

Abstract
The prefrontal cortex (PFC) is among the most evolved brain regions, contributing to our highest order cognitive abilities. It regulates behavior, thought, and emotion using working memory. Many cognitive disorders involve impairments of the PFC. A century of discoveries at Yale Medical School has revealed the neurobiology of PFC cognitive functions, as well as the molecular needs of these circuits. This work has led to the identification of therapeutic targets to treat cognitive disorders. Recent research has found that the noradrenergic α2A agonist guanfacine can improve PFC function by strengthening PFC network connections via inhibition of cAMP-potassium channel signaling in postsynaptic spines. Guanfacine is now being used to treat a variety of PFC cognitive disorders, including Tourette’s Syndrome and Attention Deficit Hyperactivity Disorder (ADHD). This article reviews the history of Yale discoveries on the neurobiology of PFC working memory function and the identification of guanfacine for treating cognitive disorders.
Keywords: prefrontal cortex, norepinephrine, alpha-2A-adrenergic receptors, working memory, ADHD, Tourette’s Syndrome, traumatic brain injury, substance abuse, schizophrenia, autism, aging
Introduction
Cognitive disorders are among the most challenging and disturbing ailments. They can alter who a person is, limit his or her success in school or work, interfere with friendships, impair the ability to care for themselves and others, and disrupt the lives of families and loved ones. Even worse, cognitive impairment can extend to loss of insight and judgment ― the patient denies that anything is wrong ― making the situation even more difficult to treat. Cognitive disorders involve dysfunction of the most highly evolved cortical regions, the association cortices, with particular vulnerabilities in the prefrontal cortex (PFC). Thus, understanding the biology of these disorders is a daunting yet fascinating task. Almost a century of research at Yale School of Medicine has revealed the neurobiology of PFC cognitive operations and has begun to reveal the molecular needs of these circuits, with the goal of identifying potential therapeutic targets. Recent research has found that the noradrenergic α2A agonist guanfacine can strengthen PFC network connections and improve PFC regulation of behavior, thought, and emotion. Based on these discoveries in animals, guanfacine is now being used to treat a variety of cognitive disorders that benefit from strengthened PFC function.
The highly evolved prefrontal cortex
The PFC is one of the most evolved regions of the brain (Figure 1A). The human neocortex consists of the basic sensory and motor areas, as well as the association areas that subserve perception and cognition. The neocortex contains six layers of neurons (Figure 1A), which are intricately interconnected to form neural networks. The association cortices expand tremendously in the primate brain, and, of these, the PFC subserves the highest order cognitive functions. The PFC guides thought, actions, and emotion using representational knowledge [1,2], allowing us to marry the past to the future using working memory [3]. PFC circuits hold information “in mind” to provide the foundation for abstract thought and mental manipulation, what is often referred to as our “mental sketch pad.” This represented information serves as goals and plans for action, the basis for the executive functions [4,5]. The PFC allows us to organize and plan for long-term ambitions (career plans, carrying out large projects) as well as very short-term goals (holding together the beginning and end of a sentence) to provide directed meaning and a purposeful life [6]. The PFC protects goal-directed behavior from distractions and compulsions [7] and thus is essential for self-control, generating the informed inhibition that is central to civilized behavior [8]. It is key for high order decision-making [9] and meta-cognition, e.g., remembering to remember as well as self-awareness (knowing what you know and what you don’t know [10], knowing what others are thinking [11], and moral conscience [12]). The PFC accomplishes these cognitive feats in a topographically organized manner, with dorsal and lateral PFC circuits regulating attention, thought, and action [13] and the more ventral and medial PFC circuits regulating emotion and physiological state [14]. There are likely other organizational topographies as well, e.g., increasingly abstract operations being performed by increasingly rostral regions of PFC [15]. It is remarkable that the neurobiology of these highest order cognitive functions has begun to be understood, and much of this success has arisen based on the primary studies at Yale School of Medicine.
Figure 1.

Prefrontal cortical (PFC) circuits mediating higher cognitive operations. A. The PFC expands tremendously in brain evolution, comprising a very small proportion of the brain in rodents such as the rat and increasing dramatically in primates, with special prominence in the human brain. The PFC is highlighted in blue. An inset of the human dlPFC is shown at the right; this Nissl-stained section shows the six layers of dlPFC. B. The dlPFC microcircuits subserving working memory, discovered by Goldman-Rakic (1995) [1]. Pyramidal cells in deep layer III receive visuospatial information from the parietal association cortex. Pyramidal cells with similar spatial inputs excite each other through connections on dendritic spines to maintain persistent firing throughout the delay period. The spatial tuning of the neuron’s response is sharpened by lateral inhibition from parvalbumin-containing GABAergic interneurons, such as the Basket cell (B) shown in this figure. Note that chandelier cells also serve this function (not shown). The red rectangle highlights an axo-spinous synapse enlarged in C and D. These dlPFC microcircuits are the ones most afflicted in schizophrenia, where there is loss of neuropil (including loss of dendritic spines) in deep layer III [100], and reduced parvalbumin GABAergic function [101]. C. A working model of the cAMP-potassium channel signaling mechanisms in spines that dynamically weaken synaptic efficacy and gate out network inputs to the neuron. cAMP directly opens HCN channels, while cAMP activation of PKA signaling increases the open state of KCNQ channels. cAMP generated by calcium build up, e.g., feedback fatigue via NMDA or mGluR1/5, or actively generated by stress exposure, e.g., via D1 or β1 receptor stimulation. D. NE or guanfacine stimulation of α2A receptors on spines inhibits cAMP production and closes HCN and KCNQ channels, strengthening network connectivity, increasing neuronal PFC firing, and thus improving PFC regulation of behavior, thought and emotion. See Figure 5 for data supporting the model. C and D artistically adapted from Arnsten et al., 2010 [71].
The essential role of the prefrontal cortex in working memory: The original discovery by Jacobsen and Fulton
The first groundbreaking studies of the PFC’s role in cognition began in the 1930s at Yale School of Medicine. John Fulton arrived at Yale in 1930 to serve as Chair of the new Department of Physiology. He had been trained as a neurosurgeon by Harvey Cushing at Harvard and was able to perform complex surgeries that opened new avenues of research. Together with Carlyle Jacobsen (Figure 2A), they examined the effects of specific brain lesions on behavior in primates. Although they are widely known for their work relevant to lobotomies, i.e., that very large PFC lesions made aggressive animals calmer, their most important breakthrough is rarely cited: the discovery that the dorsolateral region of the PFC is essential for abstract thought [16]. Jacobsen published this finding in 1936 (citing the key role of Fulton’s neurosurgical skills). He had designed a definitive study comparing the effects of specific cortical lesions on a series of cognitive tasks during which the information needed to solve the problem was either available in the environment or had to be held in working memory. He found that monkeys with bilateral lesions to the dorsolateral PFC (dlPFC), but not other cortical lesions, were markedly impaired on problems requiring working memory and that this impairment was permanent (Figure 2B-C). He wrote, “The animal without the frontal association area learns and retains sensory-motor habits and visual discriminations but it is unable to remember for even a few seconds under which of two cups a piece of food is concealed . . . It is as if ‘out of sight, out of mind’ were literally applicable” [16].
Figure 2.

The discovery of the key role of the dlPFC in working memory by Jacobsen in 1936 [16]. A. Carlyle Jacobsen, in about 1940, who later became the first President of SUNY Upstate Medical Center. Photo courtesy of the F.W. Kent Collection of Photographs, University of Iowa Archives, University of Iowa Libraries. B. An artistic rendering based on a figure from Jacobsen’s paper showing the bilateral dlPFC lesion that gravely impaired spatial working memory. Unilateral PFC lesions or lesions of the same size elsewhere in the cortex had much less effect on performance. C. Reconstruction of a portion of a table from Jacobsen’s paper showing the marked impairment on the delayed response spatial working memory task and preserved performance on the visual discrimination problems that did not require working memory to solve.
World War II interrupted this extraordinary research, but the work continued in the 1950s and 1960s, when researchers such as Karl Pribram and Mortimer Mishkin performed lesion studies at Yale and further defined the roles of PFC subregions in cognitive performance [17], including work showing the importance of PFC for resisting distraction [18] and the key roles of the sensory association cortices for object perception [19].
The Neurobiology of Thought: the pioneering work of Goldman-Rakic
Patricia Goldman-Rakic began her career in neuroscience performing developmental and lesion studies at the National Institute of Mental Health (NIMH), but then moved to Yale, where she illuminated the physiology, micro-circuitry ,and neuromodulation of the brain networks underlying spatial working memory (Figure 3A). Early in her career at the NIMH, she further refined Jacobsen’s work and defined the essential PFC subregion needed for visuospatial working memory: the caudal two-thirds of the principal sulcal dlPFC [20,21]. She then went on to study the circuitry, physiology, and modulatory needs of this PFC region, performing most of this work at Yale. She found parallel, reciprocal circuits between the dlPFC and the sensory association cortices, where projections from visuospatial, visual feature, auditory spatial, and auditory feature association cortices occupied distinct domains in the dlPFC, with the spatial information residing more dorsally and the feature information more ventrally [13]. In particular, the visuospatial projections from parietal area 7 terminated in the subregion of dlPFC area 46 that she showed to be essential for visuospatial working memory [22]. These data indicated a specific topography of anatomical projections even for the highest order cognitive abilities.
Figure 3.
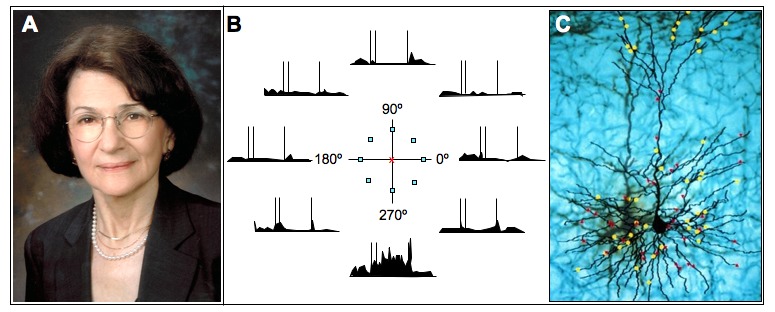
The discovery of the neurobiology of thought by Patricia Goldman-Rakic. A. Patricia Goldman-Rakic about 2000. B. A dlPFC Delay Cell whose firing patterns represent a precise portion of visual space. The neuron fired throughout the delay period if the cue had occurred at 270º but did not fire if the cue had occurred at other spatial locations. Goldman-Rakic [1] discovered the microcircuitry underlying the two key physiological features needed for working memory: 1) persistent firing throughout the delay period, generated by recurrent excitation in pyramidal cell microcircuits (Figure 1B), and 2) spatial tuning, sculpted in part via lateral inhibition from GABAergic, parvalbumin-containing interneurons such as the Basket cell depicted in Figure 1B. Artistically rendered based on Funahashi et al., 1989 [24]. C. Dopamine inputs onto a subset of spines of a monkey dlPFC neuron. The yellow dots represent tyrosine-hydroxylase positive axon terminals; red dots indicate dopamine D1 receptors. An original Goldman-Rakic figure based on Krimer et al., 1997 [102].
Goldman-Rakic’s group adapted an earlier task [23] “to explore the full perimetry of visual space” and reveal the cellular basis of visuospatial working memory [24]. They recorded from area 46 of the dlPFC while monkeys performed an oculomotor spatial working memory task that required them to remember one of eight ever-changing locations over a brief delay. Earlier studies had shown that dlPFC neurons show persistent firing across the delay period while the monkey remembers the spatial position. This new task allowed Goldman-Rakic to see that dlPFC Delay Cells are able to represent visuospatial position, e.g., a neuron that continued to fire throughout the delay period if the cue had appeared at 270º but not other locations, for example, the neural representation of visual space (Figure 3B). Goldman-Rakic then uncovered the dlPFC microcircuits that create this neural representation (reviewed in [1]). She showed that persistent firing across the delay period is generated by columns of pyramidal cells in deep layer III that excite each other to maintain firing in the absence of visual stimulation (Figure 1B), while the precise spatial tuning (e.g., firing to represent 270º but not 90º) is sculpted by lateral inhibition from parvalbumin-containing GABAergic interneurons, i.e., basket and chandelier cells (Figure 1B). These layer III microcircuits are the ones that expand most in primate evolution [25] and are the focus of neuropil loss in schizophrenia [26].
Goldman-Rakic also made the landmark discovery that dopamine (DA) inputs onto PFC neurons (Figure 3C) have a critical modulatory influence on dlPFC spatial working memory function and that depletion of DA [27] or blockade of DA D1 receptors [28] in the dlPFC produced spatial working memory deficits as profound as ablation of the cortex itself. This was the first indication that dlPFC was absolutely dependent on the correct neurochemical environment. Arnsten and Goldman-Rakic then discovered that very high levels of DA release, as occurs during stress [29] or with drugs of abuse [30], was as detrimental to working memory function as was too little DA, the DA “inverted U” [31-34]. As DA signaling is known to be dysregulated in schizophrenia [35,36], changes in DA modulation combined with altered layer III microcircuits likely underlie the profound PFC cognitive deficits observed in this illness [37,38].
Although DA plays key roles in PFC circuits, Arnsten and Goldman-Rakic discovered that norepinephrine (NE) is equally important for proper PFC function via its actions at post-synaptic, α2A adrenergic receptors [39] and that this receptor may be especially amenable as a therapeutic target.
Parallel breakthrough by Aghajanian and Cohen: The therapeutic effects of clonidine
While Goldman-Rakic explored the effects of DA in primate dlPFC, George Aghajanian studied α2 adrenergic receptor actions in the brains of opiate-dependent rats (Figure 4A). He observed that the NE cells of the locus coeruleus (LC) in the brainstem (Figure 4B) became overactive during opiate withdrawal and that the α2 receptor agonist clonidine reduced its firing through actions at presynaptic α2 receptors on LC neurons (Figure 4C) [40-42]. The LC provides NE to most of the brain and spinal cord (Figure 4B); thus, clonidine’s ability to reduce LC cell firing and NE release has a pervasive effect on the nervous system. This landmark finding continues to guide most research on α2 receptors to this day. Based on these physiological data in rats, clonidine and other α2 agonists are still being used to treat opiate withdrawal in humans [43]. However, this initial finding also led to a breakthrough treatment for Tourette’s Syndrome [44].
Figure 4.

The discoveries of clonidine’s effects on LC neuronal firing by George Aghajanian that led to its therapeutic effects in Tourette’s Syndrome by Donald Cohen. A. George Aghajanian, Professor of Psychiatry and Pharmacology at Yale. Photo courtesy of Dr. Aghajanian. B. A schematic drawing showing the position of the locus coeruleus in the human brainstem, with arrows representing its widespread noradrenergic projections throughout the brain and spinal cord. C. The α2 agonist, clonidine (C), reduced LC firing in opiate-withdrawing rats. Artistically rendered based on Svensson et al., 1975 [40]. D. Donald Cohen, Director of the Yale Child Study Center until 2001. Photo courtesy of Yale University. E. Clonidine treatment in a boy with Tourette’s Syndrome reduced the number of tics compared to baseline. The number of clinician-rated measures of Overall Severity of symptoms, including motor and phonic tics, decreased with continued clonidine treatment. Artistically rendered based on Cohen et al., 1980 [44].
Donald Cohen was a child psychiatrist (and soon to be Director) of the Yale Child Study Center (Figure 4D). He was treating children with Tourette’s Syndrome, a disorder in which unwanted movements or utterances (tics) repeatedly disrupt normal behavior and even endanger patients when the movements are violent. Cohen brought Aghajanian with him on rounds to see one boy with particularly debilitating, self-destructive tics. Aghajanian saw a similarity to the dysregulated movements of opiate withdrawal and suggested they try clonidine [45]. The medication was successful in relieving the tics (Figure 4E), and clonidine became one of the first effective treatments for Tourette’s [44]. Later, Hunt and Cohen tried clonidine in another disinhibited disorder, Attention Deficit Hyperactivity Disorder (ADHD), again with general success [46]. Clonidine had significant sedative side effects, but it was assumed that the sedation was key to its therapeutic effects and that clonidine acted by reducing LC firing to lower arousal in “hyperaroused” patients [47]. But research in monkeys was about to reveal that α2 receptors also have powerful effects through actions in the PFC.
Guanfacine: An unexpected mechanism
Arnsten and Goldman-Rakic set out to study the cognitive-enhancing effects of DA agonists in aged monkeys with naturally occurring DA depletion, but the compound that produced the most dramatic improvement in their cognitive performance was not a DA drug, but clonidine. Following clonidine administration, the aged monkeys were almost asleep and yet performed near perfectly [39]. However, these beneficial cognitive effects were not due to the expected presynaptic effects of clonidine on LC neurons, but rather arose from actions at post-synaptic receptors in the dlPFC [39]. Indeed, destruction of the presynaptic sites only made clonidine’s effects more potent [39,48]. Further research revealed that the α2A-adrenoceptor subtype was essential for these actions [49] and that the α2A-preferring agonist guanfacine can improve working memory [50,51], attention regulation (Figure 5A, top) [52,53], and behavioral inhibition [54] independent of its sedative actions. Guanfacine is more selective for the α2A receptor subtype than is clonidine, which also binds with high affinity to α2B, α2C, and imidazoline receptors [55,56]. Guanfacine is weaker than clonidine in producing hypotension and sedation [50] and has weaker presynaptic actions in the brain, i.e., it is 10 times less effective in reducing LC firing and decreasing NE release [57]. However, guanfacine is more potent than clonidine in enhancing PFC working memory function in aged monkeys, suggesting greater efficacy at post-synaptic sites in PFC [50]. Most recently, guanfacine has been shown to improve impulse control in monkeys performing a delayed discounting task, i.e., increasing the ability to resist an immediate, small reward and instead wait for a larger reward [58]. The PFC is the site of beneficial drug actions, as guanfacine improves cognitive performance when infused directly into the rat or monkey PFC (Figure 5A, bottom) [59-61]. Indeed, guanfacine’s enhancing effects can even be observed at the cellular level, where application of drug directly onto dlPFC neurons increases the delay-related neuronal firing needed for working memory [62] (Figure 5B). Conversely, blocking α2A receptors in the monkey dlPFC markedly impairs working memory [63] and behavioral inhibition [64,65] and greatly reduces persistent neuronal firing [62,66], demonstrating that endogenous NE stimulation of α2A receptors is essential for PFC regulation of behavior, thought, and emotion.
Figure 5.

The α2A adrenoceptor agonist guanfacine improves PFC neuronal firing and cognitive function through actions at α2A receptors on spines in layer III dlPFC neurons. A. Top: Systemic administration of guanfacine to aged monkeys improves working memory performance and is particularly effective in protecting performance from the deleterious effects of distracters presented during the delay period. Artistically rendered based on Arnsten and Contant, 1992 [52]. Bottom: Infusion of guanfacine directly into the rat PFC improved performance of a working memory task (similar effects were seen with guanfacine infusions into monkey dlPFC by the Li lab in China [103]). Guanfacine’s enhancing effects were blocked by co-infusion of the cAMP analog, Sp-cAMPS, demonstrating actions through cAMP signaling pathways. Artistically rendered based on Ramos et al., 2006 [60]. B. Iontophoresis of guanfacine directly onto dlPFC neurons in monkeys performing a working memory task significantly increased delay-related firing. Firing was suppressed when Sp-cAMPS was co-applied with the guanfacine. Similar enhancing effects were observed with the HCN channel blocker ZD7288, which reversed the suppressive effects of the α2 blocker, yohimbine (not shown). Artistically rendered based on Wang et al., 2007 [62]. C. Double-label immunogold electron microscopy by Dr. Constantinos Paspalas demonstrating α2A-receptors co-localized with HCN channels in the spines of layer III dlPFC network synapses.
Ionic regulation of prefrontal microcircuits: Vulnerabilities and opportunities
Further research identified the molecular basis of guanfacine’s enhancing effects in dlPFC, one that was “upside down and backwards” from actions elsewhere in brain. α2A receptors are coupled to Gi proteins that inhibit cAMP signaling. In most brain circuits, e.g., in hippocampus, cAMP strengthens synaptic connections [67]. However, in PFC, cAMP weakens persistent firing and impairs working memory [34,62,68-70]. These seemingly opposite effects arise from cAMP actions on ion channels that dynamically alter the strength of PFC network connections [71]. The pyramidal cell microcircuits in layer III of dlPFC interconnect on dendritic spines via NMDA receptor synapses, exciting each other to keep information “in mind” [71]. Immunoelectron microscopy has revealed α2A receptors on these spines, situated next to ion channels that can gate network connections [62] (Figure 5C). Of special interest are the potassium channels that are opened by cAMP signaling: HCN cation channels (Hyperpolarization-activated Cyclic Nucleotide gated) that are directly influenced by cAMP and KCNQ channels that are opened indirectly by cAMP activation of protein kinase A (PKA) (Figure 1C). Opening these channels by high levels of cAMP signaling, e.g., during stress exposure or with α2A receptor blockade, weakens PFC network connections and reduces persistent firing (Figure 1C), while blockade of HCN channels restores firing [34,62]. In this way, exposure to a stressor can rapidly take PFC “off-line” to switch control of behavior to more primitive brain circuits that mediate stress reflexes, such as freezing or fight or flight habitual responses [72]. This mechanism has survival value when faced with danger but may be counterproductive when stressors require thoughtful PFC responses, e.g., during public speaking or when needing to make a complex decision.
In contrast, NE or guanfacine stimulation of α2A receptors on PFC spines strengthens PFC network connections by inhibiting cAMP signaling, closing HCN channels, and increasing delay-related firing (Figure 1D [62]). As can be seen in Figure 5, the enhancing effects of guanfacine on both PFC persistent firing [62] and working memory performance [60] can be reversed by the cAMP analog, Sp-cAMPS. Recently, guanfacine has also been shown to restore persistent firing in the aged monkey dlPFC via inhibition of cAMP-HCN or KCNQ channel signaling [73]. Taken together, the basic data show that guanfacine inhibits cAMP opening of potassium channels on spines, which increases PFC network firing and strengthens PFC cognitive control of behavior. This process engages PFC circuits to protect goals from distraction, overcome inappropriate habits, and resist impulses to allow civilized, purposeful behavior. It is remarkable that these highest order cognitive operations can be understood at the level of ion channels.
The intricate molecular mechanisms needed to precisely regulate PFC network connectivity are exceptionally vulnerable to disruption from both environmental and genetic insults. For example, cAMP-PKA signaling becomes dysregulated with advancing age due to loss of inhibitory influences on cAMP, and increased cAMP-K+ channel actions lead to reduced persistent firing and impaired working memory [73,74]. A remarkable number of genetic insults in cAMP signaling pathways are associated with mental illness. For example, a taq1 polymorphism in the promoter region of the gene encoding for dopamine β hydroxylase (DβH) leads to reduced DβH expression, reduced NE synthesis, weaker PFC executive abilities [75,76], and often a diagnosis of ADHD [77-79]. Genetic insults to cAMP intracellular signaling pathways are increasingly associated with schizophrenia, including the scaffolding protein DISC1 (Disrupted In Schizophrenia) that tethers the phosphodiesterases PDE4A, PDE4B, and PDE4D to regulate cAMP concentrations [80,81], as well as insults to the phosphodiesterases themselves [82,83], all of which are found in the spines of layer III dlPFC neurons [84] (and Paspalas and Arnsten, unpublished). There are also genetic insults in receptors regulating cAMP synthesis (mGluR3, VIPR2) and potassium channels regulated by cAMP (KCNH2). Alterations in these cAMP-regulating proteins may weaken layer III microcircuits and lead to the PFC dysfunction which characterizes this illness.
The therapeutic effects of guanfacine in prefrontal cognitive disorders
Based on the research in animals, guanfacine is now in widespread use for the treatment of a variety of PFC cognitive disorders. For example, weaker PFC function is a hallmark of ADHD, particularly deficits in the right inferior PFC that is specialized for inhibiting inappropriate actions [85]. This PFC subregion normally enlarges as a child matures, but fails to do so in those with ADHD [86]. Immediate-release guanfacine was first tested in children with ADHD by Hunt, based on the successful animal data and his previous experience with clonidine [87]. It was also tested open label in ADHD patients with tics at the Yale Child Study Center [88]. Although guanfacine has a long half-life in adults, it is rapidly metabolized by children, and thus an extended release formulation was developed (Intuniv™) and approved by the FDA in 2009 for the treatment of pediatric ADHD. Guanfacine helps ADHD patients control their own behavior and inhibit inappropriate distractions and impulses [89,90]. Guanfacine also allows inhibition of inappropriate aggressive impulses [91], likely through its actions in ventral PFC. Guanfacine is also in widespread use “off label” to inhibit inappropriate motor and vocal tics in patients with Tourette’s Syndrome and in children with ADHD and tics who often cannot take stimulant medications [92]. It is also being tested in autism spectrum disorders to treat the disinhibited behaviors that often accompany the social deficits in these disorders [93,94].
Immediate-release guanfacine is in experimental use for a broader number of adult disorders involving PFC dysfunction. Importantly, it is helping patients who have traumatic brain injury to the PFC for whom there is great need of treatment [95]. Guanfacine also has been shown to improve working memory in patients with schizotypal disorder with cognitive deficits resembling those in schizophrenia [96], and it improves PFC function and metabolism in patients with some forms of epilepsy [97]. Guanfacine is being used to treat attentional neglect caused by stroke [98], which may be related to its attention-enhancing effects in normal subjects [99]. Studies from the Yale Stress Center are finding that guanfacine can strengthen PFC self-control during stress and help people quit smoking (S. McKee, Yale Dept. Psychiatry, personal communication), and these beneficial effects may extend to other drugs of abuse as well (R. Sinha, Yale Dept. Psychiatry, personal communication). Finally, very low doses of guanfacine are currently being tested in the elderly to see if they will ameliorate age-related cognitive decline in humans as they do in monkeys (C. van Dyck, Yale Dept. Psychiatry and Neurology, personal communication). Thus, guanfacine may be useful for a breadth of PFC cognitive disorders.
Conclusions and Outlook
Many scientists have thought that understanding the neurobiological basis of our highest order cognitive functions was beyond the realm of scientific inquiry, but research at Yale has shown that it is possible to reveal the microcircuitry of cognition even at the molecular level. It is noteworthy that many of these key discoveries have been made through studies of monkeys engaged in working memory tasks. Given the great expansion of the PFC in brain evolution, especially in the microcircuits of layer III, these breakthroughs may not have occurred without this invaluable resource. More research is needed to understand how genetic insults lead to alterations in layer III microcircuits, so that we may develop informed interventions for cognitive disorders.
Abbreviations
- PFC
prefrontal cortex
- DA
dopamine
- dlPFC
dorsolateral prefrontal cortex
- NE
norepinephrine
- LC
locus coeruleus
- cAMP
cyclic adenosine monophosphate
- HCN channel
Hyperpolarization-activated Cycle Nucleotide gated cation channel
- PKA
protein kinase A
- DISC1
Disrupted In Schizophrenia
- PDE4
phosphodiesterase 4
- NIMH
National Institute of Mental Health
References
- Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995;14:477–485. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. The “psychic” neuron of the cerebral cortex. Ann NY Acad Sci. 1999;868:13–26. doi: 10.1111/j.1749-6632.1999.tb11270.x. [DOI] [PubMed] [Google Scholar]
- Fuster JM. In: Cerebral Cortex. Peters A, Jones EG, editors. Plenum Press; 1984. The prefrontal cortex and temporal integrations. [Google Scholar]
- Robbins TW. Dissociating executive functions of the prefrontal cortex. Phil Trans R Soc London. 1996;351:1463–1471. doi: 10.1098/rstb.1996.0131. [DOI] [PubMed] [Google Scholar]
- Wallis JD, Anderson KC, Miller EK. Single neurons in prefrontal cortex encode abstract rules. Nature. 2001;411:953–956. doi: 10.1038/35082081. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. In: Psychopathology and the Brain. Carroll BJ, Barrett JE, editors. New York: Raven Press; 1991. Prefrontal cortical dysfunction in schizophrenia: The relevance of working memory; pp. 1–23. [Google Scholar]
- Thompson-Schill SL, Jonides J, Marshuetz C, Smith EE, D’Esposito M, Kan IP. et al. Effects of frontal lobe damage on interference effects in working memory. Cogn Affect Behav Neurosci. 2002;2:109–120. doi: 10.3758/cabn.2.2.109. [DOI] [PubMed] [Google Scholar]
- Aron AR, Robbins TW, Poldrack RA. Inhibition and the right inferior frontal cortex. Trends Cogn Sci. 2004;8:170–177. doi: 10.1016/j.tics.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Lee D, Rushworth MF, Walton ME, Watanabe M, Sakagami M. Functional specialization of the primate frontal cortex during decision making. J Neurosci. 2007;27:8170–8173. doi: 10.1523/JNEUROSCI.1561-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado MA, Junqué C, Vendrell P, Treserras P, Grafman J. Overestimation and unreliability in “feeling-of-doing” judgements about temporal ordering performance: impaired self-awareness following frontal lobe damage. J Clin Exp Neuropsychol. 1998;20:353–364. doi: 10.1076/jcen.20.3.353.816. [DOI] [PubMed] [Google Scholar]
- Gilbert SJ, Williamson ID, Dumontheil I, Simons JS, Frith CD, Burgess PW. Distinct regions of medial rostral prefrontal cortex supporting social and nonsocial functions. Soc Cogn Affect Neurosci. 2007;2:217–226. doi: 10.1093/scan/nsm014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SW, Bechara A, Damasio H, Tranel D, Damasio AR. Impairment of social and moral behavior related to early damage in human prefrontal cortex. Nat Neurosci. 1999;2:1032–1037. doi: 10.1038/14833. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. In: Handbook of Physiology, The Nervous System, Higher Functions of the Brain. Plum F, editor. Bethesda: American Physiological Society; 1987. Circuitry of the primate prefrontal cortex and the regulation of behavior by representational memory; pp. 373–417. [Google Scholar]
- Price JL, Carmichael ST, Drevets WC. Networks related to the orbital and medial prefrontal cortex; a substrate for emotional behavior? Prog Brain Res. 1996;107:523–536. doi: 10.1016/s0079-6123(08)61885-3. [DOI] [PubMed] [Google Scholar]
- Badre D, D’Esposito M. Functional magnetic resonance imaging evidence for a hierarchical organization of the prefrontal cortex. J Cogn Neurosci. 2007;19:2082–2099. doi: 10.1162/jocn.2007.19.12.2082. [DOI] [PubMed] [Google Scholar]
- Jacobsen CF. Studies of cerebral function in primates. Comp Psychol Monogr. 1936;13:1–68. [Google Scholar]
- Pribram KH, Mishkin M, Rosvold HE, Kaplan SJ. Effects on delayed response performance of dorsolateral and ventromedial frontal cortex in baboons. J Comp Physiol Psychol. 1952;45:565–575. doi: 10.1037/h0061240. [DOI] [PubMed] [Google Scholar]
- Grueninger WE, Pribram KH. Effects of spatial and nonspatial distractors on performance latency of monkeys with frontal lesions. J Comp Physiol Psychol. 1969;68:203–209. doi: 10.1037/h0027498. [DOI] [PubMed] [Google Scholar]
- Pribram KH, Mishkin M. Simultaneous and successive visual discrimination by monkeys with inferotemporal lesions. J Comp Physiol Psychol. 1955;48:198–202. doi: 10.1037/h0049140. [DOI] [PubMed] [Google Scholar]
- Goldman PS, Rosvold HE. Localization of function within the dorsolateral prefrontal cortex of the rhesus monkey. Exp Neurol. 1970;27:291–304. doi: 10.1016/0014-4886(70)90222-0. [DOI] [PubMed] [Google Scholar]
- Goldman PS, Rosvold HE, Vest B, Galkan TW. Analysis of the delayed-alternation deficit produced by dorsolateral prefrontal lesions in the rhesus monkey. J Comp Phys Psych. 1971;77:212–220. doi: 10.1037/h0031649. [DOI] [PubMed] [Google Scholar]
- Cavada C, Goldman-Rakic PS. Posterior parietal cortex in rhesus monkey: II. Evidence for segregated corticocortical networks linking sensory and limbic areas with the frontal lobe. J Comp Neurol. 1989;287:422–445. doi: 10.1002/cne.902870403. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, Wurtz RH. Visual and oculomotor functions of monkey substantia nigra pars reticulata. III. Memory-contingent visual and saccade responses. J Neurophysiol. 1983;49:1268–1284. doi: 10.1152/jn.1983.49.5.1268. [DOI] [PubMed] [Google Scholar]
- Funahashi S, Bruce CJ, Goldman-Rakic PS. Mnemonic coding of visual space in the monkey’s dorsolateral prefrontal cortex. J Neurophysiol. 1989;61:331–349. doi: 10.1152/jn.1989.61.2.331. [DOI] [PubMed] [Google Scholar]
- Elston GN. Cortex, cognition and the cell: new insights into the pyramidal neuron and prefrontal function. Cereb Cortex. 2003;12:1124–1138. doi: 10.1093/cercor/bhg093. [DOI] [PubMed] [Google Scholar]
- Arnsten AF. Prefrontal cortical network connections: key site of vulnerability in stress and schizophrenia. Int J Dev Neurosci. 2011;29:215–223. doi: 10.1016/j.ijdevneu.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brozoski T, Brown RM, Rosvold HE, Goldman PS. Cognitive deficit caused by regional depletion of dopamine in prefrontal cortex of rhesus monkey. Science. 1979;205:929–931. doi: 10.1126/science.112679. [DOI] [PubMed] [Google Scholar]
- Sawaguchi T, Goldman-Rakic PS. D1 dopamine receptors in prefrontal cortex: Involvement in working memory. Science. 1991;251:947–950. doi: 10.1126/science.1825731. [DOI] [PubMed] [Google Scholar]
- Deutch AY, Roth RH. The determinants of stress-induced activation of the prefrontal cortical dopamine system. Prog in Brain Res. 1990;85:367–403. doi: 10.1016/s0079-6123(08)62691-6. [DOI] [PubMed] [Google Scholar]
- Bradberry CW, Rubino SR. Phasic alterations in dopamine and serotonin release in striatum and prefrontal cortex in response to cocaine predictive cues in behaving rhesus macaques. Neuropsychopharmacology. 2004;29:676–685. doi: 10.1038/sj.npp.1300386. [DOI] [PubMed] [Google Scholar]
- Arnsten AFT, Goldman-Rakic PS. Stress impairs prefrontal cortex cognitive function in monkeys: role of dopamine. Soc Neurosci Abstr. 1990;16:164. [Google Scholar]
- Arnsten AFT, Goldman-Rakic PS. Noise stress impairs prefrontal cortical cognitive function in monkeys: Evidence for a hyperdopaminergic mechanism. Arch Gen Psychiatry. 1998;55:362–369. doi: 10.1001/archpsyc.55.4.362. [DOI] [PubMed] [Google Scholar]
- Arnsten AFT. The biology of feeling frazzled. Science. 1998;280:1711–1712. doi: 10.1126/science.280.5370.1711. [DOI] [PubMed] [Google Scholar]
- Vijayraghavan S, Wang M, Birnbaum SG, Bruce CJ, Williams GV, Arnsten AFT. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci. 2007;10:376–384. doi: 10.1038/nn1846. [DOI] [PubMed] [Google Scholar]
- Akil M, Pierri JN, Whitehead RE, Edgar CL, Mohila C, Sampson AR. et al. Lamina-specific alterations in the dopamine innervation of the prefrontal cortex in schizophrenic subjects. Am J Psychiatry. 1999;156:1580–1589. doi: 10.1176/ajp.156.10.1580. [DOI] [PubMed] [Google Scholar]
- Abi-Dargham A, Mawlawi O, Lombardo I, Gil R, Martinez D, Huang Y. et al. Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci. 2002;22:3708–3719. doi: 10.1523/JNEUROSCI.22-09-03708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger DR, Berman KF, Zec RF. Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia. I. Regional cerebral blood flow evidence. Archives General Psychiatry. 1986;43:114–124. doi: 10.1001/archpsyc.1986.01800020020004. [DOI] [PubMed] [Google Scholar]
- Perlstein WM, Carter CS, Noll DC, Cohen JD. Relation of prefrontal cortex dysfunction to working memory and symptoms in schizophrenia. Am J Psychiatry. 2001;158:1105–1113. doi: 10.1176/appi.ajp.158.7.1105. [DOI] [PubMed] [Google Scholar]
- Arnsten AFT, Goldman-Rakic PS. Alpha-2 adrenergic mechanisms in prefrontal cortex associated with cognitive decline in aged nonhuman primates. Science. 1985;230:1273–1276. doi: 10.1126/science.2999977. [DOI] [PubMed] [Google Scholar]
- Svensson TH, Bunney BS, Aghajanian GK. Inhibition of both noradrenergic and serotonergic neurons in brain by the alpha-adrenergic agonist clonidine. Brain Res. 1975;92:291–306. doi: 10.1016/0006-8993(75)90276-0. [DOI] [PubMed] [Google Scholar]
- Cedarbaum JM, Aghajanian GK. Catecholamine receptors on locus coeruleus neurons: pharmacological characterization. Eur J Pharmacol. 1977;44:375–385. doi: 10.1016/0014-2999(77)90312-0. [DOI] [PubMed] [Google Scholar]
- Aghajanian GK. Central noradrenergic neurons: a locus for the functional interplay between alpha-2 adrenoceptors and opiate receptors. J Clin Psychiatry. 1982;43:20–24. [PubMed] [Google Scholar]
- Gold MS, Redmond DEJ, Kleber HD. Clonidine in opiate withdrawal. Lancet. 1978;29:929–930. doi: 10.1016/s0140-6736(78)90699-2. [DOI] [PubMed] [Google Scholar]
- Cohen DJ, Detlor J, Young JG, Shaywitz BA. Clonidine ameliorates Gilles de la Tourette syndrome. Arch Gen Psychiatry. 1980;37:1350–1357. doi: 10.1001/archpsyc.1980.01780250036004. [DOI] [PubMed] [Google Scholar]
- Cohen DJ. Sterling Lecture, February 27, 2001 “Into Life: Autism, Tourette’s Syndrome and the Community of Clinical Research”. Israel Journal of Psychiatry. 2001;38:226–234. [PubMed] [Google Scholar]
- Hunt RD, Mindera RB, Cohen DJ. Clonidine benefits children with Attention Deficit Disorder and Hyperactivity: Reports of a double-blind placebo-crossover therapeutic trial. J Amer Acad Child Psychiatry. 1985;24:617–629. doi: 10.1016/s0002-7138(09)60065-0. [DOI] [PubMed] [Google Scholar]
- Hunt RD, Capper L, O’Connell P. Clonidine in child and adolescent psychiatry. Journal of Child and Adolescent Psychiatry. 1990;1:87–102. doi: 10.1089/cap.1990.1.87. [DOI] [PubMed] [Google Scholar]
- Arnsten AFT, Cai JX. Post-synaptic alpha-2 receptor stimulation improves working memory in aged monkeys: Indirect effects of yohimbine vs. direct effects of clonidine. Neurobiol Agin. 1993;14:597–603. doi: 10.1016/0197-4580(93)90044-c. [DOI] [PubMed] [Google Scholar]
- Franowicz JS, Kessler L, Dailey-Borja CM, Kobilka BK, Limbird LE, Arnsten AFT. Mutation of the alpha2A-adrenoceptor impairs working memory performance and annuls cognitive enhancement by guanfacine. J Neurosci. 2002;22:8771–8777. doi: 10.1523/JNEUROSCI.22-19-08771.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT, Cai JX, Goldman-Rakic PS. The alpha-2 adrenergic agonist guanfacine improves memory in aged monkeys without sedative or hypotensive side effects. J Neurosci. 1988;8:4287–4298. doi: 10.1523/JNEUROSCI.08-11-04287.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rama P, Linnankoski I, Tanila H, Pertovaara A, Carlson S. Medetomidine, atipamezole, and guanfacine in delayed response performance of aged monkeys. Pharmacol Biochem Behav. 1996;54:1–7. doi: 10.1016/s0091-3057(96)00111-6. [DOI] [PubMed] [Google Scholar]
- Arnsten AFT, Contant TA. Alpha-2 adrenergic agonists decrease distractability in aged monkeys performing a delayed response task. Psychopharmacology. 1992;108:159–169. doi: 10.1007/BF02245302. [DOI] [PubMed] [Google Scholar]
- O’Neill J, Fitten LJ, Siembieda DW, Ortiz F, Halgren E. Effects of guanfacine on three forms of distraction in the aging macaque. Life Sciences. 2000;67:877–885. doi: 10.1016/s0024-3205(00)00681-0. [DOI] [PubMed] [Google Scholar]
- Steere JC, Arnsten AFT. The alpha-2A noradrenergic agonist, guanfacine, improves visual object discrimination reversal performance in rhesus monkeys. Behav Neurosci. 1997;111:1–9. doi: 10.1037//0735-7044.111.5.883. [DOI] [PubMed] [Google Scholar]
- van Zwieten PA, Chalmers JP. Different types of centrally acting antihypertensives and their targets in the central nervous system. Cardiovasc Drugs Ther. 1994;8:787–799. doi: 10.1007/BF00877397. [DOI] [PubMed] [Google Scholar]
- Uhlén S, Muceniece R, Rangel N, Tiger G, Wikberg JE. Comparison of the binding activities of some drugs on alpha 2A, alpha 2B and alpha 2C-adrenoceptors and non-adrenergic imidazoline sites in the guinea pig. Pharmacol Toxicol. 1995;76:353–364. doi: 10.1111/j.1600-0773.1995.tb00161.x. [DOI] [PubMed] [Google Scholar]
- Engberg G, Eriksson E. Effects of alpha-2-adrenoceptor agonists on locus coeruleus firing rate and brain noradrenaline turnover in EEDQ-treated rats. Naunyn-Schmiedebergs Arch Pharmacol. 1991;343:472–477. doi: 10.1007/BF00169548. [DOI] [PubMed] [Google Scholar]
- Kim S, Bobeica I, Gamo NJ, Arnsten AF, Lee D. Effects of α-2A adrenergic receptor agonist on time and risk preference in primates. Psychopharmacology (Berl) 2012;219(2):363–375. doi: 10.1007/s00213-011-2520-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT, Li B-M. Neurobiology of executive functions: Catecholamine influences on prefrontal cortical function. Biological Psychiatry. 2005;57:1377–1384. doi: 10.1016/j.biopsych.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Ramos B, Stark D, Verduzco L, van Dyck CH, Arnsten AFT. Alpha-2A-adrenoceptor stimulation improves prefrontal cortical regulation of behavior through inhibition of cAMP signaling in aging animals. Learning and Memory. 2006;13:770–776. doi: 10.1101/lm.298006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Tang ZX, Li BM. Enhanced visuomotor associative learning following stimulation of alpha 2A-adrenoceptors in the ventral prefrontal cortex in monkeys. Brain Res. 2004;1024:176–182. doi: 10.1016/j.brainres.2004.07.062. [DOI] [PubMed] [Google Scholar]
- Wang M, Ramos B, Paspalas C, Shu Y, Simen A, Duque A. et al. Alpha2A-adrenoceptor stimulation strengthens working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129:397–410. doi: 10.1016/j.cell.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Li B-M, Mei Z-T. Delayed response deficit induced by local injection of the alpha-2 adrenergic antagonist yohimbine into the dorsolateral prefrontal cortex in young adult monkeys. Behav Neural Biol. 1994;62:134–139. doi: 10.1016/s0163-1047(05)80034-2. [DOI] [PubMed] [Google Scholar]
- Ma C-L, Qi X-L, Peng J-Y, Li B-M. Selective deficit in no-go performance induced by blockade of prefrontal cortical alpha2-adrenoceptors in monkeys. Neuroreport. 2003;14:1013–1016. doi: 10.1097/01.wnr.0000070831.57864.7b. [DOI] [PubMed] [Google Scholar]
- Ma C-L, Arnsten AFT, Li B-M. Locomotor hyperactivity induced by blockade of prefrontal cortical alpha-2-adrenoceptors in monkeys. Biol Psychiatry. 2005;57:192–195. doi: 10.1016/j.biopsych.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Li B-M, Mao Z-M, Wang M, Mei Z-T. Alpha-2 adrenergic modulation of prefrontal cortical neuronal activity related to spatial working memory in monkeys. Neuropsychopharmacology. 1999;21:601–610. doi: 10.1016/S0893-133X(99)00070-6. [DOI] [PubMed] [Google Scholar]
- Frey U, Huang Y-Y, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- Taylor JR, Birnbaum SG, Ubriani R, Arnsten AFT. Activation of cAMP-dependent protein kinase A in prefrontal cortex impairs working memory performance. J Neurosci. 1999;19(18):RC23. doi: 10.1523/JNEUROSCI.19-18-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runyan JD, Dash PK. Distinct prefrontal molecular mechanisms for information storage lasting seconds versus minutes. Learn Mem. 2005;12:232–238. doi: 10.1101/lm.92405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT, Ramos B, Birnbaum SB, Taylor JR. Protein kinase A as a therapeutic target for memory disorders: Rationale and challenges. Trends Mol Med. 2005;11:121–128. doi: 10.1016/j.molmed.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Arnsten AFT, Paspalas CD, Gamo NJ, Yang Y, Wang M. Dynamic Network Connectivity: A new form of neuroplasticity. Trends Cog Sci. 2010;14:365–375. doi: 10.1016/j.tics.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT. Stress signaling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. 2009;32:267–287. doi: 10.1038/nrn2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Gamo NJ, Yang Y, Jin LE, Wang XJ, Laubach M. et al. Neuronal basis of age-related working memory decline. Nature. 2011;476:210–213. doi: 10.1038/nature10243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos B, Birnbaum SB, Lindenmayer I, Newton SS, Duman R, Arnsten AFT. Dysregulation of protein kinase A signaling in the aged prefrontal cortex: New strategy for treating age-related cognitive decline. Neuron. 2003;40:835–845. doi: 10.1016/s0896-6273(03)00694-9. [DOI] [PubMed] [Google Scholar]
- Greene CM, Birnbaum MA, Gill M, Robertson IH. Noradrenergic genotype predicts lapses in sustained attention. Neuropsychologia. 2009;47:591–594. doi: 10.1016/j.neuropsychologia.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Hess C, Reif A, Strobel A, Boreatti-Hümmer A, Heine M, Lesch KP. et al. A functional dopamine-beta-hydroxylase gene promoter polymorphism is associated with impulsive personality styles, but not with affective disorders. J Neural Transm. 2009;116:121–130. doi: 10.1007/s00702-008-0138-0. [DOI] [PubMed] [Google Scholar]
- Roman T, Schmitz M, Polanczyk GV, Eizirik M, Rohde LA, Hutz MH. Further evidence for the association between attention-deficit/hyperactivity disorder and the dopamine-beta-hydroxylase gene. Am J Med Genet. 2002;114(2):154–158. doi: 10.1002/ajmg.10194. [DOI] [PubMed] [Google Scholar]
- Kopecková M, Paclt I, Goetz P. Polymorphisms of dopamine-beta-hydroxylase in ADHD children. Folia Biol (Praha) 2006;52:194–210. [PubMed] [Google Scholar]
- Kieling C, Genro JP, Hutz MH, Rohde LA. The -1021 C/T DBH polymorphism is associated with neuropsychological performance among children and adolescents with ADHD. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:485–490. doi: 10.1002/ajmg.b.30636. [DOI] [PubMed] [Google Scholar]
- Millar Jk, Mackie S, Clapcote SJ, Murdoch H, Pickard BS, Christie S. et al. Disrupted in schizophrenia 1 and phosphodiesterase 4B: towards an understanding of psychiatric illness. J Physiol. 2007;584:401–405. doi: 10.1113/jphysiol.2007.140210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch H, Mackie S, Collins DM, Hill EV, Bolger GB, Klussmann E. et al. Isoform-selective susceptibility of DISC1/phosphodiesterase-4 complexes to dissociation by elevated intracellular cAMP levels. J Neurosci. 2007;27:9513–9524. doi: 10.1523/JNEUROSCI.1493-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, King DP, Reutiman TJ, Folsom TD, Laurence JA, Lee S. et al. PDE4B polymorphisms and decreased PDE4B expression are associated with schizophrenia. Schizophr Res. 2008;101:36–49. doi: 10.1016/j.schres.2008.01.029. [DOI] [PubMed] [Google Scholar]
- Deng X, Takaki H, Wang L, Kuroki T, Nakahara T, Hashimoto K. et al. Positive association of phencyclidine-responsive genes, PDE4A and PLAT, with schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:850–858. doi: 10.1002/ajmg.b.31233. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick B, Xu L, Cascella N, Ozeki Y, Sawa A, Roberts RC. et al. DISC1 immunoreactivity at the light and ultrastructural level in the human neocortex. J Comp Neurol. 2006;497:436–450. doi: 10.1002/cne.21007. [DOI] [PubMed] [Google Scholar]
- Rubia K, Overmeyer S, Taylor E, Brammer M, Williams SCR, Simmons A. et al. Hypofrontality in Attention Deficit Hyperactivity Disorder during higher-order motor control: A study with functional MRI. Am J Psychiatry. 1999;156:891–896. doi: 10.1176/ajp.156.6.891. [DOI] [PubMed] [Google Scholar]
- Shaw P, Lalonde FM, Lepage C, Rabin C, Eckstrand K, Sharp W. et al. Development of cortical asymmetry in typically developing children and its disruption in attention-deficit/hyperactivity disorder. Arch Gen Psychiatry. 2009;66:888–896. doi: 10.1001/archgenpsychiatry.2009.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RD, Arnsten AFT, Asbell MD. An open trial of guanfacine in the treatment of attention deficit hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 1995;34:50–54. doi: 10.1097/00004583-199501000-00013. [DOI] [PubMed] [Google Scholar]
- Chappell PB, Riddle MA, Scahill L, Lynch KA, Schultz R, Arnsten A. et al. Guanfacine treatment of comorbid attention deficit hyperactivity disorder and Tourette’s Syndrome: Preliminary clinical experience. J Am Acad Child Adolesc Psychiatry. 1995;34:1140–1146. doi: 10.1097/00004583-199509000-00010. [DOI] [PubMed] [Google Scholar]
- Biederman J, Melmed RD, Patel A, McBurnett K, Konow J, Lyne A. et al. A randomized, double-blind, placebo-controlled study of guanfacine extended release in children and adolescents with attention-deficit/hyperactivity disorder. Pediatrics. 2008;121:e73–e84. doi: 10.1542/peds.2006-3695. [DOI] [PubMed] [Google Scholar]
- Sallee FR, McGough JJ, Wigal T, Donahue J, Lyne A, Biederman J. et al. Guanfacine Extended Release in Children and Adolescents With Attention-Deficit/Hyperactivity Disorder: A Placebo-Controlled Trial. J Am Acad Child Adolesc Psychiatry. 2009;48:155–165. doi: 10.1097/CHI.0b013e318191769e. [DOI] [PubMed] [Google Scholar]
- Connor DF, Findling RL, Kollins SH, Sallee F, López FA, Lyne A. et al. Effects of guanfacine extended release on oppositional symptoms in children aged 6-12 years with attention-deficit hyperactivity disorder and oppositional symptoms: a randomized, double-blind, placebo-controlled trial. CNS Drugs. 2010;24:755–768. doi: 10.2165/11537790-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Scahill L, Chappell PB, Kim YS, Schultz RT, Katsovich L, Shepherd E. et al. Guanfacine in the treatment of children with tic disorders and ADHD: A placebo-controlled study. Amer J Psychiatry. 2001;158:1067–1074. doi: 10.1176/appi.ajp.158.7.1067. [DOI] [PubMed] [Google Scholar]
- Scahill L, Aman MG, McDougle CJ, McCracken JT, Tierney E, Dziura J. et al. A prospective open trial of guanfacine in children with pervasive developmental disorders. J Child Adolesc Psychopharmacol. 2006;16:589–598. doi: 10.1089/cap.2006.16.589. [DOI] [PubMed] [Google Scholar]
- McCracken JT, Aman MG, McDougle CJ, Tierney E, Shiraga S, Whelan F. et al. Possible influence of variant of the P-glycoprotein gene (MDR1/ABCB1) on clinical response to guanfacine in children with pervasive developmental disorders and hyperactivity. J Child Adolesc Psychopharmacol. 2006;20:1–5. doi: 10.1089/cap.2009.0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister TW, McDonald BC, Flashman LA, Ferrell RB, Tosteson TD, Yanofsky NN. et al. Alpha-2 adrenergic challenge with guanfacine one month after mild traumatic brain injury: Altered working memory and BOLD response. Int J Psychophysiol. 2011;82(1):107–114. doi: 10.1016/j.ijpsycho.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure MM, Barch DM, Romero MJ, Harvey PD, Siever LJ. The effects of guanfacine on context processing abnormalities in schizotypal personality disorder. Biol Psychiatry. 2007;61:1157–1160. doi: 10.1016/j.biopsych.2006.06.034. [DOI] [PubMed] [Google Scholar]
- Swartz BE, Kovalik E, Thomas K, Torgersen D, Mandelkern MA. The effects of an alpha-2 adrenergic agonist, guanfacine, on rCBF in human cortex in normal controls and subjects with focal epilepsy. Neuropsychopharmacology. 2000;23:263–275. doi: 10.1016/S0893-133X(00)00101-9. [DOI] [PubMed] [Google Scholar]
- Singh-Curry V, Malhotra P, Farmer SF, Husain M. Attention deficits following ADEM ameliorated by guanfacine. J Neurol Neurosurg Psychiatry. 2011;82:688–690. doi: 10.1136/jnnp.2009.195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerkin SM, Schulz KP, Halperin JM, Newcorn JH, Ivanov I, Tang CY. et al. Guanfacine potentiates the activation of prefrontal cortex evoked by warning signals. Biol Psychiatry. 2009;66:307–312. doi: 10.1016/j.biopsych.2009.04.013. [DOI] [PubMed] [Google Scholar]
- Selemon LD, Goldman-Rakic PS. The reduced neuropil hypothesis: a circuit based model of schizophrenia. Biol Psychiatry. 1999;45:17–25. doi: 10.1016/s0006-3223(98)00281-9. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Gonzalez-Burgos GR. Pathophysiologically based treatment interventions in schizophrenia. Nat Med. 2006;12:1016–1022. doi: 10.1038/nm1478. [DOI] [PubMed] [Google Scholar]
- Krimer LS, Jakab RL, Goldman-Rakic PS. Quantitative three-dimensional analysis of the catecholaminergic innervation of identified neurons in the macaque prefrontal cortex. J Neurosci. 1997;17:7450–7461. doi: 10.1523/JNEUROSCI.17-19-07450.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z-M, Arnsten AFT, Li B-M. Local infusion of alpha-1 adrenergic agonist into the prefrontal cortex impairs spatial working memory performance in monkeys. Biol Psychiatry. 1999;46:1259–1265. doi: 10.1016/s0006-3223(99)00139-0. [DOI] [PubMed] [Google Scholar]