

Abstract
Inbred mouse strains MRL and LG share the ability to fully heal ear hole punches with the full range of appropriate tissues without scarring. They also share a common ancestry, MRL being formed from a multi-strain cross with two final backcrosses to LG before being inbred by brother-sister mating. Many gene-mapping studies for healing ability have been performed using these two strains, resulting in the location of about 20 quantitative trait loci (QTLs). Here, we combine two of these crosses (N=638), MRL/lpr × C57BL/6NTac and LG/J × SM/J, in a single combined cross analysis to increase the mapping power, decrease QTL support intervals, separate multiple QTLs and establish allelic states at individual QTL. The combined cross analysis located 11 QTLs, 6 affecting only one cross (5 LG × SM and 1 MRL × B6) and 5 affecting both crosses, approximately the number of common QTLs expected given strain SNP similarity. Amongst the five QTLs mapped in both crosses, three had significantly different genetic effects, additive in one cross and over or underdominant in the other. It is possible that allelic states at these three loci are different in SM and B6 because they lead to differences in dominance interactions with the LG and MRL alleles. QTL support intervals are 40% smaller in the combined cross analysis than in either of the single crosses. Combined cross analysis was successful in enhancing the interpretation of earlier QTL results for these strains.
Keywords: wound healing, quantitative trait loci, mouse, combined cross, tissue regeneration
Introduction
Ear hole closure in mammals was first identified in rabbits, where tissue replacement consisted of architecturally perfect tissue, including the re-growth of cartilage (Goss & Grimes, 1975; Labandeira-Garcia and Guerra-Seijas, 1986; Urist et al., 1997). Ear hole closure in the mouse, however, is not a general characteristic and appears to be unique to one strain of mouse, the MRL mouse, both MRL/lpr and MRL/MpJ (Desquenne-Clark et al., 1998), and its ancestral parent the LG/J mouse (Kench et al., 1999; Li et al., 2001). This type of healing has characteristics that are similar to amphibian limb regeneration. First, rapid re-epithelialization is seen, a breakdown in the basement membrane follows, and then the formation of a blastema, a key tissue structure in limb outgrowth in the regenerative response, begins (Gourevich et al., 2003). Unusual healing properties in this mouse strain have been reported in other tissues as well including heart (Leferovich et al., 2001; Naseem et al., 2007), cornea (Ueno et al., 2005), digit (Chadwick et al., 2007; Gourevitch et al., 2009), and articular joint cartilage (Fitzgerald et al., 2008).
Genetic mapping studies of ear hole closure have been carried out using crosses between MRL/lpr × C57BL/6NTac (McBrearty et al., 1998; Blankenhorn et al., 2003), MRL/MpJ × SJL/J (Masinde et al., 2001; Yu et al., 2007), MRL/MpJ × CAST/Ei (Heber-Katz et al., 2004; Yu et al., 2005), and LG/J × SM/J (Blankenhorn et al., 2009). Quantitative trait loci (QTL) have been identified using various analyses including composite interval mapping (Zeng, 1994) and Bayesian shrinkage estimation (Yu et al., 2007). More than 20 QTL were identified using these techniques. The healing response was shown to be sexually dimorphic with females closing ear holes more completely, and with some QTL effects restricted to males or females (Blankenhorn et al., 2003). All of these crosses utilized either MRL or LG/J and thus the results can be compared across studies.
In the current study, we combine data from the MRL/lpr × C57BL/6NTac (Blankenhorn et al., 2003) and LG/J × SM/J (Blankenhorn et al., 2009) crosses in a combined cross analysis (Li et al., 2005) to determine the extent to which mapped QTLs are common across studies. Li et al., 2005 have shown that combined cross analysis can increase the power to detect QTLs, narrow QTL-support intervals, separate multiple linked QTLs and establish allelic states for haplotype analysis. Given that MRL/lpr and LG/J share 91% of their genome identical by descent (http://cgd.jax.org/straincomparison/), we expect that they will share some QTL map positions when crossed to the unrelated C57BL/6NTac and SM/J strains.
Materials and methods
Animals
MRL/lpr (MRL), LG/J (LG) and SM/J (SM) male and female mice were obtained from the Jackson Laboratories (Bar Harbor, ME, USA). C57BL/6NTac (B6) mice were obtained from Taconic Laboratories, (Hudson, NY, USA). (MRL × B6) F1 and F2 crosses were generated at the Wistar Institute, whereas (LG × SM) F1 crosses were generated at Washington University, St Louis, and F2 mice were generated at the Wistar Institute. The sample consists of 638 F2 mice, 356 from the MRL × B6 cross and 282 from the LG × SM cross. DNA was extracted from liver and microsatellite typing was carried out at the Wistar Institute and Drexel University and SNP typing was carried out using Illumina arrays by the Wistar Genomics Core Facility. Ear pinnae were punched (2 mm deficit) bilaterally in 6-week-old mice and read on day 15 and day 30 post-injury, as described in McBrearty et al. (1998).
Mapping analysis
The genotypes from each cross were combined in a common map using R/QTL (Browman and Sen, 2009) as shown in Supplementary Table 1. Genotypes and wound healing phenotypes are provided for all individuals in the Dryad repository: doi:10.5061/dryad.fh2hp7k4, with genotypes imputed at 1-c intervals using the method of Haley and Knott (1992). All mapping analyses used the same combined genetic map. Three separate analyses were performed: in the LG × SM cross alone, in the MRL × B6 cross alone, and in the combined cross. The model for analyzing the separate crosses is
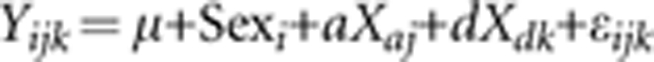 |
where Yijk is the size of the hole at 30 days, Sexi is male or female, ‘a' is the additive genotypic value (half the difference between the homozygotes), ‘d' is the dominance genotypic value (deviation of the heterozygotes from the midpoint between the homozygotes), and ɛijk is the residual. Additive genotype scores (Xa) were +1 for the LG/LG and MRL/MRL homozygotes, 0 for the LG/SM and MRL/B6 heterozygotes, and –1 for the SM/SM and B6/B6 homozygotes. Dominance genotype scores (Xd) are 0 for homozygotes and +1 for heterozygotes.
After analysis of the pooled-sex model given above, we tested for genotype by sex interactions within each of the crosses using,
where ‘as' and ‘ds' are the additive and dominance genotype by sex interaction coefficients. Sex interaction's significance was determined using the difference in −2ln (likelihood) between the two models distributed as χ2 with 2 degrees of freedom. Significant interactions indicate significant differences in genetic effects across the sexes. When such variation is present, we mapped the genes separately by sex within each cross.
We performed a combined-cross genome scan using F2 intercross animals from the cross of LG × SM and the cross of MRL × B6 together. Mapping was performed on the combined cross using the following model:
where Yijk is the size of the hole at 30 days, Crossi specifies the LG × SM cross or the MRL × B6 cross, ‘ac' is the coefficient for the additive genotypic value by cross interaction, ‘dc' is the coefficient for the dominance genotypic value by cross interaction, and ɛijk is the residual. The sexes were pooled in this combined analysis. Significant interaction terms indicate that the genetic effects are significantly different in the two crosses.
Statistical significance was determined using the Bonferroni correction based on the number of independent tests in the genome scan (Cheverud, 2001; Li and Ji, 2005). Probabilities are presented as logarithm of probability (LPR) scores (=−log10(probability)), corresponding to the usual logarithm of odds (LOD) scores. The genome-wide LPR significance threshold is 3.29. We also generated chromosome-specific thresholds to avoid accumulating type 1 errors (Chen and Storey, 2006). In a genome scan using chromosome-specific thresholds, we expect one false positive chromosome by chance when there are no QTL effects in the population. Chromosome-specific thresholds (see Supplementary Table 1) range from 2.194 down to 1.895, becoming smaller for the shorter chromosomes. Confidence regions were defined by a one LPR drop on either side of the peak LPR score.
Results
LG × SM intercross QTL
There were 12 QTLs mapped to chromosomes (chr) 2, 4, 6, 8, 9, 10 (3 loci), 11, 14, 18 and 19 in the LG × SM intercross (see Table 1; see Figure 1a). The LG allele significantly improves healing on chromosomes 2, 6, 9, 11 and 18, whereas at the three chr 10 QTLs, the SM allele improves healing. The average absolute additive genotypic value for QTLs with significant additive (a) effects is small, 0.15 mm, representing a 0.30 mm difference in extent of healing between LG and SM homozygotes at a single QTL locus. On an s.d. scale, significant differences between homozygotes range from 0.25 to 0.66 s.d. units. The average QTL support interval is 23 c.
Table 1. Healing QTLs discovered in the LG/J × SM/J F2 intercross.
| Chr | cM | −CI | +CI | SEX | Rsq | Full_LPR | a (LPR) | d (LPR) | Int | Int (s.e.) | a | a (s.e.) | d | d (s.e.) | a/s.d. | d/s.d. | LL | LS | SS | Name |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 11 | 0 | 40 | M | 0.057 | 2.12 | 2.04 | 1.34 | 0.97 | 0.05 | −0.13 | 0.05 | −0.18 | 0.09 | −0.34 | −0.48 | 0.84 | 0.79 | 1.10 | Heal15 |
| 4 | 88 | 72 | 95 | B | 0.024 | 1.85 | 0.17 | 2.46 | 0.69 | 0.04 | 0.01 | 0.03 | 0.16 | 0.05 | 0.04 | 0.42 | 0.70 | 0.85 | 0.67 | Heal8 |
| 6 | 71 | 28 | 87 | B | 0.030 | 2.27 | 2.81 | 0.30 | 0.76 | 0.03 | −0.09 | 0.03 | 0.03 | 0.04 | −0.25 | 0.07 | 0.66 | 0.78 | 0.85 | New |
| 8 | 51 | 34 | 72 | B | 0.020 | 1.49 | 0.20 | 2.05 | 0.85 | 0.04 | 0.02 | 0.03 | −0.14 | 0.05 | 0.04 | −0.37 | 0.86 | 0.71 | 0.83 | Heal1 |
| 9 | 58 | 39 | 64 | B | 0.171 | 14.06 | 14.75 | 0.49 | 0.79 | 0.03 | −0.23 | 0.03 | −0.04 | 0.04 | −0.61 | −0.11 | 0.55 | 0.74 | 1.02 | Sth8−9 |
| 30 | 24 | 66 | M | 0.144 | 5.58 | 5.60 | 0.78 | 0.91 | 0.04 | −0.20 | 0.04 | −0.08 | 0.06 | −0.53 | −0.20 | 0.71 | 0.83 | 1.11 | Sth8a | |
| 57 | 54 | 63 | F | 0.226 | 9.92 | 9.96 | 1.42 | 0.73 | 0.04 | −0.25 | 0.04 | −0.12 | 0.06 | −0.66 | −0.31 | 0.47 | 0.61 | 0.98 | Sth9b | |
| 10 | 29 | 24 | 37 | B | 0.068 | 5.29 | 5.89 | 0.51 | 0.75 | 0.03 | 0.15 | 0.03 | 0.05 | 0.04 | 0.39 | 0.12 | 0.89 | 0.79 | 0.60 | Heal16 |
| 10 | 47 | 41 | 53 | B | 0.067 | 5.21 | 5.28 | 1.52 | 0.72 | 0.03 | 0.13 | 0.03 | 0.09 | 0.04 | 0.35 | 0.24 | 0.86 | 0.81 | 0.59 | Chr10b |
| 10 | 63 | 62 | 67 | B | 0.060 | 4.68 | 5.36 | 0.01 | 0.75 | 0.03 | 0.12 | 0.03 | 0.00 | 0.04 | 0.32 | 0.00 | 0.88 | 0.76 | 0.63 | New |
| 11 | 51 | 45 | 56 | B | 0.060 | 4.63 | 4.46 | 1.33 | 0.73 | 0.03 | −0.13 | 0.03 | 0.09 | 0.04 | −0.34 | 0.23 | 0.60 | 0.82 | 0.86 | Heal10 |
| 14 | 17 | 10 | 24 | B | 0.038 | 2.88 | 0.50 | 3.33 | 0.70 | 0.03 | −0.03 | 0.03 | 0.15 | 0.04 | −0.08 | 0.39 | 0.67 | 0.85 | 0.73 | Heal12 |
| 18 | 24 | 7 | 38 | B | 0.031 | 2.33 | 2.84 | 0.29 | 0.76 | 0.03 | −0.09 | 0.03 | 0.03 | 0.04 | −0.24 | 0.07 | 0.67 | 0.79 | 0.85 | Heal9 |
| 19 | 0 | 0 | 4 | B | 0.039 | 3.00 | 0.07 | 3.50 | 0.73 | 0.03 | 0.00 | 0.03 | 0.25 | 0.07 | 0.01 | 0.66 | 0.74 | 0.98 | 0.73 | New |
Abbreviations: a, additive; Chr, chromosome; (−) and (+) CI, proximal and distal confidence interval limits; cM, QTL centiMorgan location; d, dominance; Full, LPR for full model; Int, intercept; LL, LS, SS, mean phenotypes for the respective genotypes with L for LG/J and S for SM/J; Logarithm of Probability (LPR), −log10(Prob); Name, Previous designation or new if the QTL was unreported before; Rsq, r-squared; Sex, (M)ale, (F)emale, (B)oth.
Significant results are in bold face font.
Figure 1.

LOD scores from the genome-wide QTL scan for the (a) LG × SM cross, (b) MRL × B6 cross, and (c) combined cross. Constant genome-wide and variable chromosome-specific significance thresholds are indicated by horizontal lines.
QTLs on chromosomes 6, 9, proximal 10, distal 10 and 18 are co-dominant, whereas the chr 2 and mid-chr 10 QTLs are LG/J dominant and the chr 11 QTL is SM/J dominant. The better-healing phenotype is recessive at mid-chr 10 and chr 11, and dominant on chr 2. Four QTLs (chromosomes 4, 8, 14 and 19) are only significant for the dominance term, three being underdominant (chromosomes 4, 14, 19), the heterozygote healing relatively poorly and one overdominant (chr 8), the heterozygotes healing more fully than the homozygotes. The absolute size of significant dominance effects are similar to those observed for additive effects.
Two chromosomes (chr 2 with LPR=2.695 and chr 9 with LPR 1.76) had significant genotype by sex interactions. Chr 2 was significant only in males and chr 9, although significant in both males and females, exhibited sex-specific peaks at 30 and 57 c, respectively. The confidence interval for the females on chr 9 is quite refined, from 54 to 63 c, whereas the confidence region for the males is broad (24 to 66 c) and is likely due to more than one quantitative trait gene within the interval. The chr 9 effect is also stronger in females than in males, including both additive and dominance genetic components. Certainly, the genetics of chromosome 9 are complex and will require further recombination to resolve. In addition, the LPRs for females were greater than for males for all three chromosome 10 QTLs whereas the reverse was true for the chromosome 11 QTL, even though additive and dominance effect estimates were quite similar across the sexes.
MRL × B6 intercross QTL
The MRL × B6 intercross produced 10 QTLs on chromosomes 1, 4, 8, 9, 11, 12, 13, 16 and 18 (2 QTLs) (see Table 2; Figure 1b). Alleles from the healing strain, MRL, improved healing at four loci (chromosomes 4, 11, 13 and proximal 18) whereas the B6 allele promoted healing on chromosomes 8, 12 and 16. The two QTLs on chr 18, labeled as proximal and distal, are only 20 c apart and have overlapping confidence regions and thus should only be considered provisionally distinct. However, the genetic effects are quite different at the two locations, in that the proximal QTL affects both sexes and has a strong additive effect whereas the more distal location only affects females and has an overdominant effect with heterozygotes healing well. The average absolute additive genotypic value for QTLs with significant additive (a) effects is only 0.11 mm, representing a 0.22 mm difference in extent of healing between MRL and B6 homozygotes at individual QTLs. On a s.d. scale, significant differences between homozygotes range from 0.20 to 0.43 s.d. units. QTL support intervals averaged 25 c.
Table 2. Healing QTLs discovered in the MRL/lpr × C57BL/6NTac F2 intercross.
| Chr | cM | −CI | +CI | SEX | Rsq | Full_LPR | a (LPR) | d (LPR) | Int | Int (s.e.) | a | a (s.e.) | d | d (s.e.) | a/s.d. | d/s.d. | MM | MB | BB | Name |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 101 | 93 | 106 | F | 0.074 | 2.36 | 0.10 | 2.91 | 0.57 | 0.04 | −0.01 | 0.04 | 0.22 | 0.07 | −0.03 | 0.56 | 0.56 | 0.79 | 0.58 | New |
| 4 | 66 | 62 | 79 | B | 0.049 | 3.07 | 3.09 | 0.90 | 0.73 | 0.03 | −0.11 | 0.03 | 0.08 | 0.05 | −0.28 | 0.22 | 0.63 | 0.82 | 0.84 | Heal8 |
| 8 | 51 | 42 | 73 | B | 0.060 | 3.74 | 4.44 | 0.14 | 0.77 | 0.03 | 0.11 | 0.03 | 0.01 | 0.04 | 0.30 | 0.04 | 0.88 | 0.78 | 0.65 | Heal1 |
| 9 | 60 | 48 | 74 | M | 0.077 | 2.32 | 0.10 | 2.95 | 0.74 | 0.04 | −0.01 | 0.04 | 0.24 | 0.07 | −0.03 | 0.62 | 0.73 | 0.98 | 0.75 | Sth9b |
| 11 | 55 | 46 | 72 | M | 0.116 | 3.57 | 4.28 | 0.07 | 0.84 | 0.04 | −0.17 | 0.04 | 0.01 | 0.05 | −0.43 | 0.03 | 0.67 | 0.85 | 1.00 | Heal10 |
| 12 | 13 | 0 | 19 | B | 0.033 | 2.01 | 2.26 | 0.57 | 0.79 | 0.03 | 0.08 | 0.03 | −0.04 | 0.04 | 0.20 | −0.11 | 0.87 | 0.75 | 0.71 | New |
| 13 | 30 | 21 | 37 | B | 0.067 | 4.22 | 3.81 | 1.48 | 0.81 | 0.03 | −0.10 | 0.03 | −0.08 | 0.04 | −0.27 | −0.21 | 0.71 | 0.73 | 0.91 | Heal7 |
| 16 | 12 | 3 | 22 | M | 0.082 | 2.48 | 1.74 | 1.86 | 0.77 | 0.04 | 0.10 | 0.04 | 0.17 | 0.07 | 0.26 | 0.44 | 0.87 | 0.94 | 0.67 | Heal11 |
| 18 | 24 | 0 | 36 | B | 0.022 | 1.35 | 1.84 | 0.33 | 0.79 | 0.03 | −0.07 | 0.03 | −0.03 | 0.05 | −0.19 | −0.09 | 0.72 | 0.76 | 0.87 | Heal9 |
| 18 | 43 | 0 | 47 | F | 0.067 | 2.13 | 0.01 | 2.74 | 0.77 | 0.04 | 0.00 | 0.04 | −0.18 | 0.06 | 0.00 | −0.47 | 0.77 | 0.58 | 0.77 | Heal9 |
Abbreviations: a, additive; Chr, chromosome; (−) and (+) CI, proximal and distal confidence interval limits; cM, QTL centiMorgan location; d, dominance; Full, LPR for full model; Int, intercept; MM, MB, BB, mean phenotype for the respective genotypes with M for MRL allele and B for C57BL/6 allele; Logarithm of Probability (LPR), −log10(Prob); Name, Previous designation or new if the QTL was unreported before; Rsq, r-squared; Sex, (M)ale, (F)emale, (B)oth.
Significant results are in bold face font.
Four of the loci (chromosomes 4, 8, 11 and 12) are co-dominant with healing intermediate in the heterozygotes, whereas chr 1 and chr 9 are underdominant, the heterozygotes healing poorly relative to both homozygotes, and distal chr 18 is overdominant, the heterozygotes healing more completely than either homozygote. On chr 13 the heal-promoting MRL allele is dominant to the B6 allele and on chr 16 the non-healing MRL allele is dominant to the B6 allele. Absolute dominance genotypic values range from 0.08 to 0.24 mm or 0.2 to 0.6 s.d. units.
Half of the QTLs in the MRL/J by C57BL/6J cross affect both sexes (chromosomes 4, 8, 12, 13 and proximal 18) whereas the other half are sex-specific (chr 1 (LPR for sex interaction=1.94), chr 9 (LPR=2.95), chr 11 (LPR=2.06), chr 16 (LPR=1.54) and distal chr 18 (LPR=2.76)), three with male-specific effects (chromosomes 9, 11 and 16) and two with female-specific effects (chr 1 and distal chr 18).
Combined cross-analysis
The combined cross-genome scan identified 11 QTLs on chromosomes 4, 8, 9, 10, 11, 13, 14, 18 and 19, including three distinct peaks on chr 10 (see Table 3; Figure 1c). Five of these 11 were significant at the genome-wide level whereas 6 were significant at the chromosome-specific level. The average support interval is 15 c. Of these 11 QTLs, 9 had significant cross by genotype interactions indicating differential mapping in the two crosses. Five of these nine were loci that mapped in the LG × SM cross but not in the MRL × B6 cross, three on chr 10, and one each on chr 14 and chr 19, one that mapped only in the MRL × B6 cross (chr 13), and three (chromosomes 4, 8 and 9) that mapped in both crosses to the same locations but displayed different patterns of genetic effects in the two intercrosses. The significance of the differences in effects on chromosomes 4, 8 and 9 is indicated by their significant interaction LPR scores (‘ac' and ‘dc'). On chromosomes 4 and 8 the MRL × B6 intercross displayed additive effects whereas LG × SM intercross displayed underdominance and overdominance, respectively. On chr 9 the effect was additive in the LG × SM intercross and under-dominant in the MRL × B6 intercross. Loci were also mapped in the separate crosses that were not significant in the combined-cross analysis, including QTLs on chromosomes 2 and 6 in the LG × SM intercross and QTLs in the MRL × B6 intercross on chromosomes 1, 12, 16 and distal 18.
Table 3. Healing QTLs discovered in the Combined F2 intercross.
| Chr | Pop | cM | −CI | +CI | Rsq | Full LPR | a (LPR) | d (LPR) | ac (LPR) | dc (LPR) | Int | Int (s.e.) | a | a (s.e.) | d | d (s.e.) | ac | ac (s.e.) | dc | dc (s.e.) | Name |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 | B* | 88 | 79 | 94 | 0.034 | 3.16 | 1.22 | 1.34 | 1.99 | 0.48 | −0.13 | 0.07 | 0.14 | 0.07 | 0.24 | 0.12 | −0.12 | 0.05 | −0.08 | 0.08 | Heal8 |
| 8 | B* | 51 | 43 | 68 | 0.034 | 3.27 | 0.57 | 2.17 | 1.52 | 1.68 | 0.16 | 0.07 | −0.08 | 0.07 | −0.30 | 0.11 | 0.10 | 0.04 | 0.16 | 0.07 | Heal1 |
| 9 | B* | 58 | 54 | 63 | 0.115 | 14.35 | 10.22 | 1.12 | 4.93 | 1.34 | 0.08 | 0.06 | −0.42 | 0.06 | −0.18 | 0.10 | 0.19 | 0.04 | 0.13 | 0.07 | Sth9b |
| 10 | L | 29 | 23 | 36 | 0.043 | 4.39 | 4.70 | 0.59 | 2.66 | 0.47 | −0.07 | 0.07 | 0.30 | 0.07 | 0.12 | 0.11 | −0.15 | 0.05 | −0.08 | 0.08 | Heal16 |
| 10 | L | 47 | 41 | 53 | 0.043 | 4.33 | 4.11 | 1.13 | 2.30 | 0.64 | −0.09 | 0.06 | 0.25 | 0.06 | 0.17 | 0.09 | −0.12 | 0.04 | −0.08 | 0.06 | Chr10b |
| 10 | L | 63 | 62 | 67 | 0.041 | 4.03 | 3.28 | 0.02 | 1.40 | 0.03 | −0.03 | 0.06 | 0.21 | 0.06 | −0.01 | 0.09 | −0.08 | 0.04 | 0.01 | 0.06 | New |
| 11 | B | 51 | 45 | 56 | 0.052 | 5.51 | 1.71 | 0.83 | 0.27 | 0.37 | −0.07 | 0.07 | −0.16 | 0.07 | 0.14 | 0.10 | 0.03 | 0.04 | −0.05 | 0.06 | Heal10 |
| 13 | M | 30 | 18 | 40 | 0.027 | 2.37 | 1.40 | 1.18 | 2.33 | 1.47 | −0.09 | 0.07 | 0.14 | 0.07 | 0.17 | 0.09 | −0.12 | 0.04 | −0.13 | 0.06 | Heal7 |
| 14 | L | 17 | 10 | 26 | 0.024 | 2.03 | 0.54 | 3.20 | 0.44 | 2.21 | −0.16 | 0.06 | −0.07 | 0.06 | 0.32 | 0.09 | 0.04 | 0.04 | −0.17 | 0.06 | Heal12 |
| 18 | B | 24 | 6 | 37 | 0.027 | 2.43 | 1.12 | 0.46 | 0.19 | 0.46 | −0.04 | 0.06 | −0.11 | 0.06 | 0.09 | 0.09 | 0.02 | 0.04 | −0.06 | 0.06 | Heal9 |
| 19 | L | 0 | 0 | 4 | 0.026 | 2.23 | 0.23 | 3.71 | 0.31 | 3.08 | −0.09 | 0.06 | 0.03 | 0.06 | 0.52 | 0.14 | −0.03 | 0.04 | −0.26 | 0.08 | New |
Abbreviations: a, additive; ac, additive by cross interaction; Chr, chromosome; (−) and (+) CI, proximal and distal confidence interval limits; cM, QTL centiMorgan location; d, dominance; dc, dominance by cross interaction; Full, LPR for full model; Int, intercept; MM, MB, BB, mean phenotype for the respective genotypes with M for MRL allele and B for C57BL/6 allele; Logarithm of Probability (LPR), −log10(Prob); Name, Previous designation or new if the QTL was unreported before; Pop, population (L=LG × SM intercross specific, M=MRL × B6 intercross specific, B, both intercrosses); Rsq, r-squared; Sex, (M)ale, (F)emale, (B)oth. Significant results are in bold face font.
*Significantly different genetic architecture in the two intercrosses.
Overall, in the three analyses there were 17 QTLs discovered with 7 specific to the LG/J by SM/J cross, 5 to the MRL by C57BL/6J cross, and five locations where QTLs mapped in both crosses, although genetic patterns of variation differed between the crosses at three of these common localities.
Discussion
Each of the two crosses contain a similar number of QTLs, 12 in the LG × SM cross and 10 in the MRL × B6 cross. Five QTLs are shared between strains. Estimates based on SNP genotypes for inbred strains scored for the 623 000 SNPs on the Mouse Genetic Diversity Array (Yang et al., 2009) indicate that approximately 67% of the genome is identical by descent among unrelated strains. LG and MRL share 91% of their genomes identical by descent: 100% in identical regions directly shared due to genealogy (75% of the genome) and 67% identity in the remaining 25% of the genome (1.00 × 0.75+0.67 × 0.25=0.91). Given these values, we expect the two crosses to share about 41% (=0.91 × 0.67 × 0.67) of their QTLs. With 11 QTLs in the combined cross analysis, we expect 4.5 shared QTLs and we observe five shared positions. This result is approximately what would be expected, given the proportions of shared and distinct SNPs among the strains in these crosses. Even then, the genetic effects at three of these five shared locations (chromosomes 4, 8 and 9) differ raising the possibility that they represent the effects of different linked loci. Using the 13 377 Oxford/CTC SNPs (http://www.well.ox.ac.uk/mouse/INBREDS/) scored in LG/J and MRL/MpJ, it is possible to determine whether there is potential polymorphism between LG and MRL within the confidence regions of these shared QTLs with different effects. There are no polymorphic SNPs in the confidence regions of the chr 4 (140–155 Mb) and chr 9 (84–118 Mb) QTLs so differences in genetic effects at these loci are unlikely to be due to different linked genes mapping in the two crosses. The chr 8 QTL is polymorphic over 40% of its confidence region, with polymorphic blocks present ranging from 61 to 83.7 Mb and from 109 to 128 Mb, and monomorphic over the remaining 60% of the interval. The chr 8 peak is near 96.5 Mb, within the monomorphic interval. Given that LG and MRL carry the same genotypes at these three QTLs, the observed differences in genetic effects in the two crosses could be due to different alleles in the B6 and SM strains that interact differently with the common LG/MRL allele or epistatic interactions with other loci that are different in the two crosses.
Overall, both LG and MRL alleles usually promote healing, although there are several QTL in both crosses where the healing allele comes from the alternate strain. Dominance is variable with most loci having co-dominant alleles. When there is dominance, it is most typical for the healing allele to be recessive with heterozygotes healing poorly relative to the midpoint between the parental strains. There are also several loci for which the heterozygote heals either better than or worse than both homozygotes.
Effects found in each strain vary from 0.2 to 0.6 s.d. units and percentage effects because of individual QTL vary from 2 to 17%, whereas overall about 65% of the phenotypic variance in each cross is accounted for by the QTLs. Thus each cross is approximately as powerful as the other in detecting QTL. These values are probably an overestimate of phenotypic variance and allelic effects because some QTLs are linked, such as those on chromosome 10 in the LG/J by SM/J cross, and sample sizes in the separate crosses, whereas substantial, can still lead to an inflation of allelic effects and of variance explained (Beavis, 1994). Together, we find 50–100% more QTLs than are found in each single cross alone showing the value of mapping in multiple, related crosses. Each cross can bring out different healing alleles depending on which loci happen to be the variable between the parental strains tested. Furthermore, QTL support intervals average 37.5% narrower in the combined cross (15 c) than in the individual cross analyses (24 c). Even then, the reduced intervals contain large numbers of genes. To identify the genes responsible for these QTL more recombination is required. Ongoing studies in the LG, SM Advanced Intercross Line will serve this purpose.
Overall, the results of the two crosses are similar. However, one clear difference is that there are many more genetic variations in sexual dimorphism in the MRL × B6 cross with five sex-specific QTLs, than in the LG/J by SM/J cross with only one or two sex-specific effects. The sexes are dimorphic in both crosses (Blankenhorn et al., 2003, 2009), but only the MRL × B6 cross harbors much genetic variation in sexual dimorphism. This difference between strains likely indicates differences in the interactions between steroid hormones and variable loci in the two crosses.
The results for chr 9 seem very complex. It is most likely that there are at least two loci, one with larger effects in males than in females in the mid-portion of the chromosome and another with larger effects in females than in males on the distal portion of the chromosome. Chr 10 is also complex. Three peaks, with non-overlapping confidence regions, occur at 20 c intervals along the mid-section of the chromosome. All three QTL show the same additive effect, with the SM/J allele promoting healing. The proximal and distal locations have co-dominant effects, whereas at the middle QTL the non-healing LG/J allele is dominant. There is not enough precision in this F2 population to securely locate these closely linked QTLs relative to one another. We expect that the accumulation of recombination in later generations of the LG by SM intercross will clarify these results.
Additional support exists for the likely identity of two genes underlying the QTLs on chr 11 and chr 18 in the combined cross analysis. Both of these QTL share similar dominance and additivity, and with the exception that there is a more pronounced male-specific effect in the MRLxB6 cross for Heal10 on chr 11. Indeed, there is a significant male-preference of the QTL in the LG × SM cross when assessed by composite interval mapping (Blankenhorn et al., 2009) and in an MRL/MpJ × CAST/Ei cross (Heber-Katz et al., 2004). Taken together, evidence points to two closely linked QTL on chr11, a relatively strong one that is male-specific and an additional QTL that is weaker and acts in mice of both sexes. Heal9, on proximal chr18, shares all major genetic parameters in both crosses. Such concordance will make it possible to perform comparative haplotype association mapping studies (Burgess-Herbert et al., 2008) to further narrow the list of candidate genes in the supported intervals to those that share alleles between LG and MRL vs CAST/Ei, SM/J and C57BL/6J.
Most of the QTLs identified here have been previously reported in single cross analyses (see Tables 1, 2, 3; McBrearty et al., 1998; Masinde et al., 2001; Blankenhorn et al., 2003, 2009; Heber-Katz et al., 2004; Yu et al., 2007). However, several new results are also reported. These new results are most likely due to differences in the analytical methods, significance thresholds and sample sizes applied. We identified a QTL on chr 6 (LPR=2.27) in the LG × SM cross as significant at the chromosome-wide level that was sub-significant according to the criteria used by Blankenhorn et al. (2009), a third more distal QTL on chr 10 (LPR=4.68) that is in linkage disequilibrium with the two more proximal QTLs on chr 10 reported by Blankenhorn et al. (2009), and a new QTL on chr 19 (LPR=3.00). The chromosome 10 and 19 QTLs were also significant in the combined-cross analysis with cross by genotype interactions, bolstering support for these locations. Two new locations on chr 1 (LPR=2.36) and chr 12 (LPR=2.01) that are significant at the chromosome-wide level are also reported for the MRL × B6 cross. Although other healing QTLs have been reported on these chromosomes, Sth1 and Heal5, respectively, the locations reported here are distant from their positions. These new results are most likely due to differences in the analytical methods and significance thresholds applied. For example, Blankenhorn et al. (2009), utilized composite interval mapping (Zeng, 1994) and determined significance at genome-wide α=0.32, 0.10, 0.05 and 0.01 levels utilizing permutation analysis (Wang et al., 2009), rather than using interval mapping and chromosome-wide thresholds as done here.
In the combined cross analysis we have provided novel results, not evident in earlier analyses of these crosses (Blankenhorn et al., 2003, 2009): (1) We confirmed that many QTL mapped in the LG × SM cross are not also present in the MRL × B6 cross and vice versa, as evinced by significant cross by genotype interactions; (2) QTL support intervals are 37.5% smaller in the combined cross than in the separate crosses (15 vs 24 c); (3) Even when QTLs map to the same location in the two crosses, there can be significant differences in gene effects at the loci suggesting different alleles in SM and B6 at these loci or different epistatic interactions with the background; (4) Loci on chr 11 and chr 18 have the same allelic contrasts in the two crosses; and (5) Two new QTL loci, distal chr 10 and chr 19, were identified by using different analytical and significance testing approaches.
Data archiving
Data have been deposited at Dryad: doi:10.5061/dryad.fh2hp7k4.
Acknowledgments
This study was supported by a grant from NIH, NIGMS (GM073226).
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on Heredity website (http://www.nature.com/hdy)
Supplementary Material
References
- Beavis W. The power and deceit of QTL experiments: lessons from comparative QTL studies. Proc Corn Sorghum Ind Res Conf. 1994;49:250–266. [Google Scholar]
- Blankenhorn EP, Bryan G, Kossenkov AV, Desquenne Clark L, Zhang X-M, Celia Chang C, et al. Loci that regulate healing and regeneration in LG/J and SM/J mice. Mamm Genome. 2009;20:720–733. doi: 10.1007/s00335-009-9216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenhorn EP, Troutman S, Clark LD, Zhang XM, Chen P, Heber-Katz E. Sexually dimorphic genes regulate healing and regeneration in MRL mice. Mamm Genome. 2003;14:250–260. doi: 10.1007/s00335-002-2222-3. [DOI] [PubMed] [Google Scholar]
- Browman KW, Sen Ś. A Guide to QTL Mapping with R/qtl. Springer: New York; 2009. [Google Scholar]
- Burgess-Herbert SL, Cox A, Tsaih S-W, Paigen B. Practical applications of the bioinformatics toolbox for narrowing quantitative trait loci. Genetics. 2008;180:2227–2235. doi: 10.1534/genetics.108.090175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick RB, Bu L, Yu H, Wergedal JE, Mohan S, Baylink DJ. Digit tip regrowth and differential gene expression in MRL/Mpj, DBA/2, and C57BL/6 mice. Wound Repair Regen. 2007;15:275–284. doi: 10.1111/j.1524-475X.2007.00216.x. [DOI] [PubMed] [Google Scholar]
- Chen L, Storey LD. Relaxed significance criteria for linkage analysis. Genetics. 2006;173:2371–2381. doi: 10.1534/genetics.105.052506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheverud JM. A simple correction for multiple comparisons in interval mapping genome scans. Heredity. 2001;87:52–58. doi: 10.1046/j.1365-2540.2001.00901.x. [DOI] [PubMed] [Google Scholar]
- Desquenne-Clark L, Clark R, Heber-Katz E. A new model for mammalian wound repair and regeneration. Clin Immun Immunopath. 1998;88:35–45. doi: 10.1006/clin.1998.4519. [DOI] [PubMed] [Google Scholar]
- Fitzgerald J, Rich C, Burkhardt D, Allen J, Herzka AS, Little CB. Evidence for articular cartilage regeneration in MRL/MpJ mice. Osteoarthritis Cartilage. 2008;16:1319–1326. doi: 10.1016/j.joca.2008.03.014. [DOI] [PubMed] [Google Scholar]
- Goss RJ, Grimes LN. Epidermal downgrowths in regenerating rabbit ear holes. J Morphol. 1975;146:533–542. doi: 10.1002/jmor.1051460408. [DOI] [PubMed] [Google Scholar]
- Gourevitch D, Clark L, Bedelbaeva K, Leferovich J, Heber-Katz E. Dynamic changes after murine digit amputation: The MRL mouse digit shows waves of tissue remodeling, growth, and apoptosis. Wound Repair Regen. 2009;17:447–455. doi: 10.1111/j.1524-475X.2009.00492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourevich D, Clark L, Chen P, Seitz A, Samulewicz S, Heber-Katz E. Matrix Metalloproteinase Activity Correlates with Blastema Formation in the Regenerating MRL Ear Hole Model. Dev Dyn. 2003;226:377–387. doi: 10.1002/dvdy.10243. [DOI] [PubMed] [Google Scholar]
- Haley C, Knott S. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity. 1992;69:315–324. doi: 10.1038/hdy.1992.131. [DOI] [PubMed] [Google Scholar]
- Heber-Katz E, Chen P, Clark L, Zhang XM, Troutman S, Blankenhorn EP. Regeneration in MRL mice: further genetic loci controlling the ear hole closure trait using MRL and M.m. castaneus mice. Wound Repair Regen. 2004;12:384–392. doi: 10.1111/j.1067-1927.2004.012308.x. [DOI] [PubMed] [Google Scholar]
- Kench JA, Russell DM, Fadok VA, Young SK, Worthen GS, Jones-Carson J, et al. Aberrant wound healing and TGF-beta production in the autoimmune-prone MRL/+ mouse. Clin Immunol. 1999;92:300–310. doi: 10.1006/clim.1999.4754. [DOI] [PubMed] [Google Scholar]
- Labandeira-Garcia J, Guerra-Seijas M. Intracellular lipids in rabbit ear cartilage during tissue regeneration. Acta Anat (Basel) 1986;127:249–254. doi: 10.1159/000146294. [DOI] [PubMed] [Google Scholar]
- Leferovich J, Bedelbaeva K, Samulewicz S, Xhang X-M, Zwas DR, Lankford EB, et al. Heart regeneration in adult MRL mice. Proc Natl Acad Sci USA. 2001;98:9830–9835. doi: 10.1073/pnas.181329398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ji L. Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity. 2005;95:221–227. doi: 10.1038/sj.hdy.6800717. [DOI] [PubMed] [Google Scholar]
- Li R, Lyons MA, Wittenburg H, Paigen B, Churchill GA. Combining data from multiple inbred line crosses improves the power and resolution of quantitative trait mapping. Genetics. 2005;169:1699–1709. doi: 10.1534/genetics.104.033993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Gu W, Masinde G, Hamilton-Ulland M, Xu S, Mohan S, et al. Genetic control of the rate of wound healing in mice. Heredity. 2001;86:668–674. doi: 10.1046/j.1365-2540.2001.00879.x. [DOI] [PubMed] [Google Scholar]
- Masinde GL, Li X, Gu W, Davidson H, Mohan S, Baylink DJ. Identification of wound healing/regeneration quantitative trait loci (QTL) at multiple time points that explain seventy percent of variance in (MRL/MpJ and SJL/J) mice F2 population. Genome Res. 2001;11:2027–2033. doi: 10.1101/gr.203701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBrearty BA, Clark LD, Zhang XM, Blankenhorn EP, Heber-Katz E. Genetic analysis of a mammalian wound-healing trait. Proc Natl Acad Sci USA. 1998;95:11792–11797. doi: 10.1073/pnas.95.20.11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naseem RH, Meeson AP, DiMaio JM, White MD, Kallhoff J, Humphries C, et al. Reparative myocardial mechanisms in adult C57BL/6 and MRL mice following injury. Physiol Genomics. 2007;30:44–52. doi: 10.1152/physiolgenomics.00070.2006. [DOI] [PubMed] [Google Scholar]
- Ueno M, Lyons BL, Burzenski LM, Gott B, Shaffer DJ, Roopenian DC, et al. Accelerated wound healing of alkali-burned corneas in MRL mice is associated with a reduced inflammatory signature. Invest Ophthalmol Vis Sci. 2005;46:4097–4106. doi: 10.1167/iovs.05-0548. [DOI] [PubMed] [Google Scholar]
- Urist MR, Raskin K, Goltz D, Merickel K. Endogenous bone morphogenetic protein: immunohistochemical localization in repair of a punch hole in the rabbit's ear. Plast Reconstr Surg. 1997;99:1382–1389. doi: 10.1097/00006534-199704001-00028. [DOI] [PubMed] [Google Scholar]
- Wang S, Basten CJ, Zeng ZB. Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University: Raleigh, NC; 2009. [Google Scholar]
- Yang H, Ding Y, Hutchins LN, Szatkiewicz J, Bell TA, Paigen B, et al. A customized and versatile high-density genotyping array for the mouse. Nat Methods. 2009;6:663–666. doi: 10.1038/nmeth.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Baylink DJ, Masinde GL, Li R, Nguyen B, Davidson HM, et al. Mouse chromosome 9 quantitative trait loci for soft tissue regeneration: congenic analysis and fine mapping. Wound Repair Regen. 2007;15:922–927. doi: 10.1111/j.1524-475X.2007.00317.x. [DOI] [PubMed] [Google Scholar]
- Yu H, Mohan S, Masinde GL, Baylink DJ. Mapping the dominant wound healing and soft tissue regeneration QTL in MRL × CAST. Mamm Genome. 2005;16:918–924. doi: 10.1007/s00335-005-0077-0. [DOI] [PubMed] [Google Scholar]
- Zeng ZB. Precision mapping of quantitative trait loci. Genetics. 1994;136:1457–1468. doi: 10.1093/genetics/136.4.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.