

Abstract
Mass spectrometry-based investigation of clinical samples enables the high-throughput identification of protein biomarkers. We provide an overview of mass spectrometry-based proteomic techniques that are applicable to the investigation of clinical samples. We address sample collection, protein extraction and fractionation, mass spectrometry modalities, and quantitative proteomics. Finally, we examine the limitations and further potential of such technologies. Liquid chromatography fractionation coupled with tandem mass spectrometry is well suited to handle mixtures of hundreds or thousands of proteins. Mass spectrometry-based proteome elucidation can reveal potential biomarkers and aid in the development of hypotheses for downstream investigation of the molecular mechanisms of disease.
Keywords: chronic pancreatitis, biomarkers, pancreas, mass spectrometry
Introduction
Proteomics entails the characterization of the complete set of proteins encoded by the genome of a given organism in a given state [1]. A cornerstone of proteomics is the ability to perform sensitive mass spectrometric analysis on a complex mixture of proteins and peptides. Whereas traditional techniques have focused on only a few targeted proteins per analysis, proteomics attempts to conduct the comprehensive study of complex protein mixtures and can identify hundreds or thousands of proteins for future investigation [2]. Proteomics can address challenges that cannot be approached by genomic analysis, namely, relative abundance of the protein products, post-translational modifications, compartmentalization, and turnover, as well as protein interactions and protein function. In addition, the proteomic analysis of human body fluids and tissues can be a valuable approach in the search for diagnostic and prognostic biomarkers. For example, the acquisition of body fluids has potential advantages of being relatively noninvasive, economical, and easily collected. If readily available, proximal body fluids, which bathe the organ of interest, also have the benefit of being rich in secreted proteins, which are likely to include markers of diseases affecting that particular organ.
One gene may produce more than one protein, such as a genome of 30,000 genes can produce more than 100,000 proteins, when alternative splicing is considered [3]. In addition, post-translational modifications occurring in cells, such as phosphorylation and glycosylation, further expand the number of possible protein isoforms to be identified [4,5]. Challenges related to the complexity of clinical samples to be investigated may be overcome by the development of standardized sample preparation and handling protocols. Modern peptide mass spectrometry, performed following liquid chromatography fractionation, is very well suited for handling mixtures of hundreds or thousands of proteins.
Below, we provide an overview of various mass spectrometry-based proteomic techniques that are applicable to the study of translational medicine (Figure 1). We begin with a qualitative assessment of the proteins in a particular sample, covering the principles of sample collection, protein extraction, and fractionation. We then follow the innovations through the current state of quantitative proteomics. Finally, we comment on the potential of mass spectrometry-based proteomic technologies to investigate clinical samples.
Figure 1.
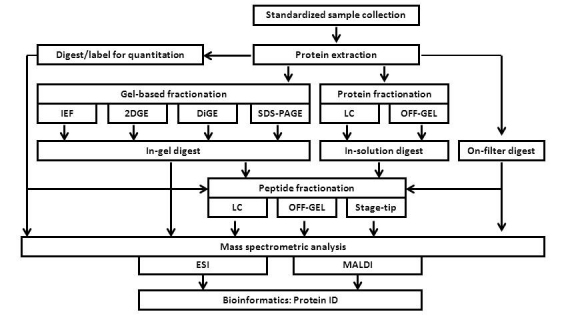
General workflow for mass spectrometry sample processing. Human body fluids may be processed for mass spectrometry-based proteomic analysis using approaches including, but not limited to, those listed in the workflow diagram.
GENERAL WORKFLOW
Sample collection
Consistent sample processing, collection, and analysis strategies must be established to develop reproducible clinical proteomics assays. Often in translational research, there are insufficient standardized protocols regarding specimen collection, storage, and processing. Significant changes in the proteomic profile also may be introduced during sample preparation if consensus methodology is not in place. We realize that variability cannot be entirely avoided; however, it may be kept to a minimum by careful and standardized sample handling.
Protein extraction
As the collected samples are to be prepared for proteomic analysis, proteins should be extracted from lipids, metabolites, and other non-proteinaceous compounds, which may interfere with downstream procedures. In general, salt removal is accomplished via methods such as dialysis (spin, micro) [6], ultrafiltration [7,8], gel filtration/electrophoresis, precipitation with acid or organic solvents, and/or solid-phase extraction. Various chemical precipitation methods are available for protein isolation; these include acetone, trichloroacetic acid (TCA), ethanol, isopropanol, chloroform/methanol, and ammonium sulfate. The efficiency of protein precipitation varies among different organic solvents. For example, acetone has been determined to precipitate more acidic and hydrophilic proteins, whereas ultracentrifugation fractionates more basic, hydrophobic, and membrane proteins [9]. Alternatively, chloroform methanol extraction has been used to successfully extract hydrophobic proteins [10]. It is important that precipitation strategies be optimized for a particular sample type.
Protein fractionation
Following protein extraction, various approaches can be used to fractionate proteins prior to mass spectrometry analysis. Fractionation can be performed either at the protein level prior to proteolytic digestion or at the peptide level following proteolytic digestion. Below, we describe several methods that can be used for fractionation of proteins from clinical samples prior to digestion. All methods described in this section have the advantage of removing additional non-proteinacious compounds that remain after protein extraction and may interfere with downstream analysis while simultaneously simplifying the protein mixture to be further processed. These methods can be implemented individually or in tandem, according to sample complexity and the depth of analysis required.
Gel-based fractionation
One-dimensional SDS-PAGE. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) fractionates proteins according to electrophoretic mobility, an approximation of the molecular weight of the proteins. The proteins are denatured first by SDS, a strong ionic detergent, and heat. Disulfide bonds are reduced (commonly with DTT (dithiothreitol) or TCEP (tris(2-carboxyethyl)phosphine)) and then alkylated (typically with iodoacetamide or acrylamide) to prevent bond reformation. This solubilized protein mixture is loaded onto the gel, and upon application of an electric field, proteins travel through the polyacrylamide matrix forming distinct bands. These bands can be visualized by conventional stains such as Coomassie brilliant blue [11] and silver stain [12], or fluorescent staining such as SYPRO Ruby [13] and Deep Purple Total Protein Stain [14]. Mass spectrometry-based techniques coupled with in-gel tryptic digestion can identify proteins present in Coomassie- or silver-stained gel bands. For this approach, it is advantageous to have the fewest number of proteins in a sample. Sample complexity may be reduced prior to, or following, electrophoresis by further sample fractionation.
Isoelectric focusing. Proteins (or peptides) may be fractionated according to their isoelectric points (pI). The most common method of pI fractionation involves isoelectric focusing (IEF), which separates molecules by their intrinsic charge differences. In IEF, proteins are loaded onto a medium on which a pH gradient has been created by amphoteric molecules (ampholytes) that have both acidic and basic groups. An electric current is passed through the medium, producing positively and negatively charged ends. Negatively charged proteins migrate through the pH gradient toward the positive end (anode), while positively charged proteins move toward the negative end (cathode) of the medium. The proteins are ultimately focused around the pH that is equal to their respective pI, at which point the protein has no net charge and thus no longer migrates in an electric field. Proteins now can be stained or identified using gel-based mass spectrometry techniques or separated further according to mass by overlaying the IEF medium onto an SDS-PAGE slab [15].
Alternatively, recently developed OFF-GEL fractionation (Agilent Technologies) allows for the collection of peptides and proteins in liquid fractions, which does not require protein/peptide extraction from the gel matrix for downstream applications [16,17]. During OFF-GEL fractionation, samples are added into buffer compartments over an IPG strip. Peptides then migrate through the gel strip and can be collected in solution from the appropriate buffer compartment without the need of extracting from the gel strip. Similar to standard IEF, OFF-GEL fractions can be analyzed further by separation in a second dimension or digested in-solution for mass spectrometry analysis.
Similarly, capillary electrophoresis (CE) or capillary zone electrophoresis (CZE) offers an alternative gel-free method to separate peptides and proteins. CE can separate ionic species by their charge, hydrodynamic radius, and frictional forces [18]. Essentially, CE can be used in place of traditional reversed-phase liquid chromatography for protein (as well as peptide) separation prior to measurement by mass spectrometry. Similar to a traditional set-up with a reversed-phase column, CE can be directly coupled with mass spectrometers in which the capillary outlet is introduced into an ESI ion source, resulting in ions than can be analyzed by the mass spectrometer. Such analyses can be performed for both proteins and peptides [19,20].
Two-dimensional gel electrophoresis. Two-dimensional gel electrophoresis (2DGE) is used often to separate highly complex protein samples. In the first dimension, the proteins are typically separated by isoelectric point. Traditionally, a polyacrylamide tube gel is cast and a pH gradient is formed using carrier ampholytes. More recently, immobilized pH gradients on a polyacrylamide matrix supported by a plastic backing have been substituted for tube gels [21]. Unlike the tube gels, which must be extruded from their glass casting tubes, immobilized pH gradient (IPG) strips have mechanical stability and are less likely to break or stretch. In addition, IPG strips can accommodate a wider range of pH values, have improved reproducibility, and are not as prone to pH drift (due to electro-osmotic flow) as IEF tube gels. In the second dimension, 2DGE typically separates proteins based on mass via SDS-PAGE.
Although sample complexity is decreased compared to 1D SDS-PAGE as the result of a second dimension of separation in 2DGE, some pitfalls of this technique persist. Disadvantages of 2DGE include (i) spots may contain multiple proteins; (ii) poor spot resolution at high pI values; (iii) very acidic and very basic proteins not being well represented; (iv) very small or very large proteins not being resolved on a standard gel; and (v) irreproducibility of gels [22]. Two-dimensional gel electrophoresis of membrane proteins is additionally hampered by the potential insolubility of the hydrophobic portions of these proteins in IEF focusing buffer, as well as the propensity for precipitation at the isoelectric point of the protein [23]. In addition, it has been determined that the technical variability of 2DGE due to sample preparation, reagent sources, experimenter variability, and staining methods accounts for a 20 percent to 30 percent coefficient of variability [24]. The use of IPG, standardized buffers, and large format 2D gels are among the improvements that have overcome some of these pitfalls, allowing for the continued use of 2DGE in proteomics [25].
Difference gel electrophoresis. Difference gel electrophoresis (DiGE) resolves some of the caveats associated with 2DGE. DiGE eliminates gel-to-gel variation as two or three samples can be conjugated to a different fluorescent dye (Cy2, Cy3, or Cy5) and simultaneously analyzed on a single gel [26]. For example, to compare the protein profile of two samples, one sample can be labeled with Cy5 dye and the second with Cy3 dye. An optional third sample control may consist of the two aforementioned samples combined and labeled with Cy2 dye, which acts as an internal standard. The Cy-series of dyes generally binds to lysine residues, although cysteine-conjugating dyes and other chemistries are available [27-29]. All three samples are combined and analyzed on an IPG strip for the first dimension, and then separated by SDS-PAGE in the second dimension. Following excitation at different wavelengths (640 nm for Cy5 and 550 nm for Cy3), the Cy5-labeled proteins can be visualized at a particular wavelength (670 nm, red) and Cy3 can be visualized under a second wavelength (570 nm, green) [30]. Images taken at each emission wavelength are then superimposed and differences can be qualitatively (presence or absence) and quantitatively (fluorescent signal intensity) determined.
Gel spots corresponding to proteins unique to one sample, or to proteins that overlap in both samples, can then be excised. These gels spots can be in-gel enzymatically digested and analyzed by mass spectrometry. The sensitivity and dynamic range of DiGE is relatively high compared to other staining methods. Quantitatively, a DiGE experiment can detect 0.5-1 fmol [31] of a single protein and has a dynamic range of five orders of magnitude, as opposed to silver stain, which has a detection limit of 1 ng and a dynamic range of only two orders of magnitude [25]. In summary, DiGE allows for enhanced comparison of the protein profiles of two or three samples and is compatible with analysis by mass spectrometry.
Liquid chromatography-based fractionation
Liquid chromatography can be used to separate proteins on the basis of hydrophobicity, ionic charge, or size, as well as to remove substances that may interfere with downstream analyses. The method most commonly used to fractionate proteins by their hydrophobicity uses reversed-phase C4 or C8 resin. A similar chemistry using beads with longer hydrocarbon chains, C18, is used to resolve different populations of peptides. Elution with volatile organic solvents makes this mode of chromatography ideal for mass spectrometric analysis [32]. However, these resins are incompatible with certain detergents that can compete for binding sites on the stationary phase (e.g., Triton X-100). Ion exchange chromatography can be used for the removal of detergent from samples. Resins can consist of strong cation exchange (SCX), weak cation exchange (WCX), strong anion exchange (SAX), weak anion exchange (WAX), or mixed-bed, such as SCX-WAX [33]. Of these, SCX is commonly used upstream of reversed-phase HPLC-MS/MS. The stationary phase of SCX columns is negatively charged and attracts positive ions (e.g., protonated peptides). An increase in salt concentration or pH of the mobile phase will promote elution of the bound peptides.
In some instances, size exclusion chromatography can also be used to fractionate further a complex protein sample [34]. Affinity chromatography methods also may be useful when attempting to isolate specific proteins or complexes of interest [35]. For liquid chromatography fractionation of proteins, caution should be taken that proteolytic enzymes are properly deactivated with the appropriate protease inhibitors prior to performing liquid chromatography, ensuring sample integrity over prolonged periods during chromatographic processing.
Proteolytic digestion
Mass spectrometry can be performed on either intact proteins (“top-down”) or on peptides that originate from proteins digested by a specific protease (“bottom-up”). To date, proteomics of clinical samples has mainly focused on bottom-up approaches. Although top-down proteomics may be a viable alternative for clinical sample analysis, we will only focus on bottom-up approaches for the purpose of this review. Several excellent reviews are available which explore top-down proteomics [36-38].
Using the bottom-up approach, proteins can either be digested in-gel or in-solution, by a variety of strategies. When performing in-gel digestions, gel slices or spots are excised and washed with an ammonium bicarbonate/acetonitrile solution to remove SDS and buffer molecules from the gel slices. The gel slices/spots are then dried and re-hydrated with an ammonium bicarbonate solution containing the appropriate protease [39].
Trypsin is typically the protease of choice as it has a high specificity, a low price per unit, and is inherently stable under a wide range of conditions, including 40 percent acetonitrile and 2 M urea [40,41]. Additionally, it cleaves at the C-terminal of arginine and lysine residues, placing a highly basic residue at this terminus, resulting in multiply charged, or protonated, peptides. This phenomena is important for larger species, as the mass-to-charge ratio value (m/z) may be decreased enough to be within the mass range of the instrument [40]. Similarly, Lys-C, which cleaves only at lysine residues, is used often for both in-gel and in-solution digests [42]. For proteins having a low arginine and lysine content, for example, those with multiple membrane spanning regions, other proteases may be substituted for trypsin. Chymotrypsin, for example, can be used without any modifications to many standard trypsin-based protocols [43]. Chymotrypsin cleaves peptides at the carboxyl group of tryptophan, tyrosine, phenylalanine, leucine, and methionine. This protease has been used successfully where trypsin had failed to give an adequate spectra [44].
After digestion, peptides are extracted, vacuum dried, and ready for analysis by mass spectrometry. Alternatively, protocols may include in-solution digestion in lieu of GeLC, resulting in peptides that are fractionated in a single dimension (reversed-phase, strong cation exchange, or isoelectric focusing) [45] or via MUDPIT (multidimensional protein identification technology) [46,47]. A novel method, filter-aided sample preparation (FASP), in which filter units are used to remove mass spectrometry-incompatible substances, may also be a promising alternative to in-gel and in-solution digests [48,49].
Peptide fractionation
Techniques similar to those utilized at the protein level can be used to reduce sample complexity at the peptide level. Peptide fractionation is particularly necessary when in-solution digestions are performed, but can also be valuable for in-gel or FASP protocols. Orthogonal methods of separation include high pH reversed-phase chromatography, ion exchange chromatography, and/or isoelectric focusing prior to MALDI or LC-MS/MS analysis. Several recent studies have compared the various fractionation methods mentioned above [50-54]. However, such studies investigated specific questions, and their particular strengths and weaknesses may not apply to all study designs. When comparing methods among laboratories, bias may also be introduced as a result of experimenter expertise and familiarity with a frequently used method. As with upstream sample preparation techniques, optimization may be needed to establish standardized peptide fractionation approaches for a particular workflow.
Following proteolytic digests and peptide fractionation, substances that interfere with downstream mass spectrometric acquisition must be removed. Desalting is commonly accomplished using C18 resin, which elutes peptides in a volatile solvent, or by using strong cation exchange (SCX) resin, particularly for detergent-containing samples, which can elute peptides with changes in buffer pH. Desalting can be performed in a spin column or in pipettes packed with the aforementioned resin using, for example, StageTips [55] or Zip-Tips [56]. Sample clean-up can enhance signal, prevent contaminants from entering the mass spectrometer, and prolong the longevity of analytical columns which elute peptides into the mass spectrometer.
Quantitative proteomics
Although DiGE does overcome some of the limitations of 2DGE, it still carries over biases against small proteins and hydrophobic proteins, many of which are of great importance in biomarker discovery. In such cases, quantitative proteomics methods may provide an unbiased approach to comprehensive proteome analysis. In cell cultures, stable isotope labels may be introduced during cell growth to attain up to 100 percent labeling efficiency, as is the case when using SILAC (stable isotope labeling by amino acids in cell culture) [57]. Here, two cell states can be prepared, with one state grown in media with heavy isotope labeled arginine and/or lysine. The two states are combined and chromatographically separated to assess relative differences and similarities in protein content.
Such methods are not practical when investigating body fluids or tissue directly collected from humans, rather they are primarily applicable to cell culture-based systems and particularly those investigating the cellular secretomes. Among the simplest in vitro peptide labeling is 18O labeling, in which heavy 18O-water is incorporated into the peptides during directed proteolysis, i.e., with trypsin [58]. Alternatively, the isotope-coded affinity tag (ICAT) technique can be used, whereby samples are labeled at thiol groups with either of two reagents; the heavy reagent contains eight deuterium atoms instead of eight hydrogen atoms, as in the light form of the label. A more robust method involves multiplexed isobaric tag labeling (e.g., isobaric tag for relative and absolute quantitation: iTRAQ [59] or tandem mass tag: TMT [60]) that can quantitatively analyze up to eight diseased states, time points, or samples in a single experiment. These samples are then pooled and fractionated by liquid chromatography prior to tandem mass spectrometry analysis. The fragmentation of the attached tag generates low molecular mass reporter ions that can be used to relatively quantify peptides and the proteins from which they originate.
In the absolute quantitation (AQUA) method, a synthetic heavy-isotope-labeled standard peptide is introduced into cell lysates at a known concentration and selected reaction monitoring is used to detect and quantitate the peptide of interest [61-63]. These quantitative methods allow detection of subtle changes in the proteome, which would not have been possible using only qualitative identification methods (i.e., presence vs. absence).
Label-free methods for quantitation represent attractive alternatives [64], particularly for experiments that have been previously performed without labeling. With the exception of SILAC, any of the aforementioned quantitative mass spectrometric techniques may be used to directly analyze clinical samples.
Technological overview of mass spectrometry
Mass spectrometry has become an indispensable analytical tool for the study of proteomics. Using various bottom-up proteomic approaches, specific proteins or protein complexes can be isolated, digested to peptides, and identified by mass spectrometry. Known proteins can be validated, and unknown proteins can be discovered with this technique. Classical techniques such as co-immunoprecipitation, reciprocal western blotting and yeast-two-hybrid assays can typically provide information on binary, or at most ternary, interactions. Mass spectrometry, however, can discover protein-protein networks without any a priori suspicion of interaction.
Mass spectrometric modalities
Various mass spectrometric modalities are available for use in proteomic analysis (Figure 2). The three main variable components of a mass spectrometer are the ion source, the mass analyzer, and the ion detector. The ion source produces the ions that are subsequently introduced into the gas phase. The mass analyzer measures the mass-to-charge (m/z) ratio of the ionized peptides. The two ionization techniques commonly used for mass spectrometric analyses of proteins and peptides are electrospray ionization (ESI) and matrix-assisted laser desorption/ionization (MALDI). ESI and MALDI can be coupled with various types of mass analyzers, some of which require an additional detector. The mass detector records the abundance of ions at each mass-to-charge value. As the number of ions leaving the mass analyzer at a given time is small, amplification, usually by electron multipliers, is needed to acquire a measurable signal [65]. The following sections provide an overview of technological aspects of mass spectrometry and current application thereof to proteomic analysis.
Figure 2.
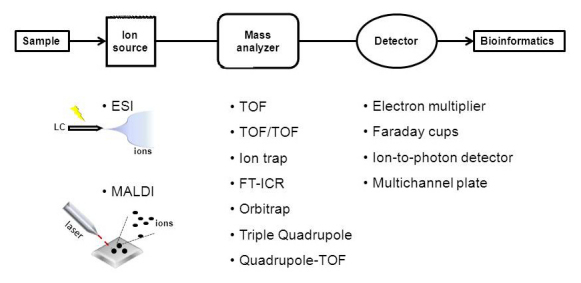
Mass spectrometer modalities commonly used in proteomic research. A mass spectrometer consists of three modules: 1) an ion source (ESI or MALDI), which ionizes the peptides to be analyzed, 2) a mass analyzer (or a combination of analyzers), which can be used as a collision cell for fragmentation and/or sort the ions by their mass-to-charge ratio, and 3) a detector which amplifies and quantifies the resulting signal.
Tandem MS/MS and peptide sequencing
A mass spectrometer also can be used to identify proteins by sequencing peptides using a procedure called tandem MS or MS/MS. This process involves recording a full mass spectrum and selecting a peak or several peaks to be further fragmented. The selection process is usually automated as a threshold is set and MS/MS is performed on any peak with an intensity greater than that value. Peptide fragmentation can occur anywhere along the peptide backbone. Fragments produced from a single bond fragmentation are denoted as a, b, c, x, y, and z, depending on the covalent bonds which are broken [66]. The ions y and b have prevalence for collision-induced dissociation (CID). Internal fragments and immonium ions, as well as side chain fragmentation, often also occur. Ions are selected and fragmented within a single quadrupole in instruments such as a linear ion trap [67]. CID is the most common method of generating tandem mass spectra [68]. Modern mass spectrometric instrumentation allows for automated MS/MS analysis. For example, a linear trap quadrupole instrument (LTQ) coupled to either a MALDI or ESI source and an Orbitrap or FT-ICR analyzer can select a defined number of the most intense ion peaks of a full scan MS-spectrum. These ions are subsequently isolated and fragmented to obtain sequence identification. A classic LTQ can record these spectra at a rate of greater than five scans per second, making this instrument ideal for high throughput assays [69]. In addition, multistage fragmentation (MSn) can reveal post-translational modifications, such as phosphorylation, as well as structural characterization. Proteomic analysis of clinical samples is not limited to a single mass spectrometric modality for biomarker discovery.
The mass accuracy and resolution of the mass spectrometer are important factors that enhance the signal-to-noise-ratio and lower the ion detection limit [22]. When optimized, these factors result in a considerable reduction of false positive and false negative mass identifications, which is essential when investigating data sets composed of many different proteins with unknown identities. Mass accuracy is the measurement of the exactness of a recorded peptide mass to the theoretical mass computed by in silico enzymatic digests on the primary sequence of a target protein. The units of this measure are often either Daltons or parts per million (ppm). For comparison, time-of-flight mass spectrometers provide within 2 to 5 ppm mass accuracy, while quadrupole ion traps provide accuracy in the range of 100 ppm and above. However, modern mass analyzers, such as the QTOF (quadrupole time-of-flight), Orbitrap, or FT-ICR (Fourier transform ion cyclotron resonance) instruments can achieve sub-ppm mass accuracy [70]. When analyzing samples of unknown identity, high mass accuracy is recommended [67]. Resolution in mass spectrometry is defined as the m/z value divided by the peak width at half-maximum height [66]. In other words, it is the ability of the mass spectrometer to differentiate between two peaks and, thus, two different ions or fragments, which is particularly useful for complex samples [69]. In essence, reliable data can be attained using an instrument with high mass accuracy and high resolution. In addition, high sensitivity is important for low-abundance peptides. Data derived from the fragmentations of peptide ions (MS/MS) can be used to identify amino acid sequences. However, the unambiguous identification of proteins from large data sets is not trivial.
Several computational algorithms have been developed to sequence proteins by mass spectrometry, as the sheer number of tandem MS/MS spectra precludes manual interpretation of all spectra. Software packages and associated algorithms, such as SEQUEST [71], Mascot [72], Phenyx [73], ProteinPilot [74], X!Tandem [75], and ProteinProspector [76], search a given sequence database for peptides with theoretical spectra best matching the observed spectra and subsequently assign these peptides to the corresponding proteins. Both peptide and proteins are scored, and thresholds can be determined to estimate the quality of the data [77,78]. False discovery rates (FDR) are calculated typically to estimate erroneously identified proteins [79-81]. The authors direct the reader to the references listed above for more technical details concerning the aforementioned software packages.
AN APPLICATION OF MASS SPECTROMETRY-BASED PROTEOMICS
Our laboratory uses mass spectrometry-based proteomics to study diseases of the exocrine pancreas, such as chronic pancreatitis [82]. For a broader view of other methods used in the proteomics of pancreatic disease, we refer the reader to our recent review of pancreatic fluid-based proteomics [82]. Also for those readers interested in clinical samples of different origins, much detail is available in a recently published book [83], which includes a chapter on body fluids [84]. We have utilized several of the techniques described herein to investigate chronic pancreatitis using secretin-stimulated, ePFT (endoscopic pancreatic function test)-collected pancreatic fluid [85]. Figure 3 illustrates our general methodology for pancreatic fluid proteomic analysis. Standardized sample preparation conditions were first established to minimize protein degradation, thus maximizing yield [86,87]. Protein degradation of pancreatic fluid samples were of considerable concern due to the high concentration of various proteases. In our hands, one-dimensional GeLC-MS/MS (SDS-PAGE gel coupled with liquid chromatography tandem mass spectrometry)-based methods produced more robust results than DiGE analysis [88]. Using analogous GeLC-MS/MS techniques, we have recently investigated the proteomes of a pancreatic stellate cell culture line [89]. In addition, we have analyzed pancreas formalin-fixed paraffin embedded tissue using in-solution digestions [90]. Following our established protocols, trypsin-digested samples were subjected to nanoESI, after which an LTQ-FTICR mass spectrometer was used to measure peptide and fragment masses. Data was primarily analyzed with MASCOT, although other software packages (e.g., ProteomeDiscoverer and ProteinPilot) can also be used. We commonly use Scaffold, GO (gene ontology) analysis, and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways to extract biological information from our data. Using our GeLC-MS/MS strategy, we have identified and classified more than 1,000 proteins in pancreatic fluid (manuscript in preparation). Further experimentation using targeted assays, such as those described above, will allow for the validation of our biomarker discovery study. In summary, mass spectrometry-based proteomics offers robust and high throughput methods for biomarker discovery and the development of hypotheses for downstream investigations.
Figure 3.
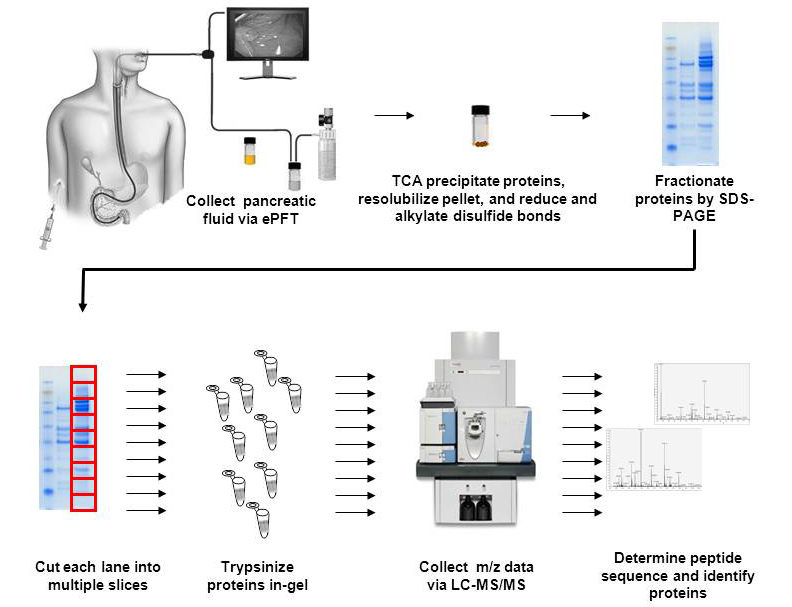
Optimized workflow for proteomic analysis of pancreatic fluid. Pancreatic fluid is collected via ePFT. After particulates are removed by centrifugation, proteins are extracted from the remaining supernatant with TCA. The protein pellet is reconstituted in Laemmli buffer, alkylated, and reduced prior to analysis by SDS-PAGE. GeLC-MS/MS analysis is performed, in which gel lanes are divided into smaller segments, which are individually in-gel tryptically digested. Digested peptides are eluted from a nanoflow C18 reversed-phased column into a mass spectrometer for accurate mass analysis. The resulting mass spectra are processed to determine the peptides and eventually the proteins from which these peptides originate.
LIMITATIONS OF MASS SPECTROMETRY-BASED PROTEOMICS FOR TRANSLATIONAL RESEARCH
Careful and consistent sample handling is essential for reproducible and robust mass spectrometry results. The decision to use protease inhibitors and/or acidification must be determined for a particular experiment or assay, as the use of such reagents may result in irreversible modifications and may be detrimental to downstream analyses. For example, several small molecule inhibitors, such as PMSF (phenylmethylsulfonyl fluoride) and AEBSF (4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride), have been shown to form covalent bonds with proteins [91], thereby changing pI and electrophoretic mobility [92]. In addition, many protease inhibitor cocktails contain small molecule or peptide inhibitors, which can interfere with subsequent peptide ionization [93].
Standardization is a necessary component of sample collection and mass spectrometry analysis. Only through standardized sample preparation approaches and mass spectrometry procedures can results be reproduced. Although not currently feasible for this relatively novel technology, future standardization of mass spectrometry techniques will substantially improve the accuracy and precision of diagnosing and following the progression of diseases among individuals. Some factors, however, cannot be controlled in the laboratory. Confounders, such as age, alcohol consumption, etiology, gender, race, and smoking, must also be taken into consideration as such differences may result in additional variability. Cohorts should be chosen carefully and matched as closely as possible.
Similarly, another aspect of sample preparation that requires further investigation is the depletion of abundant proteins. Many human body fluids and tissue, particularly those with blood, have mg/mL levels of serum albumin and α2-macroglobulin. High concentrations of these and other proteins may necessitate depletion, possibly with antibody-conjugated microspheres, to achieve the sensitivity needed to detect low abundance proteins; many important differentially expressed proteins may be present at very low levels in the cells. For examples, cytokines are generally on the order of pg/mL in human body fluids [94-97]. Targeted mass spectrometry assays, such as selected/multiple reaction monitoring (SRM/MRM) assays, may be required to detect such proteins and identify differences in protein content between the normal and diseased states. In addition, differential protein analysis may require quantitative methods, as there may be basal levels of expression of a targeted protein regardless of the disease.
OUTLOOK
The combination of high-resolution separation techniques and powerful mass spectrometric analysis enables the acquisition of previously unattainable information about the proteome of diseased patients and healthy controls. The comprehensive analysis of protein mixtures and the identification of hundreds or thousands of proteins are possible for clinical applications due to recent developments in high-throughput mass spectrometry [98-101]. Proteomics facilitates the elucidation of proteins that regulate the pathogenesis of disease and facilitate the discovery of clinically relevant biomarkers. However, the quality of proteomic results depends heavily on the methodology by which samples are prepared. Variations in methods may introduce discrepancies that can impede the progress of mass spectrometry proteomics in translational research. Further standardization of methods can maximize protein extraction and minimize the heterogeneity of samples by reducing protein degradation.
Quantitative proteome profiling is key for comparative analysis of proteins from normal and diseased patients, as similar proteins may be present in both states but at significantly different concentrations. Without quantitative information, the value of these differentially abundant proteins as biomarkers may be overlooked. In comparative proteomics, sample preparation is of utmost importance as minor differences in experimental and control samples may be instrumental to understanding the mechanisms that underlie a particular disease. Techniques, such as those outlined herein, may be useful in the study of quantitative differences between diseased and non-diseased cohorts. Whereas the genome is relatively stable and identical in all cells, the proteome varies by organ, cell, subcellular location, temporally, and due to stimuli, such as changes in health, diet, and environment. In-depth quantitative proteomic interrogation of bodily fluids and tissue can assist in determining if such variations are indeed a result of a particular disease state.
Once biomarkers and molecular pathways of disease have been elucidated, efforts must be focused on validation of these mass spectrometry-based data. For example, disease-specific biomarkers may be validated on large numbers of patients and controls, while longitudinal studies can be developed that examine the appearance, disappearance, or modulation of expression over the course of disease. In addition, biomolecular pathways that may be altered during the course of disease progression can be investigated further using the more controlled environment of cell culture and/or animal models. With further methodological and technological advances, the mass spectrometry-based proteome analysis of human body fluids and tissues offers phenomenal potential for the detection, prevention and treatment of diseases.
Acknowledgments
We would like to thank the Burrill family for their generous support through the Burrill Research Grant. We would also like to thank members of the Steen Laboratory at Children’s Hospital Boston, in particular John F.K. Sauld and Dominic Winter, for their technical assistance and critical reading of the manuscript. In addition, we thank members of the Center for Pancreatic Disease at Brigham and Women’s Hospital, particularly Shadeah L. Suleiman and Scott A. Brizard, for their technical assistance.
Glossary
- TCA
Trichloroacetic acid
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- DTT
dithiothreitol
- TCEP
tris(2-carboxyethyl)phosphine)
- CE
capillary electrophoresis
- CZE
capillary zone electrophoresis
- 2DGE
two-dimensional gel electrophoresis
- IPG
immobilized pH gradient
- IEF
isoelectric focusing
- DiGE
difference gel electrophoresis
- SCX
strong cation exchange
- WCX
weak cation exchange
- SAX
strong anion exchange
- WAX
weak anion exchange
- HPLC
high performance liquid chromatography
- MudPIT
multidimensional protein identification technology
- MALDI
matrix-assisted laser desorption/ionization
- ESI
electrospray ionization
- ICAT
isotope-coded affinity tag
- AQUA
absolute quantitation
- SILAC
stable isotope labeling by amino acids in cell culture
- CID
collision-induced dissociation
- QTOF
parts per million, ppm, quadrupole time-of-flight
- FT-ICR
Fourier transform ion cyclotron resonance
- ePFT
endoscopic pancreatic function test
- GO
gene ontology
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- PMSF
phenylmethylsulfonyl fluoride
- AEBSF
4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride
- SRM/MRM
selected/multiple reaction monitoring
- FASP
filter-aided sample preparation
- GeLC-MS/MS
gel coupled with liquid chromatography tandem mass spectrometry
- iTRAQ
isobaric tag for relative and absolute quantitation
- TMT
tandem mass tag
Footnotes
Funds were provided by the following NIH grants: 1 F32 DK085835-01A1) (JP), 1 R21 DK081703-01A2 (DC) and 5 P30 DK034854-24 (Harvard Digestive Diseases Center; DC).
References
- James P. Protein identification in the post-genome era: the rapid rise of proteomics. Q Rev Biophys. 1997;30(4):279–331. doi: 10.1017/s0033583597003399. [DOI] [PubMed] [Google Scholar]
- Din S, Lennon AM, Arnott ID, Hupp T, Satsangi J. Technology insight: the application of proteomics in gastrointestinal disease. Nat Clin Pract Gastroenterol Hepatol. 2007;4(7):372–385. doi: 10.1038/ncpgasthep0872. [DOI] [PubMed] [Google Scholar]
- Roberts GC, Smith CW. Alternative splicing: combinatorial output from the genome. Curr Opin Chem Biol. 2002;6(3):375–383. doi: 10.1016/s1367-5931(02)00320-4. [DOI] [PubMed] [Google Scholar]
- Wilkins MR, Gasteiger E, Gooley AA, Herbert BR, Molloy MP, Binz PA. et al. High-throughput mass spectrometric discovery of protein post-translational modifications. J Mol Biol. 1999;289(3):645–657. doi: 10.1006/jmbi.1999.2794. [DOI] [PubMed] [Google Scholar]
- Banks RE, Dunn MJ, Hochstrasser DF, Sanchez JC, Blackstock W, Pappin DJ. et al. Proteomics: new perspectives, new biomedical opportunities. Lancet. 2000;356(9243):1749–1756. doi: 10.1016/S0140-6736(00)03214-1. [DOI] [PubMed] [Google Scholar]
- Manabe T, Miyamoto H, Inoue K, Nakatsu M, Arai M. Separation of human cerebrospinal fluid proteins by capillary isoelectric focusing in the absence of denaturing agents. Electrophoresis. 1999;20(18):3677–3683. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3677::AID-ELPS3677>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Fountoulakis M, Juranville JF, Jiang L, Avila D, Roder D, Jakob P. et al. Depletion of the high-abundance plasma proteins. Amino Acids. 2004;27(3-4):249–259. doi: 10.1007/s00726-004-0141-1. [DOI] [PubMed] [Google Scholar]
- Jiang L, He L, Fountoulakis M. Comparison of protein precipitation methods for sample preparation prior to proteomic analysis. J Chromatogr A. 2004;1023(2):317–320. doi: 10.1016/j.chroma.2003.10.029. [DOI] [PubMed] [Google Scholar]
- Thongboonkerd V, McLeish KR, Arthur JM, Klein JB. Proteomic analysis of normal human urinary proteins isolated by acetone precipitation or ultracentrifugation. Kidney Int. 2002;62(4):1461–1469. doi: 10.1111/j.1523-1755.2002.kid565.x. [DOI] [PubMed] [Google Scholar]
- Stark M, Jornvall H, Johansson J. Isolation and characterization of hydrophobic polypeptides in human bile. Eur J Biochem. 1999;266(1):209–214. doi: 10.1046/j.1432-1327.1999.00845.x. [DOI] [PubMed] [Google Scholar]
- Wilson CM. Staining of proteins on gels: comparisons of dyes and procedures. Methods Enzymol. 1983;91:236–247. doi: 10.1016/s0076-6879(83)91020-0. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68(5):850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Lopez MF, Berggren K, Chernokalskaya E, Lazarev A, Robinson M, Patton WF. A comparison of silver stain and SYPRO Ruby Protein Gel Stain with respect to protein detection in two-dimensional gels and identification by peptide mass profiling. Electrophoresis. 2000;21(7):3673–3683. doi: 10.1002/1522-2683(200011)21:17<3673::AID-ELPS3673>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Chevalier F, Rofidal V, Vanova P, Bergoin A, Rossignol M. Proteomic capacity of recent fluorescent dyes for protein staining. Phytochemistry. 2004;65(11):1499–1506. doi: 10.1016/j.phytochem.2004.04.019. [DOI] [PubMed] [Google Scholar]
- Wittmann-Liebold B, Graack HR, Pohl T. Two-dimensional gel electrophoresis as tool for proteomics studies in combination with protein identification by mass spectrometry. Proteomics. 2006;6(17):4688–4703. doi: 10.1002/pmic.200500874. [DOI] [PubMed] [Google Scholar]
- Michel PE, Reymond F, Arnaud IL, Josserand J, Girault HH, Rossier JS. Protein fractionation in a multicompartment device using Off-Gel isoelectric focusing. Electrophoresis. 2003;24(1-2):3–11. doi: 10.1002/elps.200390030. [DOI] [PubMed] [Google Scholar]
- Heller M, Michel PE, Morier P, Crettaz D, Wenz C, Tissot JD. et al. Two-stage Off-Gel isoelectric focusing: protein followed by peptide fractionation and application to proteome analysis of human plasma. Electrophoresis. 2005;26(6):1174–1188. doi: 10.1002/elps.200410106. [DOI] [PubMed] [Google Scholar]
- Burgi D, Smith AJ. Capillary electrophoresis of proteins and peptides. Curr Protoc Protein Sci. 2001 doi: 10.1002/0471140864.ps1009s02. Chapter 10: Unit 10.9. [DOI] [PubMed] [Google Scholar]
- Elhamili A, Wetterhall M, Arvidsson B, Sebastiano R, Righetti PG, Bergquist J. Rapid capillary electrophoresis time-of-flight mass spectrometry separations of peptides and proteins using a monoquaternarized piperazine compound (M7C4I) for capillary coatings. Electrophoresis. 2008;29(8):1619–1625. doi: 10.1002/elps.200700737. [DOI] [PubMed] [Google Scholar]
- Stroink T, Ortiz MC, Bult A, Lingeman H, de Jong GJ, Underberg WJ. On-line multidimensional liquid chromatography and capillary electrophoresis systems for peptides and proteins. J Chromatogr A. 2005;817(1):49–66. doi: 10.1016/j.jchromb.2004.11.057. [DOI] [PubMed] [Google Scholar]
- Righetti PG, Gianazza E. Isoelectric focusing in immobilized pH gradients: theory and newer methodology. Methods Biochem Anal. 1987;32:215–278. doi: 10.1002/9780470110539.ch4. [DOI] [PubMed] [Google Scholar]
- Tannu NS, Hemby SE. Two-dimensional fluorescence difference gel electrophoresis for comparative proteomics profiling. Nat Protoc. 2006;1(4):1732–1742. doi: 10.1038/nprot.2006.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CC, Yates JR 3rd. The application of mass spectrometry to membrane proteomics. Nat Biotechnol. 2003;21(3):262–267. doi: 10.1038/nbt0303-262. [DOI] [PubMed] [Google Scholar]
- Molloy MP, Brzezinski EE, Hang J, McDowell MT, VanBogelen RA. Overcoming technical variation and biological variation in quantitative proteomics. Proteomics. 2003;3(10):1912–1919. doi: 10.1002/pmic.200300534. [DOI] [PubMed] [Google Scholar]
- Lilley KS, Friedman DB. All about DIGE: quantification technology for differential-display 2D-gel proteomics. Expert Rev Proteomics. 2004;1(4):401–409. doi: 10.1586/14789450.1.4.401. [DOI] [PubMed] [Google Scholar]
- Unlu M, Morgan ME, Minden JS. Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis. 1997;18(11):2071–2077. doi: 10.1002/elps.1150181133. [DOI] [PubMed] [Google Scholar]
- Mujumdar RB, Ernst LA, Mujumdar SR, Lewis CJ, Waggoner AS. Cyanine dye labeling reagents: sulfoindocyanine succinimidyl esters. Bioconjug Chem. 1993;4(2):105–111. doi: 10.1021/bc00020a001. [DOI] [PubMed] [Google Scholar]
- Southwick PL, Ernst LA, Tauriello EW, Parker SR, Mujumdar RB, Mujumdar SR. et al. Cyanine dye labeling reagents--carboxymethylindocyanine succinimidyl esters. Cytometry. 1990;11(3):418–430. doi: 10.1002/cyto.990110313. [DOI] [PubMed] [Google Scholar]
- Ernst LA, Gupta RK, Mujumdar RB, Waggoner AS. Cyanine dye labeling reagents for sulfhydryl groups. Cytometry. 1989;10(1):3–10. doi: 10.1002/cyto.990100103. [DOI] [PubMed] [Google Scholar]
- Mujumdar RB, Ernst LA, Gupta RK, Mujumdar SR, Waggoner AS. Cyanine dye labeling reagents containing isothiocyanate groups. Cytometry. 1989;10(1):11–19. doi: 10.1002/cyto.990100104. [DOI] [PubMed] [Google Scholar]
- Gong L, Puri M, Unlu M, Young M, Robertson K, Viswanathan S. et al. Drosophila ventral furrow morphogenesis: a proteomic analysis. Development. 2004;131(3):643–656. doi: 10.1242/dev.00955. [DOI] [PubMed] [Google Scholar]
- Molnar I, Horvath C. Reverse-phase chromatography of polar biological substances: separation of catechol compounds by high-performance liquid chromatography. Clin Chem. 1976;22(9):1497–1502. [PubMed] [Google Scholar]
- Fritz JS, Gjerde DT. Ion chromatography. 3rd edition. New York: Wiley-VCH; 2000. 254 p [Google Scholar]
- Porath J, Flodin P. Gel filtration: a method for desalting and group separation. Nature. 1959;183(4676):1657–1659. doi: 10.1038/1831657a0. [DOI] [PubMed] [Google Scholar]
- Zachariou M. Affinity chromatography: methods and protocols. 2nd edition. Totowa, NJ: Humana Press; 2008. 343 p. [DOI] [PubMed] [Google Scholar]
- McLafferty FW, Breuker K, Jin M, Han X, Infusini G, Jiang H. et al. Top-down MS, a powerful complement to the high capabilities of proteolysis proteomics. FEBS J. 2007;274(24):6256–6268. doi: 10.1111/j.1742-4658.2007.06147.x. [DOI] [PubMed] [Google Scholar]
- Scherperel G, Reid GE. Emerging methods in proteomics: top-down protein characterization by multistage tandem mass spectrometry. Analyst. 2007;132(6):500–506. doi: 10.1039/b618499p. [DOI] [PubMed] [Google Scholar]
- Kelleher NL. Top-down proteomics. Anal Chem. 2004;76(11):197A–203A. [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1(6):2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- Olsen JV, Ong SE, Mann M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol Cell Proteomics. 2004;3(6):608–614. doi: 10.1074/mcp.T400003-MCP200. [DOI] [PubMed] [Google Scholar]
- van Montfort BA, Canas B, Duurkens R, Godovac-Zimmermann J, Robillard GT. Improved in-gel approaches to generate peptide maps of integral membrane proteins with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J Mass Spectrom. 2002;37(3):322–330. doi: 10.1002/jms.288. [DOI] [PubMed] [Google Scholar]
- Jeno P, Mini T, Moes S, Hintermann E, Horst M. Internal sequences from proteins digested in polyacrylamide gels. Anal Biochem. 1995;224(1):75–82. doi: 10.1006/abio.1995.1010. [DOI] [PubMed] [Google Scholar]
- Salplachta J, Marchetti M, Chmelik J, Allmaier G. A new approach in proteomics of wheat gluten: combining chymotrypsin cleavage and matrix-assisted laser desorption/ionization quadrupole ion trap reflectron tandem mass spectrometry. Rapid Commun Mass Spectrom. 2005;19(18):2725–2728. doi: 10.1002/rcm.2092. [DOI] [PubMed] [Google Scholar]
- Hemling ME, Carr SA, Capiau C, Petre J. Structural characterization of recombinant hepatitis B surface antigen protein by mass spectrometry. Biochemistry. 1988;27(2):699–705. doi: 10.1021/bi00402a031. [DOI] [PubMed] [Google Scholar]
- Manadas B, English JA, Wynne KJ, Cotter DR, Dunn MJ. Comparative analysis of OFFGel, strong cation exchange with pH gradient, and RP at high pH for first-dimensional separation of peptides from a membrane-enriched protein fraction. Proteomics. 2009;9(22):5194–5198. doi: 10.1002/pmic.200900349. [DOI] [PubMed] [Google Scholar]
- Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res. 2003;2(1):43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- Washburn MP, Wolters D, Yates JR 3rd. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19(3):242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Mann M. Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J Proteome Res. 2009;8(12):5674–5678. doi: 10.1021/pr900748n. [DOI] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nature Methods. 2009;6(5):359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Delmotte N, Lasaosa M, Tholey A, Heinzle E, Huber CG. Two-dimensional reversed-phase x ion-pair reversed-phase HPLC: an alternative approach to high-resolution peptide separation for shotgun proteome analysis. J Proteome Res. 2007;6(11):4363–4373. doi: 10.1021/pr070424t. [DOI] [PubMed] [Google Scholar]
- Dwivedi RC, Spicer V, Harder M, Antonovici M, Ens W, Standing KG. et al. Practical implementation of 2D HPLC scheme with accurate peptide retention prediction in both dimensions for high-throughput bottom-up proteomics. Anal Chem. 2008;80(18):7036–7042. doi: 10.1021/ac800984n. [DOI] [PubMed] [Google Scholar]
- Elschenbroich S, Ignatchenko V, Sharma P, Schmitt-Ulms G, Gramolini AO, Kislinger T. Peptide separations by on-line MudPIT compared to isoelectric focusing in an off-gel format: application to a membrane-enriched fraction from C2C12 mouse skeletal muscle cells. J Proteome Res. 2009;8(10):4860–4869. doi: 10.1021/pr900318k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald WH, Ohi R, Miyamoto DT, Mitchison TJ, Yates JR. Comparison of three directly coupled HPLC MS/MS strategies for identification of proteins from complex mixtures: single-dimension LC-MS/MS, 2-phase MudPIT, and 3-phase MudPIT. International Journal of Mass Spectrometry. 2002;219(1):245–251. [Google Scholar]
- Motoyama A, Yates JR 3rd. Multidimensional LC separations in shotgun proteomics. Anal Chem. 2008;80(19):7187–7193. doi: 10.1021/ac8013669. [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem. 2003;75(3):663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- Baczek T. Fractionation of peptides in proteomics with the use of pI-based approach and ZipTip pipette tips. J Pharm Biomed Anal. 2004;34(5):851–860. doi: 10.1016/j.jpba.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Ong S-E, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A. et al. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol Cell Proteomics. 2002;1(5):376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Stewart II, Thomson T, Figeys D. 18O labeling: a tool for proteomics. Rapid Commun Mass Spectrom. 2001;15(24):2456–2465. doi: 10.1002/rcm.525. [DOI] [PubMed] [Google Scholar]
- Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G. et al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75(8):1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S. et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3(12):1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- Gerber SA, Kettenbach AN, Rush J, Gygi SP. The absolute quantification strategy: application to phosphorylation profiling of human separase serine 1126. Methods Mol Biol. 2007;359:71–86. doi: 10.1007/978-1-59745-255-7_5. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick DS, Gerber SA, Gygi SP. The absolute quantification strategy: a general procedure for the quantification of proteins and post-translational modifications. Methods. 2005;35(3):265–273. doi: 10.1016/j.ymeth.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci USA. 2003;100(12):6940–6945. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgren DH, Hwang SI, Wu L, Han DK. Role of spectral counting in quantitative proteomics. Expert Rev Proteomics. 2010;7(1):39–53. doi: 10.1586/epr.09.69. [DOI] [PubMed] [Google Scholar]
- Dubois F, Knochenmuss R, Zenobi R. Optimization of an ion-to-photon detector for large molecules in mass spectrometry. Rapid Commun Mass Spectrom. 1999;13(19):1958–1967. doi: 10.1002/(SICI)1097-0231(19991015)13:19<1958::AID-RCM738>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Steen H, Mann M. The ABC’s (and XYZ’s) of peptide sequencing. Nat Rev Mol Cell Biol. 2004;5(9):699–711. doi: 10.1038/nrm1468. [DOI] [PubMed] [Google Scholar]
- Olsen JV, de Godoy LM, Li G, Macek B, Mortensen P, Pesch R. et al. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol Cell Proteomics. 2005;4(12):2010–2021. doi: 10.1074/mcp.T500030-MCP200. [DOI] [PubMed] [Google Scholar]
- Aebersold R, Goodlett DR. Mass spectrometry in proteomics. Chem Rev. 2001;101(2):269–295. doi: 10.1021/cr990076h. [DOI] [PubMed] [Google Scholar]
- Scigelova M, Makarov A. Orbitrap mass analyzer — overview and applications in proteomics. Proteomics. 2006;6(Suppl 2):16–21. doi: 10.1002/pmic.200600528. [DOI] [PubMed] [Google Scholar]
- Hu Q, Noll RJ, Li H, Makarov A, Hardman M, Graham Cooks R. The Orbitrap: a new mass spectrometer. J Mass Spectrom. 2005;40(4):430–443. doi: 10.1002/jms.856. [DOI] [PubMed] [Google Scholar]
- Eng JK, Mccormack AL, Yates JR. An Approach to Correlate Tandem Mass-Spectral Data of Peptides with Amino-Acid-Sequences in a Protein Database. J Am Soc Mass Spectrom. 1994;5(11):976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Colinge J, Masselot A, Giron M, Dessingy T, Magnin M. OLAV: towards high-throughput tandem mass spectrometry data identification. Proteomics. 2003;3(8):1454–1463. doi: 10.1002/pmic.200300485. [DOI] [PubMed] [Google Scholar]
- Shilov IV, Seymour SL, Patel AA, Loboda A, Tang WH, Keating SP. et al. The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol Cell Proteomics. 2007;6(9):1638–1655. doi: 10.1074/mcp.T600050-MCP200. [DOI] [PubMed] [Google Scholar]
- Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20(9):1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- Clauser KR, Baker P, Burlingame AL. Role of accurate mass measurement (+/- 10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal Chem. 1999;71(14):2871–2882. doi: 10.1021/ac9810516. [DOI] [PubMed] [Google Scholar]
- Choi H, Nesvizhskii AI. False discovery rates and related statistical concepts in mass spectrometry-based proteomics. J Proteome Res. 2008;7(1):47–50. doi: 10.1021/pr700747q. [DOI] [PubMed] [Google Scholar]
- Kall L, Storey JD, MacCoss MJ, Noble WS. Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J Proteome Res. 2008;7(1):29–34. doi: 10.1021/pr700600n. [DOI] [PubMed] [Google Scholar]
- Renard BY, Timm W, Kirchner M, Steen JA, Hamprecht FA, Steen H. Estimating the confidence of peptide identifications without decoy databases. Anal Chem. 2010;82(11):4314–4318. doi: 10.1021/ac902892j. [DOI] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol Biol. 2010;604:55–71. doi: 10.1007/978-1-60761-444-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4(3):207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- Paulo JA, Lee LS, Wu B, Banks PA, Steen H, Conwell DL. Mass spectrometry-based proteomics of endoscopically collected pancreatic fluid in chronic pancreatitis research. Proteomics Clin Appl. 2011;5(3-4):109–120. doi: 10.1002/prca.201000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Lazarev A. Sample Preparation in Biological Mass Spectrometry. New York: Springer; 2011. 1118 p [Google Scholar]
- Paulo J, Vaezzadeh A, Conwell D, Lee R, Steen H. In: Sample Preparation in Biological Mass Spectrometry. Ivanov A, Lazarev A, editors. New York: Springer; 2011. Sample Handling of Body Fluids for Proteomics. [Google Scholar]
- Paulo JA, Lee LS, Wu B, Repas K, Mortele KJ, Banks PA. et al. Identification of pancreas-specific proteins in endoscopically (endoscopic pancreatic function test) collected pancreatic fluid with liquid chromatography-tandem mass spectrometry. Pancreas. 2010;39(6):889–896. doi: 10.1097/MPA.0b013e3181cf16f4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo JA, Lee LS, Wu B, Repas K, Banks PA, Conwell DL. et al. Optimized sample preparation of endoscopic collected pancreatic fluid for SDS-PAGE analysis. Electrophoresis. 2010;31(14):2377–2387. doi: 10.1002/elps.200900762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo JA, Lee LS, Wu B, Repas K, Banks PA, Conwell DL. et al. Proteomic analysis of endoscopically (endoscopic pancreatic function test) collected gastroduodenal fluid using in-gel tryptic digestion followed by LC-MS/MS. Proteomics Clin Appl. 2010;4(8-9):715–725. doi: 10.1002/prca.201000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo JA, Lee LS, Banks PA, Steen H, Conwell DL. Difference gel electrophoresis identifies differentially expressed proteins in endoscopically collected pancreatic fluid. Electrophoresis. 2011;32(15):1939–1951. doi: 10.1002/elps.201100203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo JA, Urrutia R, Banks PA, Conwell DL, Steen H. Proteomic Analysis of an Immortalized Mouse Pancreatic Stellate Cell Line Identifies Differentially-Expressed Proteins in Activated vs Nonproliferating Cell States. J Proteome Res. 2011;10(10):4835–4844. doi: 10.1021/pr2006318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo JA, Lee L, Banks PA, Conwell DL, Steen H. Proteomic analysis of formalin-fixed paraffin-embedded pancreatic tissue using liquid chromatography tandem mass spectrometry (LC-MS/MS) Pancreas. doi: 10.1097/MPA.0b013e318227a6b7. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnie C, Svensson B. Proteolysis during the isoelectric focusing step of two-dimensional gel electrophoresis may be a common problem. Anal Biochem. 2002;311(2):182–186. doi: 10.1016/s0003-2697(02)00389-5. [DOI] [PubMed] [Google Scholar]
- Rai AJ, Gelfand CA, Haywood BC, Warunek DJ, Yi J, Schuchard MD. et al. HUPO Plasma Proteome Project specimen collection and handling: towards the standardization of parameters for plasma proteome samples. Proteomics. 2005;5(13):3262–3277. doi: 10.1002/pmic.200401245. [DOI] [PubMed] [Google Scholar]
- Marshall J, Kupchak P, Zhu W, Yantha J, Vrees T, Furesz S. et al. Processing of serum proteins underlies the mass spectral fingerprinting of myocardial infarction. J Proteome Res. 2003;2(4):361–372. doi: 10.1021/pr030003l. [DOI] [PubMed] [Google Scholar]
- Masamune A, Kikuta K, Watanabe T, Satoh K, Hirota M, Hamada S. et al. Fibrinogen induces cytokine and collagen production in pancreatic stellate cells. Gut. 2009;58(4):550–559. doi: 10.1136/gut.2008.154401. [DOI] [PubMed] [Google Scholar]
- Talukdar R, Tandon RK. Pancreatic stellate cells: new target in the treatment of chronic pancreatitis. J Gastroenterol Hepatol. 2008;23(1):34–41. doi: 10.1111/j.1440-1746.2007.05206.x. [DOI] [PubMed] [Google Scholar]
- Shimizu K, Kobayashi M, Tahara J, Shiratori K. Cytokines and peroxisome proliferator-activated receptor gamma ligand regulate phagocytosis by pancreatic stellate cells. Gastroenterology. 2005;128(7):2105–2118. doi: 10.1053/j.gastro.2005.03.025. [DOI] [PubMed] [Google Scholar]
- Mews P, Phillips P, Fahmy R, Korsten M, Pirola R, Wilson J. et al. Pancreatic stellate cells respond to inflammatory cytokines: potential role in chronic pancreatitis. Gut. 2002;50(4):535–541. doi: 10.1136/gut.50.4.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Aslanian A, Yates JR 3rd. Mass spectrometry for proteomics. Curr Opin Chem Biol. 2008;12(5):483–490. doi: 10.1016/j.cbpa.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsangaris GT. From proteomics research to clinical practice. Expert Rev Proteomics. 2009;6(3):235–238. doi: 10.1586/epr.09.14. [DOI] [PubMed] [Google Scholar]
- Yates JR, Ruse CI, Nakorchevsky A. Proteomics by mass spectrometry: approaches, advances, and applications. Annu Rev Biomed Eng. 2009;11:49–79. doi: 10.1146/annurev-bioeng-061008-124934. [DOI] [PubMed] [Google Scholar]
- Latterich M, Abramovitz M, Leyland-Jones B. Proteomics: new technologies and clinical applications. Eur J Cancer. 2008;44(18):2737–2741. doi: 10.1016/j.ejca.2008.09.007. [DOI] [PubMed] [Google Scholar]