

Abstract
Metabolic cytometry is a form of chemical cytometry wherein metabolic cascades are monitored in single cells. We report the first example of metabolic cytometry where two different metabolic pathways are simultaneously monitored. Glycolipid catabolism in primary rat cerebella neurons was probed by incubation with tetramethylrhodamine-labeled GM1 (GM1-TMR). Simultaneously, both catabolism and anabolism were probed by co-incubation with BODIPY-FL labeled LacCer (LacCer-BODIPY-FL). In a metabolic cytometry experiment, single cells were incubated with substrate, washed, aspirated into a capillary, and lysed. The components were separated by capillary electrophoresis equipped with a two-spectral channel laser-induced fluorescence detector. One channel monitored fluorescence generated by the metabolic products produced from GM1-TMR and the other monitored the metabolic products produced from LacCer-BODIPY-FL. The metabolic products were identified by comparison with the mobility of a set of standards. The detection system produced at least six orders of magnitude dynamic range in each spectral channel with negligible spectral crosstalk. Detection limits were 1 zmol for BODIPY-FL and 500 ymol for tetramethylrhodamine standard solutions.
Glycosphingolipids are the most abundant glycolipids in neuronal membranes, and their metabolic pathways are well characterized.1–4 Briefly, ceramide is synthesized in the endoplasmic reticulum, transported to the Golgi where glycosyltransferases are responsible for the stepwise addition of monosaccharides, generating structures of remarkable complexity. The newly formed glycolipids are transported to the plasma membrane to fulfill their functional roles. Likewise, through endocytosis, the lipids are recycled back into the cell interior, and undergo degradation by stepwise removal of the sugars by glycosidases present in the lysosome and plasma membrane.5 Defects in sphingolipid metabolism are associated with a number of devastating lysosomal storage diseases including Niemann Pick type A,6 Gaucher,7 Tay-Sachs,8 and Krabbe diseases.9
We are interested in probing the metabolism of glycolipids within single cells. Immunostaining experiments have revealed that the distribution of lipids in a neuronal population is highly heterogeneous.10, 11 Characterizing glycolipid metabolism in single cells has the potential of unveiling functional differences in these neurons, which may provide insight into the nature of the lysosomal storage diseases and therefore leading to innovative therapies.
We coined the term metabolic cytometry to refer to the use of modern chemical instrumentation to monitor metabolism in single cells.12–14 In our experiments, we incubate cells with a fluorescence substrate, which is taken up and metabolized. A single cell is aspirated within a capillary and lysed. The fluorescence products are separated by capillary electrophoresis and detected with laser-induced fluorescence. As long as those metabolites retain their fluorescent tag, they many be detected with exquisite sensitivity using high sensitivity laser-induced fluorescence.
To date, our efforts have primarily employed a tetramethylrhodamine (TMR)-labeled glycolipid as substrate, which is taken up by cells and is catabolized.13–16 We have recently described the use of a BODIPY-labeled glycolipid as substrate;17 this substrate undergoes both catabolism and anabolism.18–19 In metabolic cytometry, a cell is aspirated within a capillary and lysed by contact with a surfactant. The contents are separated by capillary electrophoresis equipped with an ultrasensitive laser-induced fluorescence detector.
Our instruments have been designed to monitor fluorescence in one spectral channel, which allows use of a single labeled substrate. We have observed a large cell-to-cell variation in uptake of TMR-labeled substrates for both cultured PC12 cells and primary neurons, and significant cell-to-cell variation in metabolism for primary neurons.12–14 To address this very wide variation in uptake and metabolic activity, we have created a set of fluorescence detectors with six to nine orders of magnitude dynamic range.15–16 These instruments have revealed that cultured PC12 cells tend to generate very similar expression profiles of fluorescent glycolipids, which provides confidence in the reproducibility of the analytical method. In constrast, neurons provide a quite diverse population of metabolic products, undoubtedly reflecting differences in their function.
In this paper, we expand our technology to simultaneously monitor glycolipid metabolism in two different pathways. catabolism and anabolism. Our new system employed GM1-TMR and BODIPY-FL-labeled LacCer, which also undergoes anabolism, as substrates. Primary rat cerebella neurons were incubated with these substrates. Single cells were aspirated into a capillary and lysed. Their contents were separated by capillary electrophoresis coupled with a two-spectral channel laser-induced fluorescence detector. One spectral channel was tuned to detect tetramethylrhodamine-labeled metabolites, while the other detected BODIPY-FL labeled metabolites.
Experimental Section
Reagents
5-carboxyl-tetramethylrhodamine (TMR), boron dipyrromethene difluoride (BODIPY-FL), TMR-succinimidyl ester, and BODIPY-FL-succinimidyl ester were from Invitrogen. All other reagents, unless noted, are from Sigma.
Cerebellar cells
Primary cells from the rat cerebellum were isolated and cultured as described previously,20 except that a supplement (NS21)21 was used in place of serum. Briefly, cerebella were dissected from postnatal day 5–6 rats, meninges were removed and the tissue was treated with papain. Tissue was dissociated into single cells by triturating, the cells were collected and washed by centrifugation and then re-suspended in growth medium (Neurobasal supplemented to 25 mM KCl, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin). A mixture of neuron-supportive factors (NS21) was added to the growth medium as described previously.21 Cells were plated on polylysine-coated tissue culture dishes at a density of 2 × 106 cells in 2 ml NS21-containing medium on each 35-mm dish, and then were incubated at 37°C, 5% CO2 in a humidified incubator for 7 days. The medium was changed every 2 days.
Metabolic cytometry
GM1-TMR was synthesized as described,22 and LacCer-BODIPY was synthesized in identical manner, except that BODIPY-FL succinimidyl ester was used as the fluorescent label.17 Equimolar amounts of each lipid and fatty acid free bovine serum albumin (BSA) were pre-complexed in ethanol-water (2:1) and then diluted to 5 μM in NS21-free growth medium. Seven-day cell cultures were washed with NS21-free growth medium, then simultaneously incubated for 2 h with 5 μM of each of the lipid /BSA complexes in the same medium, followed by 22 h incubation in serum-free medium. Cells were then collected using 2.5 mg/mL trypsin/0.9 mM EDTA in phosphate buffer (Invitrogen), followed by 2.5 mg/ml soybean trypsin inhibitor. Released cells were washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde in PBS, and then suspended in PBS containing 10 mM glycine until analysis. In some experiments, for comparison with single cell metabolomic studies, PBS-washed cell pellets were lysed in 0.5 ml of 1% sodium dodecyl sulfate in water without fixation. Cell cultures were also incubated with individual lipid/BSA complex to serve as control experiments to the co-incubation experiment. Cells were selected based on their size and injected using our standard protocol.26
Fluorescent lipid substrates
Two fluorescent exogenous lipids were selected for this study: GM1-TMR and LacCer-BODIPY-FL. GM1-TMR has been used by our group to monitor catabolism in single cells.12 LacCer-BODIPY was chosen because the small substrate also undergoes anabolism.17–19 In addition, the spectral properties of BODIPY-FL (excitation/emission maxima at 504/512 nm respectively) are significantly different from TMR (556/580 nm), which allows discrimination of the two pathways.
To evaluate the figures of merit of the instrument, standard solutions of the free dyes were analyzed individually, using a 10 mM borate buffer. The limits of detection for both channels were evaluated using these solutions in the picomolar concentration range, while the linear dynamic range was established using solutions from the picomolar to the micromolar concentration range, by appropriately scaling the attenuated signal from the photodiode cascade.15–16
Capillary electrophoresis
The capillary electrophoresis instrument is a modification of the design previously reported in our group.24–27 The instrument employed a high voltage power supply (Spellman, NY) to establish an electric field across a 39 cm long, 20 μm ID bare-fused silica capillary (Polymicro, Phoenix, AZ). The lipid standards and cell homogenate samples were injected electrokinetically (1 kV for 1s), while single cells were injected hydrodynamically.26 On-column cell lysis was performed by injecting a plug of Triton X-100 (Sigma) before and after the cell injection; the application of the separation voltage (18 kV) produced cell lysis. The separation mechanism used was micellar electrokinetic capillary chromatography, with a separation buffer comprising 10 mM sodium tetraborate, 35 mM sodium deoxycholate and 5 mM methyl-β-cyclodextrin.26 The data were acquired by a custom Labview program using two National Instruments cards (6035E and 6602, Austin, TX) at a rate of 50 Hz. The signal was recorded in a PC, corrected for the photodiode dead-time,28 and passed through a 5 point median filter to remove spikes from the signal. The data were then convoluted with a Gaussian function with a 5-point standard deviation. The median signal was subtracted, and the data from the diode cascades in each spectral channel was used to generate a high-dynamic range signal.15–16
Excitation light sources and emission collection optics
The post-column laser-induced fluorescence detector is based on a sheath-flow cuvette. The two-spectral channel detector employed a 15 mW solid-state laser operating at 473 nm (Lasermate) and a 20 mW solid-state laser operating at 532 nm (Lasermate), Figure 1. The beams were combined with a dichroic beam splitter with a 380 – 490 nm reflection bandwidth and a 520 – 700 nm transmission bandwidth (Thorlabs), and focused with a 6.3×, 0.20 NA microscope objective (Melles Griot). The beams were aligned so that the 532 nm laser was focused approximately 50-μm downstream from the separation capillary's exit and the 473 nm beam was focused approximately 20-um further downstream. By offsetting the beams, the Raman scatter generated by the blue beam will not interfere with the TMR fluorescence excited by the green beam, and the Rayleigh scattered green beam will not interfere with the BODIPY-FL fluorescence. This offset introduces a few millisecond offset in the signals from the two beams as analyte traverses the distance between them. This design is reminiscent of that employed by Block engineering in an early multiple color flow cytometer.29
Figure 1.
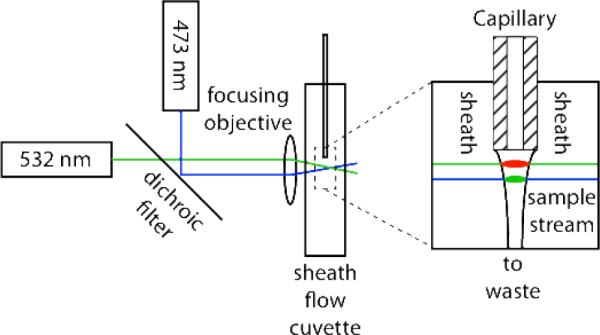
Schematic diagram of the excitation channels for the two-color fluorescence detector.
The fluorescence emission was collected by a 0.7 N.A microscope objective (Universe Kogaku) and imaged through a dichroic beam splitter (Omega Optical), which transmits red (TMR) and reflects green (BODIPY-FL), Figure 2. The red channel was equipped with a 580DF30 bandpass filter while the green channel was equipped with a 510DF10 bandpass filter (Thorlabs). The fluorescence in each channel was imaged onto its own GRIN-lens coupled fiber-optic. Each fiber optic, in turn, was attached to a high dynamic-range detector array constructed from two fiber-optic beamsplitters (Timbercon, Inc) placed in series.15–16 These beam splitter cascades divided the fluorescence intensity into three channels with successively higher attenuation. Each channel was monitored by an avalanche photodiode single-photon counter (PerkinElmer), with a dead time of 55 ns.
Figure 2.

Schematic diagram of the emission channels for the two-color fluorescence detector. The high dynamic range detector array is described in ref (15–16).
Results and Discussion
Two channel metabolic cytometry
Glycosphingolipid metabolism is quite complex, dividing into lacto-, neolacto-, globo-, and ganglio-series. These series consist of dozens of compounds that differ in their oligosaccharide groups; Figure S2 the supporting information for this manuscript presents a small portion of the metabolic pathway for gangliosphingolipids. These compounds are continually remodeled through anabolism and catabolism by the action of many dozens of enzymes, and the balance of synthetic and degradative pathways is important in generating stable membrane composition. We have developed instruments and reagents that allow us to monitor metabolism associated with a single substrate,12–16 which provides a snapshot of metabolism in a selected region of the pathway.
This paper expands our earlier work by developing an instrument and reagents that allow us to simultaneously monitor metabolism in two regions of the metabolic pathway. We describe a two-color fluorescence capillary electrophoresis detector that is tuned to monitor TMR and BODIPY-FL with high sensitivity and modest crosstalk. We also report the use of two fluorescent substrates (GM1-TMR and LacCer-BODIPY-FL), which are taken up by primary neurons and converted to sets of metabolic products corresponding to overlapping portions of the metabolic pathways.
Instrument characterization
There is a long history of the use of two-excitation channels for laser-induced fluorescence in capillary electrophoresis,30 including generation of internal standards for capillary sieving electrophoresis of proteins, for two-color and four-color DNA sequencing,31- and for study of dyes encapsulated in lyposomes. The two-color laser-induced fluorescence detector used in this experiment was based on previous versions from this group.30–31 Unlike those earlier instruments, we now offset the two beams by roughly 20-μm within the cuvette. The 532 nm excitation beam is placed closest to the capillary to first excite TMR-labeled compounds; BODIPY-FL has negligible absorbance at this wavelength. TMR has a modest absorbance at 476 nm; by aligning the lasers so that the TMR is partially photobleached before reaching the blue laser, we reduce its contribution to the BODIPY background.
Figures of merit (linear dynamic range and limit of detection)
A calibration curve was constructed for both peak height and area for dye concentration ranging from 10−10 M to 10−5 M using both BODIPY-FL and TMR. The results are summarized in Table 1. The detection limits were in the low zeptomole to high yoctomole of dye injected onto the capillary. Log-log calibration curves based on peak height and peak area both gave slopes of 1.01 with reduced X2 below 1, which implies a linear calibration curve across the five orders of magnitude in analyte concentration employed in constructing the curve. Separation efficiency was consistently greater than 500,000 theoretical plates. BODIPY-FL generated a modest crosstalk (~1%) in the TMR spectral channel. In contrast, TMR generated a negligible signal in the BODIPY-FL spectral channel (~4 ppm). As we show below, the emission spectrum of BODIPY-FL conjugate with glycolipids undergoes a red shift, increasing its contribution to the TMR channel.
Table 1.
calibration curve results
| dye | *Peak area reduced x2 | *Peak area slope | *Peak height reduced x2 | *Peak height slope | Concentration detection limit | Mass detection limit | Average theoretical plate count | Spectral cross-talk |
|---|---|---|---|---|---|---|---|---|
| BODIPY | 0.10 | 1.01 ± 0.01 | 0.10 | 1.01 ± 0.03 | 20 ± 7 pM | 1.4 ± 0.4 zmol | 5.0 ± 0.4 × 105 | 1.4% |
| tetramethyl-rhodamine | 0.94 | 1.02 ± 0.01 | 0.88 | 1.02 ± 0.02 | 10 ± 2 pM | 600 ± 100 ymol | 6.0 ± 1.0 × 105 | 0.0004% |
Reduced X2 and slope are from a plot of log concentration vs. log signal. In calculating the reduced X2 statistic, propagation of errors was used to estimate the uncertainty in the signal, starting with the standard deviation of the mean of three replicate injections.
Capillary dimensions: 40 cm length and 20-μm inner diameter. Separation voltage =18 kV. Injection voltage = 1 kV. Injection time = 1 s. Injection volume ~10 pL. Analyte concentration ranged from 50 pM to 10 μM for tetramethylrhodamine and from 100 pM to 10 μM for BODIPY.
Microscopy
We incubated a cell culture prepared from primary rat cerebral cells with both TMR and BODIPY-FL labeled glycolipids. Fluorescence microscopy revealed a significant difference in uptake and distribution, Figure 3. The tetramethylrhodamine labeled substrate and products were found primarily in non-neuronal cells, and the reagents may prove to be a valuable stain for such purpose. When present within neurons, they formed puncta within the cellular body. In contrast, BODIPY-FL labeled product were more diffusely distributed throughout the cell body and axons.
Figure 3.

Primary cerebellar granule neuron cultures 24 hours after co-incubation with 5 μM (each) of BODIPY-LacCer and TMR-GM1. Small round nerve cell bodies and associated axons are well labeled with BODIPY (left), whereas larger non-neuronal cells are intensely stained with TMR (center). Scale bar = 50 μm. Left panel. Green spectral channel. Center panel. Red spectral channel. Right panel. Combined image.
Metabolism of GM1-TMR and LacCer-BODIPY in cerebellar granule neurons
Metabolic cytometry was used to monitor the products from the co-incubation of cerebellar granule cells with GM1-TMR and LacCer-BODIPY, Figure 4. Note that the BODIPY-FL labeled metabolites have a lower net mobility than the TMR-labeled metabolites. GD1a, GM2, GM3, glucosylceramide (GlcCer), and ceramide (Cer) were produced from catabolism of GM1-TMR in these primary cells, Figure 4 and 5.
Figure 4.

Two-color laser-induced fluorescence detection of a cell lysate prepared from the co-incubation of cerebellar granule neurons with TMR-GM1 (Green Trace) and LacCer- BODIPY (blue trace). The green trace was multiplied by a factor of three before plotting. Peaks are identified by comparison with the migration of standard compounds.
Figure 5.

Capillary electrophoresis analysis seven granule neurons incubated with both GM1-TMR and LacCer-BODIPY-FL. Data shown for the TMR spectral channel. A – full scale. B – Expanded scale. Unknown components are marked with ?. The peak marked with the asterisk (*) is an artifact peak due to incomplete compensation of the LacCer-BODIPY signal.
The metabolism of LacCer-BODIPY by cerebellar granule neurons follows the catabolic as well as the anabolic pathways. GlcCer and Cer are the major degradation products. All other peaks migrated faster than the LacCer peak and presumably originate from anabolism of the LacCer substrate, suggesting the presence of appropriate glycosyltransferases within the subcellular compartments being sampled. Among these fast-migrating products, GM1, GD1a, and GM3 have been identified based on the mobility of standard compounds, Figure 4 and 6. LacCer-BODIPY can therefore be used effectively to sample cellular compartments other than the lysosome, yielding glycolipids with higher complexity than the starting material.
Figure 6.

Capillary electrophoresis analysis of the same seven granule neurons used in Figure 5. Data shown for the BODIPY spectral channel. A – full scale. B – Expanded scale. Unknown components are marked with ?.
There was no need to correct the blue channel for spectral crosstalk; the emission of TMR labeled glycolipids did not contribute a noticeable signal in the blue channel. However, we observed that the BODIPY-labeled compounds generated a larger crosstalk into the red channel compared with the pure dyes used in the construction of the calibration curve. The dye's spectral characteristics are known to vary depending upon the polarity of its surroundings,17 so that this spectral shift upon conjugation to a polar lipid is not surprising. To correct for this crosstalk, the TMR signal had 5% of the blue spectral channel subtracted, which corrected for most of the crosstalk. A small amount of crosstalk remained, which varied with the details of the system's alignment.
After protocol optimization using cellular homogenates, single neuron analysis was performed. The data from the co-incubation of LacCer-BODIPY and GM1-TMR with the neurons are presented in Figures 5–6. The metabolic products showed a large cell-to-cell variation in amplitude. The amplitude of one cell, plotted at the top-trace, was particularly large, reflecting larger uptake than the other cells. This cell may have been non-neuronal. All cells tended to show more unmetabolized LacCer than was observed in the lysate. One fast migrating component was noted at roughly 5 minute migration time; that component appears to be associated with the dye itself.
The cells were simultaneously analyzed for metabolites of the TMR-GM1, Figure 6. As in the LacCer-Bodipy channel, one cell generated an anomalously large signal. That cell's signal was dominated by an intense ceramide peak. A relatively large amount of GM1 and GM2 were produced.
The separation efficiency was remarkably high for the metabolic cytometry experiments; efficiency typically fell in the 400,000–600,000 theoretical plate range, which is similar to the results from the calibration curve. Cell lysis in this experiment did not contribute significantly to peak dispersion.
Supplementary Material
Acknowledgment
We acknowledge funding from the National Health Institutes (R01NS061767).
Footnotes
SUPPORTING INFORMATION AVAILABLE Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
Literature Cited
- (1).Merrill AH., Jr. J. Biol. Chem. 2002;277:25843–25846. doi: 10.1074/jbc.R200009200. [DOI] [PubMed] [Google Scholar]
- (2).Tettamanti G. Glycoconj. J. 2004;20:301–317. doi: 10.1023/B:GLYC.0000033627.02765.cc. [DOI] [PubMed] [Google Scholar]
- (3).Kolter T, Proia RL, Sandhoff K. J. Biol. Chem. 2002;277:25859–25862. doi: 10.1074/jbc.R200001200. [DOI] [PubMed] [Google Scholar]
- (4).Sandhoff K, van Echten G. Prog. Brain Res. 1994;101:17–29. doi: 10.1016/s0079-6123(08)61937-8. [DOI] [PubMed] [Google Scholar]
- (5).Miyagi T, Wada T, Yamaguchi K, Hata K, Shiozaki K. J. Biochem. 2008;144:279–285. doi: 10.1093/jb/mvn089. [DOI] [PubMed] [Google Scholar]
- (6).Graber D, Salvayre R, Levade T. J. Neurochem. 1994;63:1060–1068. doi: 10.1046/j.1471-4159.1994.63031060.x. [DOI] [PubMed] [Google Scholar]
- (7).Ringden O, Groth CG, Erikson A, Granqvist S, Mansson JE, Sparrelid E. Transplantation. 1995;59:864–870. [PubMed] [Google Scholar]
- (8).Platt FM, Butters TD. Biochem. Pharmacol. 1998;56:421–430. doi: 10.1016/s0006-2952(98)00115-4. [DOI] [PubMed] [Google Scholar]
- (9).Pagano RE, Puri V, Dominguez M, Marks DL. Traffic. 2000;1:807–815. doi: 10.1034/j.1600-0854.2000.011101.x. [DOI] [PubMed] [Google Scholar]
- (10).Heinemann M, Zenobi R. Curr. Opin. Biotechnol. 2010;22:26–31. doi: 10.1016/j.copbio.2010.09.008. [DOI] [PubMed] [Google Scholar]
- (11).Gong Y, Tagawa Y, Lunn MP, Laroy W, Heffer-Lauc M, Li CY, Griffin JW, Schnaar RL, Sheikh KA. Brain. 2002;125:2491–2506. doi: 10.1093/brain/awf258. [DOI] [PubMed] [Google Scholar]
- (12).Krylov SN, Zhang Z, Chan NW, Arriaga E, Palcic MM, Dovichi NJ. Cytometry. 1999;37:14–20. doi: 10.1002/(sici)1097-0320(19990901)37:1<14::aid-cyto2>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- (13).Whitmore CD, Hindsgaul O, Palcic MM, Schnaar RL, Dovichi NJ. Anal. Chem. 2007;79:5139–5142. doi: 10.1021/ac070716d. [DOI] [PubMed] [Google Scholar]
- (14).Cohen D, Dickerson JA, Whitmore CD, Turner EH, Palcic MM, Hindsgaul O, Dovichi NJ. Annu. Rev. Anal. Chem. (Palo Alto Calif) 2008;1:165–190. doi: 10.1146/annurev.anchem.1.031207.113104. [DOI] [PubMed] [Google Scholar]
- (15).Whitmore CD, Essaka D, Dovichi NJ. Talanta. 2009;80:744–748. doi: 10.1016/j.talanta.2009.07.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dada OO, Essaka DC, Hindsgaul O, Palcic MM, Prendergast J, Schnaar RL, Dovichi NJ. Anal. Chem. 2011;83:2748–2753. doi: 10.1021/ac103374x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sarver SA, Keithley RB, Essak DC, Tanaka H, Yoshimura Y, Palcic MM, Hindsgaul O, Dovichi NJ. J. Chromatogr. A. doi: 10.1016/j.chroma.2012.01.031. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Pagano RE, Martin OC, Kang HC, Haugland RP. J. Cell. Biol. 1991;113:1267–1279. doi: 10.1083/jcb.113.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Daikoku S, Ono Y, Ohtake A, Hasegawa Y, Fukusaki E, Suzuki K, Ito Y, Goto S, Kanie O. Analyst. 2011;136:1046–1050. doi: 10.1039/c0an00715c. [DOI] [PubMed] [Google Scholar]
- (20).Mehta NR, Lopez PH, Vyas AA, Schnaar RL. J. Biol. Chem. 2007;282:27875–27886. doi: 10.1074/jbc.M704055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Chen Y, Stevens B, Chang J, Milbrandt J, Barres BA, Hell JW. J. Neurosci. Methods. 2008;171:239–247. doi: 10.1016/j.jneumeth.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Larsson EA, Olsson U, Whitmore CD, Martins R, Tettamanti G, Schnaar RL, Dovichi NJ, Palcic MM, Hindsgaul O. Carbohydr. Res. 2007;342:482–489. doi: 10.1016/j.carres.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Le X, Scaman C, Zhang Y, Zhang J, Dovichi NJ, Hindsgaul O, Palcic MM. J. Chromatogr. A. 1995;716:215–220. doi: 10.1016/0021-9673(95)00645-4. [DOI] [PubMed] [Google Scholar]
- (24).Dovichi NJ, Hu S. Curr. Opin. Chem. Biol. 2003;7:603–608. doi: 10.1016/j.cbpa.2003.08.012. [DOI] [PubMed] [Google Scholar]
- (25).Krylov SN, Starke DA, Arriaga EA, Zhang Z, Chan NW, Palcic MM, Dovichi NJ. Anal. Chem. 2000;72:872–877. doi: 10.1021/ac991096m. [DOI] [PubMed] [Google Scholar]
- (26).Whitmore CD, Olsson U, Larsson EA, Hindsgaul O, Palcic MM, Dovichi NJ. Electrophoresis. 2007;28:3100–3104. doi: 10.1002/elps.200700202. [DOI] [PubMed] [Google Scholar]
- (27).Turner EH, Lauterbach K, Pugsley HR, Palmer VR, Dovichi NJ. Anal. Chem. 2007;79:778–781. doi: 10.1021/ac061778r. [DOI] [PubMed] [Google Scholar]
- (28).Curbelo R, Schildkraut ER, Hirschfeld T, Webb RH, Block MJ, Shapiro HM. J. Histochem. Cytochem. 1976;24:388–395. doi: 10.1177/24.1.1254933. [DOI] [PubMed] [Google Scholar]
- (29).Craig DB, Polakowski RM, Arriaga E, Wong JC, Ahmadzadeh H, Stathakis C, Dovichi NJ. Electrophoresis. 1998;19:2175–2178. doi: 10.1002/elps.1150191222. [DOI] [PubMed] [Google Scholar]
- (30).Chen DY, Harke HR, Dovichi NJ. Nucleic Acids Res. 1992;25:4873–4880. doi: 10.1093/nar/20.18.4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Dovichi NJ, Zhang J. Angew Chem Int Ed Engl. 2000;39:4463–4468. doi: 10.1002/1521-3773(20001215)39:24<4463::aid-anie4463>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- (32).Fuller RR, Sweedler JV. Cytometry. 1996;25:144–155. doi: 10.1002/(SICI)1097-0320(19961001)25:2<144::AID-CYTO3>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.