochem. Biophys. Res. Commun. 1995;209:974–980. doi: 10.1006/bbrc.1995.1593. [DOI] [PubMed] [Google Scholar]
- [17].Zhou YT, Shimabukuro M, Wang MY, Lee Y, Higa M, Milburn JL, Newgard CB, Unger RH. Role of peroxisome proliferator-activated receptor alpha in disease of pancreatic beta cells. Proc. Natl. Acad. Sci. USA. 1998;95:8898–8903. doi: 10.1073/pnas.95.15.8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Muellenbach EA, Diehl CJ, Teachey MK, Lindborg KA, Archuleta TL, Harrell NB, Andersen G, Somoza V, Hasselwander O, Matuschek M, Henriksen EJ. Interactions of the advanced glycation end product inhibitor pyridoxamine and the antioxidant alpha-lipoic acid on insulin resistance in the obese Zucker rat. Metabolism. 2008;57:1465–1472. doi: 10.1016/j.metabol.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tong X, Hou X, Jourd'heuil D, Weisbrod RM, Cohen RA. Upregulation of Nox4 by TGF{beta}1 oxidizes SERCA and inhibits NO in arterial smooth muscle of the prediabetic Zucker rat. Circ. Res. 2010;107:975–983. doi: 10.1161/CIRCRESAHA.110.221242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Henriksen EJ, Diamond-Stanic MK, Marchionne EM. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic. Biol. Med. 2011;51:993–999. doi: 10.1016/j.freeradbiomed.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Oltman CL, Coppey LJ, Gellett JS, Davidson EP, Lund DD, Yorek MA. Progression of vascular and neural dysfunction in sciatic nerves of Zucker diabetic fatty and Zucker rats. Am. J. Physiol. Endocrinol. Metab. 2005;289:E113–E122. doi: 10.1152/ajpendo.00594.2004. [DOI] [PubMed] [Google Scholar]
- [22].Obrosova IG, Ilnytska O, Lyzogubov VV, Pavlov IA, Mashtalir N, Nadler JL, Drel VR. High-fat diet induced neuropathy of pre-diabetes and obesity: effects of “healthy” diet and aldose reductase inhibition. Diabetes. 2007;56:2598–2608. doi: 10.2337/db06-1176. [DOI] [PubMed] [Google Scholar]
- [23].Vincent AM, Hayes JM, McLean LL, Vivekanandan-Giri A, Pennathur S, Feldman EL. Dyslipidemia-induced neuropathy in mice: the role of oxLDL/LOX-1. Diabetes. 2009;58:2376–2385. doi: 10.2337/db09-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Davidson EP, Coppey LJ, Calcutt NA, Oltman CL, Yorek MA. Diet-induced obesity in Sprague-Dawley rats causes microvascular and neural dysfunction. Diabetes Metab. Res. Rev. 2010;26:306–318. doi: 10.1002/dmrr.1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Watcho P, Stavniichuk R, Ribnicky DM, Raskin I, Obrosova IG. High-fat diet-induced neuropathy of prediabetes and obesity: effect of PMI-5011, an ethanolic extract of Artemisia dracunculus L. Mediators Inflamm. 2010;2010:268547. doi: 10.1155/2010/268547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shertzer HG, Schneider SN, Kendig EL, Clegg DJ, D'Alessio DA, Genter MB. Acetaminophen normalizes glucose homeostasis in mouse models for diabetes. Biochem. Pharmacol. 2008;75:1402–1410. doi: 10.1016/j.bcp.2007.12.003. [DOI] [PubMed] [Google Scholar]
- [27].Hoehn KL, Salmon AB, Hohnen-Behrens C, Turner N, Hoy AJ, Maghzal GJ, Stocker R, Van Remmen H, Kraegen EW, Cooney GJ, Richardson AR, James DE. Insulin resistance is a cellular antioxidant defense mechanism. Proc. Natl. Acad. Sci. USA. 2009;106:17787–17792. doi: 10.1073/pnas.0902380106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kamanna VS, Kashyap ML. Mechanism of action of niacin. Am. J. Cardiol. 2008;101:20B–26B. doi: 10.1016/j.amjcard.2008.02.029. [DOI] [PubMed] [Google Scholar]
- [29].Jacobson EL, Kim H, Kim M, Jacobson MK. Niacin: vitamin and antidyslipidemic drug. Subcell. Biochem. 2012;56:37–47. doi: 10.1007/978-94-007-2199-9_3. [DOI] [PubMed] [Google Scholar]
- [30].Ikemura M, Nishikawa M, Hyoudou K, Kobayashi Y, Yamashita F, Hashida M. Improvement of insulin resistance by removal of systemic hydrogen peroxide by PEGylated catalase in obese mice. Mol. Pharm. 2010;7:2069–2076. doi: 10.1021/mp100110c. [DOI] [PubMed] [Google Scholar]
- [31].Elam MB, Hunninghake DB, Davis KB, Garg R, Johnson C, Egan D, Kostis JB, Sheps DS, Brinton EA. Effect of niacin on lipid and lipoprotein levels and glycemic control in patients with diabetes and peripheral arterial disease: the ADMIT study: A randomized trial. Arterial Disease Multiple Intervention Trial. JAMA. 2000;284:1263–1270. doi: 10.1001/jama.284.10.1263. [DOI] [PubMed] [Google Scholar]
- [32].Shih KC, Kwok CF, Hwu CM, Hsiao LC, Li SH, Liu YF, Ho LT. Acipimox attenuates hypertriglyceridemia in dyslipidemic noninsulin dependent diabetes mellitus patients without perturbation of insulin sensitivity and glycemic control. Diabetes Res. Clin. Pract. 1997;36:113–119. doi: 10.1016/s0168-8227(97)00039-9. [DOI] [PubMed] [Google Scholar]
- [33].Montecucco F, Bertolotto M, Vuilleumier N, Franciosi U, Puddu A, Minetti S, Delrio A, Quercioli A, Bergamini E, Ottonello L, Pende A, Lenglet S, Pelli G, Mach F, Dallegri F, Viviani GL. Acipimox reduces circulating levels of insulin and associated neutrophilic inflammation in metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 2011;300:E681–E690. doi: 10.1152/ajpendo.00527.2010. [DOI] [PubMed] [Google Scholar]
- [34].Bae JH, Bassenge E, Kim KB, Kim YN, Kim KS, Lee HJ, Moon KC, Lee MS, Park KY, Schwemmer M. Postprandial hypertriglyceridemia impairs endothelial function by enhanced oxidant stress. Atherosclerosis. 2001;155:517–523. doi: 10.1016/s0021-9150(00)00601-8. [DOI] [PubMed] [Google Scholar]
- [35].Padilla A, Descorbeth M, Almeyda AL, Payne K, De Leon M. Hyperglycemia magnifies Schwann cell dysfunction and cell death triggered by PA-induced lipotoxicity. Brain Res. 2011;1370:64–79. doi: 10.1016/j.brainres.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Li F, Drel VR, Szabó C, Stevens MJ, Obrosova IG. Low-dose poly(ADP-ribose) polymerase inhibitor-containing combination therapies reverse early peripheral diabetic neuropathy. Diabetes. 2005;54:1514–1522. doi: 10.2337/diabetes.54.5.1514. [DOI] [PubMed] [Google Scholar]
- [37].Obrosova IG, Xu W, Lyzogubov VV, Ilnytska O, Mashtalir N, Vareniuk I, Pavlov IA, Zhang J, Slusher B, Drel VR. PARP inhibition or gene deficiency counteracts intraepidermal nerve fiber loss and neuropathic pain in advanced diabetic neuropathy. Free Radic. Biol. Med. 2008;44:972–981. doi: 10.1016/j.freeradbiomed.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Drel VR, Lupachyk S, Shevalye H, Vareniuk I, Xu W, Zhang J, Delamere NA, Shahidullah M, Slusher B, Obrosova IG. New therapeutic and biomarker discovery for peripheral diabetic neuropathy: PARP inhibitor, nitrotyrosine, and tumor necrosis factor-{alpha} Endocrinology. 2010;151:2547–2555. doi: 10.1210/en.2009-1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lupachyk S, Shevalye H, Maksimchyk Y, Drel VR, Obrosova IG. PARP inhibition alleviates diabetes-induced systemic oxidative stress and neural tissue 4-hydroxynonenal adduct accumulation: correlation with peripheral nerve function. Free Radic. Biol. Med. 2011;50:1400–1409. doi: 10.1016/j.freeradbiomed.2011.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Szabó C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- [42].Szabó C, Mabley JG, Moeller SM, Shimanovich R, Pacher P, Virag L, Soriano FG, Van Duzer JH, Williams W, Salzman AL, Groves JT. Part I: pathogenetic role of peroxynitrite in the development of diabetes and diabetic vascular complications: studies with FP15, a novel potent peroxynitrite decomposition catalyst. Mol Med. 2002;8:571–580. [PMC free article] [PubMed] [Google Scholar]
- [43].Pacher P, Obrosova IG, Mabley JG, Szabó C. Role of nitrosative stress and peroxynitrite in the pathogenesis of diabetic complications. Emerging new therapeutical strategies. Curr. Med. Chem. 2005;12:267–275. doi: 10.2174/0929867053363207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lehmann HC, Höke A. Schwann cells as a therapeutic target for peripheral neuropathies. CNS Neurol Disord Drug Targets. 2010;9:801–806. doi: 10.2174/187152710793237412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Obrosova IG, Drel VR, Pacher P, Ilnytska O, Wang ZQ, Stevens MJ, Yorek MA. Oxidative-nitrosative stress and poly(ADP-ribose) polymerase (PARP) activation in experimental diabetic neuropathy: the relation is revisited. Diabetes. 2005;54:3435–3441. doi: 10.2337/diabetes.54.12.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Stevens MJ, Li F, Drel VR, Abatan OI, Kim H, Burnett D, Larkin D, Obrosova IG. Nicotinamide reverses neurological and neurovascular deficits in streptozotocin diabetic rats. J. Pharmacol. Exp. Ther. 2007;320:458–464. doi: 10.1124/jpet.106.109702. [DOI] [PubMed] [Google Scholar]
- [47].Askwith T, Zeng W, Eggo MC, Stevens MJ. Oxidative stress and dysregulation of the taurine transporter in high-glucose-exposed human Schwann cells: implications for pathogenesis of diabetic neuropathy. Am. J. Physiol. Endocrinol. Metab. 2009;297:620–628. doi: 10.1152/ajpendo.00287.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Stavniichuk R, Drel VR, Shevalye H, Vareniuk I, Stevens MJ, Nadler JL, Obrosova IG. Role of 12/15-lipoxygenase in nitrosative stress and peripheral prediabetic and diabetic neuropathies. Free Radic. Biol. Med. 2010;49:1036–1045. doi: 10.1016/j.freeradbiomed.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Askwith T, Zeng W, Eggo MC, Stevens MJ. Taurine reduces nitrosative stress and nitric oxide synthase expression in high glucose-exposed human Schwann cells. Exp. Neurol. 2011 doi: 10.1016/j.expneurol.2011.09.010. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Drel VR, Xu W, Zhang J, Kador PF, Ali TK, Shin J, Julius U, Slusher B, El-Remessy AB, Obrosova IG. Poly(ADP-ribose)polymerase inhibition counteracts cataract formation and early retinal changes in streptozotocin-diabetic rats. Invest. Ophthalmol. Vis. Sci. 2009;50:1778–1790. doi: 10.1167/iovs.08-2191. [DOI] [PubMed] [Google Scholar]
- [51].Fraser DA, Thoen J, Rustan AC, Førre O, Kjeldsen-Kragh J. Changes in plasma free fatty acid concentrations in rheumatoid arthritis patients during fasting and their effects upon T-lymphocyte proliferation. Rheumatology (Oxford) 1999;38:948–952. doi: 10.1093/rheumatology/38.10.948. [DOI] [PubMed] [Google Scholar]
- [52].Kalyanaraman B, Darley-Usmar V, Davies KJ, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ, 2nd, Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med. 2011 Oct 2; doi: 10.1016/j.freeradbiomed.2011.09.030. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lan H, Cheng CC, Kowalski TJ, Pang L, Shan L, Chuang CC, Jackson J, Rojas-Triana A, Bober L, Liu L, Voigt J, Orth P, Yang X, Shipps GW, Jr., Hedrick JA. Small-molecule inhibitors of FABP4/5 ameliorate dyslipidemia but not insulin resistance in mice with diet-induced obesity. J. Lipid Res. 2011;52:646–656. doi: 10.1194/jlr.M012757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Obrosova IG, Mabley JG, Zsengellér Z, Charniauskaya T, Abatan OI, Groves JT, Szabó C. Role for nitrosative stress in diabetic neuropathy: evidence from studies with a peroxynitrite decomposition catalyst. FASEB J. 2005;19:401–403. doi: 10.1096/fj.04-1913fje. [DOI] [PubMed] [Google Scholar]
- [55].Drel VR, Pacher P, Vareniuk I, Pavlov IA, Ilnytska O, Lyzogubov VV, Bell SR, Groves JT, Obrosova IG. Evaluation of the peroxynitrite decomposition catalyst Fe(III) tetra-mesitylporphyrin octasulfonate on peripheral neuropathy in a mouse model of type 1 diabetes. Int. J. Mol. Med. 2007;20:783–792. [PMC free article] [PubMed] [Google Scholar]
- [56].Obrosova IG, Drel VR, Oltman CL, Mashtalir N, Tibrewala J, Groves JT, Yorek MA. Role of nitrosative stress in early neuropathy and vascular dysfunction in streptozotocin-diabetic rats. Am. J. Physiol. Endocrinol. Metab. 2007;293:E1645–E1655. doi: 10.1152/ajpendo.00479.2007. [DOI] [PubMed] [Google Scholar]
- [57].Vareniuk I, Pavlov IA, Obrosova IG. Inducible nitric oxide synthase gene deficiency counteracts multiple manifestations of peripheral neuropathy in a streptozotocin-induced mouse model of diabetes. Diabetologia. 2008;51:2126–2133. doi: 10.1007/s00125-008-1136-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Stavniichuk R, Drel VR, Shevalye H, Maksimchyk Y, Kuchmerovska TM, Nadler JL, Obrosova IG. Baicalein alleviates diabetic peripheral neuropathy through inhibition of oxidative-nitrosative stress and p38 MAPK activation. Exp. Neurol. 2011;230:106–113. doi: 10.1016/j.expneurol.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Nagamatsu M, Nickander KK, Schmelzer JD, Raya A, Wittrock DA, Tritschler H, Low PA. Lipoic acid improves nerve blood flow, reduces oxidative stress, and improves distal nerve conduction in experimental diabetic neuropathy. Diabetes Care. 1995;18:1160–1167. doi: 10.2337/diacare.18.8.1160. [DOI] [PubMed] [Google Scholar]
- [60].Sagara M, Satoh J, Wada R, Yagihashi S, Takahashi K, Fukuzawa M, Muto G, Muto Y, Toyota T. Inhibition of development of peripheral neuropathy in streptozotocin-induced diabetic rats with N-acetylcysteine. Diabetologia. 1996;39:263–269. doi: 10.1007/BF00418340. [DOI] [PubMed] [Google Scholar]
- [61].Cameron NE, Cotter MA, Horrobin DH, Tritschler HJ. Effects of alpha-lipoic acid on neurovascular function in diabetic rats: interaction with essential fatty acids. Diabetologia. 1998;41:390–399. doi: 10.1007/s001250050921. [DOI] [PubMed] [Google Scholar]
- [62].Cameron NE, Tuck Z, McCabe L, Cotter MA. Effect of the hydroxyl radical scavenger, dimethylthiourea, on peripheral nerve tissue perfusion, conduction velocity and nociception in experimental diabetes. Diabetologia. 2001;44:1161–1169. doi: 10.1007/s001250100626. [DOI] [PubMed] [Google Scholar]
- [63].Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Lund DD, Yorek MA. Effect of antioxidant treatment of streptozotocin-induced diabetic rats on endoneurial blood flow, motor nerve conduction velocity, and vascular reactivity of epineurial arterioles of the sciatic nerve. Diabetes. 2001;50:1927–1937. doi: 10.2337/diabetes.50.8.1927. [DOI] [PubMed] [Google Scholar]
- [64].Schmeichel AM, Schmelzer JD, Low PA. Oxidative injury and apoptosis of dorsal root ganglion neurons in chronic experimental diabetic neuropathy. Diabetes. 2003;52:165–171. doi: 10.2337/diabetes.52.1.165. [DOI] [PubMed] [Google Scholar]
- [65].Wang Y, Schmeichel AM, Iida H, Schmelzer JD, Low PA. Ischemia-reperfusion injury causes oxidative stress and apoptosis of Schwann cell in acute and chronic experimental diabetic neuropathy. Antioxid. Redox Signal. 2005;7:1513–1520. doi: 10.1089/ars.2005.7.1513. [DOI] [PubMed] [Google Scholar]
- [66].Ziegler D, Ametov A, Barinov A, Dyck PJ, Gurieva I, Low PA, Munzel U, Yakhno N, Raz I, Novosadova M, Maus J, Samigullin R. Oral treatment with alpha-lipoic acid improves symptomatic diabetic polyneuropathy: the SYDNEY 2 trial. Diabetes Care. 2006;29:2365–2370. doi: 10.2337/dc06-1216. [DOI] [PubMed] [Google Scholar]
- [67].Ziegler D, Low PA, Litchy WJ, Boulton AJ, Vinik AI, Freeman R, Samigullin R, Tritschler H, Munzel U, Maus J, Schütte K, Dyck PJ. Efficacy and safety of antioxidant treatment with {alpha}-lipoic acid over 4 years in diabetic polyneuropathy: The NATHAN 1 trial. Diabetes Care. 2011;34:2054–2060. doi: 10.2337/dc11-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Bierhaus A, Haslbeck KM, Humpert PM, Liliensiek B, Dehmer T, Morcos M, Sayed AA, Andrassy M, Schiekofer S, Schneider JG, Schulz JB, Heuss D, Neundörfer B, Dierl S, Huber J, Tritschler H, Schmidt AM, Schwaninger M, Haering HU, Schleicher E, Kasper M, Stern DM, Arnold B, Nawroth PP. Loss of pain perception in diabetes is dependent on a receptor of the immunoglobulin superfamily. J. Clin. Invest. 2004;114:1741–1751. doi: 10.1172/JCI18058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wada R, Yagihashi S. Role of advanced glycation end products and their receptors in development of diabetic neuropathy. Ann. N Y Acad. Sci. 2005;1043:598–604. doi: 10.1196/annals.1338.067. [DOI] [PubMed] [Google Scholar]
- [70].Wang Y, Schmeichel AM, Iida H, Schmelzer JD, Low PA. Enhanced inflammatory response via activation of NF-kappaB in acute experimental diabetic neuropathy subjected to ischemia-reperfusion injury. J. Neurol. Sci. 2006;247:47–52. doi: 10.1016/j.jns.2006.03.011. [DOI] [PubMed] [Google Scholar]
- [71].Cameron NE, Cotter MA. Pro-inflammatory mechanisms in diabetic neuropathy: focus on the nuclear factor kappa B pathway. Curr. Drug Targets. 2008;9:60–67. doi: 10.2174/138945008783431718. [DOI] [PubMed] [Google Scholar]
- [72].Nukada H, McMorran PD, Baba M, Ogasawara S, Yagihashi S. Increased susceptibility to ischemia and macrophage activation in STZ-diabetic rat nerve. Brain Res. 2011;1373:172–182. doi: 10.1016/j.brainres.2010.11.084. [DOI] [PubMed] [Google Scholar]
- [73].Haslbeck KM, Schleicher E, Bierhaus A, Nawroth P, Haslbeck M, Neundörfer B, Heuss D. The AGE/RAGE/NF-(kappa)B pathway may contribute to the pathogenesis of polyneuropathy in impaired glucose tolerance (IGT) Exp. Clin. Endocrinol. Diabetes. 2005;113:288–291. doi: 10.1055/s-2005-865600. [DOI] [PubMed] [Google Scholar]
- [74].Stevens MJ, Obrosova I, Cao X, Van Huysen C, Greene DA. Effects of DL-alpha-lipoic acid on peripheral nerve conduction, blood flow, energy metabolism, and oxidative stress in experimental diabetic neuropathy. Diabetes. 2000;49:1006–1015. doi: 10.2337/diabetes.49.6.1006. [DOI] [PubMed] [Google Scholar]
- [75].Obrosova IG, Van Huysen C, Fathallah L, Cao X, Stevens MJ, Greene DA. Evaluation of alpha(1)-adrenoceptor antagonist on diabetes-induced changes in peripheral nerve function, metabolism, and antioxidative defense. FASEB J. 2000;14:1548–1558. doi: 10.1096/fj.14.11.1548. [DOI] [PubMed] [Google Scholar]
- [76].Toth C, Rong LL, Yang C, Martinez J, Song F, Ramji N, Brussee V, Liu W, Durand J, Nguyen MD, Schmidt AM, Zochodne DW. Receptor for advanced glycation end products (RAGEs) and experimental diabetic neuropathy. Diabetes. 2008;57:1002–1017. doi: 10.2337/db07-0339. [DOI] [PubMed] [Google Scholar]
- [77].Chinen I, Shimabukuro M, Yamakawa K, Higa N, Matsuzaki T, Noguchi K, Ueda S, Sakanashi M, Takasu N. Vascular lipotoxicity: endothelial dysfunction via fatty-acid-induced reactive oxygen species overproduction in obese Zucker diabetic fatty rats. Endocrinology. 2007;148:160–165. doi: 10.1210/en.2006-1132. [DOI] [PubMed] [Google Scholar]
- [78].Poduslo JF, Curran GL. Increased permeability across the blood-nerve barrier of albumin glycated in vitro and in vivo from patients with diabetic polyneuropathy. Proc. Natl. Acad. Sci. U S A. 1992;89:2218–2222. doi: 10.1073/pnas.89.6.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Schmelzer JD, Zochodne DW, Low PA. Ischemic and reperfusion injury of rat peripheral nerve. Proc. Natl. Acad. Sci. U S A. 1989;86:1639–1642. doi: 10.1073/pnas.86.5.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Thornalley PJ. The potential role of thiamine (vitamin B1) in diabetic complications. Curr. Diabetes Rev. 2005;1:287–298. doi: 10.2174/157339905774574383. [DOI] [PubMed] [Google Scholar]
- [81].Jolivalt CG, Mizisin LM, Nelson A, Cunha JM, Ramos KM, Bonke D, Calcutt NA. B vitamins alleviate indices of neuropathic pain in diabetic rats. Eur. J. Pharmacol. 2009;612:41–47. doi: 10.1016/j.ejphar.2009.04.028. [DOI] [PubMed] [Google Scholar]
- [82].Mizukami H, Ogasawara S, Yamagishi S, Takahashi K, Yagihashi S. Methylcobalamin effects on diabetic neuropathy and nerve protein kinase C in rats. Eur. J. Clin. Invest. 2011;41:442–450. doi: 10.1111/j.1365-2362.2010.02430.x. [DOI] [PubMed] [Google Scholar]
- [83].Hotta N, Kakuta H, Fukasawa H, Koh N, Sakakibara F, Komori H, Sakamoto N. Effect of niceritrol on streptozocin-induced diabetic neuropathy in rats. Diabetes. 1992;41:587–591. doi: 10.2337/diab.41.5.587. [DOI] [PubMed] [Google Scholar]
- [84].Hotta N, Nakamura J, Kakuta H, Fukasawa H, Koh N, Sakakibara F, Mori K, Sakamoto N. Niceritrol prevents the decrease in red blood cell 2,3-diphosphoglycerate and neuropathy in streptozotocin-induced diabetic rats. J. Diabetes Complications. 1995;9:133–139. doi: 10.1016/1056-8727(95)00049-8. [DOI] [PubMed] [Google Scholar]
- [85].Hotta N, Sugimura K, Tsuchida I, Sano T, Koh N, Matsumae H, Sakamoto N. Use of the C64 quantitative tuning fork and the effect of niceritrol in diabetic neuropathy. Clin. Ther. 1994;16:1007–1015. [PubMed] [Google Scholar]
- [86].Nakamura S, Li H, Adijiang A, Pischetsrieder M, Niwa T. Pyridoxal phosphate prevents progression of diabetic nephropathy. Nephrol. Dial. Transplant. 2007;22:2165–2174. doi: 10.1093/ndt/gfm166. [DOI] [PubMed] [Google Scholar]
- [87].Karachalias N, Babaei-Jadidi R, Rabbani N, Thornalley PJ. Increased protein damage in renal glomeruli, retina, nerve, plasma and urine and its prevention by thiamine and benfotiamine therapy in a rat model of diabetes. Diabetologia. 2010;53:1506–1516. doi: 10.1007/s00125-010-1722-z. [DOI] [PubMed] [Google Scholar]
- [88].Sugimoto K, Rashid IB, Shoji M, Suda T, Yasujima M. Early changes in insulin receptor signaling and pain sensation in streptozotocin-induced diabetic neuropathy in rats. J. Pain. 2008;9:237–245. doi: 10.1016/j.jpain.2007.10.016. [DOI] [PubMed] [Google Scholar]