

Abstract
19F nuclear Overhauser effects (NOEs) between fluorine labels on the cytoplasmic domain of rhodopsin solubilized in detergent micelles are reported. Previously, high-resolution solution 19F NMR spectra of fluorine-labeled rhodopsin in detergent micelles were described, demonstrating the applicability of this technique to studies of tertiary structure in the cytoplasmic domain. To quantitate tertiary contacts we have applied a transient one-dimensional difference NOE solution 19F NMR experiment to this system, permitting assessment of proximities between fluorine labels specifically incorporated into different regions of the cytoplasmic face. Three dicysteine substitution mutants (Cys-140–Cys-316, Cys-65–Cys-316, and Cys-139–Cys-251) were labeled by attachment of the trifluoroethylthio group through a disulfide linkage. Each mutant rhodopsin was prepared (8–10 mg) in dodecylmaltoside and analyzed at 20°C by solution 19F NMR. Distinct chemical shifts were observed for all of the rhodopsin 19F labels in the dark. An up-field shift of the Cys-316 resonance in the Cys-65–Cys-316 mutant suggests a close proximity between the two residues. When analyzed for 19F-19F NOEs, a moderate negative enhancement was observed for the Cys-65–Cys-316 pair and a strong negative enhancement was observed for the Cys-139–Cys-251 pair, indicating proximity between these sites. No NOE enhancement was observed for the Cys-140–Cys-316 pair. These NOE effects demonstrate a solution 19F NMR method for analysis of tertiary contacts in high molecular weight proteins, including membrane proteins.
Keywords: G protein-coupled receptors, signal transduction, site-directed 19F labeling, NMR structure contacts, membrane protein
A conformational change in the cytoplasmic domain of rhodopsin upon light activation, and in G protein coupled receptors in general upon agonist binding, is the first event in the initiation of signal transduction in this superfamily of cell receptors. A molecular description of this conformational change is a long-range goal of studies on signal transduction. Previous work on rhodopsin has given some insights into the tertiary structure in the cytoplasmic domain and its change upon light activation (1–4). Cysteine scanning mutagenesis, the determination of proximity relationships by disulfide bond formation between pairs of cysteines, and EPR spin label studies have been instrumental in this work. However, these approaches have not provided detailed resolution of the structures.
An important recent advance has been the solution of the crystal structure of rhodopsin in the dark state (5). This work has provided high-resolution descriptions of the transmembrane and intradiscal domains as well as portions of the cytoplasmic tertiary structure. The crystal structure has been useful in confirming more than a decade of biochemical results from experiments designed to probe the structure of rhodopsin (5). Although the availability of this high-resolution dark-state structure is of great significance to the G protein coupled receptor field in general, its usefulness is limited by the lack of insight it provides into the conformational change and activated tertiary structure of the molecule upon illumination.
Solution and solid-state NMR now present alternatives to the low-resolution biochemical experiments as well as the crystal x-ray techniques in analyses of dynamics and conformational changes in proteins (6). In recent studies we have developed methods for large-scale preparation of rhodopsin that have enabled NMR experiments (7). In the first application of NMR the protonated retinylidene Schiff base nitrogen was studied by solid-state MAS NMR using 15N-lysine- and 13C-glycine-labeled rhodopsin (8). More recently, the applicability of 19F solution NMR in structural work has been demonstrated (9). Mono-cysteine mutants of rhodopsin, derivatized with 19F containing trifluoroethylthio (TET) groups (Fig. 1) in different regions of the cytoplasmic face showed resolved chemical shifts in the dark and position specific changes in chemical shifts and line widths upon illumination. This study indicated that solution 19F NMR can be used to obtain tertiary structure information even in very large, slowly tumbling membrane proteins in conformationally fluctuating detergent micelles.
Figure 1.

Schematic representation of a cysteine side chain attached via a disulfide bond to the 19F TET label.
We have now applied solution 19F NMR to TET derivatives of rhodopsin mutants containing cysteine pairs in the cytoplasmic face (Fig. 2). These include the wild type (WT) pair (Cys-140–Cys-316) as well as two mutants (Cys-65–Cys-316 and Cys-139–Cys-251), the latter two being chosen based on proximity relationships indicated previously by disulfide bond formation between these sites, and spin-spin interactions observed in EPR spectra of spin-labeled derivatives (10–12). Distinct chemical shifts were observed for all of the rhodopsin TET labels in the dark. An up-field shift of the Cys-316 resonance in the Cys-65–Cys-316 mutant is indicative of the close proximity of these two residues. Homonuclear 19F nuclear Overhauser effect (NOE) between the fluorine-labeled cysteines were analyzed by a transient one-dimensional (1D) difference NOE solution NMR experiment. Negative enhancements were observed for the Cys-65–Cys-316 and Cys-139–Cys-251 pairs, whereas no NOE enhancement was observed for the Cys-140–Cys-316 pair. These NOE effects are consistent with proximities determined by biochemical methods (10–12) as well as distances observed in the rhodopsin crystal structure (5).
Figure 2.

WT (A) and two dicysteine rhodopsin mutants (B and C), Cys-65–Cys-316 and Cys-139–Cys-251, are highlighted in secondary structural models of bovine rhodopsin. WT cysteines and mutations to cysteine residues are highlighted with circles. Deleted WT cysteines, replaced with serine, are highlighted with squares.
Materials and Methods
Frozen retinas were purchased from J. A. Lawson (Lincoln, NE). 11-cis, Retinal was prepared from all-trans-retinal after a published procedure (13). 4,4′-Dithiodipyridine (4-PDS) and DTT were purchased from Sigma, and dodecylmaltoside (DM) was from Anatrace (Maumee, OH). Antirhodopsin mAb 1D4 (14) was purified from a myeloma cell line provided by R. S. Molday (University of British Columbia, Edmonton). Sepharose 4B (Sigma) was activated with cyanogen bromide as according to the procedure of Kumel et al. (15). 1D4 was coupled to the cyanogen bromide-activated Sepharose 4B as described (14), at a level of ≈10 mg/ml of swollen Sepharose beads. The nonapeptide (Massachusetts Institute of Technology Biopolymers Laboratory) corresponding to the C-terminal sequence of rhodopsin, the antibody epitope, was used to elute rhodopsin from the 1D4-Sepharose. The sources of all reagents for cell culture have been described (7). Sodium butyrate was purchased from Fluka.
Buffers used were as follows: buffer A, 137 mM NaCl/2.7 mM KCl/1.8 mM KH2PO4/10 mM Na2HPO4, pH 7.2; buffer B, buffer A + 1% DM; buffer C, buffer A + 0.05% DM; buffer D, 2 mM NaH2PO4/Na2HPO4, pH 6; buffer E, buffer D + 0.05% DM; buffer F, buffer E + 100 μM nonapeptide; buffer Q, 20 mM NaH2PO4/Na2HPO4 (pH 6) in 99.9% D2O (Cambridge Isotope Laboratories, Andover, MA).
UV-Visible Absorption Spectroscopy.
UV-visible absorption spectra were recorded by using a Perkin–Elmer λ6 spectrophotometer. The molar extinction value used for rhodopsin was 40,600 M−1−cm−1 (16).
19F NMR Spectroscopy.
19F NMR spectra were recorded on a Varian INOVA 500 spectrometer (frequency 470.668 MHz). Data acquisition was carried out by using VNMR 6.1 software (Varian). Samples were locked on deuterium. A line broadening of 20 Hz was used for data processing. Samples were referenced to an internal standard, trifluoroacetate (TFA). Acquisition times, number of scans averaged, and specific parameters were as indicated in the text or legends.
Transient 1D difference NOE experiments were performed by using a selective longitudinal relaxation (T1) inversion-recovery method, in which a relaxation delay (RD) is followed by an 180o shaped pulse (iBurp2) and a variable relaxation delay (t) before collection of the free induction decay (17, 18). The selective inversion (iBurp2) pulse was 20–30 ms in length. RD between scans was set to five times T1. t, following the selectively inverting 180o pulse was varied from 0.0 to 2.0 s. NOE enhancements in neighboring nuclei, arising due to cross relaxation of the perturbed signal over the course of t, were detected by subtraction of control spectra according to the difference NOE definition:
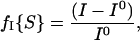 |
1 |
where fI{S} is the fractional change in the intensity of resonance I upon saturating resonance S, and I0 is the equilibrium intensity of I (19). Control spectra were collected independently at identical values of RD and intervals of t, but with selective irradiation at a blank region of the spectrum set an equal distance but on the opposite side of the unirradiated signal from the selectively perturbed signal. Proton decoupling was not used. Nonselective T1 relaxation time constants were estimated by using the same inversion recovery method with RD set to a minimum of 5 s and an exponential fit of the recovering peak intensities versus t. All NMR data were processed by using the VNMR 6.1 software (Varian).
Isolation of WT Rhodopsin.
Rod outer segment (ROS) membranes were prepared, and WT rhodopsin was solubilized from the ROS according to standard procedures (20).
Cloning, Expression, and Reconstitution of Dicysteine Rhodopsin Mutants.
Synthetic bovine opsin genes containing double cysteine substitution codons were first created in the vector pMT4 (21). Construction of the Cys-65–Cys-316 (H65C-C140S) mutant has been described (11). Construction of the Cys-139–Cys-251 (V139C-T251C-C140S-C316S) mutant occurred as reported for the V139C/T251C mutant (12). The mutant opsin genes then were excised from pMT4 as EcoRI/NotI DNA fragments and inserted into the expression plasmid pACHrhoC as described (7). The vector pACHEnc was kindly provided by A. Shafferman (Israel Institute for Biological Research, Ness-Ziona).
HEK293S cells were transfected by the method of Chen and Okayama (22) as modified by O'Mahoney and Adams (23). Stable cell lines expressing the opsin genes at high levels were selected by using elevated concentrations of geneticin (2–3 mg/ml) (7). Cells then were grown in suspension cultures and harvested, and the opsin was constituted with 11-cis-retinal as described, except 5 mM sodium butyrate was added 2 days before cell harvesting (7, 9, 24).
Purification and TET Labeling of WT Rhodopsin and Mutants.
Cells were solubilized in buffer B (10 ml/flask), and rhodopsin was bound to 1D4-Sepharose beads and washed as described except 10 mM DTT was added to the 1D4-Sepharose beads/cell lysate suspension and nutated at room temperature for 1 h immediately before column packing and washing (9). After the DTT reduction, all washing and labeling buffers were at 4°C and saturated with argon. The column chromatography also was performed under argon. TET labeling was carried out essentially as described with minor modification (9). The total beads containing ≈10 mg of bound rhodopsin were resuspended in 40 ml of buffer E and 4-PDS was added to a final concentration of 10 mM and incubated for 10 min at room temperature. Excess reagent was removed by multiple washing with 40 ml of buffer E under slight argon pressure. A total of at least 100 column volumes of buffer E was used. Complete removal of 4-PDS was tested spectrophotometrically (9). The beads then were resuspended in 30 ml of buffer E and 1 mM TET (3 μl of 11.2 M stock) overnight (8–12 h) at room temperature. Excess reagent was removed as described above for 4-PDS removal. Preparation of TET-labeled WT rhodopsin was as described above except 4-PDS was added to a final concentration of 50 μM and the incubation was carried out for 3 h at room temperature. The TET-labeled rhodopsins then were eluted from the 1D4 Sepharose by using buffer F at a flow rate of ≈0.3 ml/min.
Preparation of NMR Samples.
Fractions from 1D4 immunoaffinity chromatography containing >0.2 μM rhodopsin (A280/A500, 1.6–2.0) were pooled and concentrated by using Centricon 30 membrane filtration devices (Amicon). A total of 6–10 mg of rhodopsin was concentrated to a final volume of 0.1 ml and then exchanged into buffer Q by the addition of 1.0 ml buffer Q followed by membrane filtration to 0.1 ml again. This later step was repeated three times. The final volume then was adjusted to 0.3 ml with buffer Q. TFA was added from a 50 mM stock in D2O to give a final concentration of 0.2 mM. The samples were transferred to Shigemi NMR tubes.
Structural Modeling.
Coordinates of the rhodopsin crystal structure were kindly provided by K. Palczewski (University of Washington, Seattle). Distances between side chains were determined by using version 1.5 of the noncovalent bond finder software (E. Martz, University of Massachusetts, Boston). Cysteine modeling was carried out on a SGI terminal with insightii software (Molecular Simulations, Waltham, MA).
Results
High-Resolution Solution 19F NMR Spectra in the Dark of TET-Labeled Dicysteine Mutants of Rhodopsin.
1D 19F NMR spectra, recorded in the dark, of TET derivatives of the three dicysteine mutants (Fig. 2) are shown in Fig. 3. In all cases, two distinct NMR resonances are observed, one for each TET label incorporated into the sample. This observation extends the applicability of the previously reported procedure to obtain 19F NMR spectra of monocysteine mutants (9) to dicysteine mutants. The chemical shifts observed for the respective TET-labeled cysteines are indicated in the figure. These are relative to TFA, which was present as an internal standard and set to 0.0 ppm in all of the samples. The integration intensity of the signals relative to that of TFA, which was present at 0.2 mM concentration, indicated 70–90% labeling efficiency. Approximately 2,000 scans were averaged per spectrum.
Figure 3.

Solution 19F NMR spectra in the dark of WT and the two dicysteine mutants. The cysteine residues were derivatized with the TET group as described in the text. All samples contained 0.2 mM TFA as an internal reference, which was set to 0.0 ppm. Approximately 2,000 scans were averaged per spectrum. (A) WT Cys-140–Cys-316. (B) Cys-65–Cys-316. (C) Cys-139–Cys-251.
The WT chemical shifts correspond with data collected previously for singly labeled samples, permitting assignment of the resonances as Cys-140 at 10.6 ppm and Cys-316 at 10.0 ppm (9). In contrast Cys-65–Cys-316 resonances were detected at 9.7 ppm and 9.2 ppm. Clearly an up-field shift of the Cys-316 resonance has occurred, likely as a result of a change in the local environment due to the H65C mutation. Unambiguous assignment of the specific resonances for these pairs is not required for the NOE analysis. Therefore, no further attempts were carried out to specifically assign the main resonances in the dicysteine mutants Cys-65–Cys-316 and Cys-139–Cys-251. As with monocysteine derivatives (9), secondary peaks at 9.2 ppm and 9.5 ppm were observed in addition to the main resonances. These correspond to minor contamination of the preparations by oxidized TET and the mixed disulfide, thiopyridinyl-TET (9).
19F NMR Longitudinal T1 Measurements.
The resonances from the three dicysteine mutants were sufficiently resolved and intense that measurements of their T1 values could be made. A simple inversion recovery method (17) was applied and values were obtained by direct exponential analysis of the irradiated peak recovery with respect to relaxation time t. T1 values for all samples are compiled in Table 1 and are in the range of 0.3 to 0.4 s. These correspond to the value obtained previously (0.32 s) for fluorotyrosine incorporated into the M13 coat membrane protein and studied in solution in lipid vesicles (25). These values were used to set the RD time in the NOE experiments to five times T1.
Table 1.
19F longitudinal relaxation time constants for TET-labeled dicysteine rhodopsin mutants
| Mutant | Line, ppm* | T1, sec† |
|---|---|---|
| WT | 10.6 | 0.34 ± 0.01 |
| WT | 10.0 | 0.39 ± 0.01 |
| Cys-65–Cys-316 | 9.7 | 0.31 ± 0.02 |
| Cys-65–Cys-316 | 9.2 | 0.30 ± 0.02 |
| Cys-139–Cys-251 | 9.8 | 0.28 ± 0.03 |
| Cys-139–Cys-251 | 11.1 | 0.38 ± 0.02 |
Chemical shifts are relative to TFA at 0.0 ppm.
T1 values and errors were determined by using vnmr 6.1 software.
Homonuclear 19F Solution NMR NOEs Between TET-Labeled Rhodopsin Dicysteine Pairs.
Homonuclear 19F NOEs were observed by application of a transient 1D difference NOE solution NMR experiment. Due to limitations in the availability of a suitable probe, 1H was not decoupled. The experiment was based on a selective inversion-recovery (17) where NOE enhancements in neighboring nuclei, arising due to cross relaxation of the perturbed signal over the course of t were detected by subtraction of control spectra. An example is shown in Fig. 4 for the selective inversion of the 9.2-ppm resonance of the Cys-65–Cys-316 mutant including control and difference spectra. The latter show a change in peak intensity (NOE) at 9.7 ppm. NOEs at nonirradiated neighboring resonances detected in the difference spectra were normalized relative to relaxed peak intensities and plotted as % NOE versus t (Fig. 5). Negative NOE enhancements developed and then decayed over time for the Cys-65–Cys-316 (moderate, maximum at 0.06 s) and the Cys-139–Cys-251 (strong, maximum at 0.5 s) pairs. The characteristic transient NOE signal was not observed for the Cys-140–Cys-316 control pair as expected. Minor background T1 relaxation (5%) was detected in this latter experiment.
Figure 4.

(A) Selective 19F inversion experiment on Cys-65–Cys-316 by the progressive inversion-recovery method (17). Shown is the recovery of the 9.2-ppm resonance line in the Cys-65–Cys-316 1D-19F solution NMR spectrum after selective inversion by a 180o pulse. The relaxation delay times (t) were (i) 0.0, (ii) 0.02, (iii) 0.04, (iv) 0.06, (v) 0.1, and (vi) 1.5 s. (B) Control spectra for Cys-65–Cys-316 collected at identical values of t as in A, after selective irradiation of a blank region (show with a bar) of the NMR spectrum. (C) Difference spectra for Cys-65–Cys-316 resulting from subtraction of control spectra (as shown in B) from inverted spectra (as shown in A). The NOE enhancement at 9.7 ppm is marked with an arrow. Approximately 2,000 scans were averaged for each spectrum in A and B.
Figure 5.

NOE enhancements are plotted against t compared with WT (●) for (A) Cys-65–Cys-316 (■) and (B) Cys-139–Cys-251 (▴). Each point is the average of three measurements. The error given is the SEM.
Discussion
The dicysteine pairs used in these studies include the native cysteine pair (Cys-140–Cys-316) and two mutants Cys-65–Cys-316 and Cys-139–Cys-251. The latter two mutants were chosen based on proximity relationships indicated in previous biochemical work (10–12). Application of EPR to estimate the Cys-65–Cys-316 proximity indicated movement apart upon light activation, with distances of ≈10 Å in the dark, going to ≈15 Å in the light (11). Similar studies on Cys-139–Cys-251 indicated an estimated EPR distance of 12–14 Å in the dark, going to >20 Å upon illumination (10). However, biochemical data indicated that these mutants form disulfide bonds between helices I and VII and helices III and VI in the dark state (10–12). Although the disulfide bonding data cannot provide a discrete distance value, the geometric requirements for disulfide formation imply distances that are shorter (Cα-Cα distance of 4.4–6.8 Å) than those suggested by EPR (26). Current limitations in EPR methods lead to a systematic overestimation of distances lower than 8 Å (27). Here we demonstrate that these shorter distances can be better approximated by solution 19F NOE effects, providing a method for proximity determination complementary to EPR analysis. One facilitating factor is the very small and unobtrusive size of the TET label compared with fluorescent and EPR-sensitive labels that contain large ring structures (Fig. 1).
The negative quality of the NOE enhancements observed herein is consistent with what is expected for large molecules and very viscous samples. Rhodopsin itself is a large molecule by NMR standards at 348 aa. Furthermore, the presence of the detergent-mixed micelles dramatically increases the effective molecular mass to ≈100 kDa, slowing down the molecular tumbling rate. As well, the effect of concentrating the sample results in a 5% (wt/vol) DM solution that is highly viscous. These conditions lead to preferential zero quantum transitions during cross relaxation, such that saturation of an NMR signal is expected to cause a decrease in the intensity of resonances from proximal nuclei.
Theoretical rates of cross relaxation (σ) in a two-spin 19F-19F system can be determined by plotting the fractional change in signal intensity (I/I0) as a function of t. The initial rate is proportional to 0 and the overall dependence can be described by Eq. 2 (28).
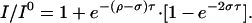 |
2 |
The theoretical curves did not describe the experimentally obtained curves very well, suggesting that the rhodopsin 19F NOEs are not arising in a simple two-spin system. Spin diffusion between fluorine and proximal protons is likely a major contributing factor to this lack of correlation. As such, discrete distance limitations cannot be applied. However, there are distinctive NOE buildup curves represented for the three different fluorine pairs, which are reflective of the different distances expected based on the biochemical data and the dark-state crystal structure of rhodopsin.
Cys-140–Cys-316.
Although a 5% shift over time is observed in the WT Cys-140–Cys-316 NOE plot, this is attributed to background T1 relaxation, arising due to the slight overlap between the resonance lines in the 1D spectrum. The nonirradiated peak was partially saturated by the selective irradiation pulse, causing up to 30% background T1 relaxation in the NOE plot (data not shown). Subtraction of control spectra, acquired as described herein, decreased this background longitudinal relaxation to <5%. The lack of any NOE enhancement confirms that the WT rhodopsin cysteines are not in close proximity. Previously, spin-label EPR studies showed no dipole-dipole interactions between these sites, indicating distances >18 Å (29). Our results are in agreement with this as well as a recently published crystal structure of the dark state of rhodopsin, which included portions of the cytoplasmic domain (5). The closest distance between the Cys-140 and Cys-316 side chains according to the x-ray crystal structure is 29.8 ± 0.5 Å (5). The WT molecule presents an excellent negative control in these NOE experiments.
Cys-65–Cys-316.
The 1D 19F NMR spectra presented here show an up-field shift of the Cys-316 resonance in the Cys-65–Cys-316 spectrum as compared with WT. This shift likely arises due to the change in environment resulting from the histidine to cysteine mutation at position 65. The effect is qualitatively indicative of the close proximity of the two side chains. This proximity is confirmed by the NOE enhancement reported herein. According to the crystalline structure, the closest distance between His-65 and Cys-316 is 3.9 ± 0.5 Å with a possible hydrogen bond between the side chains (5). Substitution of a cysteine side chain at the His-65 position in the model increased the distance to 4.2 ± 0.5 Å. These values correlate well with the intermediate intensity of the NOE enhancement observed for this pair.
Cys-139–Cys-251.
Proximity between the Cys-139 and Cys-251 residues is confirmed by the observation of an NOE enhancement. According to the crystalline model, the closest distance between the Val-139 and Thr-251 side chains is 3.5 ± 0.5 Å (5). Substitution of cysteines into the model at these sites decreased the distance to 3.0 ± 0.5 Å. These short distances are in line with the strong NOE enhancement observed for this pair.
Conclusion.
We have presented a 19F NMR method for analysis of tertiary contacts in the cytoplasmic domain of rhodopsin. Although discrete distance limitations cannot be applied under these experimental conditions, assessment of the NOE enhancement intensities indicated correlation with expected distances based on the x-ray crystal structure. Overall, the high sensitivity and lack of background 19F makes this method applicable to many membrane and soluble protein systems that are too large for study by conventional NMR techniques.
Acknowledgments
We gratefully acknowledge Prof. P. Wright at Scripps Institute (San Diego, CA) and Prof. B. Sykes at the University of Alberta (Edmonton, Canada) for discussions and critical reviews of this work. We thank Prof. K. Palczewski of the University of Washington (Seattle) for early release of the rhodopsin crystal coordinates. We have benefited greatly from discussions with Prof. U. L. RajBhandary and all of our colleagues at the Massachusetts Institute of Technology. We also thank the staff of the Instrumentation Facility in the Department of Chemistry (Massachusetts Institute of Technology) for technical advice on NMR experiments. Research reported here was supported by National Institutes of Health Grants GM28289 and EY11716 (H.G.K.), a Massachusetts Institute of Technology Startup Grant, and the Karl-Winnacher Foundation (H.S.). M.C.L. is the recipient of a Natural Sciences and Engineering Research Council of Canada fellowship. J.K.-S. is the recipient of a Howard Hughes Medical Institute predoctoral fellowship. E.V.G. is the recipient of an Association for Research in Vision and Ophthalmology/Japan National Society for the Prevention of Blindness Research fellowship.
Abbreviations
- WT
wild type
- NOE
nuclear Overhauser effect
- DM
dodecylmaltoside
- 4-PDS
4,4′-dithiodipyridine
- TET
trifluoroethylthio
- TFA
trifluoroacetate
- T1
longitudinal relaxation time
- RD
relaxation delay
- 1D
one-dimensional
Footnotes
See commentary on page 4819.
References
- 1.Cai K, Klein-Seetharaman J, Farrens D, Zhang C, Altenbach C, Hubbell W L, Khorana H G. Biochemistry. 1999;38:7925–7930. doi: 10.1021/bi9900119. [DOI] [PubMed] [Google Scholar]
- 2.Altenbach C, Cai K, Khorana H G, Hubbell W L. Biochemistry. 1999;38:7931–7937. doi: 10.1021/bi9900121. [DOI] [PubMed] [Google Scholar]
- 3.Altenbach C, Klein-Seetharaman J, Hwa J, Khorana H G, Hubbell W L. Biochemistry. 1999;38:7945–7949. doi: 10.1021/bi990014l. [DOI] [PubMed] [Google Scholar]
- 4.Klein-Seetharaman J, Hwa J, Cai K, Altenbach C, Hubbell W L, Khorana H G. Biochemistry. 1999;38:7938–7944. doi: 10.1021/bi990013t. [DOI] [PubMed] [Google Scholar]
- 5.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox B A, Le Trong I, Teller D C, Okada T, Stenkamp R E. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 6.Wüthrich K. NMR in Structural Biology. Singapore: World Scientific; 1995. [Google Scholar]
- 7.Reeves P J, Thurmond R L, Khorana H G. Proc Natl Acad Sci USA. 1996;93:11487–11492. doi: 10.1073/pnas.93.21.11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eilers M, Reeves P J, Ying W, Khorana H G, Smith S O. Proc Natl Acad Sci USA. 1999;96:487–492. doi: 10.1073/pnas.96.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klein-Seetharaman J, Getmanova E V, Loewen M C, Reeves P J, Khorana H R. Proc Natl Acad Sci USA. 1999;96:13744–13749. doi: 10.1073/pnas.96.24.13744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farrens D L, Altenbach C, Yang K, Hubbell W L, Khorana H G. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 11.Yang K, Farrens D L, Altenbach C, Farahbakhsh Z T, Hubbell W L, Khorana H G. Biochemistry. 1996;35:14040–14046. doi: 10.1021/bi962113u. [DOI] [PubMed] [Google Scholar]
- 12.Cai K, Klein-Seetharaman J, Hwa J, Hubbell W L, Khorana H G. Biochemistry. 1999;38:12893–12898. doi: 10.1021/bi9912443. [DOI] [PubMed] [Google Scholar]
- 13.Knowles A, Priestley A. Vision Res. 1978;18:115–116. doi: 10.1016/0042-6989(78)90086-x. [DOI] [PubMed] [Google Scholar]
- 14.Oprian D D, Molday R S, Kaufman R J, Khorana H G. Proc Natl Acad Sci USA. 1987;84:8874–8878. doi: 10.1073/pnas.84.24.8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumel G, Daus H, Mauch H. J Chromatogr. 1979;172:221–226. doi: 10.1016/s0021-9673(00)90957-9. [DOI] [PubMed] [Google Scholar]
- 16.Wald G, Brown P K. J Gen Physiol. 1953;37:189–200. doi: 10.1085/jgp.37.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vold R L, Waugh J S, Klein M P, Phelps D E. J Chem Phys. 1968;48:3831–3832. [Google Scholar]
- 18.Geen H, Freeman R. J Magn Reson. 1991;93:93. [Google Scholar]
- 19.Neuhaus D, Williamson M P. The Nuclear Overhauser Effect in Structural and Conformational Analysis. New York: Wiley; 2000. [Google Scholar]
- 20.Papermaster D S. Methods Enzymol. 1982;81:48–52. doi: 10.1016/s0076-6879(82)81010-0. [DOI] [PubMed] [Google Scholar]
- 21.Franke R R, Sakmar T P, Oprian D D, Khorana H G. J Biol Chem. 1988;263:2119–2122. [PubMed] [Google Scholar]
- 22.Chen C, Okayama H. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Mahoney J V, Adams T E. DNA Cell Biol. 1994;13:1227–1232. doi: 10.1089/dna.1994.13.1227. [DOI] [PubMed] [Google Scholar]
- 24.Reeves P J, Klein-Seetharaman J, Getmanova E V, Eilers M, Loewen M C, Smith S O, Khorana H G. Biochem Soc Trans. 1999;27:950–955. doi: 10.1042/bst0270950. [DOI] [PubMed] [Google Scholar]
- 25.Hagen D S, Weiner J H, Sykes B D. Biochemistry. 1978;17:3860–3866. doi: 10.1021/bi00611a028. [DOI] [PubMed] [Google Scholar]
- 26.Richardson J S. Adv Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 27.Langen R, Oh K-J, Cascio D, Hubbell W L. Biochemistry. 2000;39:8396–8405. doi: 10.1021/bi000604f. [DOI] [PubMed] [Google Scholar]
- 28.Cavanagh J, Fairbrother W J, Palmer A G, III, Skelton N J. Protein NMR Spectroscopy: Principles, and Practice. Boston: Academic; 1996. [Google Scholar]
- 29.Farahbakhsh Z T, Hideg K, Hubbell W L. Science. 1993;262:1416–1419. doi: 10.1126/science.8248781. [DOI] [PubMed] [Google Scholar]