

Abstract
The polyisoprenylation pathway incorporates a reversible step that metabolizes polyisoprenylated methylated proteins from the ester to the carboxylate form. Polyisoprenylated protein methyl transferase (PPMTase) catalyses the esterification whereas polyisoprenylated methylated protein methyl esterase (PMPMEase) hydrolyzes them. Significant changes in the balance between the two enzymes may alter polyisoprenylated protein function possibly resulting in disease. Previous studies show that PMPMEase is the serine hydrolase, Sus scrofa carboxylesterase. Its susceptibility to the nonspecific serine hydrolase inhibitor, phenylmethylsulfonyl fluoride (PMSF) paved the way for its use as a prototypical compound to design and synthesize a series of putative high affinity specific inhibitors of PMPMEase. Pseudo first-order kinetics revealed an over 680-fold increase in kobs/[I] values from PMSF (6 M−1s−1), S-phenyl (L-50, 180 M−1s−1), S-benzyl (L-51, 350 M−1s−1), S-trans, trans-farnesyl (L-28, 2000 M−1s−1), to S-trans-geranylated (L-23, 4100 M−1s−1) 2-thioethanesulfonyl fluorides. C10 S-alkyl substitution revealed a kobs/[I] value (1800 M−1s−1) that was 298 times greater than that for PMSF. The compounds induced the degeneration of human neuroblastoma SH-SY5Y cells with EC50 values of 49, 130 and >1000 μM for L-28, L-23 and PMSF, respectively. The increased affinity with the polyisoprenyl derivatization is consistent with the observed substrate specificity and the reported hydrophobic nature of the active site. These results suggest that (1) PMPMEase is a key enzyme for polyisoprenylated protein metabolism, (2) regulation of its activity is essential for maintaining normal cell viability, (3) abnormal activities may be involved in degenerative diseases and cancers and (4) its specific inhibitors may be useful in combating cancers.
Keywords: Cell Death, Esterase Inhibitors, Methyl Esterase, Molecular Docking, Neuroblastoma, Polyisoprenyltion, Psuedo-first order kinetics, Sulfonyl Fluorides
Introduction
Polyisoprenylated methylated protein methyl esterase (PMPMEase) is a serine hydrolase also known as Sus scrofa carboxylesterase [1, 2]. X-ray crystallographic studies of the human isoform, human carboxylesterase 1 (hCE1) revealed a hydrophobic active site [3, 4]. Docking analyses show that polyisoprenyl groups such as those in polyisoprenylated protein substrates would interact with the hydrophobic active site. Although the catalytic triad of amino acids is common to numerous serine esterases, proteases and peptidases [5], the hydrophobic binding site for PMPMEase distinguishes it from the other serine hydrolases. These differences can be exploited in the design of specific inhibitors of PMPMEase with minimal interactions to other enzymes. Substrate kinetics analysis using various S-alkylated cysteinyl substrates [1, 2, 6] suggests this may be achieved by incorporating polyisoprenyl moieties into the inhibitors as the targeting moiety. This is likely to have the effect of improving the affinity and selectivity towards PMPMEase.
Successful approaches to the design of serine hydrolase inhibitors have often exploited the catalytic mechanism of the enzymes to improve their effectiveness [5, 7]. During catalysis, the histidine and aspartate residues interact to transiently abstract the proton from the hydroxyl group of the catalytic serine, promoting its nucleophilic attack on the carbonyl carbon of the ester or amide/peptide bond resulting in the temporary acylation of the catalytic serine residue [5]. Water is a strong enough nucleophile that rapidly reverses the acylation resulting in rapid enzyme recovery. However, compounds in which the carbonyl group is replaced with sulfonyl and phosphonyl moieties result in exceedingly more stable active site adducts and consequently poor enzyme recovery rates [7]. The compounds thus serve as pseudo-substrates or irreversible inhibitors of the enzymes [5]. PMPMEase is susceptible to phenylmethylsulfonylfluoride (PMSF) [1] as well as various organophosphorus compounds [1, 6, 8]. We thus hypothesized that substituting the carboxylmethyl ester group of the high affinity substrates with the sulfonyl ester moiety would result in highly effective and more selective inhibitors of PMPMEase than PMSF. We further opined that such compounds may have effects on cell viability that would be dependent on PMPMEase inhibition. This is supported by numerous reports linking polyisoprenylation pathway defects to either degenerative disorders or cancers [9–11]. On the other extreme of the cell viability spectrum are the estimated 30% of cancers that are linked to mutated, constitutively active Ras or overexpressed and thus hyperactive Rab [12]. Given that farnesylation is an essential element for the functions of these monomeric G-proteins, farnesyl transferase inhibitors have been developed as potential anti-cancer drugs [13, 14].
In the current study, the potential role of PMPMEase as anti-cancer target was evaluated through the synthesis and analysis of sulfonyl fluorides as putative irreversible inhibitors. The polyisoprenylated analogs were the most effective at inhibiting PMPMEase activity and induction of cultured human neuroblastoma cells death. The results suggest that PMPMEase may constitute a valuable target for anticancer drug development.
Materials and Methods
Materials
Phosphorus tribromide, trans-geraniol, and trans, trans-farnesol were from Sigma-Aldrich (St. Louis, MO). 2-Chloroethanesulfonyl chloride was purchased from TCI America (Portland, OR).
Synthesis
All experiments were conducted under argon. Flame-dried glassware was used when necessary. Solvents and liquid reagents for the synthesis of thiols were deoxygenated by passing a stream of argon over them for 30 min. Unless otherwise stated, NMR spectra were obtained on a Varian Mercury 300 (300 MHz) in CDCl3. Chemical shifts (δ) are given in ppm relative to TMS. Uncorrected melting points were determined on a Melt-Temp 3.0 apparatus. Elemental analyses were conducted at Atlantic Microlab, Norcross, GA, and the accepted values are 0.6–3.5% of the theoretical values except otherwise stated.
trans-Geranyl bromide 2a
A solution of PBr3 (6 ml, 63.2 mmol) in anhydrous ether (10 mL) was added drop-wise to trans-geraniol (21.7 g, 141 mmol) and anhydrous ether (250 mL) at −15 - −10°C. When the addition was complete, the mixture was kept at 0°C for 10 min and then at 20°C for 30 min. The organic phase was decanted, washed with cold 5% NaHCO3, brine, dried over MgSO4 and the solvent removed at 40°C/3 mm Hg, giving 2a as a colorless liquid (22.2 g, 73%). 1H NMR: 1.60 (s, CH3), 1.69 (s, CH3), 1.74 (s, CH3), 1.95–2.20 (m, 2 CH2), 4.03 (d, CH2Br, J=8.4 Hz), 5.09 (m, C=CH), 5.54 (m, C=CHCBr); 13C NMR: 16.2, 17.9, 25.9, 26.4, 29.9, 39.8, 120.8, 123.8, 132.2, 143.8.
trans, trans-Farnesyl bromide 2b
A solution of PBr3 (6 mL, 63.2 mmol) in anhydrous ether (10 mL) was added drop-wise to trans, trans-farnesol (20.47 g, 92 mmol) and anhydrous ether (250 mL) at −15 - −10°C. When the addition was complete, the mixture was kept at 0°C for 10 min and then at 20°C for 30 min. The organic phase was decanted, washed with cold 5% NaHCO3, brine, dried over MgSO4 and the solvent removed at 40°C/3 mm Hg, giving 2b as a pale-yellow liquid (24.43 g, 93%). 1H NMR: 1.60 (s, 2 CH3), 1.67 (s, CH3), 1.72 (s, CH3), 1.95–2.15 (m, 4 CH2), 4.16 (d, CH2Br, J=8.4 Hz), 5.09 (m, 2 × C=CH), 5.54 (m, C=CHCBr); 13C NMR: 16.2, 16.3, 17.9, 25.9, 26.3, 26.9, 29.7, 39.7, 39.9, 120.8, 123.6, 124.5, 131.5, 135.8, 143.7.
S-trans-Geranylisothiuronium bromide 3a
Thiourea (3.8 g, 50 mmol) was dissolved in warm dry i-PrOH (150 mL) and the solution quickly cooled with water after which trans-geranyl bromide (10.85 g, 50 mmol) was added in one portion. The reaction mixture was left overnight at RT. Its volume was reduced to about 50 mL and dry ether (150 mL) was added to precipitate the salt. Filtration of the solid yielded the first fraction of S-trans-geranylisothiuronium bromide (3.0 g). Addition of a further portion of dry ether (150 mL) to the filtrate followed by filtration afforded the second fraction of trans-geranylisothiuronium bromide (2.8 g). Evaporation of the mother liquor and washing of the residue with dry ether (2 × 100 mL) resulted in the third fraction (5.2 g) of the product. After drying at 40°C/3 mm Hg, all fractions melted within the narrow range of 116–119°C. These were combined (total yield 11.4 g, 74%) and used for the next step without further purification. 1H NMR (CD3OD): 1.61 (s, CH3), 1.68 (s, CH3), 1.76 (s, CH3), 2.05–2.20 (m, 2 CH2), 3.86 (d, CH2S, J=7.5 Hz), 5.07 (m, C=CH), 5.30 (m, C=CHCS); 13C NMR (CD3OD): 15.3, 16.7, 24.8, 26.1, 29.5, 39.3, 115.2, 123.5, 131.7, 144.6, 171.9.
S-trans, trans-Farnesylisothiuronium bromide 3b
trans, trans-Farnesyl bromide (5.71 g, 20 mmol) was added in one portion to a solution of thiourea (1.52 g, 20 mmol) in dry i-PrOH (30 mL). The reaction was left overnight at RT. The solvent was removed in vacuum and the solid residue washed with hexane (4 × 50 mL) and dried at 40°C/3 mm Hg. Crude S-trans-farnesylisothiuronium bromide was obtained in practically quantitative yield and was used for the next step without further purification. After crystallization of a small sample from i-PrOH, it melted at 119–120°C (sealed capillary) [100–101°C lit.]. 1H NMR (CD3OD): 1.60 (s, 2 CH3), 1.66 (s, CH3), 1.76 (s, CH3), 1.95–2.40 (m, 4 CH2), 3.87 (d, CH2S, J=8.1 Hz), 5.09 (m, 2 C=CH), 5.31 (m, C=CHCS); 13C NMR (CD3OD): 15.0, 15.1, 16.6, 24.7, 26.1, 26.6, 29.5, 39.3, 39.6, 115.1, 123.5, 124.2, 130.0, 135.5, 144.7, 172.0.
trans-Geranylthiol 4a
S-trans-geranylthioronium bromide (9.27 g, 32 mmol) was refluxed with a solution of NaOH (8.32 g, 208 mmol) in water (40 mL) for 1.5 h. After allowing to cool, the mixture was brought to pH 4 with 6.1 M H2SO4. This was extracted with hexane and the organic phase dried over MgSO4. After removal of solvent, the crude thiol (4.56 g, 84%) was distilled at 78–80°C/4 mm Hg to yield the pure product. 1H NMR: 1.38 (t, SH, J=6.8 Hz), 1.58 (s, CH3), 1.64 (s, CH3), 1.66 (s, CH3), 1.90–2.10 (m, 2 CH2), 3.14 (t, CH2S, J=7.4 Hz), 5.07 (m, C=CH), 5.32 (m, C=CHCS); 13C NMR: 16.0, 17.9, 22.3, 25.9, 26.6, 39.9, 123.6, 124.1, 131.8, 137.6.
trans, trans-Farnesylthiol 4b
S-trans, trans-Farnesylthioronium bromide (15.36 g, 42 mmol) was refluxed with a solution of NaOH (8.5 g, 212 mmol) in water (35 mL) and PEG-400 (0.5 g) for 20 h. After allowing the mixture to cool, it was brought to pH 4 with conc. HCl and extracted with hexane. The organic phase was washed with brine (2 × 35 mL) and dried over MgSO4. After solvent removal, the crude thiol was distilled at 137–145°C/4 mm Hg, giving (5.59 g, 55%) of trans, trans-farnesylthiol. 1H NMR: 1.39 (t, SH, J=7.0 Hz), 1.60 (s, 2 CH3), 1.65 (s, CH3), 1.67 (s, CH3), 1.90–2.15 (m, 4 CH2), 3.15 (t, CH2S, J=7.5 Hz), 5.09 (m, 2 C=CH), 5.33 (m, C=CHCS); 13C NMR: 16.0, 16.3, 17.9, 22.4, 26.0, 26.5, 26.9, 39.6, 39.9, 123.6, 124.0, 124.6, 131.5, 135.5, 137.7.
Vinylsulfonyl fluoride3 7
2-Chlorosulfonyl chloride (25 g, 0.153 mol) was gently heated with KF (63 g, 1.1 mol) and water (40 mL) and a two-phase distillate was collected. The lower layer was dried over CaCl2 and distilled through a Vigreux column, collecting the 103–110°C fraction (6.53 g, 38%). 1H NMR: 6.7 (m, 2H), 6.4 (m, 1H); 13C NMR: 130.0 (d, JCSO2F = 110 Hz), 135.9 (d, JC=CSO2F = 11 Hz).
2-trans-Geranylthioethanesulfonyl fluoride 8a (L-23)
trans-Geranylthiol (222 mg, 1.3 mmol) was reacted at RT for 12h with 154 mg (1.39 mmol) of vinylsulfonyl fluoride in the presence of a 1 M solution of TBAF (0.14 mmol) in THF. After solvent removal, the oil was dissolved in ether, washed with water, brine and dried over CaCl2. The ether was removed and the residue was flash-distilled in Kugelrohr at 0.03 mm Hg and an external temperature of 200°C to afford 248 mg (68%) of the product (L-23). 1H NMR: 1.60 (s, CH3), 1.68 (s, 2 CH3), 2.05–2.15 (m, 2 CH2), 2.93 (m, SCH2), 3.22 (d, CH2S, J=7.5 Hz), 3.57 (m, CH2SO2), 5.06 (m, C=CH), 5.21 (m, C=CHCS); 13C NMR: 16.4, 18.09, 23.8, 25.9, 26.6, 29.9, 39.8, 51.4 (d, JCSO2F = 57 Hz), 119.4, 123.8, 132.3, 140.9. Anal. Calcd for C12H21FO2S2·0.14 THF: C, 49.61; H, 7.29; S, 22.07. Found: C, 49.56; H, 7.1; S, 22.87.
2-trans, trans-Farnesylthioethanesulfonyl fluoride 8b (L-28)
trans, trans-Farnesylthiol (454 mg, 1.9 mmol) was reacted at RT for 20 h with 347 mg (3.88 mmol) of vinylsulfonyl fluoride in the presence of 1 M solution of TBAF (0.35 mmol) in THF. After solvent removal, the oil was flash-distilled in a Kugelrohr at 0.1 mm Hg and an external temperature 190°C giving 380 mg (57%) of ca. 90% pure (1H and 13C NMR) product. 1H NMR: 1.53 (s, 2 CH3), 1.61 (s, 2 CH3), 1.95–2.05 (m, 4 CH2), 2.86 (m, SCH2), 3.16 (d, CH2S, J=8.1 Hz), 3.50 (m, CH2SO2), 5.02 (m, 2 C=CH), 5.15 (m, C=CHCS); 13C NMR: 16.3, 16.4, 17.9, 24.0, 26.0, 26.5, 26.9, 30.0, 39.8, 39.9, 51.5 (d, JCSO2F = 57 Hz), 119.4, 123.7, 124.5, 131.6, 135.9, 141.0. Anal. Calcd for C17H29FO2S2·0.14 THF·0.11 H2O: C, 56.62; H, 8.17; S, 17.78. Found: C, 56.63; H, 8.18; S, 17.77.
Phenylthioethanesulfonyl fluoride (L-50)
Phenylthiol (601 mg, 5.45 mmol) was reacted at RT for 15 h with 667 mg (6.06 mmol) of vinylsulfonyl fluoride in the presence of a 1 M solution of TBAF (0.15 mmol) in THF. After solvent removal, the oil was dissolved in ether, washed with water, brine and dried over CaCl2. The ether was removed and the residue was flash-distilled in Kugelrohr at 0.03 mm Hg and an external temperature of 160°C to afford 1.20 g (98%) of the product (L-50). 1H NMR: 3.30–3.34 (t, CH2S, J=8.1 Hz), 3.35–3.60 (t, CH2SO2F), 7.30–7.40 (m, 5 CH J=6.0 Hz); 13C NMR: 27.8, 50.7, 128.4, 129.9, 131.6, 132.6. Anal. Calcd for C8H9FO2S2: C, 43.62; H, 4.12; S, 29.11. Found: C, 44.18; H, 4.06; S, 29.11.
Benzylthioethanesulfonyl fluoride (L-51)
Benzylthiol (663 mg, 5.53 mmol) was reacted at RT for 15 h with 667 mg (6.06 mmol) of vinylsulfonyl fluoride in the presence of a 1 M solution of TBAF (0.55 mmol) in THF. After solvent removal, the oil was dissolved in ether, washed with water, brine and dried over CaCl2. The ether was removed and the residue was flash-distilled in Kugelrohr at 0.03 mm Hg and an external temperature of 160°C to afford 1.103 g (91%) of the product (L-51). 1H NMR: 2.85–2.91 (t, CH2S, J=9.0Hz), 3.32–3.37 (t, CH2SO2F), 3.77 (s, CH2), 7.25–7.40 (m, 5 CH J=9.0 Hz); 13C NMR: 24.5, 37.0, 51.2, 128.0, 129.0, 129.2, 137.2. Anal. Calcd for C9H11FO2S2: C, 46.13; H, 4.73; S, 27.37. Found: C, 46.53; H, 4.77; S, 27.11.
Decanylthioethanesulfonyl fluoride (L-52)
Decanethiol (1.106 g, 6.34 mmol) was reacted at RT for 15 h with 647 mg (5.70 mmol) of vinylsulfonyl fluoride in the presence of a 1 M solution of TBAF (0.70 mmol) in THF. After solvent removal, the oil was dissolved in ether, washed with water, brine and dried over CaCl2. The ether was removed and the residue was flash-distilled in Kugelrohr at 0.03 mm Hg and an external temperature of 190°C to afford 1.38 g (77%) of the product (L-52). 1H NMR: 0.86–0.89 (d, CH3), 1.26 (Broad, 5xCH2,), 1.31–1.42 (m, CH2 J=16.5 Hz), 1.48–1.61 (m, CH2), 2.54–2.60 (t, CH2S), 2.91–3.01 (t, CH2S J=15.0 Hz), 3.52–3.62 (t, CH2SO2F); 13C NMR: 14.3, 22.9, 25.1, 28.8, 28.9, 29.3, 29.5, 29.5, 29.7, 29.7, 32.1, 32.6, 51.3. Anal. Calcd for C12H25FO2S2: C, 50.67; H, 8.86; S, 22.54. Found: C, 51.16; H, 8.81; S, 22.33.
Gel-Filtration analysis of L-28-treated PMPMEase
To ascertain that the inhibition of PMPMEase by the sulfonyl fluorides is reversible, PMPMEase (1 mg) was pre-incubated with or without L-28 (10 μM) for 60 min in identical conditions as in the enzyme assays except that no substrate was included. These were then fractionated on a Superdex 200 gel-filtration column (2 cm ID × 90 cm), eluting with 50 mM Tris-HCl (pH 7.4) containing 0.1 % Triton X-100 and 0.5 M NaCl. Aliquots of the 4 ml fractions were then analyzed for enzyme activity using RD-PNB as the substrate.
Enzyme inhibition kinetics analysis
Enzyme assays were conducted using RD-PNB as the substrate as previously described [2]. Stock solutions of the sulfonyl fluorides were made in DMSO and stored at −20°C. To obtain the IC50 values for the sulfonyl fluorides, varying concentrations of each compound were pre-incubated in assay buffer [1, 2, 6] with PMPMEase (1 μg) for 15 min at 37°C before the addition of RD-PNB substrate. This was then followed by further incubation and analysis to determine the residual enzyme activity. To determine the pseudo-first order kinetics constants (Kobs/[I]), PMPMEase (82.5μg) was pre-incubated with a concentration of each inhibitor that inhibited about 90% of the enzyme activity and that when diluted 100-fold will result in an ineffective concentration as judged by the respective IC50 results. At time intervals from 0 to 30 min, aliquots from the incubation mixtures were diluted 100-fold with 100 mM Tris-HCl, pH 7.4. RD-PNB substrate was added and assayed for the residual enzyme activities. The residual activities were calculated as percentages of the enzyme activity at the onset of the pre-incubations with inhibitors. The natural logs of the percent residual activities were plotted against pre-incubation times and the slopes (s−1) were divided by the pre-incubation inhibitors concentrations to obtain the Kobs/[I].
Docking analysis
The crystal structure of porcine carboxylesterase (PMPMEase) used in biochemical analysis in our laboratory has not been determined. On the other hand, hCE1 shares 79% sequence identity and 88% sequence similarity to porcine liver PMPMEase [1]. It is therefore believed to be the human version of porcine liver enzyme. Therefore, in the absence of a crystal structure for the porcine enzyme, those for hCE1 with various bound hydrophobic ligands [15–17] were considered for docking studies. The structures reveal an active site that is large, flexible and hydrophobic [15, 17]. These have been suggested to be the essential elements for binding the polyisoprenes and accommodating the large, structurally varied polypeptides [1]. One such X-ray crystal structures of hCE1 [EC 3.1.1.1] is 1yah. It has an ethyl acetate, an analog of fatty acid ethyl ester (FAEE) bound to the active site [17]. The ethyl acetate was used to create a ligand-binding site for the substrates. ArgusLab Version 4.01 software was used for the docking analyses. Procedures for the docking of inhibitors were essentially as previously described [18]. Briefly, a Chemdraw structure for each compound was first saved as an MDL Molfile and opened in ArgusLab. The atoms were then set to the appropriate hybridizations (sp3, sp2, etc) followed by the addition of the hydrogen atoms. The geometry of each compound was optimized before it was made into a ligand using the “Make a Ligand Group from this Residue” option of the ArgusLab software.
The docking between the binding site and each inhibitor was performed using the “Dock a ligand” option. A maximum number of 500 poses was set in order to increase the binding precision. All the amino acid residues of the ethyl acetate binding site were defined to be part of the binding site using a cubic box measuring 18 × 18 × 22 points built to include the entire active site. All other docking analysis parameters were set as previously described [18]. Spacing between the grid points was set at 0.4 Å. “ArgusDock” and “Dock” were chosen as the docking engine for the simulations and calculation type, respectively. “Flexible” was chosen for the ligand and “Ascore” for the scoring function. The binding energies for the ligands (inhibitors) were calculated with parameters from the AScore.prm file using the AScore function. Ligands that were previously defined, from ligand setup, were then docked and the AScore energies recorded. Poses were rank-ordered by docking energy and the pose with the lowest energy was chosen as the predicted receptor-bound conformation of the ligand.
Cell culture
Human neuroblastoma SH-SY5Y cells were initially cultured in a 1:1 mixture of Dulbecco’s modified Eagle’s Medium (DMEM) and F12 Medium (Invitrogen, Carlsbad, CA), supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (Invitrogen, Carlsbad, CA), 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA) in 75 cm2 vented culture flasks. The cultures were incubated at 37°C in 5% CO2/95% humidified air. When cells had reached 80–90% confluence, they were trypsinized and seeded onto 96-well plates at a density of 105 or 24-well plates at the density of 5 × 105 and incubated at 37°C in 5% CO2/95% humidified air.
Cell viability assays
CellTiter-Blue Cell Viability Assay kit (CTB) with fluorescence readout was used to measure resazurin reduction as a marker of metabolic activity following treatment with the inhibitors. The human neuroblastoma SH-SY5Y cells in 96-well plates were exposed to varying concentrations of PMSF, L-23 or L-28 in serum-free DMEM/F12 for 24, 48 and 72 h. Resazurin (Promega, Madison, WI) was used to measure the cell viability according to the vendor instructions. Resazurin (20 μL) was added to each well and the contents gently mixed and incubated in the dark for 2 h at 37°C before measurement of the fluorescence with excitation at 560 nm and emission at 590 nm using FLx 800 Microplate Fluorescence Reader (Bio-Tek Instruments, Inc., Winooski, VM). Cell viability was expressed as the percentage of the fluorescence in the treated cells relative to that of the controls.
PMPMEase activity in sulfonyl fluoride-treated human neuroblastoma SH-SY5Y cells
Human neuroblastoma SH-SY5Y cells were seeded in 24-well plates as described above. The cells were either treated with PMSF, L-23 or L-28 (0–200 μM) and incubated for 24 h. Some of the medium (320 μL) in each well was removed and Triton–X 100 (20 μL, 1 % final concentration) was then added to the remaining 180 μL of medium. These were thoroughly mixed and aliquots of the resulting lysate were assayed for PMPMEase activity using RD-PNB as the substrate.
Statistical analysis
All results were expressed as the means ± S.E.M. The data were analyzed using one-way ANOVA. Statistical differences between control and treated groups were determined by Dunnett’s post-test comparisons. P-values of less than 0.05 were considered statistically significant. The concentrations that inhibited 50% of the activity (IC50) were calculated from a nonlinear regression curve using Graphpad Prism version 4.0 for Windows (San Diego, CA). The dose–response curves were obtained by plotting the percentage inhibition against the log of the concentrations.
Results
Polyisoprenylation potentiates the inhibition of PMPMEase by sulfonyl fluorides
Previous studies in this laboratory showed that PMPMEase is a serine hydrolase with susceptibility to inactivation by compounds such as organophosphorus compounds and PMSF [1, 6, 8]. These are pseudo-substrates that exploit the enzymes’ catalytic process for inhibition [7]. Fig. 1A illustrates the similarities between the sulfonyl fluoride inhibitor, L-28 and the high affinity PMPMEase substrate, BzGFCM [2, 6]. The sulfonyl fluoride inhibitors inhibited the enzyme activity in a concentration-dependent manner (Fig. 1B). The least effective inhibitor was the prototypical compound PMSF with an IC50 of 1800 nM. Increasing the size of the alkyl group resulted in higher potencies for the compounds as shown by the IC50 values in Table 1. Amongst the most potent compounds were the trans-geranylated (L-23) and trans, trans-farnesylated (L-28) 2-thioethane sulfonyl fluorides. Pseudo-first order kinetics analysis revealed some differences in the relative potencies between the compounds. While the Kobs/[I]for PMSF was the lowest as could be predicted from its IC50 value, the Kobs/[I] value for L-51 was over 5-fold lower than that Kobs/[I] for L-28 with a comparable IC50 (Fig. 1C and Table 1). IC50 and pseudo-first order kinetics analysis suggest L-23 and L-28 to be the most potent inhibitors of PMPMEase. Relative to the prototypical PMSF, L-23 and L-28 display IC50 values that were 56- and 37-fold lower and Kobs/[I] that were over 680- and 330-fold higher, respectively. The Kobs/[I] values are the more accurate measurement of the relative potencies of irreversible inhibitors and therefore more closely depict the vast improvement of the sulfonyl fluoride inhibitors by the polyisoprenyl groups. In addition, significant reduction in enzymatic activity was detected in L-28-treated PMPMEase samples when compared to untreated controls even after both were subjected to gel-filtration analysis (Fig. 2). This confirms the expected irreversible inhibition by the sulfonyl fluorides.
Fig. 1. Sulfonyl fluoride inhibitors of PMPMEase.
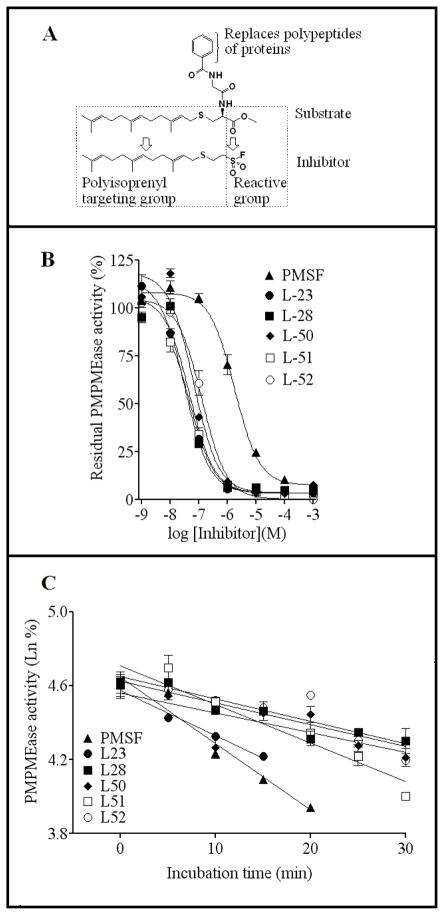
(A) Structural relationship between BzGFCM, a high affinity PMPMEase substrate [2, 6] and the potent inhibitor, L-28. (B) Concentration-dependent inhibition of PMPMEase by the sulfonyl fluoride inhibitors. Purified porcine liver PMPMEase (1 μg) was pre-incubated for 15 min with varying concentrations of each inhibitor as described in the methods section. Substrate was then added followed by incubation and analysis for the residual enzyme activity. The results are percentages of the control uninhibited reactions and are the means (± SEM, N=3). (C) Pseudo-first order kinetics analysis of PMPMEase inhibition by the sulfonyl fluoride inhibitors. PMPMEase (82.5μg) was pre-incubated with suitable concentrations of each inhibitor in the assay buffer as described methods section. At the indicated time points, aliquots were removed and diluted 100-fold in ice-cold buffer. Triplicate aliquots of the diluents were then incubated with substrate and analyzed for residual enzyme activity. The results reflect a plot of the natural logs of the percent residual enzyme activity relative to the controls against time and are the means (± SEM, N=3).
Table 1.
Physicochemical, binding and inhibition kinetics data for sulfonyl fluoride inhibitors of PMPMEase.
| Compound | Structure | ClogP | IC50 (nM) | Kobs/[I] (M−1s−1) | AScore Docking energy (kcal/mole) |
|---|---|---|---|---|---|
| PMSF |
|
1.21 | 1800 | 6 | −9.56 |
| L-23 |
|
2.81 | 32 | 4100 | −12.00 |
| L-28 |
|
4.33 | 48 | 2000 | −14.19 |
| L-50 |
|
1.71 | 66 | 180 | −10.64 |
| L-51 |
|
1.78 | 47 | 350 | −10.68 |
| L-52 |
|
3.79 | 130 | 1800 | −12.02 |
Fig. 2. Gel-filtration analysis of L-28-treated of L-28-treated PMPMEase.
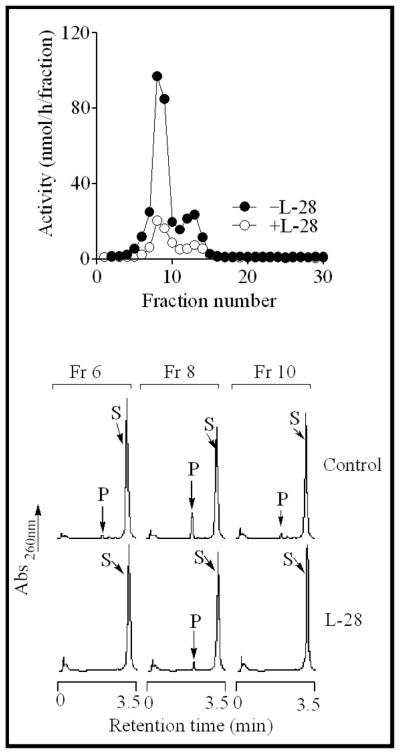
PMPMEase (1 mg) was incubated with or without L-28 (10 μM) for 60 min. These were then fractionated on a Superdex 200 gel-filtration column as described in the methods to remove unreacted L-28. Aliquots of the fractions were then analyzed for the residual enzyme activity.
Docking analysis
Docking analysis of the sulfonyl inhibitors revealed a close association and interaction of (1) the trans, trans-farnesyl group to subsite hydrophobic amino acid residues and (2) the proximity of the sulfonyl moiety of all the inhibitors to the nucleophilic oxygen atom of the catalytic Ser221 residue (Fig. 3). The AScore docking energies also reflect the degree of associations and interactions between the compounds and the active site (Table 1). L-28 which showed a higher number of interactions by virtue of its larger hydrophobic S-polyisoprenyl group had the lowest AScore docking energy of −14.19 kcal/mole. This is 4.63 kcal/mole less than that for PMSF and about 2.2 kcal/mole lower than those L-23 and L-52 (Table. 1).
Fig. 3. Sulfonyl fluoride inhibitors in the active site of hCE1.
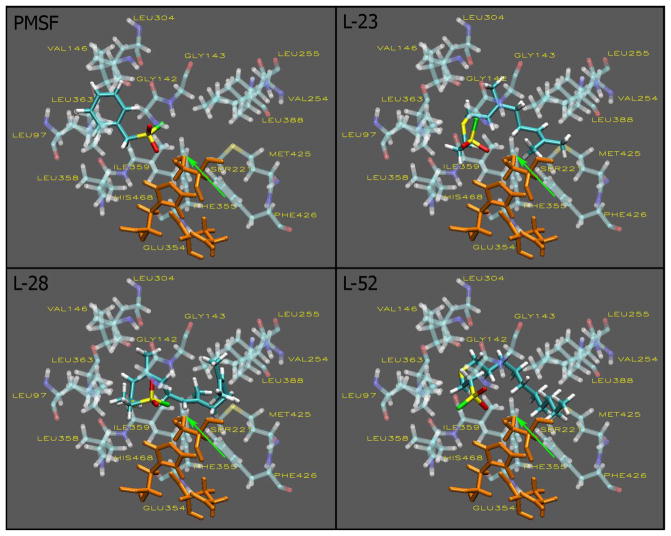
Docking to the active site of hCE1 was conducted as described in the methods section. The visualizations were created using Visual Molecular Dynamics (VMD). The active site and inhibitors are displayed in the Licorice visualization. The active site amino acids are shown with the coloring method: Name (carbon atoms in blue, oxygen in red, sulfur in yellow, nitrogen in dark blue, fluoride in green, hydrogen in white) and material: Transparent. The sulfonyl fluoride inhibitors are shown with the coloring method: Name and material: Opaque. The catalytic triad is shown in orange and the oxygen atom of ser221 is indicated by the green arrow.
Polyisoprenylation potentiates the cell degenerative effects of sulfonyl fluorides
A well studied aspect of polyisoprenylated proteins is their involvement in many cancers [12, 19]. Equally important is deficient geranylgeranylation observed in macular degeneration that is caused by defective Rab escort protein 1 [10, 20]. We thus tested the relative effects of the most potent sulfonyl fluoride inhibitors on cell viability as monomeric G-proteins are known to be polyisoprenylated and have effects on cell proliferation, differentiation and apoptosis [21]. When human neuroblastoma SH-SY5Y cells were treated with PMSF, L-23 or L-28, extensive degeneration was observed in cells exposed to L-23 and L-28 but not PMSF. As shown in Fig. 4, the degeneration was observed at significantly lower concentrations in cells treated with L-28 than with L-23. Cells treated with PMSF did not show degeneration even at 200 μM concentrations.
Fig. 4. Sulfonyl fluoride inhibitors of PMPMEase induce the degeneration of human neuroblastoma SH-SY5Y cells.

Human neuroblastoma cells were cultured and seeded in 24-well plates as described in the methods. They were then treated with varying concentrations of the respective inhibitors. Images of the cells were captured after a 24 h exposure to the inhibitors with an Olympus DP70 camera.
The viability of the treated cells decreased with increasing concentrations of L-23 and L-28, with 24 h exposure time EC50 values of 144.5 and 66.5μM, respectively (Fig. 5A). PMSF again had no significant effect on the viability of the cultured cells. The degenerative effect of L-28 increased with exposure time with more significant reduction in viability after 72 hours (EC50 value of 13.63μM) compared to 24 and 48 hours (EC50 values of 66.5 and 24.26μM, respectively) of exposure (Fig. 5B). It appears that the degenerative potencies of the compounds correlate better with the AScore docking energies than with the Kobs/[I] and the IC50 values.
Fig. 5. Sulfonyl fluoride inhibitors of PMPMEase induce the degeneration of human neuroblastoma SH-SY5Y cells.
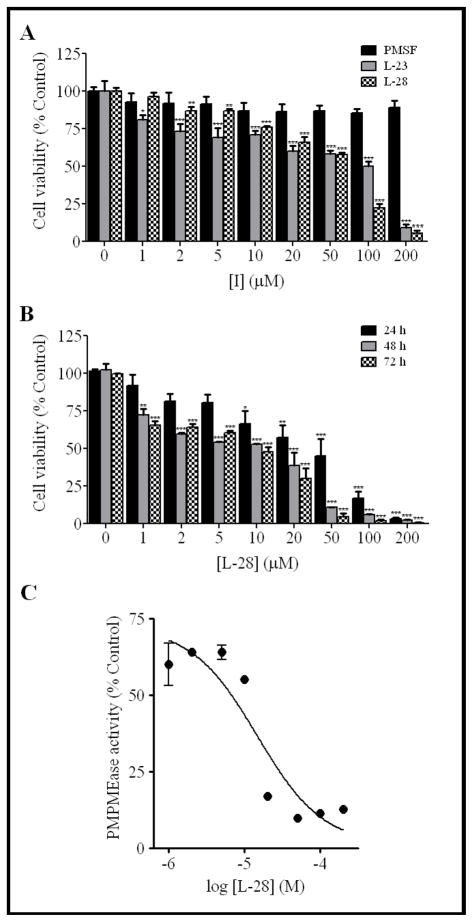
(A) Human neuroblastoma cells were cultured and seeded in 96-well plates as described in the methods. At 24 h after treatment with varying concentrations of respective compounds, cell viability was measured by fluorescence using the resazurin reduction assay. The results are expressed as the means (± SEM, N=4) relative to the control untreated cells. *P<0.05, **P<0.01 and ***P<0.001 versus untreated control cells compared by ANOVA, followed by the Dunnett’s post-test. (B) The induction of neuroblastoma cell degeneration by L-28 increases with exposure time. Cells were treated as described in the methods and analyzed for cell viability after 24, 48 and 72 hours exposure to L-28 using the resazurin reduction assay. The results are expressed as the means (± SEM, N=4) relative to the control untreated cells. *P<0.05, **P<0.01 and ***P<0.001 versus untreated control cells compared by ANOVA, followed by the Dunnett’s post-test. (C) Residual PMPMEase activity in degenerating L-28-treated human neuroblastoma SH-SY5Y cells. Human neuroblastoma cells were cultured and seeded in 24-well plates as described in the methods. They were then treated with the indicated concentrations of L-28. After 24 h, the cells were lysed as described in the methods and the PMPMEase activity determined using RD-PNB as the substrate.
The effect of L-28 on human neuroblastoma SH-SY5Y cell viability is correlated with PMPMEase inhibition
The degenerative effects of L-28 mirrored the PMPMEase activity in the lysate from cells that had been treated with L-28 for 24 h (Fig. 5C). The IC50 for the inhibition due to exposure of the cells to L-28 was 15 μM.
Discussion
Purification and identification of PMPMEase as a serine esterase [1] led us to hypothesize that it might be susceptible to inhibition by irreversible inhibitors of serine hydrolases. Its susceptibility to such serine hydrolase inhibitors as chloromethyl ketones, organophosphorus compounds and PMSF has been demonstrated [1, 6, 8]. The inhibitory aspect of these compounds is borne by their ability to covalently react with the catalytic serine residue to form an adduct that is far more difficult to reverse than the acylation that occurs during ester substrate hydrolysis. However, because the compounds inhibit PMPMEase mainly at millimolar concentrations [8], design strategies can be used to improve the affinity of the compounds for the enzyme active site. The low potencies of these compounds towards PMPMEase may be explained by the appendage structures on the reactive functional groups. Such appendages that were designed to fit the active sites of enzymes such as cholinesterases and proteolytic enzymes rather than PMPMEase may hinder close interactions with the PMPMEase active site. Studies with various S-alkylated substrates revealed an increased affinity toward PMPMEase with S-trans, trans-farnesylation and S-all trans-geranylgeranylation [2]. This suggests that the S-trans, trans-farnesylation or S-all trans-geranylgeranylation may produce more effective PMPMEase inhibitors. Therefore, replacement of the susceptible ester bond of a polyisoprenylated cysteinyl substrate with a reactive “warhead” [5] such as sulfonyl fluoride might convert a high affinity substrate to a highly effective pseudo-substrate irreversible inhibitor of PMPMEase. This is a well established approach for the development of potent inhibitors of serine hydrolases [22, 23]. The polyisoprenylation, besides increasing affinity, also renders the compounds more selective since polyisoprenylated cysteine ester substrates are resistant to hydrolysis by cholinesterases [2]. Incorporation of such moieties that are responsible for the affinity of substrates into the inhibitors promotes the formation of more stable enzyme-inhibitor complexes that precede the covalent reaction [5]. The results clearly indicate that replacing the benzyl group of PMSF with the 2-polyisoprenylthioethane group turned PMSF, a poor inhibitor, into far more potent PMPMEase inhibitors. L-28, the trans, trans-farnesylated inhibitor was expected to be relatively more potent than L-23, the trans-geranylated compound since substrate kinetics analysis revealed a higher affinity of the S-trans, trans-farnesylated substrate to PMPMEase than the S-trans-geranylated substrate [2]. The reverse observation may be due to the relative hydrophobicities of the compounds. While both compounds have no polar groups to aid in aqueous solubility, the fact that L-28 is a prenyl group larger than L-23 translates into a significantly more hydrophobic molecule than L-23 as shown by the ClogP values for both compounds. This may cause it to bind non-specifically to components of the assay mixture and vial and be unavailable for the timely reaction with the enzyme. The AScore binding energies for the compounds suggest that this is likely the case as they indicate the relative interactions with the enzyme active site and therefore the relative stabilities of the enzyme-inhibitor complexes that then proceed to covalent modifications.
The relative potencies of the sulfonyl fluorides against PMPMEase in vitro were not exactly matched by similar abilities to induce cell degeneration. Unlike the in vitro results, L-28 was more potent that L-23 in the cell culture analysis. As indicated earlier, possible adsorptive effects that might have adversely impacted L-28 during PMPMEase assays may have been minimized in cell culture where numerous solutes may block adsorptive sites. Furthermore, PMPMEase assays that last only for about 30 min to 1 h may not have allowed sufficient time for any adsorbed compounds to be desorbed to interact with PMPMEase. On the contrary, cell culture analysis that lasted for 24 to 72 hours might have provided ample time for interaction with PMPMEase in the cell.
PMSF inhibited PMPMEase in vitro but had no effect on the cultured cells. Although this may reflect the low potency for PMSF towards PMPMEase, it may also indicate that PMSF interacts with other enzymes in the cell, significantly limiting its availability to PMPMEase. The targeting effect of the polyisoprenyl group of L-28 is corroborated by the docking studies that show L-28 with more binding interactions and higher affinity and by previous studies that revealed the ineffectiveness of cholinesterase enzymes to hydrolyze polyisoprenylated substrates [2].
The induction of cell degeneration through PMPMEase inhibition is synonymous with various findings that link defective polyisoprenylated proteins and polyisoprenylation pathway enzymes to either cancers [12] or degenerative disorders as Choroideremia [10, 24]. Polyisoprenylation inhibitors have been developed to regulate the excessive activity and control the cell proliferation [25–29]. The data presented here is the first study describing inhibitors directed at any polyisoprenylation-dependent esterase and its anticancer implications. Also, diminished PMPMEase activity may result in degenerative disorders, thus underscoring the role that the polyisoprenylation pathway plays in balancing cell viability. Some PMPMEase-inhibitory organophosphorus compounds [6, 8] cause a Parkinsonian syndrome or neuropathy [30–33] and neuronal cell degeneration due to their inhibition of the serine esterase known as neuropathy target esterase [34–39]. These studies suggest that extreme PMPMEase activities may be involved in cancers and neurodegenerative disorders.
Acknowledgments
*This work was supported by NIH/NIGMS/SCORE Grant number GM 08111-35 and by the Pharmaceutical Research Center NIH/NCRR Grant number G12 RR0 3020 The sulfonyl fluorides were synthesized, analyzed by NMR and the synthesis data compiled by Dr. Leonid Koikov.
References
- 1.Oboh OT, Lamango NS. Liver prenylated methylated protein methyl esterase is the same enzyme as Sus scrofa carboxylesterase. J Biochem Mol Toxicol. 2008;22:51–62. doi: 10.1002/jbt.20214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lamango NS, Duverna R, Zhang W, Ablordeppey SY. Porcine liver carboxylesterase requires polyisoprenylation for high affinity binding to cysteinyl substrates. The Open Enzyme Inhibition Journal. 2009 doi: 10.2174/1874940200902010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bencharit S, Morton CL, Xue Y, Potter PM, Redinbo MR. Structural basis of heroin and cocaine metabolism by a promiscuous human drug-processing enzyme. Nat Struct Biol. 2003;10:349–56. doi: 10.1038/nsb919. [DOI] [PubMed] [Google Scholar]
- 4.Bencharit S, Edwards CC, Morton CL, Howard-Williams EL, Kuhn P, Potter PM, Redinbo MR. Multisite promiscuity in the processing of endogenous substrates by human carboxylesterase 1. J Mol Biol. 2006;363:201–14. doi: 10.1016/j.jmb.2006.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Powers JC, Asgian JL, Ekici OD, James KE. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem Rev. 2002;102:4639–750. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- 6.Lamango NS. Liver prenylated methylated protein methyl esterase is an organophosphate-sensitive enzyme. J Biochem Mol Toxicol. 2005;19:347–57. doi: 10.1002/jbt.20100. [DOI] [PubMed] [Google Scholar]
- 7.Berliner LJ, Wong SS. Spin-labeled sulfonyl fluorides as active site probes of protease structure. I. Comparison of the active site environments in alpha-chymotrypsin and trypsin. J Biol Chem. 1974;249:1668–77. [PubMed] [Google Scholar]
- 8.Lamango NS, Koikov L, Duverna R, Abonyo BO, Hubbard JB. Non-cholinergic organophosphorus toxicity: possible mechanism involving the protein prenylation pathway. In: Webster LR, editor. Neurotoxicity Syndromesed. New York: Nova Science Publishers, Inc; 2007. pp. 37–68. [Google Scholar]
- 9.Seabra MC, Ho YK, Anant JS. Deficient geranylgeranylation of Ram/Rab27 in choroideremia. J Biol Chem. 1995;270:24420–7. doi: 10.1074/jbc.270.41.24420. [DOI] [PubMed] [Google Scholar]
- 10.Pereira-Leal JB, Hume AN, Seabra MC. Prenylation of Rab GTPases: molecular mechanisms and involvement in genetic disease. FEBS Lett. 2001;498:197–200. doi: 10.1016/s0014-5793(01)02483-8. [DOI] [PubMed] [Google Scholar]
- 11.Seabra MC, Brown MS, Goldstein JL. Retinal degeneration in choroideremia: deficiency of rab geranylgeranyl transferase. Science. 1993;259:377–81. doi: 10.1126/science.8380507. [DOI] [PubMed] [Google Scholar]
- 12.Konstantinopoulos PA, Karamouzis MV, Papavassiliou AG. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat Rev Drug Discov. 2007;6:541–55. doi: 10.1038/nrd2221. [DOI] [PubMed] [Google Scholar]
- 13.Sebti SM, Hamilton AD. Anticancer activity of farnesyltransferase and geranylgeranyltransferase I inhibitors: prospects for drug development. Expert Opin Investig Drugs. 1997;6:1711–4. doi: 10.1517/13543784.6.11.1711. [DOI] [PubMed] [Google Scholar]
- 14.Woodward JK, Coleman RE, Holen I. Preclinical evidence for the effect of bisphosphonates and cytotoxic drugs on tumor cell invasion. Anticancer Drugs. 2005;16:11–9. doi: 10.1097/00001813-200501000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Bencharit S, Redinbo MR. Multisite Promiscuity in the Processing of Endogenous Substrates by Human Carboxylesterase 1. J Mol Biol. 2006;363:201–14. doi: 10.1016/j.jmb.2006.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imai T. Human carboxylesterase isozymes: catalytic properties and rational drug design. Drug Metab Pharmacokinet. 2006;21:173–85. doi: 10.2133/dmpk.21.173. [DOI] [PubMed] [Google Scholar]
- 17.Fleming CD, Bencharit S, Edwards CC, Hyatt JL, Tsurkan L, Bai F, Fraga C, Morton CL, Howard-Williams EL, Potter PM, Redinbo MR. Structural insights into drug processing by human carboxylesterase 1: tamoxifen, mevastatin, and inhibition by benzil. J Mol Biol. 2005;352:165–77. doi: 10.1016/j.jmb.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 18.Yanamala N, Tirupula KC, Klein-Seetharaman J. Preferential binding of allosteric modulators to active and inactive conformational states of metabotropic glutamate receptors. BMC Bioinformatics. 2008;9 (Suppl 1):S16. doi: 10.1186/1471-2105-9-S1-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perez-Sala D. Protein isoprenylation in biology and disease: general overview and perspectives from studies with genetically engineered animals. Front Biosci. 2007;12:4456–72. doi: 10.2741/2401. [DOI] [PubMed] [Google Scholar]
- 20.Seabra MC. New insights into the pathogenesis of choroideremia: a tale of two REPs. Ophthalmic Genet. 1996;17:43–6. doi: 10.3109/13816819609057869. [DOI] [PubMed] [Google Scholar]
- 21.Roskoski R., Jr Protein prenylation: a pivotal posttranslational process. Biochem Biophys Res Commun. 2003;303:1–7. doi: 10.1016/s0006-291x(03)00323-1. [DOI] [PubMed] [Google Scholar]
- 22.Powers JC, Tanaka T, Harper JW, Minematsu Y, Barker L, Lincoln D, Crumley KV, Fraki JE, Schechter NM, Lazarus GG, et al. Mammalian chymotrypsin-like enzymes. Comparative reactivities of rat mast cell proteases, human and dog skin chymases, and human cathepsin G with peptide 4-nitroanilide substrates and with peptide chloromethyl ketone and sulfonyl fluoride inhibitors. Biochemistry. 1985;24:2048–58. doi: 10.1021/bi00329a037. [DOI] [PubMed] [Google Scholar]
- 23.Ewoldt GR, Winkler U, Powers JC, Hudig D. Sulfonyl fluoride serine protease inhibitors inactivate RNK-16 lymphocyte granule proteases and reduce lysis by granule extracts and perforin. Mol Immunol. 1992;29:713–21. doi: 10.1016/0161-5890(92)90181-v. [DOI] [PubMed] [Google Scholar]
- 24.Seabra MC, Mules EH, Hume AN. Rab GTPases, intracellular traffic and disease. Trends Mol Med. 2002;8:23–30. doi: 10.1016/s1471-4914(01)02227-4. [DOI] [PubMed] [Google Scholar]
- 25.Bredel M, Pollack IF, Freund JM, Hamilton AD, Sebti SM. Inhibition of Ras and related G-proteins as a therapeutic strategy for blocking malignant glioma growth. Neurosurgery. 1998;43:124–31. doi: 10.1097/00006123-199807000-00081. discussion 31–2. [DOI] [PubMed] [Google Scholar]
- 26.Caraglia M, Budillon A, Tagliaferri P, Marra M, Abbruzzese A, Caponigro F. Isoprenylation of intracellular proteins as a new target for the therapy of human neoplasms: preclinical and clinical implications. Curr Drug Targets. 2005;6:301–23. doi: 10.2174/1389450053765833. [DOI] [PubMed] [Google Scholar]
- 27.Dinsmore CJ, Bell IM. Inhibitors of farnesyltransferase and geranylgeranyltransferase-I for antitumor therapy: substrate-based design, conformational constraint and biological activity. Curr Top Med Chem. 2003;3:1075–93. doi: 10.2174/1568026033452113. [DOI] [PubMed] [Google Scholar]
- 28.Ghobrial IM, Adjei AA. Inhibitors of the ras oncogene as therapeutic targets. Hematol Oncol Clin North Am. 2002;16:1065–88. doi: 10.1016/s0889-8588(02)00050-3. [DOI] [PubMed] [Google Scholar]
- 29.Sebti SM. Protein farnesylation: implications for normal physiology, malignant transformation, and cancer therapy. Cancer Cell. 2005;7:297–300. doi: 10.1016/j.ccr.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 30.Arima H, Sobue K, So M, Morishima T, Ando H, Katsuya H. Transient and reversible parkinsonism after acute organophosphate poisoning. J Toxicol Clin Toxicol. 2003;41:67–70. doi: 10.1081/clt-120018273. [DOI] [PubMed] [Google Scholar]
- 31.Ascherio A, Chen H, Weisskopf MG, O’Reilly E, McCullough ML, Calle EE, Schwarzschild MA, Thun MJ. Pesticide exposure and risk for Parkinson’s disease. Ann Neurol. 2006;60:197–203. doi: 10.1002/ana.20904. [DOI] [PubMed] [Google Scholar]
- 32.Bhatt MH, Elias MA, Mankodi AK. Acute and reversible parkinsonism due to organophosphate pesticide intoxication: five cases. Neurology. 1999;52:1467–71. doi: 10.1212/wnl.52.7.1467. [DOI] [PubMed] [Google Scholar]
- 33.Davis KL, Yesavage JA, Berger PA. Single case study. Possible organophosphate-induced parkinsonism. J Nerv Ment Dis. 1978;166:222–5. doi: 10.1097/00005053-197803000-00010. [DOI] [PubMed] [Google Scholar]
- 34.Glynn P. Neuropathy target esterase (NTE): molecular characterisation and cellular localisation. Arch Toxicol Suppl. 1997;19:325–9. doi: 10.1007/978-3-642-60682-3_30. [DOI] [PubMed] [Google Scholar]
- 35.Glynn P, Read DJ, Lush MJ, Li Y, Atkins J. Molecular cloning of neuropathy target esterase (NTE) Chem Biol Interact. 1999;119–120:513–7. doi: 10.1016/s0009-2797(99)00065-4. [DOI] [PubMed] [Google Scholar]
- 36.Glynn P. Neural development and neurodegeneration: two faces of neuropathy target esterase. Prog Neurobiol. 2000;61:61–74. doi: 10.1016/s0301-0082(99)00043-x. [DOI] [PubMed] [Google Scholar]
- 37.Johnson MK, Glynn P. Neuropathy target esterase (NTE) and organophosphorus-induced delayed polyneuropathy (OPIDP): recent advances. Toxicol Lett. 1995;82–83:459–63. doi: 10.1016/0378-4274(95)03495-1. [DOI] [PubMed] [Google Scholar]
- 38.Glynn P. A mechanism for organophosphate-induced delayed neuropathy. Toxicol Lett. 2006;162:94–7. doi: 10.1016/j.toxlet.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 39.Glynn P. Axonal degeneration and neuropathy target esterase. Arh Hig Rada Toksikol. 2007;58:355–8. doi: 10.2478/v10004-007-0029-z. [DOI] [PubMed] [Google Scholar]