

Abstract
Epithelial-mesenchymal transition (EMT) is implicated in the pathogenesis of lung fibrosis and cancer metastasis, two conditions associated with cigarette smoke (CS). CS has been reported to promote the EMT process. CS is the major cause of lung cancer and nearly half of lung cancer patients are active smokers. Nonetheless, the mechanism whereby CS induces EMT remains largely unknown. In this study we investigated the induction of EMT by CS and explored the underlying mechanisms in the human non-small cell lung carcinoma (H358) cell line. We demonstrate that exposure to an extract of CS (CSE) decreases E-cadherin and increases N-cadherin and vimentin, markers of EMT, in H358 cells cultured in RPMI-1640 medium with 1% fetal bovine serum. Pretreatment with N-acetylcysteine (NAC), a potent antioxidant and precursor of glutathione, abrogated changes in these EMT markers. In addition, CSE activated Src kinase (shown as increased phosphorylation of Src at Tyr418) and the Src kinase inhibitor, PP2, inhibited CS-stimulated EMT changes, suggesting that Src is critical in CSE-stimulated EMT induction. Furthermore, NAC treatment abrogated CSE-stimulated Src activation. However, co-incubation with catalase had no effect on CSE-mediated Src activation. Finally, acrolein, an unsaturated aldehyde present in CSE, caused Src activation. Taken together, these data suggest that CSE initiates EMT through Src, which is activated by CS through redox modification.
Keywords: epithelial-mesenchymal transition, Src, cigarette smoke, redox, N-acetylcysteine
Introduction
Epithelial-mesenchymal transition (EMT) is a process in which cells lose epithelial phenotype and acquire mesenchymal features. EMT has been suggested to play important roles in the development of idiopathic pulmonary fibrosis (IPF) [1, 2] and metastasis of lung cancer [3, 4] although the involvement of EMT in fibrosis is far more controversial than in metastasis. Cigarette smoke (CS) is a major risk factor for lung fibrosis [5] and a major contributor to lung carcinogenesis [6]. Considering that second hand CS is a common environmental hazard [7, 8] and that nearly half of lung cancer patients remain active smokers [9–11], it is important to understand how CS affects the EMT process so that, given the less than complete success of smoking cessation efforts, effective strategies can be developed to prevent and treat these CS-induced EMT-related diseases. Some studies have shown that CS can promote EMT process in lung alveolar cells [12]. The induction of EMT by CS was also observed in a recent study reporting that the levels of vimentin and other EMT markers increased in smokers with COPD compared with normal non-smokers [13]. Nonetheless, the mechanisms underlying induction of EMT by CS remain unclear. In this study we used a water soluble extract of CS (CSE) in cell culture. Although there are obvious limitations to extrapolating to pathology in using CSE, it provides a convenient and reasonably reproducible method for studying signaling mechanisms.
EMT can be induced by a variety of growth factors and other external stressors such as transforming growth factor beta1 (TGFβ1) [14, 15], epidermal growth factor (EGF) [16], platelet-derived growth factor (PDGF) [17], and hypoxia [18]. These EMT inducers activate or repress various intracellular signaling pathways, such as Ras/Mitogen-activated protein kinase kinase 1 and 2 (MEK)/ extracellular signal-regulated kinases 1 and 2 (ERK), phosphatidylinositol 3-kinase (PI3K)/the non-specific serine/threonine-protein kinase Akt/PKB, the non-receptor tyrosine kinase Src, and glycogen synthase kinase 3β (GSK3β), which sequentially regulate (activate or repress) transcription factors such as Mothers against decapentaplegic homologs 2 and 3 (Smad2/3), human snail homologs 1 and 2 (Snai1/2), and Twist, and cause the suppression of epithelial genes such as E-cadherin and zona occludens (ZO)-1, and increased expression of mesenchymal genes such as N-cadherin, vimentin, and smooth muscle actin A (SMA) [19, 20]. Interestingly, ERK, Akt, Src, GSK3β, and Smad2/3 have all been suggested as targets of redox activation [21–27].
Src is the prototypic member of a family of non-receptor membrane associated tyrosine kinases [28]. Studies have found that Src kinase is involved in cytoskeleton reorganization, cell migratory capacity, expression of mesenchymal proteins, and other EMT events [29–34]. Indeed, Src plays critical roles in EMT initiated by many stimulators [31–35]. Under resting conditions, most Src remains in an inactive form with phosphorylation at Tyr529 and intracellular interactions mediated by SH2 and SH3 domains. In response to agonists, Src undergoes dephosphorylation at Tyr529 and then conformation change that results in autophosphorylation at Tyr418 and its full activation [36]. Recently it has been found that Src activation by many stimuli is redox dependent [37–40], and can be activated directly by various oxidants such as peroxynitrite and H2O2 [24, 25, 41].
Accumulating evidence suggests that a diverse array of EMT inducers share a common redox dependent mechanism in initiating EMT [22, 27, 42–45]. N-acetylcysteine (NAC), a potent antioxidant and precursor of glutathione (GSH), inhibits EMT initiated by TGFβ and many other agonists [27, 42]. It has been reported that reactive oxygen species can directly induce or promote EMT [31, 46]. CS contains over 4000 compounds including several highly reactive α,β-unsaturated carbonyls, nitrogen oxides and H2O2. Given the facts that Src is critical for EMT initiation and redox regulated, and that CS contains strong oxidants and electrophiles, in the current study we test the hypothesis that CS causes EMT through CS-induced activation of Src kinase.
Methods and materials
Chemicals and reagents
Unless otherwise noted, all chemicals were from Sigma (St. Louis, MO). All antibodies were from Santa Cruz (Santa Cruz, CA). M-PER Mammalian Protein Extraction Reagent was from Thermal Fisher Scientific Inc. (Thermal Fisher, Rockford, IL). All chemicals used were at least analytical grade.
Preparation of cigarette smoke extracts [47]
CSE was an extract of mainstream cigarette smoke. Briefly, the smoke from one filtered cigarette (Camel regular) containing 1.2 mg of nicotine and 18 mg of tar according to the manufacturer's report was drawn through an experimental apparatus with a constant airflow driven by vacuum. The smoke was bubbled through 25 ml of RPMI-1640 medium without fetal bovine serum (FBS) in 2 min and the solution was used as the stock (100%) for further dilutions. After adjusting the pH to 7.2, the obtained CSE was filtered through a 0.22-μm filter (Millipore, Bedford, MA) for sterilization and diluted for use within 20 min after the preparation.
Cell culture and treatment
A human non-small cell lung carcinoma cell line (H358) was used. H358 cells were cultured in RPMI-1640 medium with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin, in a humidified incubator containing 5% CO2 at 37°C. Cells were treated with CSE when about 70% confluent in RPMI-1640 medium with 1% FBS. NAC was added to culture medium 4 hours before cells being exposed to CSE, and the NAC medium was replaced every 24 hours thereafter. In an experiment to detemine the extent of H2O2 involvement, catalase (final concentration is 400 U/ml) was added to the culture medium immediately before H2O2 or CSE exposure. In another set of experiments, cells were exposed to 15 μM acrolein, a non-toxic dose.
Western Analysis
Briefly, cell lysate was extracted with M-PER and 15 μg protein was heated for 15 min at 95°C in 2X loading buffer containing SDS (Tris base, pH 6.5, glycerol, DTT, and pyronin Y), electrophoresed on a 4–20% Tris-glycine acrylamide gel (Invitrogen, Carlsbad, CA), and then electroblotted onto polyvinylidene difluoride (PVDF) membranes (Millipore corporation, Bedford, MA). Membranes were blocked with 5% fat-free milk and then incubated overnight at 4°C with primary antibody in 5% milk in Tris-buffered saline (TBS). After being washed with TBS containing 0.05% Tween 20 (TTBS), membranes were incubated with secondary antibody at room temperature for 2 h. After TTBS washing, membranes were treated with an enhanced chemiluminescence reagent mixture (ECL Plus; Amersham, Arlington Heights, IL) for 5 min. The target bands were imaged on a Kodak Image Station 2000R (Kodak Company, Rochester, New York).
Statistical analysis
Densitometry data for Western Blots are expressed as mean ± standard error. SigmaStat 3.5 was used for statistical analysis and statistical significance was accepted when p < 0.05. The one-way ANOVA and Tukey test were used for comparison of protein levels.
Results
CSE induced EMT in lung epithelial H358 cells
EMT involves phenotypic changes such as shape change (from epithelial round-shaped to mesenchymal spindle-like), loss of epithelial marker proteins, including E-cadherin, and induction of mesenchymal marker proteins, including N-cadherin and vimentin [19, 20]. To examine whether CS induced EMT in human lung epithelial H358 cells, cell cultures at 70% confluence were exposed to variable concentrations of CSE for 72h. Using western Blots, cell lysates were then analyzed for the expression of epithelial and mesenchymal markers including E-cadherin, N-cadherin, and vimentin. As shown in Figure 1, treatment of cells with CSE caused a significant decrease in E-cadherin, and an increase in N-cadherin and vimentin proteins compared to untreated controls. The change of these proteins in response to CSE was dose-dependent and the effect was observed with 5% CSE exposure, and at a concentration of 20%, CSE caused a 40% decrease in E-cadherin, and a 60% and 50% increase in N-cadherin and vimentin respectively. Under the microscope, it is also observed that H358 cells became spindle-shaped and that some cells moved (detached) from the cell clusters upon CSE exposure, in contrast to rounded shape of the non-exposed cells (data not shown). This evidence suggests that CSE decreased epithelial proteins and increased mesenchymal changes in H358 cells, indicating the induction of EMT.
Figure 1.


CSE induced changes in epithelial and mesenchymal proteins in H358 cells. H358 cells were treated with different concentrations of CSE in 1% FBS medium for 72 h, and cell lysates were assayed for EMT proteins by western analysis. *, P<0.05 compared with control, N=3.
NAC inhibited CS-induced EMT markers
Some studies suggest that a redox mechanism is involved in EMT induction by various stimulators [22, 27, 42–45]. Since CS contains strong oxidants and other electrophiles and can produce additional oxidants in cells, we hypothesize that CS stimulated EMT via an oxidant-mediated mechanism. Generally, chemists consider oxidants as a subclass of electrophiles that remove electrons rather than share them with their target, as do other electrophiles. For biologists, oxidative stress also includes reactions in which electrophiles add to other molecules. Thus, Michael addition to compounds by α,β-unsaturated carbonyls, such as acrolein, a component of CSE that is formed by oxidative processes, are considered an oxidative mechanism. Therefore to test the hypothesis that this broadly defined oxidative mechanism was involved in CSE-induced EMT, H358 cells were pretreated with different concentrations of NAC for 4h and then exposed to 10% CSE for 72 h, followed by determining EMT marker proteins. As shown in Figure 2, 2 mM NAC pretreatment prevented the CSE-induced decrease in E-cadherin, and simultaneously suppressed up-regulation of vimentin by CSE. But, when cells were washed after the incubation with NAC and then CSE was added, the NAC was ineffective. These data suggest that CSE induced EMT via an oxidative mechanism (see above for the meaning of this oxidative mechanism in this context) that was prevented when NAC was present during the addition of CSE. NAC can supply cysteine, which is a limiting component for the synthesis of glutathione, a major antioxidant in vivo. Nonetheless, as NAC was only effective when in the solution to which CSE was added, it is more likely that NAC reacted directly with the oxidants in the extracellular medium. NAC, unlike antioxidants such as vitamin E, is a two electron donor that can either reduce oxidants or add to electrophiles by Michael addition. Although these reactions are slow compared with enzymatic removal of oxidants, in the extracellular environment they can be quite significant.
Figure 2.


NAC pretreatment abrogated changes in epithelial and mesenchymal proteins caused by CSE. *, P<0.05 compared with control, N=3.
Src is involved in CS-induced EMT
The tyrosine kinase Src has been reported to be important in EMT initiation by many stimulators [31–35]. To investigate whether Src was involved in CSE-caused EMT, we first studied Src activation by determining its autophosphorylation at Tyr418 (pTyr418-Src). H358 cells were treated with or without 10% CSE for various times and the cell lysates were analyzed for pTyr418-Src. Src phosphorylation at Tyr418 was increased at as early as 5 min and reached a maximum at 15 min of CSE treatment. By 1 h after CSE treatment, phosphorylation at Tyr418 had started to decrease to baseline levels (Fig. 3).
Figure 3.

Src activation by CSE. H358 cells were exposed to 10% CSE for different times. Phosphorylation of Src at Tyr418 was determined by western analysis.
To examine if Src was involved in CSE-induced EMT, H358 cells were pretreated with or without PP2 for 1 h, followed by treatment with 10% CSE for 72 h, and then EMT markers were determined. As shown in Fig. 4, treatment with 2 μM PP2 could suppress the E-cadherin decrease caused by CSE and inhibit the increase in vimentin, two characteristics of the EMT process. These data indicate that Src kinase plays a critical role in the CSE-induced EMT process.
Figure 4.
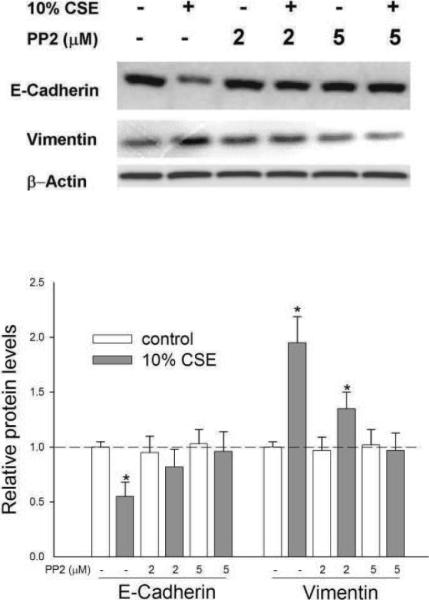
Inhibition of Src suppressed changes in epithelial and mesenchymal proteins by CSE. H358 cells were pretreated with or without PP2 for 1 h and then exposed to 10% CSE for 72 h. The cell lysates were assayed for EMT-related proteins by western analysis. *, P<0.05 compared with control, N=3.
NAC abrogated Src activation by CS
Previous reports have shown that Src activity could be oxidatively regulated [37–40]. As we observed that CSE induction of EMT was NAC-inhibitable and that Src was involved, we investigated whether Src activation by CSE might also be inhibited by NAC. H358 cells were pretreated with 2 mM NAC for 4h and then exposed to 10% CSE for 15 min. As shown in Fig. 5, NAC pretreatment inhibited phosphorylation of Src at Tyr418, suggesting that Src activation by CSE was dependent upon some component of CSE that was antagonized by NAC.
Figure 5.

NAC inhibited CSE-mediated Src activation. H358 cells were pretreated with/without 2mM NAC for 4 h and then exposed to 10% CSE for 15 min. Phosphorylation of Src at Tyr418 and total Src were determined with Western blots. *, P<0.05 compared with control, N=3.
Effect of catalase on CSE-stimulated Src activation
Cigarette smoke is a mixture that contains various oxidative compounds including H2O2 and unsaturated aldehydes. To examine if H2O2 is involved in CSE-mediated Src activation, catalase, which rapidly converts H2O2 to O2 and H2O, was added to the medium before the cells were exposed to CSE. Fig. 6 shows that both H2O2 (200 μM) and CSE activated Src (phosphorylation of Src at Tyr418) at 15 min of exposure. Catalase (final concentration 400 U/ml) completely abrogated H2O2-stimulated Src activation. However, catalase had no significant effect on Src activation by CSE exposure, suggesting that oxidants/electrophiles other than H2O2 in the CSE might be responsible for CSE-mediated Src activation.
Figure 6.

Catalase effect on CSE-mediated Src activation. catalase (final concentration 400 U/ml) was added to culture medium immediately before H358 cells were exposed to 10% CSE. Phosphorylation of Src at Tyr418 and total Src were determined with Western blots. *, P<0.05 compared with control, N=3.
Acrolein activated Src
Cells were exposed to acrolein, a potent electrophilic α,β- unsaturated aldehyde present in CSE. As shown in Fig. 7, 15 μM acrolein increased Src phosphorylation at Tyr418 at 5 min and the phosphorylation remained as late as 2 hours after exposure. These data indicate that acrolein and other electrohiles in CSE are at least partly involved in CSE-mediated Src activation.
Figure 7.

Src activation by acrolein. H358 cells were exposed to acrolein (15 PM) for indicated time and the phosphorylation of Src at Tyr418 and total Src were determined with Western blots.
Discussion
EMT has been suggested to play important roles in the initiation of pulmonary fibrosis and metastasis of lung cancers, two pulmonary diseases associated with cigarette smoke [5, 6]. In this paper we found that CS induced EMT in human pulmonary epithelial cells (H358), as evidenced by a decrease in epithelial proteins such as E-cadherin and a concomitant increase in mesenchymal markers such as N-cadherin and vimentin. To explore the underlying mechanism(s), we examined whether NAC, which can directly react with many CSE components as well as potentially increase intracellular glutathione, and PP2 (4-amino-5-(4-chloro- phenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine), a relatively specific Src inhibitor [48, 49], abrogated the changes of these EMT markers. Furthermore, it was found that Src activation by CSE was also prevented by NAC. Although NAC inhibited EMT extracellularly, it was important to explore whether NAC also inhibited Src activation as some other component of CSE that did not activate EMT and did not react with NAC might still activate Src; however, that was not found to occur. These findings suggest that smoking could cause EMT through an oxidative pathway in which Src plays a critical role. It should be noted that the composition of cell culture medium used in the current study is not the same as the extracellular fluid surrounding the alveolar epithelial cells in vivo. Physiological components that are missing include mucins, while glutathione, ascorbate, and urate differ in concentrations from those present in the cell medium. Further studies using in vivo models are obviously required to confirm current findings. Nevetheless, as the lung epithelial cells cultured in the cell culture medium maintain the epithelial phenotype and change to mesenchymal phenotype upon exposure to CSE, the current cell culture model is a useful model system study EMT at cell and molecular levels with the caveat that additional components of CS as well as differences in timing and presence of other cells and components of extracellular fluids would also affect pathophysiologic processes.
EMT is characterized by changes of cell phenotype and protein expression profile [19, 20]. During this process, cells lose epithelial phenotypic characteristics such as stable cell-cell junctions and apical-basolateral polarity, and acquire mesenchymal features such as increased matrix degradation, the ability to migrate, and a lack of cellular polarity. In addition, down regulation of epithelial proteins such as E-cadherin and β-catenin, and up regulation of the mesenchymal proteins such as fibronectin, N-cadherin and vimentin, usually occur in EMT and thus these proteins are often used as EMT markers. The current findings that CSE decreased epithelial protein E-cadherin and increased mesenchymal proteins N-cadherin and vimentin suggest that CSE induced EMT in the examined cell model. This result is in agreement with previous reports that cigarette smoking or a component (nicotine) could cause EMT changes in lung cancer cells [12, 50]. Taken together, these findings reveal that cigarette smoke is a potent EMT inducer, as are other sources of oxidative stress [1, 51]. This may partly explain a mechanism through which smoking contributes to the development of IPF, a disease in which EMT appears to play a role [1, 2], and the reason why smoking enhances metastasis and phenotypic changes of cancer cells, which also has been suggested to involve EMT [3, 4]. CSE used in this study contained about 20 μM acrolein [52]. Therefore, 10% CSE exposure contained 2 μM acrolein. Considering that acrolein in the pulmonary tract lining fluid can reach as high as 80 μM during smoking of 1 cigarette [53], the CSE concentration used in current study is within the range relevant to CS exposure. Although the lining fluid contains mucins and small molecular weight compounds that can react with acrolein, there are also many other components of CS that would also react with the lining fluid components.
In the context of the extracellular milieu, NAC may be an effective antioxidant. We demonstrated that extracellular NAC abrogated CSE-induced EMT changes (Fig. 2). CS can also induce lipid peroxidation products in vivo [54–56]. We thus conclude that CS induced EMT through a redox dependent mechanism. Many other EMT agonists, including TGFβ, EGF, and others also share this common redox dependent mechanism in EMT initiation [22, 27, 42–45], as evidenced by the fact that NAC inhibited EMT initiation by these inducers [27, 42], and that oxidants could directly induce or promote EMT [31, 46]. The results here may suggest to some that NAC administration could be a potential strategy for treatment and/or prevention of CS-induced EMT; however, the concentration and conditions here while demonstrating the ability of extracellular NAC to neutralize the EMT-inducing and Src-activating compounds in CSE are far greater than one would reasonably be able to give individuals by inhalation.
Src kinase is involved in cytoskeleton reorganization, cell migratory capacity, expression of mesenchymal proteins, and other aspects of EMT [29–34]. It is a common and critical downstream signaling target of a variety of EMT agonists, including TGFβ [33], EGF [35], endoplasmic reticulum (ER) stress [34], hepatitis B virus × protein (HBx) [32], and arachidonic acid [31, 32]. Src inhibition allows recovery of E-cadherin and suppresses expression of vimentin [57] and other proteins of the mesenchymal phenotype [30, 32]. In line with these reports, our data showed that CS activated Src and its inhibition restored E-cadherin and abrogated vimentin (Fig. 3 and Fig. 4), indicating that Src activation is essential for CS-induced EMT. Although the exact mechanisms remains to be elucidated, CS might initiate EMT through additional oxidative mechanisms and indeed, pathways contributing to EMT, such as Ras/ERK, PI3K/Akt, and GSK3β, are also redox regulated [26, 58]. However, what is most notable is that inhibition of Src activation by PP2 also abrogated EMT and that Src or other members of the Src family are modulators of the ERK, Akt and GSK3β pathways [25, 35, 59–66].
Accumulating evidence suggests that Src activity could be regulated through a redox mechanism [37–40]. Consistent with this, this study showed that CS activation of Src is also redox dependent, since NAC, a powerful antioxidant inhibited Src activation by CS (Fig.5). The redox activation of Src seems to be mediated through direct modification of cysteine residues in Src protein [67–69]. Indeed, mutation of selected cysteine residues in the C-terminus makes Src insensitive to redox regulation [70]. NAC can reverse Src activation when inactivated by disulfide formation [69] but as NAC cannot reduce an alkylated cysteine, it would not reverse that type of modification. The initial activation of Src by CSE does not appear to involve H2O2, since catalase did not significantly reduce CSE-mediated Src activation while it abrogated the positive control of H2O2-caused Src activation (Fig. 6). Whole CS contains various oxidative/electrophilic compounds, including H2O2 and α,β-unsaturated aldehydes, which can modify cysteine residues in signaling proteins including Src. Although CSE can activate NADPH oxidase and producing H2O2 [47, 71], the current data shows that oxidants other than H2O2 in the CSE are responsible for CSE-mediated Src activation. The identification of the components involved requires analysis of the adducts formed with Src using mass spectrometry, which is beyond the scope of the current study.
Finally, it remains unclear whether the redox regulation of Src by CS is by directly modifying the cysteine residues in Src protein or indirectly by regulating kinases or phosphatases involved in the dephosphorylation of Tyr529 of Src. The phosphorylation status of Tyr529 is determined by the balance between C-terminal Src kinase (CSK), a constitutively expressed kinase that phosphorylates Tyr529, and protein tyrosine phosphatases (PTPs) such as PTP1B, CD45, SHP-2, and PTPα, which mediate the dephosphorylation of Tyr529 [36]. Both CSK and aforementioned PTPs are also redox regulated and their activity can be inhibited by oxidation of their active site cysteines [72–76]. These observations suggest that CS might also activate Src by regulating Tyr529 dephosphorylation in addition to being directly activated by modification of its cysteines. Thus, determining whether there are multiple mechanisms whereby Src is redox regulated by CS need further study.
Highlights
Cigarette smoke extract (CSE) activates c-Src in H358 cells.
CSE-activated Src causes epithelial-mesenchymal transition (EMT).
N-acetylcysteine inhibits CSE-induced Src activation and EMT.
The Src kinase inhibitor, PP2, also inhibits CSE-induced Src activation and EMT.
Acknowledgments
Supported by NIH grant ES05511.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Willis BC, Borok Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2007;293(3):L525–34. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 2.Gharaee-Kermani M, et al. Recent advances in molecular targets and treatment of idiopathic pulmonary fibrosis: focus on TGFbeta signaling and the myofibroblast. Curr Med Chem. 2009;16(11):1400–17. doi: 10.2174/092986709787846497. [DOI] [PubMed] [Google Scholar]
- 3.Yauch RL, et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005;11(24 Pt 1):8686–98. doi: 10.1158/1078-0432.CCR-05-1492. [DOI] [PubMed] [Google Scholar]
- 4.Soltermann A, et al. Prognostic significance of epithelial-mesenchymal and mesenchymal-epithelial transition protein expression in non-small cell lung cancer. Clin Cancer Res. 2008;14(22):7430–7. doi: 10.1158/1078-0432.CCR-08-0935. [DOI] [PubMed] [Google Scholar]
- 5.Baumgartner KB, et al. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997;155(1):242–8. doi: 10.1164/ajrccm.155.1.9001319. [DOI] [PubMed] [Google Scholar]
- 6.Biesalski HK, et al. European Consensus Statement on Lung Cancer: risk factors and prevention. Lung Cancer Panel. CA Cancer J Clin. 1998;48(3):167–76. doi: 10.3322/canjclin.48.3.167. discussion 164–6. [DOI] [PubMed] [Google Scholar]
- 7.Besaratinia A, Pfeifer GP. Second-hand smoke and human lung cancer. Lancet Oncol. 2008;9(7):657–66. doi: 10.1016/S1470-2045(08)70172-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asomaning K, et al. Second hand smoke, age of exposure and lung cancer risk. Lung Cancer. 2008;61(1):13–20. doi: 10.1016/j.lungcan.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cooley ME, et al. Smoking cessation is challenging even for patients recovering from lung cancer surgery with curative intent. Lung Cancer. 2009;66(2):218–25. doi: 10.1016/j.lungcan.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burke L, et al. Smoking behaviors among cancer survivors: an observational clinical study. J Oncol Pract. 2009;5(1):6–9. doi: 10.1200/JOP.0912001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walker MS, et al. Smoking relapse during the first year after treatment for early-stage non-small-cell lung cancer. Cancer Epidemiol Biomarkers Prev. 2006;15(12):2370–7. doi: 10.1158/1055-9965.EPI-06-0509. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y, Gao W, Zhang D. Effects of cigarette smoke extract on A549 cells and human lung fibroblasts treated with transforming growth factor-beta1 in a coculture system. Clin Exp Med. 2010;10(3):159–67. doi: 10.1007/s10238-009-0081-x. [DOI] [PubMed] [Google Scholar]
- 13.Sohal SS, et al. Reticular basement membrane fragmentation and potential epithelial mesenchymal transition is exaggerated in the airways of smokers with chronic obstructive pulmonary disease. Respirology. 2010;15(6):930–8. doi: 10.1111/j.1440-1843.2010.01808.x. [DOI] [PubMed] [Google Scholar]
- 14.Kasai H, et al. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT) Respir Res. 2005;6:56. doi: 10.1186/1465-9921-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Willis BC, et al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166(5):1321–32. doi: 10.1016/s0002-9440(10)62351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodier JM, et al. pp60c-src is a positive regulator of growth factor-induced cell scattering in a rat bladder carcinoma cell line. J Cell Biol. 1995;131(3):761–73. doi: 10.1083/jcb.131.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kong D, et al. miR-200 regulates PDGF-D-mediated epithelial-mesenchymal transition, adhesion, and invasion of prostate cancer cells. Stem Cells. 2009;27(8):1712–21. doi: 10.1002/stem.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Higgins DF, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117(12):3810–20. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nawshad A, et al. Transforming growth factor-beta signaling during epithelial-mesenchymal transformation: implications for embryogenesis and tumor metastasis. Cells Tissues Organs. 2005;179(1–2):11–23. doi: 10.1159/000084505. [DOI] [PubMed] [Google Scholar]
- 20.Cannito S, et al. Epithelial-mesenchymal transition: from molecular mechanisms, redox regulation to implications in human health and disease. Antioxid Redox Signal. 2010;12(12):1383–430. doi: 10.1089/ars.2009.2737. [DOI] [PubMed] [Google Scholar]
- 21.Torres M, Forman HJ. Activation of several MAP kinases upon stimulation of rat alveolar macrophages: role of the NADPH oxidase. Arch Biochem Biophys. 1999;366:231–239. doi: 10.1006/abbi.1999.1225. [DOI] [PubMed] [Google Scholar]
- 22.Lee YJ, Han HJ. Troglitazone ameliorates high glucose-induced EMT and dysfunction of SGLTs through PI3K/Akt, GSK-3{beta}, Snail1, and {beta}-catenin in renal proximal tubule cells. Am J Physiol Renal Physiol. 2009 doi: 10.1152/ajprenal.00475.2009. [DOI] [PubMed] [Google Scholar]
- 23.Jain AK, Jaiswal AK. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J Biol Chem. 2007;282(22):16502–10. doi: 10.1074/jbc.M611336200. [DOI] [PubMed] [Google Scholar]
- 24.Zhuang S, Schnellmann RG. H2O2-induced transactivation of EGF receptor requires Src and mediates ERK1/2, but not Akt, activation in renal cells. Am J Physiol Renal Physiol. 2004;286(5):F858–65. doi: 10.1152/ajprenal.00282.2003. [DOI] [PubMed] [Google Scholar]
- 25.Mehdi MZ, et al. H2O2-induced phosphorylation of ERK1/2 and PKB requires tyrosine kinase activity of insulin receptor and c-Src. Antioxid Redox Signal. 2005;7(7–8):1014–20. doi: 10.1089/ars.2005.7.1014. [DOI] [PubMed] [Google Scholar]
- 26.Shaw M, Cohen P, Alessi DR. The activation of protein kinase B by H2O2 or heat shock is mediated by phosphoinositide 3-kinase and not by mitogen-activated protein kinase-activated protein kinase-2. Biochem J. 1998;336(Pt 1):241–6. doi: 10.1042/bj3360241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Felton VM, Borok Z, Willis BC. N-acetylcysteine inhibits alveolar epithelialmesenchymal transition. Am J Physiol Lung Cell Mol Physiol. 2009;297(5):L805–12. doi: 10.1152/ajplung.00009.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ingley E. Src family kinases: regulation of their activities, levels and identification of new pathways. Biochim Biophys Acta. 2008;1784(1):56–65. doi: 10.1016/j.bbapap.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 29.Liu X, Feng R. Inhibition of epithelial to mesenchymal transition in metastatic breast carcinoma cells by c-Src suppression. Acta Biochim Biophys Sin (Shanghai) 2010;42(7):496–501. doi: 10.1093/abbs/gmq043. [DOI] [PubMed] [Google Scholar]
- 30.Choi YL, et al. LYN is a mediator of epithelial-mesenchymal transition and a target of dasatinib in breast cancer. Cancer Res. 2010;70(6):2296–306. doi: 10.1158/0008-5472.CAN-09-3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez-Orozco R, et al. Arachidonic acid promotes epithelial-to-mesenchymal-like transition in mammary epithelial cells MCF10A. Eur J Cell Biol. 2010;89(6):476–88. doi: 10.1016/j.ejcb.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 32.Yang SZ, et al. HBx protein induces EMT through c-Src activation in SMMC-7721 hepatoma cell line. Biochem Biophys Res Commun. 2009;382(3):555–60. doi: 10.1016/j.bbrc.2009.03.079. [DOI] [PubMed] [Google Scholar]
- 33.Cicchini C, et al. TGFbeta-induced EMT requires focal adhesion kinase (FAK) signaling. Exp Cell Res. 2008;314(1):143–52. doi: 10.1016/j.yexcr.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 34.Ulianich L, et al. ER stress is associated with dedifferentiation and an epithelial-to-mesenchymal transition-like phenotype in PC Cl3 thyroid cells. J Cell Sci. 2008;121(Pt 4):477–86. doi: 10.1242/jcs.017202. [DOI] [PubMed] [Google Scholar]
- 35.Xu KP, Yin J, Yu FS. SRC-family tyrosine kinases in wound- and ligand-induced epidermal growth factor receptor activation in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2006;47(7):2832–9. doi: 10.1167/iovs.05-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giannoni E, Taddei ML, Chiarugi P. Src redox regulation: again in the front line. Free Radic Biol Med. 2010;49(4):516–27. doi: 10.1016/j.freeradbiomed.2010.04.025. [DOI] [PubMed] [Google Scholar]
- 37.Giannoni E, et al. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol Cell Biol. 2005;25(15):6391–403. doi: 10.1128/MCB.25.15.6391-6403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meng D, et al. Insulin-like growth factor-I (IGF-I) induces epidermal growth factor receptor transactivation and cell proliferation through reactive oxygen species. Free Radic Biol Med. 2007;42(11):1651–60. doi: 10.1016/j.freeradbiomed.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, et al. Mechanical strain-induced RhoA activation requires NADPH oxidase-mediated ROS generation in caveolae. Antioxid Redox Signal. 2010;13(7):959–73. doi: 10.1089/ars.2009.2908. [DOI] [PubMed] [Google Scholar]
- 40.Yan SR, Berton G. Regulation of Src family tyrosine kinase activities in adherent human neutrophils. Evidence that reactive oxygen intermediates produced by adherent neutrophils increase the activity of the p58c-fgr and p53/56lyn tyrosine kinases. J Biol Chem. 1996;271(38):23464–71. doi: 10.1074/jbc.271.38.23464. [DOI] [PubMed] [Google Scholar]
- 41.Mallozzi C, Di Stasi AM, Minetti M. Activation of src tyrosine kinases by peroxynitrite. FEBS Lett. 1999;456(1):201–206. doi: 10.1016/s0014-5793(99)00945-x. [DOI] [PubMed] [Google Scholar]
- 42.Lee EK, et al. Decreased expression of glutaredoxin 1 is required for transforming growth factor-beta1-mediated epithelial-mesenchymal transition of EpRas mammary epithelial cells. Biochem Biophys Res Commun. 2010;391(1):1021–7. doi: 10.1016/j.bbrc.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 43.Rhyu DY, et al. Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol. 2005;16(3):667–75. doi: 10.1681/ASN.2004050425. [DOI] [PubMed] [Google Scholar]
- 44.Wu WS, et al. Reactive oxygen species mediated sustained activation of protein kinase C alpha and extracellular signal-regulated kinase for migration of human hepatoma cell Hepg2. Mol Cancer Res. 2006;4(10):747–58. doi: 10.1158/1541-7786.MCR-06-0096. [DOI] [PubMed] [Google Scholar]
- 45.Cannito S, et al. Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis. 2008;29(12):2267–78. doi: 10.1093/carcin/bgn216. [DOI] [PubMed] [Google Scholar]
- 46.Wu WS. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006;25(4):695–705. doi: 10.1007/s10555-006-9037-8. [DOI] [PubMed] [Google Scholar]
- 47.Tollefson AK, et al. Endogenous enzymes (NOX and ECSOD) regulate smoke-induced oxidative stress. Free Radic Biol Med. 2010;49(12):1937–46. doi: 10.1016/j.freeradbiomed.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hanke JH, et al. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271(2):695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 49.Bain J, et al. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371(Pt 1):199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dasgupta P, et al. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int J Cancer. 2009;124(1):36–45. doi: 10.1002/ijc.23894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhong Q, et al. Role of ER Stress in EMT of Alveolar Epithelial Cells: Effects of Misfolded Surfactant Protein. Am J Respir Cell Mol Biol. 2010 doi: 10.1165/rcmb.2010-0347OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lambert C, et al. Acrolein in cigarette smoke inhibits T-cell responses. J Allergy Clin Immunol. 2005;116(4):916–22. doi: 10.1016/j.jaci.2005.05.046. [DOI] [PubMed] [Google Scholar]
- 53.Eiserich JP, et al. Dietary antioxidants and cigarette smoke-induced biomolecular damage: a complex interaction. Am J Clin Nutr. 1995;62(6 Suppl):1490S–1500S. doi: 10.1093/ajcn/62.6.1490S. [DOI] [PubMed] [Google Scholar]
- 54.Aoshiba K, et al. Immunohistochemical evaluation of oxidative stress in murine lungs after cigarette smoke exposure. Inhal Toxicol. 2003;15(10):1029–1038. doi: 10.1080/08958370390226431. [DOI] [PubMed] [Google Scholar]
- 55.Kirkham PA, et al. Cigarette smoke triggers macrophage adhesion and activation: role of lipid peroxidation products and scavenger receptor. Free Radic Biol Med. 2003;35(7):697–710. doi: 10.1016/s0891-5849(03)00390-3. [DOI] [PubMed] [Google Scholar]
- 56.Rahman I, et al. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;166(4):490–495. doi: 10.1164/rccm.2110101. [DOI] [PubMed] [Google Scholar]
- 57.Mandal M, et al. Epithelial to mesenchymal transition in head and neck squamous carcinoma: association of Src activation with E-cadherin down-regulation, vimentin expression, and aggressive tumor features. Cancer. 2008;112(9):2088–100. doi: 10.1002/cncr.23410. [DOI] [PubMed] [Google Scholar]
- 58.Forman HJ, et al. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Arch Biochem Biophys. 2008;477(2):183–95. doi: 10.1016/j.abb.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schmitt JM, Stork PJ. Galpha and Gbeta gamma require distinct Src-dependent pathways to activate Rap1 and Ras. J Biol Chem. 2002;277(45):43024–32. doi: 10.1074/jbc.M204006200. [DOI] [PubMed] [Google Scholar]
- 60.Pierce KL, et al. Epidermal growth factor (EGF) receptor-dependent ERK activation by G protein-coupled receptors: a co-culture system for identifying intermediates upstream and downstream of heparin-binding EGF shedding. J Biol Chem. 2001;276(25):23155–60. doi: 10.1074/jbc.M101303200. [DOI] [PubMed] [Google Scholar]
- 61.Touyz RM, et al. Increased angiotensin II-mediated Src signaling via epidermal growth factor receptor transactivation is associated with decreased C-terminal Src kinase activity in vascular smooth muscle cells from spontaneously hypertensive rats. Hypertension. 2002;39(2 Pt 2):479–85. doi: 10.1161/hy02t2.102909. [DOI] [PubMed] [Google Scholar]
- 62.Bobe R, et al. Evidence for ERK1/2 activation by thrombin that is independent of EGFR transactivation. Am J Physiol Heart Circ Physiol. 2003;285(2):H745–54. doi: 10.1152/ajpheart.01042.2002. [DOI] [PubMed] [Google Scholar]
- 63.Scapoli L, et al. Src-dependent ERK5 and Src/EGFR-dependent ERK1/2 activation is required for cell proliferation by asbestos. Oncogene. 2004;23(3):805–13. doi: 10.1038/sj.onc.1207163. [DOI] [PubMed] [Google Scholar]
- 64.Li Z, et al. Src tyrosine kinase inhibitor PP2 suppresses ERK1/2 activation and epidermal growth factor receptor transactivation by X-irradiation. Biochem Biophys Res Commun. 2006;341(2):363–8. doi: 10.1016/j.bbrc.2005.12.193. [DOI] [PubMed] [Google Scholar]
- 65.Cuevas B, et al. SHP-1 regulates Lck-induced phosphatidylinositol 3-kinase phosphorylation and activity. J Biol Chem. 1999;274(39):27583–9. doi: 10.1074/jbc.274.39.27583. [DOI] [PubMed] [Google Scholar]
- 66.Li Y, et al. The c-Src tyrosine kinase regulates signaling of the human DF3/MUC1 carcinoma-associated antigen with GSK3 beta and beta-catenin. The Journal of biological chemistry. 2001;276(9):6061–4. doi: 10.1074/jbc.C000754200. [DOI] [PubMed] [Google Scholar]
- 67.Fukazawa H, Mizuno S, Uehara Y. Effects of herbimycin A and various SH-reagents on p60v-src kinase activity in vitro. Biochem Biophys Res Commun. 1990;173(1):276–82. doi: 10.1016/s0006-291x(05)81053-8. [DOI] [PubMed] [Google Scholar]
- 68.Akhand AA, et al. Nitric oxide controls src kinase activity through a sulfhydryl group modification-mediated Tyr-527-independent and Tyr-416-linked mechanism. J Biol Chem. 1999;274(36):25821–6. doi: 10.1074/jbc.274.36.25821. [DOI] [PubMed] [Google Scholar]
- 69.Krasnowska EK, et al. N-acetyl-l-cysteine fosters inactivation and transfer to endolysosomes of c-Src. Free radical biology & medicine. 2008;45(11):1566–72. doi: 10.1016/j.freeradbiomed.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 70.Oo ML, et al. Cysteine residues in the C-terminal lobe of Src: their role in the suppression of the Src kinase. Oncogene. 2003;22(9):1411–7. doi: 10.1038/sj.onc.1206286. [DOI] [PubMed] [Google Scholar]
- 71.Cheng SE, et al. Cigarette smoke extract regulates cytosolic phospholipase A2 expression via NADPH oxidase/MAPKs/AP-1 and p300 in human tracheal smooth muscle cells. J Cell Biochem. 2010 doi: 10.1002/jcb.22949. [DOI] [PubMed] [Google Scholar]
- 72.Kemble DJ, Sun G. Direct and specific inactivation of protein tyrosine kinases in the Src and FGFR families by reversible cysteine oxidation. Proc Natl Acad Sci U S A. 2009;106(13):5070–5. doi: 10.1073/pnas.0806117106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bogeski I, et al. Inhibition of protein tyrosine phosphatase 1B by reactive oxygen species leads to maintenance of Ca2+ influx following store depletion in HEK 293 cells. Cell Calcium. 2006;40(1):1–10. doi: 10.1016/j.ceca.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 74.Lee K, Esselman WJ. Inhibition of PTPs by H(2)O(2) regulates the activation of distinct MAPK pathways. Free Radic Biol Med. 2002;33(8):1121–32. doi: 10.1016/s0891-5849(02)01000-6. [DOI] [PubMed] [Google Scholar]
- 75.Boivin B, et al. A modified cysteinyl-labeling assay reveals reversible oxidation of protein tyrosine phosphatases in angiomyolipoma cells. Proc Natl Acad Sci U S A. 2008;105(29):9959–64. doi: 10.1073/pnas.0804336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mills JE, et al. A novel disulfide bond in the SH2 Domain of the C-terminal Src kinase controls catalytic activity. J Mol Biol. 2007;365(5):1460–8. doi: 10.1016/j.jmb.2006.10.076. [DOI] [PMC free article] [PubMed] [Google Scholar]