

Abstract
Since 1992, the amyloid cascade hypothesis has played the prominent role in explaining the etiology and pathogenesis of Alzheimer's disease (AD). It proposes that the deposition of β-amyloid (Aβ) is the initial pathological event in AD leading to the formation of senile plaques (SPs) and then to neurofibrillary tangles (NFTs), neuronal cell death, and ultimately dementia. While there is substantial evidence supporting the hypothesis, there are also limitations: (1) SP and NFT may develop independently, and (2) SPs and NFTs may be the products rather than the causes of neurodegeneration in AD. In addition, randomized clinical trials that tested drugs or antibodies targeting components of the amyloid pathway have been inconclusive. This paper provides a critical overview of the evidence for and against the amyloid cascade hypothesis in AD and provides suggestions for future directions.
1. Introduction
Alzheimer's disease (AD), which is characterized by progressive deterioration in cognition, function, and behavior, places a considerable burden on western societies. It is the sixth leading cause of all deaths and the fifth leading cause of death in persons aged ≥65 years. To date, an estimated 5.4 million Americans have AD, but due to the baby boom generation, the incidence in 2050 is expected to reach a million persons per year, resulting in a total estimated prevalence of 11 to 16 million affected persons.
Since the first description of presenile dementia by Alois Alzheimer in 1907 [1], senile plaques (SPs) and neurofibrillary tangles (NFTs) are considered the key pathological hallmarks of AD [2]. The identification of β-amyloid (Aβ) in SPs [3] and genetic studies that identified mutations in the amyloid precursor protein (APP) [4], presenilin 1 (PSEN1), and presenilin 2 (PSEN2) genes [5, 6] leading to the accumulation of Aβ and early-onset familial dementia [4, 5, 7], resulted in the formulation of the “Amyloid Cascade Hypothesis” (ACH; Figure 1) [8, 9]. According to the ACH, the deposition of Aβ is the initial pathological trigger in the disease, which subsequently leads to the formation of NFTs, neuronal cell death and dementia. While there is considerable evidence supporting this hypothesis, there are observations that seem to be inconsistent. This paper summarizes the current evidence for and against the amyloid cascade in AD.
Figure 1.
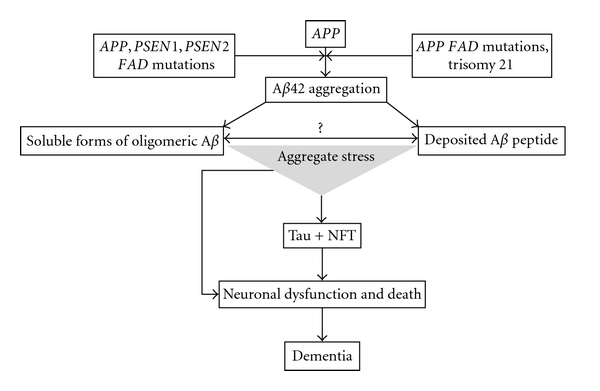
Amyloid cascade hypothesis.
2. Amyloid Cascade Hypothesis
As described above, two key observations resulted in the original formulation of the ACH (Figure 1). First, the detection of Aβ as a main constituent of the SPs [3] and second mutations of the APP [4], PSEN1, and PSEN2 genes [5, 6], which were found in families with early-onset AD (FAD, disease onset < 60 years). As a consequence of these observations, the presence of Aβ within SPs was interpreted as an effect of these mutations that subsequently leads to cell death and dementia. Since FAD has—except the earlier onset—a similar phenotype to late-onset AD, it was assumed that this amyloid deposition could explain the pathogenesis of all types of AD.
3. Evidence from Studies on the Formation of Aβ and Tau
There are two major objections regarding the ACH as originally formulated. First, SPs and NFTs may be reactive products resulting from neurodegeneration in AD rather than being its cause, and, second, it remains unclear whether and how the deposition of Aβ leads to the formation of NFTs.
3.1. Aβ and Tau as Reactive Processes
In persons who suffered from head trauma, APP is found with pathological features similar to AD in neuronal perikarya and in dystrophic neurites surrounding Aβ deposits [10]. In addition, there is evidence that neurons in the medial temporal lobe secrete APP and display increased APP immunoreactivity [11]. These findings suggest that increased expression of APP in head trauma cases may be an acute-phase response to neuronal injury [12], which in turn leads to increased Aβ deposition. This notion is supported by the observation that the different morphological forms of Aβ deposits, including diffuse, primitive, and classic deposits, contain acute phase proteins such as complement factors and α-antichymotrypsin [13]. Consequently, it has been proposed that, in AD, APP may be a reaction to the disease process in order to help maintain cell function, neuronal growth, and survival [14]. The putative neurotrophic action of APP is supported by the observation that it shares structural features with the precursor for epidermal growth factor [14]. Finally, there is also evidence that NFTs may form as a neuronal response to injury [15].
There are also findings from animal studies suggesting that the formation of Aβ and NFT may be reactive. In rats, both experimental damage or chemically induced lesions of the nucleus basalis can elevate cortical APP, and intrathecal or intraparenchymal injections of toxins can induce APP in hippocampal neurons, suggesting that the generation of APP could be a specific response to loss of functional innervation of the cortex [16, 17]. Denervation of the dopamine pathways and septal lesions affecting both the cholinergic system and γ-aminobutyric acid (GABA) neurons projecting to the dentate gyrus can result in a loss of dendritic microtubule-associated protein 2 (MAP2) and the appearance of tau-immunoreactive dentate gyrus granule cells [18]. Thus, denervation can cause transsynaptic changes in dentate gyrus neurons, and these alterations may represent an intermediate step to NFTs formation.
3.2. Relation of the Formation of NFT to Aβ
SPs and NFTs cluster in a significant proportion of cortical areas but they seem to be distributed independently of each other [19]. SP and NFTs also seem to occur temporally separated; in the entorhinal cortex the occurrence of NFTs may in fact precede the occurrence of SPs [20]. This spatial and temporal separation may suggest that they are pathogenically disconnected.
However, evidence for an effect of Aβ on the formation of NFT comes from transgenic experiments. The presence of APP mutations alone or in combination with PSEN1 mutations seems to induce Aβ deposits in normal brain and some degree of hyperphosphorylated tau in neurites [21] although it does not appear to induce tau pathology or a significant inflammatory response. These findings are consistent with studies in which fetal rat hippocampal neurons and human cortical neurons treated with fibrielar Aβ display an increased degree of tau phosphorylation [22] providing additional evidence that amyloid fibril formation might alter the phosphorylation state of tau, which in turn results in the loss of microtubule-binding capacity. Other studies showed that Aβ 25–35 can induce the aggregation of tau proteins and that a decrease in aggregation of Aβ was induced by tau peptides [23]. Thus, aggregation of tau may be associated with disassembly of Aβ, which could explain the lack of spatial correlation of the SPs and NFTs [19]. Finally, the notion of an impact of Aβ on NFT formation is supported by studies in APP-transgenic mice reporting that a reduction in endogenous levels of tau can ameliorate some of the behavioral and other deficits that are mediated by Aβ [24, 25] and by the discovery that mutations in the tau gene cause autosomal dominant frontotemporal lobe dementia with a tau pathology similar to the tau pathology seen in AD but without the appearance of Aβ plaques [26]. Both these observations seem to place tau pathology downstream of amyloid-β pathology.
4. Evidence from Genetic Studies
In particular the genes identified in the late-onset form of the disease provide support for the ACH. In general, these genes are not inherited in a Mendelian but a sporadic fashion. However, first-degree relatives of patients with late-onset AD have twice the expected life time risk of this disease compared to persons without an affected first-degree relative, and late-onset AD is more frequent among monozygotic than dizygotic cotwins, suggesting a substantial genetic contribution to this form of the disease.
The apolipoprotein E (APOE) gene, which was identified as the first susceptibility gene for late-onset AD, is the major genetic risk factor (population attributable risk: ∼20%) [27, 28]. Each APOE-ε4 allele lowers the age at onset in a dose-dependent fashion [27]. How the different APOE proteins mediate their effects in AD is not fully clarified, but there is compelling evidence by PDAPP transgenic mice models indicating that APOE mediates the clearance of amyloid-β [29], with the APOE2, APOE3, and APOE4 isoforms being increasingly less effective [30]. Consistent with this notion, the presence of an APOE-ε4 allele is associated with a higher Aβ burden in the brains of LOAD patients [31, 32], suggesting that APOE interacts with Aβ by enhancing its deposition in plaques. In various ethnic groups, two haplotypes in the sortilin-related receptor (SORL1) gene associated with LOAD were identified [33–37]. SORL1 is involved in trafficking of APP from the cell surface to the golgi-endoplasmic reticulum complex and γ-secretase processing of APP [34, 38, 39], also in line with the ACH. Recent large-scale GWA studies performed primarily in samples and populations of European ancestry detected genetic variants associated with AD in complement component (3b/4b) receptor 1 (CR1), clusterin (CLU, APOJ), bridging integrator 1 (BIN1), phosphatidylinositol-binding clathrin assembly protein (PICALM), EPH receptor A1 (EPHA1), CD33 molecule (CD33), membrane-spanning 4-domains, subfamily A, members 4 and 6E (MS4A4/MS4A6E), CD2-associated protein (CD2AP), and ATP-binding cassette, subfamily A, member 7 (ABCA7) [40–42]. While these genes remain to undergo functional validation, they are functionally plausible and also largely consistent with the ACH. Similar and additive to APOE, CLU encodes an apolipoprotein and acts as an Aβ chaperone, regulating the conversion of Aβ to insoluble forms and Aβ toxicity thereby promoting amyloid plaque formation [43]. ABCA7 is involved in the efflux of lipids from cells to lipoprotein particles, such as APOE and CLU, and in addition regulates APP processing and inhibits β-amyloid secretion [44]. There is evidence that CR1 may contribute to Aβ clearance by complement activation [45]. CD2AP, CD33, BIN1, and PICALM are involved in endocytosis (CME), and a recent study [46] showed that several of these factors involved in endocytosis modify Aβ toxicity in glutamatergic neurons of Caenorhabditis elegans and in primary rat cortical neurons. In yeast, Aβ impaired the endocytic trafficking of a plasma membrane receptor, which was ameliorated by endocytic pathway factors identified in the yeast screen also providing substantial evidence for a link between Aβ, endocytosis, and human AD [46]. In summary, convincing evidence for an Aβ-related mechanism exists for all of these identified LOAD genes, providing a substantial amount of support for the ACH in AD.
5. Evidence from Clinical Trials Targeting Aβ and Tau
The drugs currently used to treat AD (i.e., cholinesterase inhibitors, NMDA receptor antagonists, and antipsychotic drugs) have limited therapeutic value. New, potentially disease-modifying, therapeutic approaches are targeting Aβ and tau protein. Driven by the ACH, there are currently four main therapeutic approaches: (a) reducing the generation of Aβ, (b) facilitating the clearance of Aβ, (c) preventing the aggregation of Aβ and destabilizing Aβ oligomers, and (d) drugs targeting tau [47]. Drugs classes include active and passive immunization directed against Aβ, compounds that interfere with the secretases regulating Aβ generation from APP, drugs to prevent Aβ aggregation and destabilize Aβ oligomers, and drugs targeting tau protein.
5.1. Active and Passive Immunization
Active and passive immunizations were developed to inhibit generation of toxic Aβ aggregates and to remove soluble and aggregated Aβ. At least three different immune-mediated mechanisms can promote Aβ removal: solubilization by antibody binding to Aβ, phagocytosis of Aβ by microglia, and Aβ extraction from the brain by plasma antibodies.
In phase II randomized controlled trials (RCTs) of active immunization of patients with mild-to-moderate AD with the anti-Aβ vaccine AN-1792 (QS-21) most but not all participants developed significant Aβ-antibody titres [48, 49] and there was evidence of memory and function improvement and reduced CSF tau concentrations in patients with increased IgG titres [48, 49]. However, in the first trial patients immunized with AN-1792 had a greater brain atrophy rate on MRI than did patients given placebo possibly because of amyloid removal and cerebral fluid shifts. In addition, several patients developed meningoencephalitis due to a T-cell response. In the follow-up trial, brain volume loss in antibody responders was not different from that in patients receiving placebo, and no further cases of meningoencephalitis were found [49]. Responders maintained low, but detectable, anti-AN-1792 antibody titres at about 4.6 years after immunization and had significantly reduced functional decline compared with placebo-treated patients [49]. In addition, immunization with anti-AN-1792 antibody could completely remove amyloid plaques as determined by postmortem assessment although patients still had end-stage dementia symptoms before death.
In order to avoid neuroinflammation and neurotoxicity, new vaccines that selectively target B-cell epitopes have been developed. CAD-106, which consists of the immunodrug carrier Qb coupled with a fragment of the Aβ 1–6 peptide, could in animal studies induce Aβ-specific antibodies and reduce amyloid accumulation without stimulating T cells. In patients with mild-to-moderate AD, CAD-106 induced a substantial anti-Aβ IgG response and was well tolerated [50], confirmatory phase II RCTs are ongoing (NCT01097096, NCT01023685, NCT00795418, NCT00956410, and NCT00733863). ACC-001 is an Aβ 1–6 fragment derived from the N-terminal B cell epitope of Aβ and conjugated to the mutated diphtheria toxin protein CRM19. It is being studied in phase II RCTs (NCT00479557, NCT01284387, NCT01227564, NCT00498602, NCT00752232, NCT00955409, NCT01238991, NCT00960531, NCT00959192). ACI-24 is a vaccine that contains Aβ 1–15 closely apposed to the surface of the liposome. It reduced brain amyloid load and restored memory deficits in mice [51] and is entering a phase II RCT. Vaccines that are currently being tested in phase I RCTs are V-950 (NCT00464334; an aluminium-containing adjuvant with or without ISCOMATRIX (CSL Behring, PA, USA, a biological adjuvant of saponin, cholesterol, and phospholipids) and UB-311 (NCT00965588), a vaccine in which the immunogen Aβ 1–14 is associated with the UBITh peptide (United Biomedical, NY, USA) and a mineral salt suspension adjuvant [52].
Affitopes, which are short peptides mimicking parts of native Aβ 1–42, represent an alternative active immunization strategy. The affitopes AD-01 and AD-02 target the N-terminal Aβ fragment and both had disease-modifying properties in animal models of AD [53]. Results of recent phase I RCTs indicate that both are safe and well tolerated (NCT00495417, NCT00633841, and NCT00711139) [53]. Affitope AD-02 recently progressed to phase II clinical testing (NCT01117818).
Passive immunotherapy is based on monoclonal antibodies or polyclonal immunoglobulins targeting Aβ to promote its clearance. Animal studies have shown that anti-Aβ antibodies can prevent oligomer formation and reduce brain amyloid load with improvement in cognitive functions [54]. Several monoclonal antibodies are currently being tested: bapineuzumab (AAB-001), solanezumab (LY-2062430), PF-04360365, GSK-933776, R-1450 (RO-4909832), and MABT-5102A. A phase II RCT of bapineuzumab in patients with mild-to-moderate AD that had a follow-up period of longer than 18 months reported no significant effects on the primary measures of cognition or activities of daily living, as measured in prespecified within-dose cohort analyses. However, post hoc analyses of clinical and neuroimaging data from all dose cohorts showed nonsignificant improvements in cognitive endpoints and signs of efficacy in APOE ɛ 4 noncarriers [55]. Phase III studies are ongoing, including separate RCTs for APOE ɛ 4 carriers and non-carriers (NCT00574132, NCT00996918, NCT00998764, NCT00667810, NCT00575055, NCT00676143, and NCT00937352). Solanezumab, a monoclonal antibody that targets specifically soluble Aβ, promotes Aβ clearance from the brain through the blood. In a phase II RCT, there was a correlation between total plasma Aβ 1–42 after treatment (dose-dependent increase), baseline amyloid plaque burden shown by single-photon emission CT scanning, and a dose-dependent increase in unbound CSF Aβ 1–42, suggesting that solanezumab might mobilize Aβ 1–42 from plaques and might normalize soluble CSF Aβ 1–42 in patients with AD [56]. Consequently, two phase III RCTs have been initiated (NCT00905372, NCT00904683, NCT01127633). PF-04360365 is a modified IgG2 antibody that binds to the C terminus of Aβ 1–40. Preliminary results on a single-dose regimen indicate that this antibody is well tolerated in patients with AD [57]. Currently, two phase II RCTs of multiple doses are ongoing (NCT00722046 and NCT00945672). GSK-933776, R-1450 (RO-4909832), and MABT-5102A are monoclonal antibodies that target Aβ and have been tested in patients with AD in phase I and phase II trials (NCT01424436, NCT00459550, NCT01224106, NCT00531804, NCT00736775, NCT00997919, NCT01343966, and NCT01397578).
Passive immunization [58] can also be achieved by intravenous infusion of immunoglobulins (IVIg), from healthy donors, which include naturally occurring polyclonal anti-Aβ antibodies. IVIg is already approved as therapy for immune deficiency, with good safety and tolerability evidence. In two small studies, short-term immunoglobulin administration in patients with AD was well tolerated, promoted a decrease of total Aβ CSF concentrations, and increased plasma total Aβ concentrations [59, 60], with evidence of improvement or stabilization of cognitive functions. Preliminary data from a phase II RCT confirmed the positive effects on cognition [61], a phase III study is ongoing (NCT00818662). In summary, the RCTs on active and passive immunization agents consistently show an effect on amyloid clearance, and several but not all phase II RCTs show promising effects on cognition.
5.2. Drugs to Reduce Aβ Generation from APP
BACE1 (β-secretase) initiates the amyloidogenic pathway. Pioglitazone and rosiglitazone are thiazolidinediones and drugs commonly used to treat type II diabetes. They happen to act as BACE1 inhibitors through stimulating the nuclear peroxisome proliferator-activated receptor γ (PPARγ). Activation of PPARγ receptors, in turn, can suppress expression of BACE1 and APP and can promote APP degradation by increasing its ubiquitination [62]. In addition to their effects on BACE1, therapeutic effects of PPARγ agonists in AD could be caused by their effect on insulin action. Both rosiglitazone and pioglitazone increase peripheral insulin sensitivity and reduce concentrations of insulin. Insulin, in turn, competes with Aβ for degradation by the insulin-degrading enzyme [62].
There are only few phase III RCTs, which likely reflects the difficulty in development of BACE1 targeting agents. BACE1 has many substrates including several with physiologically important functions such as neuregulin-1 that is involved in myelination, and drugs must cross the blood-brain barrier in order to modulate BACE1 function. Pioglitazone can cross the blood-brain barrier although whether rosiglitazone can reach the CNS in human beings is unclear [62]. Out of the RCTs that have explored the effects of pioglitazone and rosiglitazone on cognition in patients with AD or MCI (NCT00982202, NCT00736996, NCT00550420, NCT00428090, NCT00348309, NCT00242593, NCT00265148, NCT00348140, NCT00334568, and NCT00490568), only three (NCT00982202, NCT00428090, and NCT00265148) have reported results to date, and these were negative [63]. Currently, several new β-secretase inhibitors are under investigation. Of these, CTS-21166, an orally administered compound, was well tolerated and reduced plasma Aβ concentrations in mice [64] and has proceeded to phase I clinical testing [65].
Development of drugs targeting γ-secretase, the enzyme responsible for the final step in Aβ generation, presents challenges similar to those for β-secretase inhibitors as γ-secretase is one of the main complexes involved in intramembranous cleavage of several proteins, including APP, Notch receptor, and various neuronal substrates [66]. As a consequence, adverse effects of γ-secretase inhibitors include hematological and gastrointestinal toxicity, skin reactions, and changes to hair color, mainly caused by inhibition of the Notch signaling pathway, which is involved in cell differentiation.
Phase III trials for the Notch-inhibiting drug semagacestat failed. Preliminary findings showed that semagacestat not only failed to slow disease progression, but also was associated with worsening of clinical measures of cognition and the ability to perform activities of daily living and a higher incidence of skin cancer in the treatment group than the placebo group. However, several Notch-sparing γ-secretase inhibitors (second-generation inhibitors) are currently under development: begacestat was tested in a phase I RCT (NCT00959881) and BMS-708163 in two phase II RCTs in patients with prodromal or mild-to-moderate AD (NCT00810147 and NCT00890890). Begacestat reduced Aβ concentrations in the plasma (with delayed rebound) [67] but did not substantially affect CSF Aβ 1–40, whereas BMS-708163 promoted a dose-dependent decrease of Aβ 1–40 in the CSF [68]. Results from animal studies testing PF-3084014 showed decreases in Aβ in the plasma, CSF, and brain, without a rebound effect on plasma Aβ [69]. In a subsequent small phase I study, PF-3084014 promoted a dose-dependent reduction in plasma Aβ concentrations although effects on CSF concentrations were small [70]. NIC5–15, a naturally occurring monosaccharide found in many foods, can act as a Notch-sparing γ-secretase inhibitor and insulin sensitizer (i.e., it increases the sensitivity of the tissue to insulin). It is currently being tested in patients with AD in a phase II study (NCT00470418).
γ-secretase modulators can selectively block APP proteolysis without Notch-based adverse effects. A subset of nonsteroidal anti-inflammatory drugs (NSAIDs), including ibuprofen, indomethacin, and sulindac sulfide, bind to APP and act as γ-secretase modulators, decreasing Aβ 1–40 and Aβ 1–42 production, with increased generation of Aβ 1–38 fragments. Among these compounds, known as selective β-amyloid-lowering agents (SALAs), tarenflurbil was tested in phase III RCTs in patients with mild AD but did not show clinical effects [71] possibly due to low γ-secretase modulator potency, poor CNS penetration, or inhibition of microglia-mediated Aβ clearance by residual NSAID activity. Another γ-secretase modulator, CHF-5074, reduced Aβ brain load and improved behavioral deficits in animals [72] and has reached phase II clinical testing (NCT01303744 and NCT01421056).
Upregulation of α-secretase activity, leading to non-amyloidogenic cleavage of APP, can decrease Aβ formation and increase production of a potentially neuroprotective soluble domain (sAPPα) [73]. Several drugs can stimulate α-secretase (agonists of muscarinic, glutamate, and serotonin receptors; statins; oestrogens; testosterone; protein kinase C activators) and have been tested in clinical trials, but no conclusive results are available yet [74]. These α-secretase modulators include Exebryl-1, which modulates β- and α-secretase activity causing substantial reduction of Aβ formation and accumulation in the mouse brain with memory improvements (a phase I RCT was approved in 2008) [75], Etazolate (EHT-0202), a selective GABAA receptor modulator that stimulates neuronal α-secretase and increases sAPPα production [76] and has been recently tested in a phase II RCT in patients with mild-to-moderate AD (NCT00880412) [77], and Bryostatin-1, a macrocyclic lactone that can stimulate α-secretase by activating protein kinase C and promoting sAPPα secretion [78] reducing brain Aβ 1–40 and Aβ 1–42 and improving behavioral outcomes in mouse models of AD [78] (phase II study in process (NCT00606164)).
5.3. Drugs to Prevent Aβ Aggregation and Destabilize Aβ Oligomers
Compounds that inhibit Aβ aggregation or destabilize Aβ oligomeric species can act twofold: (a) either they bind to Aβ monomers thereby preventing oligomerization and allowing Aβ elimination, or (b) they react with Aβ oligomers thereby neutralizing their toxicity and promoting their clearance. They are chemically heterogeneous and also here the challenge is to develop agents that can cross the blood brain barrier and have low toxicity.
The first generation of nonpeptidic antiaggregates failed to fulfill these criteria. Tramiprosate (3APS), which maintains Aβ in the nonfibrillar state by binding to soluble form, showed negative results in the Alphase study, a phase III RCT [79] although previous experimental and phase II trials had been promising [80]. Although there are several possible reasons for this failure, including variability among study sites, differences in the treatment and control groups because of the concomitant treatment with cognitive-enhancing drugs, and low CNS bioavailability of the drug, a European phase III RCT with tramiprosate was terminated as a consequence of the negative findings.
Clioquinol (PBT1) inhibits Aβ aggregation through interfering with interactions between Aβ, copper, and zinc. Studies in Tg2576 mice and human volunteers showed that CQ entry into the brain is limited although upon brain entry it binds to amyloid plaques [81]. PBT1 showed positive results in phase II RCTs but further phase II/III studies were halted due to manufacturing toxicity issues [82]. The second-generation inhibitor, PBT2, has a greater blood-brain barrier permeability than does clioquinol, and animal experiments showed that PBT2 prevents Aβ oligomerization, promotes Aβ oligomer clearance, reduces soluble and insoluble brain Aβ, decreases plaque burden, and has positive effects on cognition [82]. A 12-week, phase II RCT in patients with mild AD, was consistent with these findings, PBT2 reduced Aβ 1–42 CSF concentrations and improved executive function [83]. Scyllo-inositol (ELND-005) is an orally administered stereoisomer of inositol that can cross the blood-brain barrier using inositol transporters. By binding to Aβ, it modulates its misfolding, inhibits its aggregation and stimulates dissociation of aggregates. It was successful in animal studies, reducing brain concentrations of soluble and insoluble Aβ 1–40 and Aβ 1–42, plaque burden, synaptic loss, and glial inflammatory reaction and significantly improving spatial memory function [84]. It is currently being tested in phase II RCTs (NCT00568776 and NCT00934050). However, because of serious adverse events among patients in the two high-dose groups (1000 mg and 2000 mg twice daily), these doses have been removed from the RCT, and the study continues restricted to patients who are assigned the lower dose (250 mg twice daily) and placebo. Epigallocatechin-3-gallate (EGCg), a polyphenol from green tea, induces α-secretase and prevents Aβ aggregation in animals by directly binding to the unfolded peptide [85]. In addition, it modulates signal transduction pathways, expression of genes regulating cell survival and apoptosis, and mitochondrial function [85]. It is currently being tested in a phase II/III RCT in patients with early AD.
5.4. Drugs to Target Tau Protein
Tau is a cytoplasmatic protein that binds to tubulin during its polymerisation, stabilising microtubules. In AD, tau is abnormally phosphorylated, resulting in the generation of aggregates (neurofibrillary tangles) toxic to neurons. The hypothesis that tau pathology causes AD has been the main competitor of the amyloid hypothesis [86]. However, only one tau-directed compound (valproate; valproic acid) has so far reached phase III RCT, with disappointing results because there were no effects on cognition and functional status [87].
There are two main therapeutic approaches to target the tau protein: modulation of tau phosphorylation with inhibitors of tau-phosphorylating kinases and compounds that inhibit tau aggregation and/or promoting aggregate disassembly. The first approach is based on the observation that tau hyperphosphorylation and neurofibrillary tangle formation can be promoted by imbalanced activity of protein kinases (glycogen-synthase-kinase-3 (GSK3) and p70-S6-kinase) and the phosphatase PP2A [88]. GSK3 deregulation might have a role in AD pathogenesis because GSK3 is involved in tau and amyloid processing, cellular signaling, and gene transcription [88].
Both lithium and valproate, well known for the treatment of psychiatric disorders, inhibit GSK3, to reduce tau phosphorylation and prevent or reverse aspects of tauopathy in animal models [89]. Both drugs can also be neuroprotective by upregulating the antiapoptotic factor BCL2, inducing neurotrophic factors, and hindering Aβ toxicity [89]. However, a small RCT with lithium (10 weeks, including a 6-week titration phase) in patients with mild AD did not show any cognitive benefit or any change in CSF biomarkers, including phosphorylated tau, total tau, and Aβ 1–42 [90].
The AD Cooperative Study (ADCS) of valproate was designed to determine whether chronic valproate treatment could delay the onset of behavioral symptoms in outpatients with mild-to-moderate AD; a secondary aim was to test whether valproate can delay cognitive and functional decline. No effects on cognition and functional status were reported, but incidence of agitation and psychosis seemed to be reduced [89].
Several GSK3 inhibitors are under development. NP-031112 (NP-12) is a thiadiazolidinone-derived compound, a non-ATP competitive inhibitor of GSK3, which can reduce brain concentrations of phosphorylated tau and amyloid deposition and prevent neuronal death and cognitive deficits in animals [91]. This drug has been tested in patients with AD in a phase II RCT (NCT00948259); no results have yet been published.
Methylthioninium chloride (methylene blue), a widely used histology dye, acts as a tau antiaggregate [92]. This compound also has antioxidant properties, enhances mitochondrial function [93], and was effective, alone and in combination with rivastigmine, in reversing learning deficits and hyoscine-induced memory impairments in animals [94]. Different doses of methylthioninium chloride (up to 100 mg) were tested in a phase II study in patients with moderate AD. The group given the 60 mg dose had improved cognitive function and, after 1 year, evidence of slower disease progression compared with placebo [95]. The ineffectiveness in the group on the 100 mg dose was attributed to drug formulation defects, limiting release. A new formulation (leuco-methylthioninium), with a higher bioavailability, was recently announced [96], and phase III RCTs are needed to confirm its safety and clinical efficacy.
Davunetide (AL-108, NAP), an intranasally administered, eight-aminoacid peptide fragment derived from the activity-dependent neuroprotective protein, and AL-208, an intravenous formulation of Davunetide, are being developed. Davunetide has been tested in animal models of AD and tauopathy, and its neuroprotective activity includes regulation of microtubule dynamics, as well as inhibition of tau hyperphosphorylation and protection against Aβ toxicity [97, 98]. Davunetide was studied in patients with amnestic mild cognitive impairment in a 12-week, phase II RCT and was safe and well tolerated and had positive effects on cognition [99], although confirmatory studies are needed.
Nicotinamide is the biologically active form of niacin (vitamin B3) and the precursor of coenzyme NAD+. Orally administered nicotinamide can prevent cognitive deficits in a mouse model of AD and can reduce brain concentrations of a species of phosphorylated tau (Thr231) that inhibits microtubule polymerization [100]. Furthermore, nicotinamide inhibits brain sirtuin deacetylase and upregulates acetyl-α-tubulin, protein p25, and MAP2c; all these interactions are associated with increased microtubule stabilization [100]. Nicotinamide has been used in several clinical studies, including RCTs in patients with neurodegenerative disorders, and is generally safe and well tolerated; a phase II RCT is ongoing in patients with mild-to-moderate AD (NCT00580931).
What do these trials tell us? Sadly, they leave little certainty. Amyloid immunization teaches us that we can massively reduce amyloid burden, but when administered late in the disease, it is not a miracle cure. It may have clinically relevant benefits and it may lead to better outcomes if it is given early in the disease or presymptomatically but we simply do not have data to address these issues.
6. Conclusions
Overall, there is substantial evidence supporting a role of the ACH in AD. However, the available results from RCTs are not in line with previous optimistic predictions of an imminent breakthrough in development of a disease-modifying therapy. To explain the disappointing results of several RCTs, researchers have highlighted various potential issues, both in drug choice and development programs. Table 1 summarizes these and provides possible solutions. Clinical trials need to be organized in those in the very earliest stages of the disease. Whether this can be carried out genetically (e.g., by using E4 homozygotes) or by PIB imaging or some combination of both is not clear. Of course, it could be argued that even persons who show PIB signals are already too far down the disease progression for disease-modifying therapy and that treatment needs to be initiated even before this stage. Certainly, even those with mild AD have profound cell loss. In addition, it would be helpful to perform antiamyloid trials in individuals with APP and PSEN mutations or those with Down's syndrome as they provide the best test of the ACH hypothesis. Biomarker studies should be included in trial designs so that the researchers can form, as clearly as possible, informed opinions as to whether the drug has hit the proposed target.
Table 1.
Issues of RCTs of AD.
| Issue | Possible solution |
|---|---|
| Subjects | |
| Target group selection: patients with AD have various types of neuropathology (i.e., amyloid plaques, NFTs, infarcts, and Lewy bodies) | Criteria for identifying subgroups with more homogeneous biomarker evidence of AD pathology are needed to facilitate RCTs |
| In patients with mild-to-moderate AD, the disease could be too advanced for a disease-modifying effect of a specific drug (e.g., immunotherapy) | RCTs that include patients with early AD might enable detection of disease-modifying effects; investigation into which stage of the AD process a therapeutic strategy is more effective is warranted |
|
| |
| Agents | |
| Choosing the right drug: compounds with positive results in preclinical and early clinical testing failed in large phase III RCTs, with costly losses (e.g., tramiprosate) | Robust proof-of-concept studies should be mandatory Investigators should take into account class efficacy |
| Use of drug-related biomarkers in preclinical and early clinical stages can help to confirm the target engagement and to assure early withdrawal of ineffective drugs | |
| Some RCTs were likely hindered by the inability to reach a therapeutic dosage (e.g., tarenflurbil) or short treatment duration | Optimization of drug dosage and treatment duration based on pharmacokinetics |
| Genetics: polymorphisms (e.g., APOE,) might affect drug response | Personalized therapeutic approach: considering genetic polymorphisms that affect drug response can help to optimize drug dosage (e.g., increased doses for individuals with a rapid metabolism) |
|
| |
| Outcome measurements | |
| Measuring effects: many RCTs are developed according to the design of AChEI RCTs, an approach that has indicated the AChEI symptomatic effect but is not sensitive in detecting the efficacy of disease-modifying drugs, rating scales used may have low sensitivity for changes and/or the drug type assessed and these tools have a subjective component | Development and use of relevant, reliable, multidimensional measures for clinical (cognitive and functional) endpoints are key factors, as well the use of biomarkers (neuroimaging, CSF, or blood molecules) that reliably and quantitatively correlate with disease progression; collection of baseline data (clinical, biomarkers) that can be used as reference to interpret later findings is advisable; for early AD (i.e., mild cognitive impairment), self-rated and observer-rated assessments of activities of daily living, instrumental activities of daily living, and quality of life are recommended |
| Unreliable evaluation of patients by RCT raters | Adequate training and monitoring of RCT raters to maximize homogeneous recruitment of patients, reduce variance, and guarantee a more accurate rating; effective implementation of quality control on data at research sites |
|
| |
| Optimization of resources | |
| Consistency: multicenter RCTs done in several countries can have cultural and linguistic issues with assessment scales (e.g., translation, validation), as well as infrastructure problems (technological disparities between centers) | Multicenter trials should use centers of excellence that are already experienced in RCTs to minimize intersite and intercountry variability |
| Unsuccessful preclinical and clinical studies are often not published leading to repetition of unsuccessful trials or errors | More collaboration between pharmaceutical companies and clinical researchers, with information sharing, can lead to more standardized RCT protocols, reduction of errors, and decreased costs |
However, in addition to implementing new guidelines in preclinical and clinical phases of drug development, several additional issues are key to validate the ACH and successfully develop therapeutic targets. From a molecular point of view, we need a focused effort to fully understand the functions of APP and Aβ and to answer the two key questions: does Aβ in fact influence tau phosphorylation and, if yes, does tau phosphorylation in fact lead to dementia? Second, we need to understand the nature of disease propagation: is permissive templating of Aβ [101, 102] and tau [103] the reason for both the characteristic neuroanatomy of the disease [104] and the reason that the disease seems to become self-propagating once it has started [105, 106]?. Finally, it makes sense to pursue other targets beyond Aβ as there is substantial evidence for additional potential pathways increasing disease susceptibility, among these lipid metabolism and inflammatory processes [107].
Conflict of Interests
The author does not have any actual or potential conflict of interests.
Acknowledgments
This work was supported by grants from the National Institute of Health and the National Institute on Aging, R37-AG15473, P01-AG07232, the Blanchette Hooker Rockefeller Foundation, the Charles S. Robertson Gift from the Banbury Fund, and the Merrill Lynch Foundation. Dr. Reitz was further supported by a Paul B. Beeson Career Development Award (K23AG034550).
References
- 1.About a peculiar disease of the cerebral cortex. By Alois Alzheimer, 1907 (Translated by L. Jarvik and H. Greenson) Alzheimer Disease and Associated Disorders. 1987;1(1):3–8. [PubMed] [Google Scholar]
- 2.Newell KL, Hyman BT, Growdon JH, Hedley-Whyte ET. Application of the National Institute on Aging (NIA)-Reagan Institute criteria for the neuropathological diagnosis of Alzheimer disease. Journal of Neuropathology and Experimental Neurology. 1999;58(11):1147–1155. doi: 10.1097/00005072-199911000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Glenner GG, Wong CW. Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochemical and Biophysical Research Communications. 1984;122(3):1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 4.Goate A, Chartier-Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349(6311):704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 5.Levy-Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269(5226):973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 6.Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375(6534):754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 7.Giasson BI, Lee VMY, Trojanowski JQ. Interactions of amyloidogenic proteins. Neuromolecular Medicine. 2003;4(1-2):49–58. doi: 10.1385/NMM:4:1-2:49. [DOI] [PubMed] [Google Scholar]
- 8.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 9.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6(4):487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 10.Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. β-Amyloid precursor protein (βAPP) as a marker for axonal injury after head injury. Neuroscience Letters. 1993;160(2):139–144. doi: 10.1016/0304-3940(93)90398-5. [DOI] [PubMed] [Google Scholar]
- 11.McKenzie JE, Gentleman SM, Roberts GW, Graham DI, Royston MC. Increased numbers of βAPP-immunoreactive neurones in the entorhinal cortex after head injury. NeuroReport. 1994;6(1):161–164. doi: 10.1097/00001756-199412300-00041. [DOI] [PubMed] [Google Scholar]
- 12.Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. β Amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. Journal of Neurology Neurosurgery and Psychiatry. 1994;57(4):419–425. doi: 10.1136/jnnp.57.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalaria RN, Perry G. Amyloid P component and other acute-phase proteins associated with cerebellar A β-deposits in Alzheimer’s disease. Brain Research. 1993;631(1):151–155. doi: 10.1016/0006-8993(93)91202-4. [DOI] [PubMed] [Google Scholar]
- 14.Regland B, Gottfries CG. The role of amyloid β-protein in Alzheimer’s disease. The Lancet. 1992;340(8817):467–469. doi: 10.1016/0140-6736(92)91780-c. [DOI] [PubMed] [Google Scholar]
- 15.Renkawek K, de Jong WW, Merck KB, Frenken CWGM, van Workum FPA, Bosman GJ. Alpha B-crystallin is present in reactive glia in Creutzfeldt-Jakob disease. Acta Neuropathologica. 1992;83(3):324–327. doi: 10.1007/BF00296796. [DOI] [PubMed] [Google Scholar]
- 16.Wallace WC, Bragin V, Robakis NK, et al. Increased biosynthesis of Alzheimer amyloid precursor protein the in cerebral cortex of rats with lesions of the nucleus basalis of Meynert. Molecular Brain Research. 1991;10(2):173–178. doi: 10.1016/0169-328x(91)90108-a. [DOI] [PubMed] [Google Scholar]
- 17.Yan R, Blenkowski MJ, Shuck ME, et al. Membrane-anchored aspartyl protease with Alzheimer’s disease β- secretase activity. Nature. 1999;402(6761):533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- 18.Torack RM, Miller JW. Immunoreactive changes resulting from dopaminergic denervation of the dentate gyrus of the rat hippocampal formation. Neuroscience Letters. 1994;169(1-2):9–12. doi: 10.1016/0304-3940(94)90344-1. [DOI] [PubMed] [Google Scholar]
- 19.Armstrong RA, Myers D, Smith CUM. The spatial patterns of plaques and tangles in Alzheimer’s disease do not support the ‘cascade hypothesis’. Dementia. 1993;4(1):16–20. doi: 10.1159/000107291. [DOI] [PubMed] [Google Scholar]
- 20.Duyckaerts C. Looking for the link between plaques and tangles. Neurobiology of Aging. 2004;25(6):735–739. doi: 10.1016/j.neurobiolaging.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 21.Armstrong RA. Plaques and tangles and the pathogenesis of Alzheimer’s disease. Folia Neuropathologica. 2006;44(1):1–11. [PubMed] [Google Scholar]
- 22.Busciglio J, Lorenzo A, Yeh J, Yankner BA. β-Amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron. 1995;14(4):879–888. doi: 10.1016/0896-6273(95)90232-5. [DOI] [PubMed] [Google Scholar]
- 23.Perez M, Cuadros R, Benitez MJ, Jimenez JS. Interaction of Alzheimer’s disease amyloid β peptide fragment 25–35 with tau protein, and with a tau peptide containing the microtubule binding domain. Journal of Alzheimer’s Disease. 2004;6(5):461–467. doi: 10.3233/jad-2004-6501. [DOI] [PubMed] [Google Scholar]
- 24.Roberson ED, Halabisky B, Yoo JW, et al. Amyloid-β/fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. Journal of Neuroscience. 2011;31(2):700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roberson ED, Scearce-Levie K, Palop JJ, et al. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316(5825):750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 26.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 27.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 28.Slooter AJC, Cruts M, Kalmijn S, et al. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: the rotterdam study. Archives of Neurology. 1998;55(7):964–968. doi: 10.1001/archneur.55.7.964. [DOI] [PubMed] [Google Scholar]
- 29.Holtzman DM, Bales KR, Tenkova T, et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(6):2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63(3):287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rebeck GW, Reiter JS, Strickland DK, Hyman BT. Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron. 1993;11(4):575–580. doi: 10.1016/0896-6273(93)90070-8. [DOI] [PubMed] [Google Scholar]
- 32.Schmechel DE, Saunders AM, Strittmatter WJ, et al. Increased amyloid β-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(20):9649–9653. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meng Y, Lee JH, Cheng R, St George-Hyslop P, Mayeux R, Farrer LA. Association between SORL1 and Alzheimer’s disease in a genome-wide study. NeuroReport. 2007;18(17):1761–1764. doi: 10.1097/WNR.0b013e3282f13e7a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rogaeva E, Meng Y, Lee JH, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nature Genetics. 2007;39(2):168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JH, Barral S, Reitz C. The neuronal sortilin-related receptor gene SORL1 and late-onset Alzheimer’s disease. Current Neurology and Neuroscience Reports. 2008;8(5):384–391. doi: 10.1007/s11910-008-0060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee JH, Cheng R, Schupf N, et al. The association between genetic variants in SORL1 and Alzheimer disease in an urban, multiethnic, community-based cohort. Archives of Neurology. 2007;64(4):501–506. doi: 10.1001/archneur.64.4.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reitz C, Cheng R, Rogaeva E, et al. Meta-analysis of the association between variants in SORL1 and Alzheimer disease. Archives of Neurology. 2011;68(1):99–106. doi: 10.1001/archneurol.2010.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lane RF, Raines SM, Steele JW, et al. Diabetes-associated SorCS1 regulates Alzheimer’s amyloid-β metabolism: evidence for involvement of SorL1 and the retromer complex. Journal of Neuroscience. 2010;30(39):13110–13115. doi: 10.1523/JNEUROSCI.3872-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reitz C, Tokuhiro S, Clark LN, et al. SORCS1 alters amyloid precursor protein processing and variants may increase Alzheimer’s disease risk. Annals of Neurology. 2011;69(1):47–64. doi: 10.1002/ana.22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nature Genetics. 2009;41(10):1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nature Genetics. 2009;41(10):1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 42.Seshadri S, Fitzpatrick AL, Ikram MA, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. Journal of the American Medical Association. 2010;303(18):1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DeMattos RB, Cirrito JR, Parsadanian M, et al. ApoE and clusterin cooperatively suppress Aβ levels and deposition: evidence that ApoE regulates extracellular Aβ metabolism in vivo . Neuron. 2004;41(2):193–202. doi: 10.1016/s0896-6273(03)00850-x. [DOI] [PubMed] [Google Scholar]
- 44.Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nature Genetics. 2011;43(5):429–436. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wyss-Coray T, Yan F, Lin AHT, et al. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(16):10837–10842. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Treusch S, Hamamichi S, Goodman JL, et al. Functional links between Aβ troxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science. 2011;334(6060):1241–1245. doi: 10.1126/science.1213210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Alzheimer’s disease: clinical trials and drug development. The Lancet Neurology. 2010;9(7):702–716. doi: 10.1016/S1474-4422(10)70119-8. [DOI] [PubMed] [Google Scholar]
- 48.Gilman S, Koller M, Black RS, et al. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64(9):1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 49.Vellas B, Black R, Thal LJ, et al. Long-term follow-up of patients immunized with AN1792: reduced functional decline in antibody responders. Current Alzheimer Research. 2009;6(2):144–151. doi: 10.2174/156720509787602852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winblad B, Minthon L, Floesser A, et al. Results of the first-in-man study with the active Aβ immunotherapy CAD106 in Alzheimer patients. Alzheimer’s & Dementia. 2009;5(4, supplement 1):P113–P114. [Google Scholar]
- 51.Muhs A, Hickman DT, Pihlgren M, et al. Liposomal vaccines with conformation-specific amyloid peptide antigens define immune response and efficacy in APP transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(23):9810–9815. doi: 10.1073/pnas.0703137104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang CY, Finstad CL, Walfield AM, et al. Site-specific UBITh amyloid-β vaccine for immunotherapy of Alzheimer’s disease. Vaccine. 2007;25(16):3041–3052. doi: 10.1016/j.vaccine.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 53.Schneeberger A, Mandler M, Otava O, Zauner W, Mattner F, Schmidt W. Development of AFFITOPE vaccines for Alzheimer’s Disease (AD)—from concept to clinical testing. Journal of Nutrition, Health and Aging. 2009;13(3):264–267. doi: 10.1007/s12603-009-0070-5. [DOI] [PubMed] [Google Scholar]
- 54.Wilcock DM, Colton CA. Anti-amyloid-β immunotherapy in Alzheimer’s disease: relevance of transgenic mouse studies to clinical trials. Journal of Alzheimer’s Disease. 2008;15(4):555–569. doi: 10.3233/jad-2008-15404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Salloway S, Sperling R, Gilman S, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73(24):2061–2070. doi: 10.1212/WNL.0b013e3181c67808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Siemers ER, Friedrich S, Dean RA, et al. Safety, tolerability and biomarker eff ects of an Aβ monoclonal antibody administered to patients with Alzheimer’s disease. Alzheimer’s & Dementia. 2008;4(4, supplement 2):p. T774. [Google Scholar]
- 57.Bednar M, Zhao Q, Landen JW, Billing CB, Rohrbacher K, Kupiec JW. Safety and pharmacokinetics of the anti-amyloid monoclonal antibody PF-04360365 following a single infusion in patients with mild-to-modertae Alzheimer’s disease: preliminary results. Alzheimer’s & Dementia. 2009;5(4, supplement 1):p. P157. [Google Scholar]
- 58.Thies W, Bleiler L. 2011 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia. 2011;7(2):208–244. doi: 10.1016/j.jalz.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 59.Relkin NR, Szabo P, Adamiak B, et al. 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiology of Aging. 2009;30(11):1728–1736. doi: 10.1016/j.neurobiolaging.2007.12.021. [DOI] [PubMed] [Google Scholar]
- 60.Dodel RC, Du Y, Depboylu C, et al. Intravenous immunoglobulins containing antibodies against β-amyloid for the treatment of Alzheimer’s disease. Journal of Neurology, Neurosurgery and Psychiatry. 2004;75(10):1472–1474. doi: 10.1136/jnnp.2003.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsakanikas D, Shah K, Flores C, Assuras S, Relkin NR. Effects of uninterrupetd intravenous immunoglobulin treatment of Alzheimer’s disease for nine months. Alzheimer’s & Dementia. 2008;4(4, supplement 2):p. T776. [Google Scholar]
- 62.Landreth G, Jiang Q, Mandrekar S, Heneka M. PPARγ agonists as therapeutics for the treatment of Alzheimer’s disease. Neurotherapeutics. 2008;5(3):481–489. doi: 10.1016/j.nurt.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gold M, Alderton C, Zvartau-Hind M, et al. Rosiglitazone monotherapy in mild-to-moderate alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dementia and Geriatric Cognitive Disorders. 2010;30(2):131–146. doi: 10.1159/000318845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tang J, Ghosh A. Treating transgenic Alzheimer mice with a β-secretase inhibitor, what have we learned? Aging. 2011;3(1):14–16. doi: 10.18632/aging.100267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tang JJN. Beta-secretase as target for amyloid-reduction therapy. Alzheimers Dement. 2009;5(4, supplement 1):p. P74. [Google Scholar]
- 66.Tomita T. Secretase inhibitors and modulators for Alzheimer’s disease treatment. Expert Review of Neurotherapeutics. 2009;9(5):661–679. doi: 10.1586/ern.09.24. [DOI] [PubMed] [Google Scholar]
- 67.Jacobsen S, Comery TA, Kreft A, et al. GSI-953 is a potent APP-selective γ-secretase inhibitor for the treatment of Alzheimer’s disease. Alzheimer’s & Dementia. 2009;5(4, supplement 1):p. P139. [Google Scholar]
- 68.Imbimbo BP. Alzheimer’s disease: γ-secretase inhibitors. Drug Discovery Today. 2008;5(3):169–175. [Google Scholar]
- 69.Lanz TA, Wood KM, Richter KEG, et al. Pharmacodynamics and pharmacokinetics of the γ-secretase inhibitor PF-3084014. Journal of Pharmacology and Experimental Therapeutics. 2010;334(1):269–277. doi: 10.1124/jpet.110.167379. [DOI] [PubMed] [Google Scholar]
- 70.Soares H, Raha N, Sikpi M, et al. Aβ variability and effect of γ secretase inhibition on cerebrospinal fluid levels of Aβ in healthy volunteers. Alzheimer’s & Dementia. 2009;5(4, supplement 1):P252–P253. [Google Scholar]
- 71.Green RC, Schneider LS, Amato DA, et al. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. Journal of the American Medical Association. 2009;302(23):2557–2564. doi: 10.1001/jama.2009.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Imbimbo BP, Hutter-Paier B, Villetti G, et al. CHF5074, a novel γ-secretase modulator, attenuates brain β-amyloid pathology and learning deficit in a mouse model of Alzheimer’s disease. British Journal of Pharmacology. 2009;156(6):982–993. doi: 10.1111/j.1476-5381.2008.00097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van Marum RJ. Current and future therapy in Alzheimer’s disease. Fundamental and Clinical Pharmacology. 2008;22(3):265–274. doi: 10.1111/j.1472-8206.2008.00578.x. [DOI] [PubMed] [Google Scholar]
- 74.Griffiths HH, Morten IJ, Hooper NM. Emerging and potential therapies for Alzheimer’s disease. Expert Opinion on Therapeutic Targets. 2008;12(6):693–704. doi: 10.1517/14728222.12.6.693. [DOI] [PubMed] [Google Scholar]
- 75.Snow AD, Cummings J, Lake T, et al. Exebryl-1: a novel small molecule currently in human clinical trials as a disease-modifying drug for the treatment of Alzheimer’s disease. Alzheimer’s & Dementia. 2009;5(4, supplement 1):p. P418. [Google Scholar]
- 76.Marcade M, Bourdin J, Loiseau N, et al. Etazolate, a neuroprotective drug linking GABAA receptor pharmacology to amyloid precursor protein processing. Journal of Neurochemistry. 2008;106(1):392–404. doi: 10.1111/j.1471-4159.2008.05396.x. [DOI] [PubMed] [Google Scholar]
- 77.Desire L, Marcade M, Peillon H, Drouin D, Sol O, Pando M. Clinical trials of EHT 0202, a neuroprotective and procognitive alpha-secretase stimulator for Alzheimer’s disease. Alzheimer’s & Dementia. 2009;5(4, supplement 1):P255–P256. [Google Scholar]
- 78.Etcheberrigaray R, Tan M, Dewachtert I, et al. Therapeutic effects of PKC activators in Alzheimer’s disease transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(30):11141–11146. doi: 10.1073/pnas.0403921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aisen PS, Gauthier S, Ferris S. A phase III, placebo-controlled, double-blind, randomized trial of tramiprosate in the clinical management of patients with mild-to-moderate Alzheimer’s disease (the Alphase study). In: Proceedings of the 61st American Academy of Neurology Annual Meeting; May 2009; Seattle, Wash, USA. S32003. [Google Scholar]
- 80.Aisen PS, Saumier D, Briand R, et al. A phase II study targeting amyloid-β with 3APS in mild-to-moderate Alzheimer disease. Neurology. 2006;67(10):1757–1763. doi: 10.1212/01.wnl.0000244346.08950.64. [DOI] [PubMed] [Google Scholar]
- 81.Opazo C, Luza S, Villemagne VL, et al. Radioiodinated clioquinol as a biomarker for β-amyloid: Zn2+ complexes in Alzheimer’s disease. Aging Cell. 2006;5(1):69–79. doi: 10.1111/j.1474-9726.2006.00196.x. [DOI] [PubMed] [Google Scholar]
- 82.Adlard PA, Cherny RA, Finkelstein DI, et al. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Aβ . Neuron. 2008;59(1):43–55. doi: 10.1016/j.neuron.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 83.Lannfelt L, Blennow K, Zetterberg H, et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Aβ as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. The Lancet Neurology. 2008;7(9):779–786. doi: 10.1016/S1474-4422(08)70167-4. [DOI] [PubMed] [Google Scholar]
- 84.McLaurin J, Kierstead ME, Brown ME, et al. Cyclohexanehexol inhibitors of Aβ aggregation prevent and reverse Alzheimer phenotype in a mouse model. Nature Medicine. 2006;12(7):801–808. doi: 10.1038/nm1423. [DOI] [PubMed] [Google Scholar]
- 85.Mandel SA, Amit T, Kalfon L, Reznichenko L, Weinreb O, Youdim MBH. Cell signaling pathways and iron chelation in the neurorestorative activity of green tea polyphenols: special reference to epigallocatechin gallate (EGCG) Journal of Alzheimer’s Disease. 2008;15(2):211–222. doi: 10.3233/jad-2008-15207. [DOI] [PubMed] [Google Scholar]
- 86.Mudher A, Lovestone S. Alzheimer’s disease—do tauists and baptists finally shake hands? Trends in Neurosciences. 2002;25(1):22–26. doi: 10.1016/s0166-2236(00)02031-2. [DOI] [PubMed] [Google Scholar]
- 87.Tarriot P, Aisen PS, Cummings J, et al. The ADCS valproate neuroprotection trial: primary efficacy and safety results. Alzheimer’s & Dementia. 2009;5(4, supplement 1):P84–P85. [Google Scholar]
- 88.Pei JJ, Sjogren M, Winblad B. Neurofibrillary degeneration in Alzheimer’s disease: from molecular mechanisms to identification of drug targets. Current Opinion in Psychiatry. 2008;21(6):555–561. doi: 10.1097/YCO.0b013e328314b78b. [DOI] [PubMed] [Google Scholar]
- 89.Tariot PN, Aisen PS. Can lithium or valproate untie tangles in Alzheimer’s disease? Journal of Clinical Psychiatry. 2009;70(6):919–921. doi: 10.4088/jcp.09com05331. [DOI] [PubMed] [Google Scholar]
- 90.Hampel H, Ewers M, Burger K, et al. Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. Journal of Clinical Psychiatry. 2009;70(6):922–931. [PubMed] [Google Scholar]
- 91.Sereno L, Coma M, Rodriguez M, et al. A novel GSK-3β inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo . Neurobiology of Disease. 2009;35(3):359–367. doi: 10.1016/j.nbd.2009.05.025. [DOI] [PubMed] [Google Scholar]
- 92.Wischik CM, Edwards PC, Lai RYK, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(20):11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Atamna H, Nguyen A, Schultz C, et al. Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB Journal. 2008;22(3):703–712. doi: 10.1096/fj.07-9610com. [DOI] [PubMed] [Google Scholar]
- 94.Deiana S, Harrington CR, Wischik CM, Riedel G. Methylthioninium chloride reverses cognitive deficits induced by scopolamine: comparison with rivastigmine. Psychopharmacology. 2009;202(1-3):53–65. doi: 10.1007/s00213-008-1394-2. [DOI] [PubMed] [Google Scholar]
- 95.Gura T. Hope in Alzheimer’s fight emerges from unexpected places. Nature Medicine. 2008;14(9):p. 894. doi: 10.1038/nm0908-894. [DOI] [PubMed] [Google Scholar]
- 96.Wischik C. Rember: issues in design of a phase 3 disease modifying clinical trial of tau aggregation inhibitor therapy in Alzheimer’s disease. Alzheimer’s & Dementia. 2009;5(4, supplement 1):p. P74. [Google Scholar]
- 97.Gozes I, Divinski I, Piltzer I. NAP and D-SAL: neuroprotection against the β amyloid peptide (1–42) BMC Neuroscience. 2008;9(supplement 3, article S3) doi: 10.1186/1471-2202-9-S3-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shiryaev N, Jouroukhin Y, Giladi E, et al. NAP protects memory, increases soluble tau and reduces tau hyperphosphorylation in a tauopathy model. Neurobiology of Disease. 2009;34(2):381–388. doi: 10.1016/j.nbd.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 99.Schmechel DE, Gerard G, Vatakis NG, et al. A phase 2, double-blind, placebo-controlled study to evaluate the safety, tolerability, and effect on cognitive function of AL-108 after 12 weeks of intranasal administration in subjects with mild cognitive impairment. Alzheimer’s & Dementia. 2008;4(4, supplement 2):p. T483. doi: 10.1159/000348347. [DOI] [PubMed] [Google Scholar]
- 100.Green KN, Steffan JS, Martinez-Coria H, et al. Nicotinamide restores cognition in Alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. Journal of Neuroscience. 2008;28(45):11500–11510. doi: 10.1523/JNEUROSCI.3203-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hardy J. Expression of normal sequence pathogenic proteins for neurodegenerative disease contributes to disease risk: “permissive templating” as a general mechanism underlying neurodegeneration. Biochemical Society Transactions. 2005;33(4):578–581. doi: 10.1042/BST0330578. [DOI] [PubMed] [Google Scholar]
- 102.Meyer-Luehmann M, Coomaraswamy J, Bolmont T, et al. Exogenous induction of cerebral β-amyloidogenesis is governed by agent and host. Science. 2006;313(5794):1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- 103.Frost B, Ollesch J, Wille H, Diamond MI. Conformational diversity of wild-type tau fibrils specified by templated conformation change. Journal of Biological Chemistry. 2009;284(6):3546–3551. doi: 10.1074/jbc.M805627200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathology. 1991;1(3):213–216. doi: 10.1111/j.1750-3639.1991.tb00661.x. [DOI] [PubMed] [Google Scholar]
- 105.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Aβ immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43(3):321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 106.Santacruz K, Lewis J, Spires T, et al. Medicine: tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309(5733):476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Noble W, Planel E, Zehr C, et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo . Proceedings of the National Academy of Sciences of the United States of America. 2005;102(19):6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]