

Abstract
The receptor tyrosine kinase MuSK is indispensable for nerve-muscle synapse formation and maintenance. MuSK is necessary for prepatterning of the endplate zone anlage and as a signaling receptor for agrin-mediated postsynaptic differentiation. MuSK-associated proteins such as Dok7, LRP4, and Wnt11r are involved in these early events in neuromuscular junction formation. However, the mechanisms regulating synapse stability are poorly understood. Here we examine a novel role for the extracellular matrix protein biglycan in synapse stability. Synaptic development in fetal and early postnatal biglycan null (bgn−/o) muscle is indistinguishable from wild-type controls. However, by 5 weeks after birth, nerve-muscle synapses in bgn−/o mice are abnormal as judged by the presence of perijunctional folds, increased segmentation, and focal misalignment of acetylcholinesterase and AChRs. These observations indicate that previously occupied presynaptic and postsynaptic territory has been vacated. Biglycan binds MuSK and the levels of this receptor tyrosine kinase are selectively reduced at bgn−/o synapses. In bgn−/o myotubes, the initial stages of agrin-induced MuSK phosphorylation and AChR clustering are normal, but the AChR clusters are unstable. This stability defect can be substantially rescued by the addition of purified biglycan. Together, these results indicate that biglycan is an extracellular ligand for MuSK that is important for synapse stability.
Introduction
Rapid and efficient communication in the nervous system requires tightly regulated synaptic differentiation and stabilization. At the neuromuscular junction (NMJ), these processes result in the high-density clustering of AChRs in the postsynaptic membrane and the elaboration of a stable, well defined endplate architecture (Sanes and Lichtman, 2001; Burden, 2002). Several elements in the pathways mediating the formation of this synapse have been identified. However, the molecular mechanisms that underlie the maintenance and stabilization of this synapse are poorly understood.
The receptor tyrosine kinase MuSK is the core signaling element in nerve-muscle synapse formation and maintenance (Glass et al., 1996b; Burden, 2002; Kong et al., 2004; Kummer et al., 2006). Several proteins interact with MuSK and function in the early stages of synapse development including LRP4, Dok7, and Wnt11r (Weatherbee et al., 2006; Kim et al., 2008; Zhang et al., 2008; Wu et al., 2010).
The development of the neuromuscular junction spans ∼E14 to ∼P14 (Sanes and Lichtman, 2001; Witzemann, 2006). Plaque-like specializations form before the axon arrival and are innervated shortly thereafter. By P14 all the hallmarks of the mature NMJs are present. Once formed, the NMJ is remarkably stable, with the structure of individual endplates remaining essentially unchanged for at least 4 months (Lichtman et al., 1987; Balice-Gordon and Lichtman, 1990).
Biglycan is an extracellular matrix protein that is selectively enriched in postsynaptic membranes from Torpedo electric organ and is highly expressed in developing and regenerating muscle (Casar et al., 2004; Lechner et al., 2006). Two forms of biglycan are expressed in muscle, a proteoglycan that bears chondroitin or dermatan sulfate side chains and a nonglycanated version that lacks these side chains. Both forms have conventional N-linked carbohydrates. The chondroitin sulfate side chains of biglycan proteoglycan mediate its binding to the polypeptide core of α-dystroglycan (Bowe et al., 2000). The polypeptide core mediates binding to α- and γ-sarcoglycans, which are key members of the dystrophin/utrophin protein complex (Holt and Campbell, 1998; Ozawa et al., 2005; Rafii et al., 2006).
Biglycan regulates the localization of utrophin and other components of the dystrophin/utrophin protein complex at the extrasynaptic sarcolemma in immature muscle. Recombinant nonglycanated biglycan upregulates the expression of utrophin and associated proteins at the sarcolemma and improves muscle health and function in dystrophic mice (Amenta et al., 2011) Biglycan null mice also display delayed muscle regeneration and late-onset (≥6 months) osteoporosis and connective tissue defects (Ameye and Young, 2002; Casar et al., 2004).
Here we have examined the role of biglycan at the synapse. We show that synapses in biglycan null mice develop normally until 2 weeks of age but then become fragmented over the subsequent 3 weeks. AChR and AChE are focally misaligned at synapses in mature mice, suggesting that previously existing synaptic territory has been vacated. Biglycan binds MuSK and the levels of this RTK are selectively reduced at biglycan null synapses. The maturation of agrin-induced AChR clusters is defective in cultured biglycan null myotubes and can be rescued by recombinant biglycan. Together, these findings indicate that biglycan is a ligand for MuSK that is important for maintaining synapse stability.
Materials and Methods
Antibodies
Anti-MuSK antisera (29–31) directed against the N-terminal half of the MuSK cytoplasmic domain was used for immunoprecipitation; an affinity purified fraction of this antibody was used for immunostaining (Hopf and Hoch, 1998). Antisera raised against the C-terminal 20 aa of MuSK (Watty et al., 2000) and cytoplasmic amino acids 548–561 of MuSK (Ganju et al., 1995) also was used for immunostaining (generously provided by S. Burden and M. Ruegg, respectively). D. Glass generously provided that anti-MuSK used for immunoblotting (Glass et al., 1996a). Anti-rapsyn was a gift from J. Cohen; anti-α-dystrobrevin-1 and -2, and anti-α-, β1-, and β2-syntrophin were generously provided by S. Froehner. Other antibodies included: anti-phosphotyrosine mAb 4G10 (Millipore); anti-FLAG M2-Cy3 (Sigma); anti-TrkA and anti-α-dystroglycan (Millipore); anti-utrophin, anti-β-dystroglycan, and anti-α-, β-, γ-sarcoglycan (Vector Laboratories); anti-nNOS (ImmunoStar); anti-laminin α2 (Alexis Biochemicals), anti-collagen VI (Rockland Immunochemicals); anti-neurofilament SMI-31 (Sternberger Monoclonals); anti-neurofilament (Millipore Bioscience Research Reagents); SV2 (Developmental Studies Hybridoma Bank); anti-synaptophysin (Zymed). The following secondary antibodies were used: Alexa 488-goat anti-mouse IgG (Invitrogen) and Cy3-goat anti-rabbit IgG (Jackson ImmunoResearch). Rhodamine-α-bungarotoxin (Invitrogen) was used to label acetylcholine receptors. AChE was detected with Oregon Green-conjugated fasciculin 2 (Peng et al., 1999). The monoclonal anti-biglycan antibody was generated by immunizing biglycan null mice with recombinant biglycan and then selected based upon its ability to stain wild-type, but not biglycan null muscle (Mercado et al., 2006).
Recombinant biglycan
Purified, recombinant biglycan and decorin were produced by vaccinia virus infection of osteosarcoma cells (Krishnan et al., 1999) or in a 293-EBNA cell line as described previously (Mercado et al., 2006). The nonglycanated forms, which lack GAG side chains, were used for all experiments.
Biglycan null mice
These animals were created as described previously (Xu et al., 1998) and were maintained on a C57BL/6 background. Heterozygous female biglycan null mice (biglycan is X-linked) were mated with normal male mice. Male progeny were collected and genotyped. For some experiments, age-matched congenic biglycan null mice on a C3H background were used (The Jackson Laboratory). Results were comparable between strains.
Immunohistochemistry
Quadriceps femoris, sternomastoid, or diaphragm muscles from 5-week-old wild-type and biglycan null mice were isolated and flash frozen in liquid nitrogen-cooled isopentane. Immunohistochemistry was performed on 10 μm transverse cryostat sections using the Vector M.O.M. basic kit (Vector Laboratories). Sections were incubated with primary antibody overnight at 4°C and then with secondary antibody for 1 h at room temperature. Sections were mounted in Permafluor (Thermo Electron) and analyzed using confocal laser scanning microscopy (Leica TCS SP2 Acousto-Optical Beam Splitter). When comparing fluorescence levels between wild-type and knock-out sections, images were acquired using identical laser power and gain settings. Images were acquired using Leica LCS acquisition software and imported into Adobe Photoshop.
Quantification of MuSK immunoreactivity was performed using MetaMorph image analysis software (Universal Imaging). A region of interest for each neuromuscular junction analyzed was defined and the mean pixel signal intensity calculated. The mean pixel signal intensity of MuSK was normalized to the mean pixel signal intensity of utrophin (signal intensity for biglycan = 1.00) for each condition. At least 3 junctions per condition were quantified.
Quantitative real-time PCR.
RNA was isolated from P35 wild-type C3H and biglycan null mouse quadriceps femoris muscle using Trizol and treated with DNase I. cDNA was prepared using the Superscript III First-Strand Synthesis System Kit (Invitrogen). Quantitative PCRs were performed using the SYBR-Green method (Invitrogen) on an ABI PRISM 7300 real-time thermocycler (Invitrogen) with ATP synthase levels used for normalization. Data analysis was performed using the standard curve method. Primers used were as follows: MuSK forward: 5′-TGAAGCTGGAAGTGGAGGTTTT-3′; MuSK reverse: 5′-GCAGTAGGGTTACAAAGGAA-3′. ATPSase forward: 5′-TGGGAAAATCGGACT CTTTG-3′; ATPSase reverse: 5′-AGTAACCACCATGGGCTTTG-3′.
Ultrastructural analysis.
Biglycan null and congenic wild-type animals (C3H; 5 weeks old) were perfusion-fixed with 2% formaldehyde and 4% glutaraldehyde in 0.1 m phosphate buffer and postfixed in the same solution overnight. Synaptic regions were located in toluidine blue-stained semithin sections and then thin sections were prepared and viewed on an FEI Morgagni 268 electron microscope. The membrane area within 2 μm of either edge of the bouton was analyzed for extrajunctional folds. The criteria for identification as a fold were communication with the sarcolemma, a minimum depth of 0.5 μm, and the presence of basal lamina throughout their extent.
Whole mounts
Sternomastoid muscles from perfusion-fixed 5-week-old biglycan null and wild-type littermates were dissected in 4% paraformaldehyde and incubated with rh-α-bungarotoxin, antibody SMI-31 and antibody SV2 to label AChRs, neurofilaments and synaptic vesicles, respectively. Flattened stacks of 1 μm optical sections were captured at 63× with 2.5× digital zoom (Leica TCS SP2 Acousto-Optical Beam Splitter with Leica LCS acquisition software). Segments, defined as the number of discrete AChR-rich regions (delimited on all sides by AChR-poor regions; e.g., marked by arrowheads in Fig. 1C), per junction were counted (MetaMorph; Universal Imaging) and results graphed. For AChE localization, hindlimb muscles from 5-week-old biglycan null and wild-type littermates were dissected and costained for AChR with rh-α bungarotoxin and Oregon-green-fasiculin II as described previously (Peng et al., 1999). Diaphragm whole mounts were labeled as described previously (Smith et al., 2001).
Figure 1.
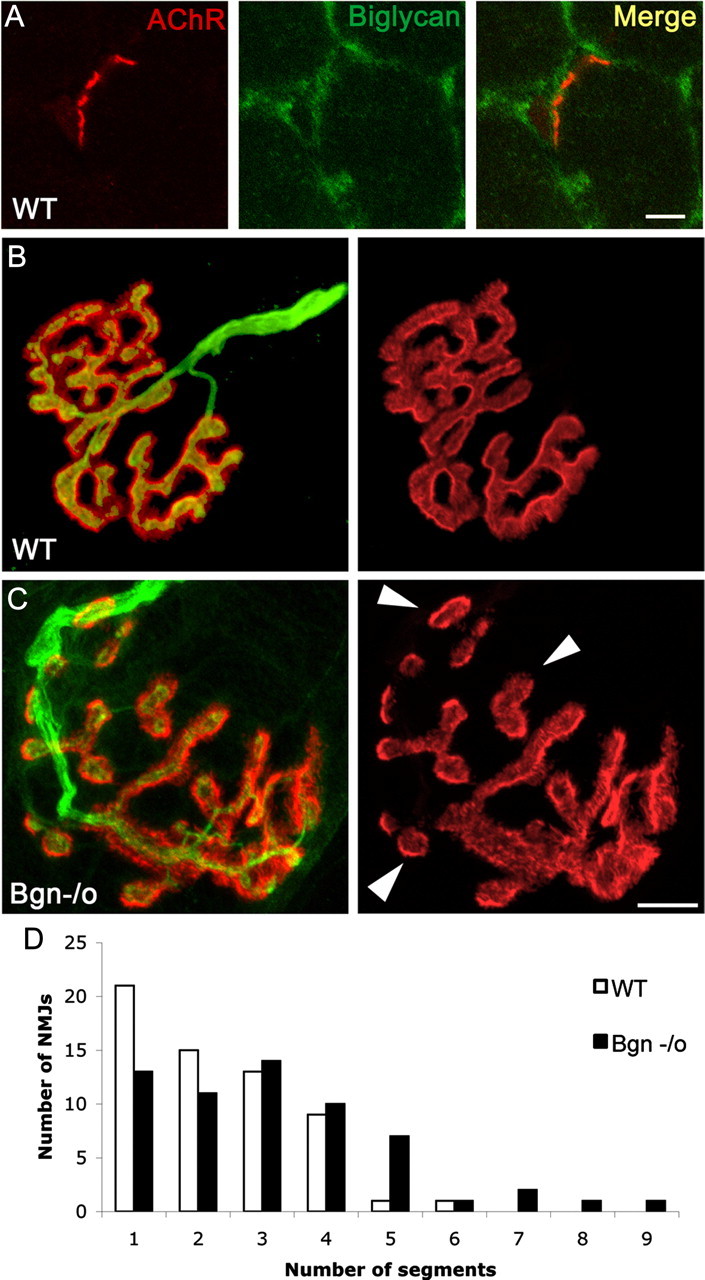
Synapses in mature biglycan null mice are abnormal. A, Biglycan is expressed at the neuromuscular junction of wild-type mice. Frozen sections of quadriceps femoris (5 weeks old) were labeled with α-bungarotoxin (red) and anti-biglycan (green). Biglycan is expressed at both the neuromuscular junctions and the noninnervated regions of the sarcolemma. No immunoreactivity is observed when sections from biglycan null muscle are stained in the same fashion (data not shown). Scale bar, 10 μm. B, C, The NMJs of biglycan null mice are hypersegmented. Sternomastoid muscle was dissected from perfusion-fixed 5-week-old wild-type (B) and biglycan null (C) littermates and then double-labeled (left) with rh-α-bungarotoxin to visualize the AChRs (red) and anti-SV/anti-neurofilament (green) to show the nerve. Merged images, left; AChR distribution only, right. Note the segmentation (arrowheads) of the biglycan null endplate in contrast to the continuous postsynaptic domains observed in the wild-type. Scale bar, 10 μm. D, The number of segments per endplate from muscles fixed and imaged as described above were scored. Endplates in biglycan null mice were significantly more segmented than those of their littermate controls (n = 60 endplates from 4 animals for each condition; Mann–Whitney U test; p < 0.01).
Biglycan binding
For cell binding assays mouse MuSK (Flag-MuSK) was subcloned in pFLAG-CMV1 between EcoRI and XbaI sites (Sigma). Deletion mutants, Flag-MuSKΔIgI (amino acids 1–108 deleted) and Flag-MuSKΔIgC6IV (amino acids 297–463 deleted), were generated using site-directed mutagenesis (Stratagene). COS7 cells were transfected with plasmids encoding full-length or mutant MuSK or wild-type trkA using Fugene (Roche Diagnostics) and incubated overnight. The cells were then rinsed with OptiMem (Invitrogen) and incubated with 20 nm biglycan polypeptide in MEM-H (Sigma) for 30 min at 4°C. To detect bound biglycan, cells were blocked with 10% horse serum in MEM-H for 10 min, incubated with anti-biglycan monoclonal antibody 4C4 for 30 min, rinsed with MEM-H, and labeled with goat-anti-mouse IgG-Alexa 488 for 30 min. To detect MuSK and TrkA, cells were blocked with 1% BSA, 5% normal mouse IgG, and incubated with either Cy3-conjugated anti-FLAG or anti-TrkA and goat-anti-rabbit-Cy3 for 30 min. All incubations and washes were conducted at 4°C. Cells were fixed with −20° methanol and mounted in Vectashield hard set (Vector Laboratories).
The extracellular domain of quail MuSK corresponding to amino acid residues 1–463 was subcloned into pCDNA-1 for expression with a His tag and a stable line of HEK-293 cells was established. This extracellular MuSK domain was purified by nickel affinity chromatography and was 90% pure as judged by Coomassie staining. For the microtiter plate assay, MuSK protein was diluted to 100 ng/100 μl and 100 μl aliquots were placed in the wells of a 96-well microtiter assay plate (Costar 3590) overnight at 4°C. Wells were blocked with 300 μl of 5 mg/ml BSA in PBS, pH 7.4, for 1 h at 4°C. A 100 μl aliquot of the appropriate dilution of biotinylated biglycan in PBS/BSA solution was then added to each well and incubated overnight at 4°C. Plates were washed for 3 × 5 min with PBS/0.05% Tween 20 followed by incubation with avidin-conjugated alkaline phosphatase (Pierce 31002; 0.2 μg/ml in 100 mm Tris pH 9.5 and 100 mm NaCl) for 1 h at RT. The plates were washed for 3 × 5 min with PBS/Tween and developed using 200 μl of p-nitrophenyl phosphate solution (Sigma; 5 mg tablet/10 ml detection buffer). The yellow reaction product was read at 415 nm in a Bio-Rad Laboratories microtiter plate spectrophotometer.
MuSK phosphorylation assay
Myotubes were incubated with or without agrin and/or biglycan 1 h. Cells were extracted with RIPA buffer (2 mm EDTA, 2 mm EGTA, 50 mm Tris pH 8, 150 mm NaCl, 10 mm tetrasodiumpyrophosphate, 100 mm NaF, 1 mm sodium orthovanadate, 10% glycerol, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 1 mm benzamidine, 1 μg/ml pepstatin, 21 μg/ml aprotinin, 1 μg/ml leupeptin, and 0.1 mg/ml PMSF), centrifuged, and soluble material precleared with protein-A beads (Bio-Rad Laboratories). Extracts were incubated with anti-MuSK antisera and protein-A beads and immunoprecipitates were electrophoresed on 5–15% gradient SDS-PAGE gels, transferred to nitrocellulose and probed for MuSK tyrosine phosphorylation (P-Tyrosine) with MAb 4G10. Blots were then stripped (Restore, Pierce) and reprobed with anti-MuSK antisera to verify equal loading. Bound antibody was detected with HRP-coupled secondary reagents and the signal was visualized using ECL plus (GE Healthcare) and quantified with Storm/Image Quant Software (GE Healthcare).
Cell culture
C2C12 cells were cultured as described previously (Gordon and Hall, 1989). Biglycan null immortalized cell line was generated as per established protocols (Morgan et al., 1994). Immortalized wild-type mouse cell lines were generously provided by Terence Partridge (Children's National Medical Center, Washington, DC).
AChR clustering assays
Wild-type and biglycan null cells were grown to ∼75% confluence on Permanox chamber slides (Nunc and Nalgene, Thermo Fisher Scientific) and differentiated for 3–5 d as described. Cells were incubated for 12 h with 1.4 nm biglycan and recombinant rat agrin 12.4.8 (5 U; Ferns et al., 1993). AChRs were labeled with rh-α-bungarotoxin for 30 min at 33°C and cells were fixed with methanol for 5 min at −20°C. AChR clusters >4 μm in length and <1000 μm2 were counted on a Nikon Eclipse 800 microscope.
Statistical analysis
All statistical analyses used Student's t test unless otherwise noted. Quantifications were performed by observers blinded to experimental condition.
Results
Biglycan is expressed at synapses
As a first step toward elucidating the function of biglycan at nerve-muscle synapses in vivo, we assessed its localization at the neuromuscular junction. In previous studies using antibodies that required in situ chondroitinase digestion, we observed that biglycan is expressed at the NMJ (Bowe et al., 2000). To confirm and extend these observations we generated monoclonal antibodies to biglycan (see Materials and Methods, Antibodies). Figure 1A shows that biglycan is expressed at both junctional as well as extrajunctional regions of the myofiber surface.
Synapses are abnormal in mature but not developing biglycan null mice
A comparison of synapse distribution in the sternomastoid muscle from P35 biglycan null mice and wild-type littermate controls showed that the overall number of synapses and the width of the endplate band were indistinguishable from controls. Moreover, AChR density and the alignment of the nerve terminals to the postsynaptic apparatus were comparable in normal and mutant synapses (AChR levels normalized to utrophin immunoreactivity, WT: 0.97 ± 0.03, bgn−/o: 0.95 ± 0.05; n = 29–31 junctions from 2 mice from each genotype, Student's unpaired t test, p > 0.8; Fig. 1B,C). However, the architecture of the endplates in the biglycan null synapses was disrupted compared with those in littermate controls. In wild-type synapses the postsynaptic domains were largely continuous, with few gaps observed. In contrast, at biglycan null endplates these AChR-rich regions were often discontinuous and segmented (compare Fig. 1B,C). Synapses from the biglycan null mice were significantly more segmented than those from littermate controls (Fig. 1D; n = 60 synapses from four animals of each genotype; Mann–Whitney U test, p < 0.01). There was a particularly striking difference in the proportion of hypersegmented synapses (≥5 segments/endplate) between the two genotypes. In wild-type mice only 3% (2/60) of synapses had ≥5 segments/endplate. In contrast, 20% (12/60) of mutant synapses were hypersegmented.
The segmentation that we observed in these P35 muscles could reflect defective stabilization of neuromuscular junctions and/or abnormalities that occurred earlier in synapse development. To address this question we studied the differentiation of synapses in developing biglycan null mice and littermate controls. To study synapse development in fetal as well as early postnatal muscle we turned to the diaphragm. Examination of E16 mice showed that endplates in mutant animals were compact and localized in a discrete band in the middle of the muscle (Fig. 2A), which is characteristic of wild-type animals of this age (Marques et al., 2000). Further, the endplates typical of P0 and P14 were indistinguishable in biglycan null and littermate controls. Double labeling for axons and presynaptic specializations showed that the disposition of the nerve terminals at the endplates was comparable in both control and biglycan-deficient synapses. Axonal sprouting was not observed in either mutant or wild-type muscle (Fig. 2B). Synapses in P14 sternomastoid were also indistinguishable in wild-type and biglycan null animals (data not shown). Thus, the structure of developing synapses in the absence of biglycan is normal as judged by the morphology of the postsynaptic specializations, their distribution across the muscle and their relationship with the axon.
Figure 2.

Synapses in developing biglycan null mice are similar to wild-type. A, Postsynaptic specializations in fetal biglycan null mice. Diaphragms were dissected from E16 biglycan null mice and labeled with rh-α-bungarotoxin to visualize the AChRs. Discrete AChR clusters are arrayed in a central band across the muscle. This morphology and localization are indistinguishable from that observed in wild-type muscle at this age (data not shown). Scale bar, 20 μm. B, Diaphragms were dissected from P0 and P14 heterozygous (top) and biglycan null (bottom) littermates and double labeled with rh-α-bungarotoxin to visualize the AChRs (red) and anti-synaptophysin/anti-neurofilament (green) to show the nerve. In each set the merged and AChR-only images are shown at left and right, respectively. No structural differences were observed in the synapses from these developing biglycan null mice compared with their normal littermates. Scale bar, 10 μm.
AChR and AChE are focally misaligned at biglycan-deficient neuromuscular junctions
The results of the developmental studies indicate that the NMJ in biglycan null mice forms normally but shows defective stabilization. In this scenario the segmentation observed in the mature animals (Fig. 1) would be due to the loss of previously occupied synaptic territory. Therefore, we looked for traces of the original, unsegmented synaptic architecture at the mutant endplates. To detect such traces we compared the localization of AChR and AChE at these junctions in P35 animals. At normal neuromuscular junctions the AChR-rich postsynaptic membrane is precisely aligned with AChE in the adjacent basal lamina (Rossi and Rotundo, 1993; Peng et al., 1999; Legay, 2000; Rotundo, 2003). However, AChE can persist in the basal lamina long after the disappearance of presynaptic and postsynaptic specializations (Marshall et al., 1977; Burden et al., 1979; Stanco and Werle, 1997). Figure 3 shows that AChE and AChR were tightly coaligned in wild-type synapses. However, there was misalignment of AChE and AChR at 80% (120/150) of the mutant synapses. We quantified the extent of this mislocalization on a per synapse basis by measuring the average areas occupied by AChR and AChE at the endplates. As expected by our qualitative observations, these areas were not significantly different at wild-type junctions (AChR 366 μm2 ± 19.43; AChE 347 μm2 ± 19.93; n = 9; Student's paired t test; p > 0.2). However, in the mutant synapses AChE occupied a greater area than that of AChR (AChR 224 μm2 ± 32.8; AChE 260 μm2 ± 41; n = 9, Student's paired t test, p < 0.02). Notably, in most cases these AChE-only domains are contiguous with the AChE/AChR-rich regions at the synapse (Fig. 3A). We note that the smaller area of the mutant synapses is likely to be a function of myofiber size, which is ∼30% reduced in biglycan null compared with wild-type animals (Mercado et al., 2006). Together, these observations suggest that these AChE-only sites represent previously occupied synaptic sites and thus provide direct evidence that synapses in biglycan null mice are unstable.
Figure 3.

Focal misalignment of AChR and AChE at synapses in biglycan null mice. A, Hindlimb muscles from 5-week-old wild-type (top) and biglycan null (bottom) littermates were dissected, fixed, and labeled with rh-α-bungarotoxin (red) and OG-fasciculin2 (green) to visualize AChR and AChE, respectively. The AChR and AChE distributions are superimposable in the WT. However, AChE domains lacking subadjacent AChR (arrows) were observed in the biglycan null junctions. Scale bar, 10 μm. B, Quantification of the number of synapses with aligned or misaligned AChR and AChE in wild-type and biglycan null muscles. In wild-type muscle complete alignment was observed at 100% (90/90) of synapses. However, only 20% of the mutant synapses (30/150) showed complete alignment. n = number of synapses scored for each genotype.
We considered the possibility that the misalignment of AChR and AChE and the increased segmentation that we observed in the 5-week-old biglycan null synapses might be due to the presence of myofibers that have either regenerated or are compromised and about to degenerate. To address this potential explanation we determined the proportion of regenerated [centrally nucleated (CN)] myofibers in P60 mice. We observed that 2.9% (±0.48; n = 11,374 fibers from 2 animals) of the fibers showed central nuclei at this age. This number is in the same range as previous measures of CN fibers at P35 (Mercado et al., 2006). Therefore, the 80% of synapses showing misalignment of AChR and AChE at P35 (Fig. 3B) and the 20% of synapses that show hypersegmentation (Fig. 1D) is far in excess of the small percentage of regenerated and/or degenerating fibers present in these mutant muscles.
The number of perijunctional folds is increased in biglycan null muscle
The fine structure of biglycan-deficient synapses also displayed features consistent with synapse instability (Fig. 4). The length of the presynaptic membrane and the number of junctional folds juxtaposed to them was equivalent in both genotypes (wild-type: 5.4 μm/bouton ± 0.45, 6.5 folds/bouton ± 0.72; n = 20; biglycan null: 5.17 μm/bouton ± 0.27, 6.56 folds/bouton ± 0.55; n = 39; Student's unpaired t test, p > 0.6). The depth of the folds was also comparable in both genotypes. Moreover, in agreement with the immunofluorescence analysis (Fig. 1B,C) all observed synaptic boutons were apposed to postsynaptic specializations. However, we detected an increase in the number of perijunctional folds flanking the synapse in the biglycan null muscle (Fig. 4B). These perijunctional folds were structurally indistinguishable from the junctional folds that were apposed to the presynaptic bouton. Statistical analysis showed that the number of perijunctional folds was significantly greater in the biglycan null compared with wild-type synapses (Kolmogorov–Smirnov test; p < 0.01). Together, these ultrastructural and light-level analyses indicate that previously established synaptic territory has been ceded in biglycan null mice.
Figure 4.
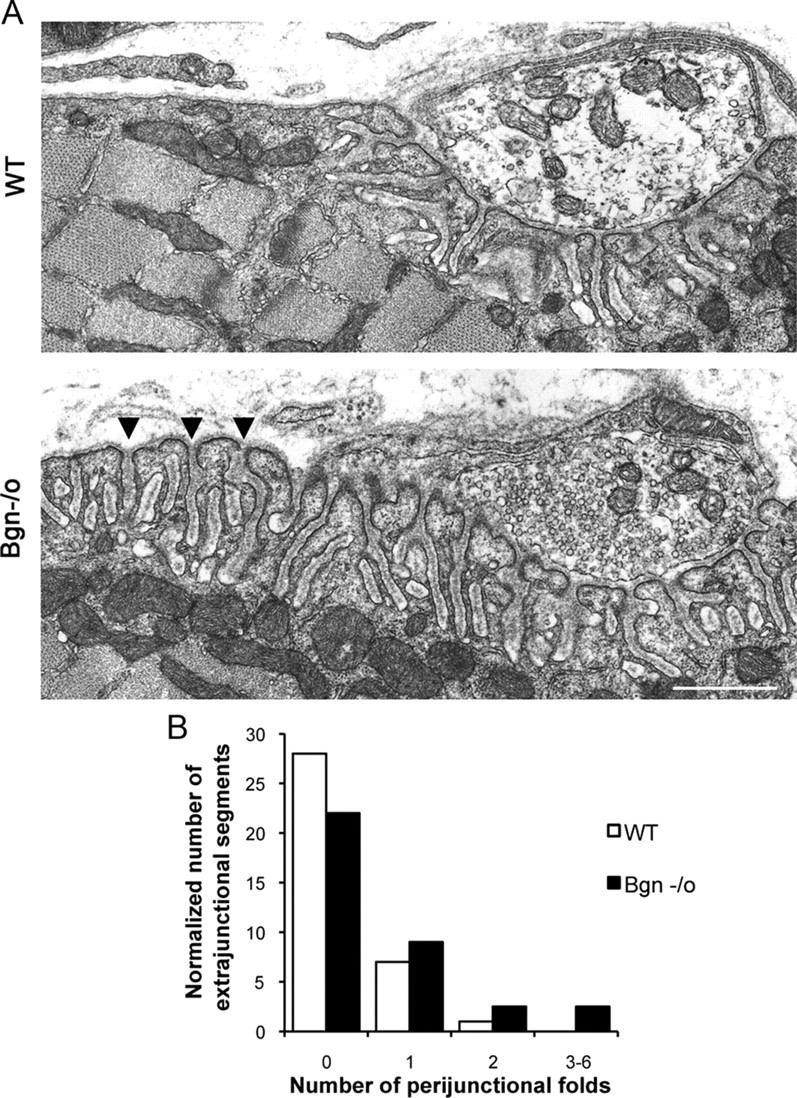
Increased number of perijunctional folds in biglycan null synapses. A, Electron micrographs of synapses in sternomastoid muscles from 5-week-old wild-type and congenic biglycan null mice. In the wild-type muscle junctional folds are largely restricted to the region directly under the synaptic bouton. However, in biglycan null animals an increased number of perijunctional folds (arrowheads) are observed flanking the synapse. Scale bar, 2 μm. B, Frequency histogram of perijunctional folds in synapses from wild-type and biglycan null mice. The number of perijunctional folds within 0.5–2 μm from the edge of nerve terminal was scored for two wild-type (n = 36 perijunctional regions) and two biglycan null muscles (n = 72 perijunctional regions). There are significantly more perijunctional folds in mutant muscle (Kolmogorov–Smirnov; p < 0.01).
MuSK expression at the postsynaptic membrane is reduced in biglycan null synapses
The results presented above indicate a function for biglycan in the stability of the neuromuscular junction. To explore the molecular basis for this role we examined the expression of 18 proteins known to be enriched at synapses, including AChR, rapsyn, MuSK, and several components of the dystrophin/utrophin-associated protein complex (Table 1). Remarkably, MuSK expression was the only component detectably altered at biglycan null synapses. Although still localized at the nerve-muscle synapses, MuSK expression in the postsynaptic membrane was reduced in quadriceps muscle from biglycan null mice compared with littermate controls (Fig. 5A). We confirmed this downregulation using three independent antibodies directed against different regions of the MuSK polypeptide (see Materials and Methods, Antibodies). We quantified this reduction at the postsynaptic apparatus by normalizing the signal of MuSK immunoreactivity to that of utrophin, which shows a virtually identical localization but which is expressed at the same levels in normal and biglycan mutant synapses. Relative MuSK levels were reduced over tenfold in the biglycan null compared with wild-type synapses (10.78 ± 3.54) and (1.00 ± 1.32) in wild-type and mutant, respectively; p < 0.02; Figure 5B. Quantitative RT-PCR revealed that the levels of MuSK mRNA (expressed as a ratio to ATP synthase transcripts) were not significantly different in wild-type compared with biglycan null muscle (1.09 ± 0.42 and 1.41 ± 0.39, respectively; Student's unpaired t test, p > 0.4). We also observed a decrease in MuSK expression at synapses in sternomastoid (Fig. 5C) as well as diaphragm (data not shown). Together, these results indicate that biglycan plays a selective role in regulating MuSK localization at the synapse.
Table 1.
Comparison of the expression of synaptically enriched proteins in biglycan null and wild-type mice
| Protein | Expression level at mutant NMJ compared with wild type |
|---|---|
| Biglycan | Not present |
| MuSK* | Decreased |
| AChE | Unchanged (focally misaligned) |
| AChR | Unchanged |
| Utrophin | Unchanged |
| Rapsyn | Unchanged |
| α-, β-Dystroglycan | Unchanged |
| α-, β-, γ-Sarcoglycan | Unchanged |
| α-Dystrobrevin-1 | Unchanged |
| α-Dystrobrevin-2 | Unchanged |
| α-Syntrophin | Unchanged |
| β1-Syntrophin | Unchanged |
| β2-Syntrophin | Unchanged |
| nNOS | Unchanged |
| Laminin-2 | Unchanged |
| Collagen VI | Unchanged |
Sections of 5-week-old quadriceps femoris from wild-type or biglycan null animals were mounted on the same slides and immunolabeled for each of the proteins indicated. To label AChRs, rh-α-bungarotoxin was applied simultaneously. The staining intensity of each protein at the neuromuscular junctions of wild-type and biglycan null animals was compared. Similar results were observed in muscles from four pairs of mutant and wild-type animals.
*Observed with three different anti-MuSK antisera; see Materials and Methods, Antibodies.
Figure 5.
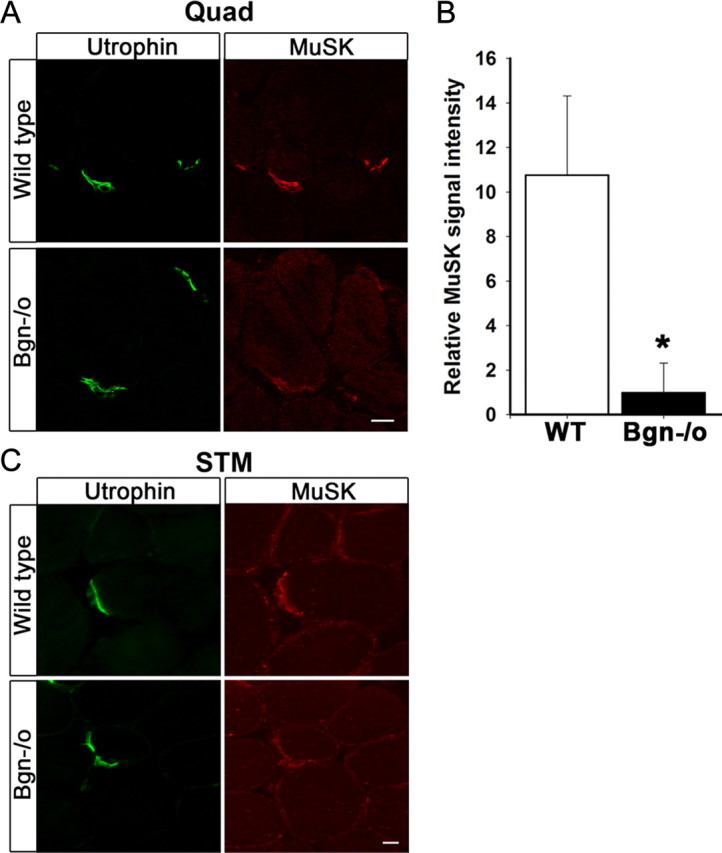
MuSK expression is decreased at synapses in biglycan null mice. A, Quadriceps femoris sections from 5-week-old wild-type and biglycan null animals were immunolabeled with a mouse anti-utrophin (green) and a rabbit anti-MuSK (affinity purified 29–31, see Materials and Methods, Antibodies; red). Utrophin is expressed at similar levels at synapses in wild-type and biglycan null muscle. However, a decrease in the intensity of MuSK staining at biglycan null neuromuscular junctions is observed. Scale bar, 10 μm. B, The mean pixel signal intensity of MuSK normalized to the mean pixel signal intensity of utrophin at the neuromuscular junctions of wild-type, 10.78 ± 3.54, and biglycan null muscle, 1.00 ± 1.32 (at least 3 synapses per condition) was determined as described in Materials and Methods. The signal intensity of MuSK was significantly higher at wild-type versus biglycan null junctions (Student's unpaired t test, *p < 0.02). C, Sternomastoid sections from 5-week-old wild-type and biglycan null mice were immunolabeled as in A. Utrophin is expressed at similar levels at the synapses in wild-type and biglycan null sternomasoid while synaptic MuSK is decreased in the biglycan null sternomastoid muscle. Scale bar, 10 μm.
Biglycan binds to MuSK
The results described above raised the possibility that biglycan interacts with MuSK. As a first test of this idea we asked whether purified recombinant biglycan binds to the purified MuSK ectodomain. We performed a solid phase binding assay where immobilized MuSK ectodomain or BSA were incubated with biotinylated biglycan or BSA followed by avidin-conjugated alkaline phosphatase. Figure 6A shows that biglycan binds to MuSK ectodomain, but not to BSA (0.45 ± 0.01 and 0.06 ± 0.01, respectively; n = 3, Student's unpaired t test, p < 0.01). Thus, biglycan binds directly to MuSK.
Figure 6.
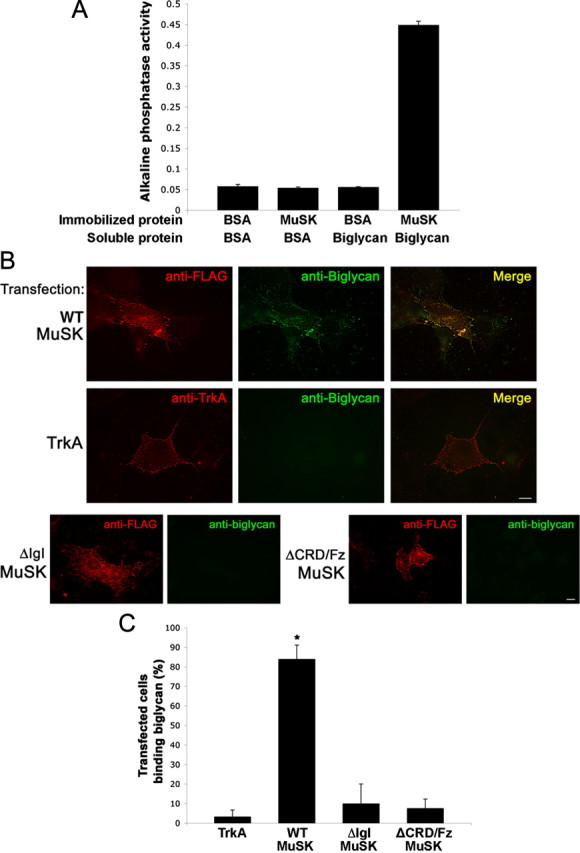
Biglycan binds to MuSK. A, Direct binding of biglycan and MuSK. Purified MuSK ectodomain or BSA was immobilized on plastic wells and then incubated with either BSA or with 500 ng of biotinylated recombinant biglycan followed by avidin-alkaline phosphatase. Biglycan binds to MuSK but not to BSA (0.45 ± 0.01 and 0.06 ± 0.01, respectively; n = 3, Student's unpaired t test, p < 0.01). B, Biglycan binds to MuSK expressed in heterologous cells. COS cells were transfected for 24 h with FLAG-tagged full-length wild-type MuSK (WT-MuSK), TrkA, or FLAG-tagged MuSK mutants lacking either the IgI (ΔIgI) or CRD/Fz domain (ΔCRD/Fz) as indicated. Live cells were incubated with purified recombinant biglycan polypeptide (20 nm) for 30 min at 4°C and bound biglycan was visualized with an anti-biglycan monoclonal antibody and Alexa488-conjugated secondary antibodies (green) and then fixed. The transfected constructs (red) were visualized with anti-FLAG (all MuSK constructs) or anti-trkA ectodomain. Note that biglycan binds to COS cells expressing full-length, wild-type MuSK (top), but not to cells expressing TrkA (middle), mutant MuSK ΔIgI, or ΔCRD/Fz (bottom). No binding was detected to untransfected cells (data not shown). Scale bars, 10 μm. C, Quantification of biglycan binding to MuSK or TrkA transfected cells. Data are expressed as the mean percentage of cells that bound biglycan per microscope field Little biglycan binding was detected on cells expressing heterologous TrkA (3.3 ± 3.3%; n = 10 fields). Cells expressing wild-type MuSK exhibit robust biglycan binding (86.3 ± 7.1%; n = 10 fields; *p < 0.001, Students unpaired t test). Biglycan binding to cells expressing MuSK-ΔIgI or ΔCRD/Fz was not significantly different from that to TrKA-expressing cells; p > 0.8, one-way ANOVA).
We next tested whether biglycan can bind to MuSK expressed on the cell surface. We expressed MuSK in COS-7 cells and then assessed the binding of recombinant biglycan to them. Figure 6B shows that cells expressing full-length MuSK exhibit robust biglycan binding (86.3 ± 7.1%; n = 10 fields, total 56 transfected cells). Further, the bound biglycan is tightly colocalized with the expressed MuSK (Fig. 6B,C). We detected no biglycan binding to either untransfected cells (data not shown) or to cells expressing heterologous TrkA (Fig. 6B,C; 3.3 ± 3.3%; n = 10 fields, total 62 transfected cells; Student's t test, p < 0.001). However, we did observe that untransfected cells adjacent to those transfected with MuSK exhibited low levels of binding (Fig. 6B), suggesting that MuSK may be shed from the transfected cells.
We also used the cell-based binding assay to determine which portions of the MuSK ecto-domain are required for biglycan binding. Since both the IgI and CRD/Fz domains have been implicated in MuSK bioactivity (Masiakowski and Yancopoulos, 1998; Zhou et al., 1999; Herbst and Burden, 2000), we tested MuSK mutants lacking either of these regions (ΔIgI and ΔCRD/Fz, respectively). Figure 6B shows that both constructs are expressed on the surface of transfected COS cells as judged by immunolabeling of unpermeabilized cells with an antibody to an epitope tag located in the ectodomain. However, no biglycan binding was detected on cells expressing either mutant MuSK (Fig. 6B). Quantification of the effect shows that biglycan binding to MuSK lacking either the IgI or the CRD/Fz domains were indistinguishable from controls (ΔIgI: 10.0% ±10.0, ΔCRD/Fz: 7.6 ± 4.7%, TrkA: 3.3 ± 3.3%; Fig. 6C). Thus, both the IgI and CRD/Fz domains of MuSK are required for biglycan binding.
Biglycan regulates agrin-induced MuSK phosphorylation
The binding of biglycan to MuSK suggested that it might regulate agrin-induced MuSK activation. To test this idea we treated cultured myotubes with agrin, biglycan, or a combination of the two and measured the level of MuSK tyrosine phosphorylation (Fig. 7A,B). In agreement with previous studies, one unit of agrin (1/2 maximal activity; Megeath and Fallon, 1998) induced MuSK phosphorylation in mouse C2C12 myotubes. No increase in MuSK phosphorylation was observed after treatment with 1.4 nm biglycan alone. However, 1.4 nm biglycan potentiated agrin-induced MuSK phosphorylation (1.5-fold increase; n = 10, Student's unpaired t test, p < 0.01). This potentiation was not observed with decorin polypeptide (1 nm–100 nm), which shares the same domain structure as biglycan and is 55% identical to it (data not shown; McEwan et al., 2006). As a further test of whether biglycan functions within the same pathway as agrin we added biglycan plus saturating levels of agrin. If these factors were acting in the same pathway, biglycan would not be expected to further increase MuSK activation under these conditions. Indeed, Figure 7C and D shows that biglycan does not superpotentiate MuSK phosphorylation induced by maximal agrin. Together, these data indicate that biglycan regulates agrin-induced MuSK activation.
Figure 7.

Biglycan regulates agrin-induced MuSK phosphorylation. A, Biglycan potentiates agrin-induced MuSK phosphorylation. C2C12 myotubes were treated for 1 h with 1U agrin, 1.4 nm of purified recombinant biglycan, or both as indicated. The levels of MuSK tyrosine phosphorylation were then assessed by probing MuSK immunoprecipitates with anti-phosphotyrosine antibody (P-Tyr). The blots were stripped and reprobed with anti-MuSK to verify equal loading. Agrin induces MuSK phosphorylation and this activity is potentiated by biglycan. B, Quantification. Western blots were scanned and analyzed with ImageQuant software (GE Healthcare) to compare relative changes in MuSK phosphorylation. Simultaneous treatment with 1U agrin and 1.4 nm biglycan induces a 1.5-fold increase in MuSK phosphorylation compared with 1U agrin alone (p < 0.001, n = 10). Treatment with saturating concentrations of agrin (5 U) alone induces a 1.6-fold increase in MuSK phosphorylation above 1U agrin (p < 0.02, n = 5). C, Biglycan does not modulate MuSK phosphorylation induced by saturating concentrations (max) of agrin. C2C12 cells were stimulated with 5 U agrin in the presence or absence of 1.4 nm biglycan. MuSK phosphorylation was assessed as in A. D, Quantification. Biglycan does not induce a statistically significant change in MuSK phosphorylation above that induced by 5 U agrin alone (p > 0.3, n = 3). E, Equivalent levels of MuSK are expressed in C2C12 and bgn−/o myotubes. Cultured C2C12 and biglycan null myotubes were extracted and MuSK was incubated with anti-MuSK antisera followed by protein-A-beads. Immunoprecipitates solubilized in sample buffer, electrophoresed, transferred to nitrocellulose and probed with an anti-MuSK antibody. F, Biglycan is not required for agrin-induced MuSK phosphorylation. Biglycan null myotubes were treated for 15 min, 30 min, or 1 h with 1U agrin. MuSK activation was determined by probing MuSK immunoprecipitates with anti-tyrosine antibody (P-Tyr). Note that agrin induces MuSK phosphorylation in biglycan null cells within 15 min of treatment and phosphorylation levels increase through 1 h of treatment.
The experiments described above used wild-type myotubes that express endogenous biglycan (Casar et al., 2004). To define the role of biglycan in the agrin pathway more precisely, we used a myoblast cell line derived from biglycan null mice (Mercado et al., 2006). These biglycan-deficient myoblasts differentiate to form myotubes and express MuSK at levels comparable to those observed in wild-type myotubes (Fig. 7E). Agrin treatment induced rapid and robust MuSK phosphorylation in these mutant myotubes (Fig. 7F; 2.0 ± 0.4-fold increase in MuSK phosphorylation after 1 h agrin incubation; n = 5, Student's unpaired t test, p < 0.05). Thus while biglycan regulates agrin-induced MuSK phosphorylation, it is not necessary for its activation.
Biglycan is necessary for the stability of agrin-induced AChR clusters
To further probe the role of biglycan in synapse stability we turned to an in vitro system based upon the ability of agrin to induce AChR clustering on cultured myotubes (Godfrey et al., 1984; Fallon et al., 1985; Nitkin et al., 1987). Discrete, compact AChR clusters form by 2 h of agrin treatment and persist for at least 12 h (Fig. 8A). AChR clustering in biglycan null myotubes treated with agrin for 2–6 h was indistinguishable from that observed in wild-type cultures. However, as shown in Figure 8A, virtually no compact AChR clusters were observed on mutant myotubes incubated with agrin for 12 h. Rather, AChRs were arrayed in abnormally large, diffuse microcluster “islands” (see below). The AChR clustering defect in the biglycan-null myotubes was specific to agrin, since biglycan null myotubes form high-density AChR clusters in response to overnight incubation with purified laminin (data not shown).
Figure 8.

Agrin-induced AChR clustering is defective in biglycan null myotubes. A, Time course of agrin-induced AChR clustering in wild-type and bgn−/o myotubes. Myotubes of indicated genotype were made from immortalized myoblast lines and were treated with 5 U agrin for 0, 2, 4, 6, 8, or 12 h. Short duration agrin treatment (2, 4, 6 h) induces small, discrete AChR clusters in both wild-type and biglycan null cells. After 12 h of agrin treatment wild-type myotubes displayed typical compact AChR clusters (<1000 μm2). In contrast, biglycan null myotubes treated for 12 h with agrin exhibited diffuse AChR microcluster islands that were abnormally large (∼1.4 × 103 to ∼1 × 104 μm2). Scale bar, 10 μm. B, Exogenous biglycan rescues the stability defects of agrin-induced AChR clusters in bgn−/o myotubes. Myotubes were incubated for 12 h with 5 U agrin with or without added biglycan (1.4 nm). Treatment with agrin and biglycan induced the formation of smaller, compact AChR clusters compared with agrin alone. Treatment with biglycan alone had no effect on AChR distribution (data not shown). Scale bar, 10 μm. C, Quantification of biglycan rescue. The number of AChR clusters (>4 μm in length and <1 × 103 μm2) per myotube segment on cultures treated with agrin alone or with agrin plus biglycan was scored (n = 70 segments/condition from 3 independent experiments; Student's unpaired t test, p < 0.03).
To demonstrate a direct role of biglycan in stabilizing AChR clusters we performed a rescue experiment where purified recombinant biglycan was added to the cultures. Biglycan null myotubes were treated with agrin alone or in combination with purified recombinant biglycan for 12 h. As noted above, typical AChR clusters are not observed on mutant myotubes after 12 h of agrin treatment. Rather, AChR are arrayed in microcluster islands (Fig. 8A). These AChR islands are composed of loosely arrayed AChR microclusters (ranging from ∼1 × 103 to ∼1 × 104 μm2). However, coincubation with 1.4 nm biglycan for 12 h increased the number of discrete, compact (<1000 μm2) AChR clusters on the myotubes surface (Fig. 8B,C; agrin: 1.07 ± 0.18, agrin/biglycan: 1.73 ± 0.22, n = 70 segments/condition from 3 independent experiments; Students unpaired t test, p < 0.03). Together, these observations indicate that biglycan is dispensable during the formation of agrin-induced AChR clusters but is required for their stabilization. Further, these findings in cultured myotubes suggest that in vivo biglycan is acting, at least in part, postsynaptically.
Discussion
In this study we introduce biglycan as an extracellular MuSK ligand that is important for synapse stability. We show that: (1) synapses in mature biglycan-deficient mice exhibit structural defects including increased numbers of perijunctional folds and focal misalignment of AChR and AChE, but synapses in developing biglycan null mice are indistinguishable from wild-type; (2) MuSK levels are selectively reduced in the postsynaptic membrane of biglycan null mice; (3) biglycan binds MuSK both in vitro and on the cell surface; (4) biglycan regulates agrin-induced MuSK phosphorylation; and (5) agrin-induced AChR clusters are unstable in cultured biglycan null myotubes and this defect can be substantially rescued by the addition of exogenous biglycan.
Our studies show that biglycan is important for synaptic stability in vivo. The early development of synapses (E16–P14) is equivalent in wild-type and biglycan null muscles (Fig. 2). However, several defects in the synaptic apparatus are observed at P35 including endplate fragmentation, increased numbers of perijunctional folds, and reduced MuSK expression. The focal misalignment of AChR and AChE at mutant synapses is likely to be an important clue to the basis of the fragmentation. AChE at the neuromuscular junction is stably associated with the synaptic basal lamina by virtue of its attachment through its collagenic tail (Hall, 1973; Lwebuga-Mukasa et al., 1976; Rossi and Rotundo, 1993; Feng et al., 1999), a relationship that persists even if the postsynaptic membrane specializations are lost (McMahan et al., 1978; Anglister and McMahan, 1985). Thus, the presence of domains in the endplate region that are rich in AChE but lack AChR indicates that postsynaptic membrane specializations have been lost. Our observation that biglycan is important for the stability but not the initial formation of agrin-induced AChR clusters in cultured myotubes is consistent with this in vivo phenotype.
In addition to the loss of synaptic territory at the biglycan-null neuromuscular junctions, we also observe defects at the level of individual boutons. While the number and depth of junctional folds per bouton were normal in these animals, there was an increase in the number of perijunctional folds (Fig. 4). We think it most likely that these folds are remnants of previous synaptic sites. However, it is also possible that they result from dysregulation of the molecular mechanisms that ensure precise apposition of the presynaptic and postsynaptic membrane.
Our studies with biglycan null myotubes give further insight into the role of biglycan in synapse stability. These myotubes express both MuSK and AChR. Brief agrin treatment induces MuSK phosphorylation in these cells (Fig. 7) and the formation of small, discrete AChR clusters (Fig. 8). However, high density, compact AChR clusters that are typically seen after prolonged (12–16 h) agrin treatment are not observed. Rather, after extended agrin treatment only low-density, abnormally large islands of AChR microclusters are observed. Importantly, the stability defect can be substantially rescued by the addition of purified biglycan polypeptide at low nanomolar concentrations. We hypothesize that biglycan promotes the stabilization of AChR clusters at least in part through its action as a MuSK scaffolding molecule. This proposal is supported by our observation that the CRD/Fz domain, which has been implicated in MuSK scaffolding activity (Zhou et al., 1999), is also required for biglycan binding (Fig. 6).
The neuromuscular junction instability in biglycan null synapses is unlikely to be the result of developmental defects or secondary to muscle degeneration. As discussed above, early synapse formation is indistinguishable in mutant and wild-type muscle. In cell culture agrin-induced AChR clusters form normally in mutant myotubes but subsequently become unstable. As discussed below, biglycan binds directly to MuSK, a protein important for synapse maintenance (Kong et al., 2004). Finally, muscle degeneration/regeneration cannot account for the observed synaptic defects: >80% of biglycan null synapses display AChR/AChE misalignment at P35, while ≤5% of myofibers showed central nuclei at this age or several weeks later.
Several lines of evidence indicate that biglycan is a ligand for MuSK. In vivo, biglycan is expressed in muscle and is localized at the neuromuscular junction (Fig. 1A). Biochemical studies show that recombinant biglycan binds to purified MuSK ectodomain (Fig. 6). Biglycan also binds to MuSK expressed on the surface of heterologous cells. The specificity of this cell-based binding is supported by the lack of binding to either untransfected COS cells or to those expressing TrkA. Moreover, biglycan does not bind to MuSK lacking either the IgI or the CRD/Fz domain, even though both of these mutant proteins are expressed at comparable levels on the cell surface. (However, it should be noted that even surface expression does not assure that a mutant protein is properly folded.) Genetic studies show that biglycan is important for the localization of MuSK at synapses in vivo (Fig. 5). Finally, biglycan modulates agrin-induced MuSK activation (Fig. 7). Together, these observations indicate that biglycan serves both scaffolding and signaling roles for MuSK.
It has been reported that ColQ can bind MuSK when expressed in COS cells, suggesting that AChE/ColQ may also play a role in MuSK localization (Cartaud et al., 2004). In the present studies, however, we did not detect MuSK in the AChR-poor regions of the biglycan mutant synapses that contain normal levels of AChE, nor have we been able to reproduce the MuSK-ColQ AChE binding using our solid phase immobilized MuSK assay (as shown in Fig. 6A; and E. Marrero and R. L. Rotundo, unpublished observations). Moreover, recent studies showed that collagen-tailed cholinesterase binds to the surface of COS cells in the absence of coexpressed MuSK (Hrabovska et al., 2010). Thus, it is unlikely that the collagen-tailed AChE plays a role in MuSK localization at the neuromuscular synapse, in contrast to biglycan where direct binding with MuSK can be demonstrated in vitro (Fig. 6A).
A large number of studies have advanced our understanding of the early events in nerve-muscle synaptogenesis, but the molecular mechanisms for maintaining these synapses remain largely obscure. For example, nerve independent prepatterning, the earliest stage of synapse formation, requires MuSK, rapsyn, Dok7, LRP4, Wnt11r, and functional DHPR Ca channels (Kim et al., 2008; Zhang et al., 2008; Jing et al., 2009; Chen et al., 2011). Agrin, MuSK, LRP4, and neural activity collaborate during the subsequent prenatal differentiation of the synapse (Witzemann, 2006; Wu et al., 2010). Our data demonstrate a role for biglycan in stabilizing synapses after P14—the time at which synapses reach their mature configuration. Consistent with this selective role in synapse stabilization, biglycan is neither necessary for synapse formation in vivo (Fig. 2) nor MuSK activation and initial AChR clustering in cell culture (Figs. 7, 8). Such a stage-specific function for biglycan in postsynaptic stabilization conceptually, and perhaps mechanistically, parallels the role of extracellular factors that stabilize presynaptic differentiation during distinct developmental epochs (Fox et al., 2007). Finally, MuSK is necessary for the maintenance of the neuromuscular junction (Kong et al., 2004; Punga et al., 2011). We therefore propose that the biglycan-mediated localization of MuSK is important for synapse stability. It will be of interest to identify additional members of the biglycan-MuSK stabilization pathway and to determine whether they play a role in diseases of the neuromuscular junction or age-associated synapse loss (Coggan et al., 2004; Vincent et al., 2004; Ryall et al., 2008).
Biglycan binds to α- and γ-sarcoglycan and regulates the expression of syntrophins, dystrobrevins, and nNOS at the sarcolemma (Mercado et al., 2006; Rafii et al., 2006). However, the synaptic expression of these molecules is unaffected in biglycan null mice (Table 1), indicating that biglycan plays distinct roles at junctional and nonjunctional regions of the muscle. In this regard biglycan is analogous to dystrobrevin and syntrophins, which also have distinct functions at synaptic and nonsynaptic regions of muscle (Grady et al., 2003; Adams et al., 2004).
This work also has implications for disease mechanisms and therapies. The loss of synaptic connections is one the earliest and most consistent events in a range of neurological diseases including spinal muscular atrophy and amyotrophic lateral sclerosis (Williams et al., 2009). Therapies directed at stabilizing synapses could slow the disease course and prolong the lifetime of these patients. Notably, recombinant biglycan can be delivered systemically in mice for months and is active in muscle (Amenta et al., 2011). As an extracellular matrix protein that acts to stabilize the neuromuscular junction, we propose that biglycan could be a protein therapeutic for motor neuron diseases such as spinal muscular atrophy and amyotrophic lateral sclerosis.
Footnotes
This work was funded by grants from the National Institutes of Health (HD23924 and RR15578 to J.R.F.; AR42826 and R44 NS045432 to D.J.M.; AG05917 to R.L.R.; T32GM007601 to H.E.C.), the Division of Intramural Research, National Institute of Dental and Craniofacial Research of the Intramural Research Program, NIH, Department of Health and Human Services (to M.F.Y.), and the Muscular Dystrophy Association (to R.L.R.). B.E.L. was a fellow of the Pediatric Scientist Development Program, HD00850. H.E.C. was supported in part by a Fulbright Fellowship. We gratefully acknowledge generous gifts of reagents from D. Glass, S. Burden, S. Froehner, and A. Ross. We thank R. Balice-Gordon and C. Cisterni for advice on whole mount preparations, and R. Creton and S. Kunz for their advice on confocal microscopy. The expert technical assistance of Carol Ayala for the electron microscopy is gratefully acknowledged.
This paper is dedicated to the memory of Werner Hoch, a generous and valued collaborator and colleague.
J.R.F. is an inventor on patents owned by Brown University related to biglycan. These patents have been licensed to Tivorsan Pharmaceuticals, in which J.R.F. is founder and shareholder.
References
- Adams ME, Kramarcy N, Fukuda T, Engel AG, Sealock R, Froehner SC. Structural abnormalities at neuromuscular synapses lacking multiple syntrophin isoforms. J Neurosci. 2004;24:10302–10309. doi: 10.1523/JNEUROSCI.3408-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amenta AR, Yilmaz A, Bogdanovich S, McKechnie BA, Abedi M, Khurana TS, Fallon JR. Biglycan recruits utrophin to the sarcolemma and counters dystrophic pathology in mdx mice. Proc Natl Acad Sci U S A. 2011;108:762–767. doi: 10.1073/pnas.1013067108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameye L, Young MF. Mice deficient in small leucine-rich proteoglycans: novel in vivo models for osteoporosis, osteoarthritis, Ehlers-Danlos syndrome, muscular dystrophy, and corneal diseases. Glycobiology. 2002;12:107R–116R. doi: 10.1093/glycob/cwf065. [DOI] [PubMed] [Google Scholar]
- Anglister L, McMahan UJ. Basal lamina directs acetylcholinesterase accumulation at synaptic sites in regenerating muscle. J Cell Biol. 1985;101:735–743. doi: 10.1083/jcb.101.3.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Lichtman JW. In vivo visualization of the growth of pre- and postsynaptic elements of neuromuscular junctions in the mouse. J Neurosci. 1990;10:894–908. doi: 10.1523/JNEUROSCI.10-03-00894.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowe MA, Mendis DB, Fallon JR. The small leucine-rich repeat proteoglycan biglycan binds to alpha-dystroglycan and is upregulated in dystrophic muscle. J Cell Biol. 2000;148:801–810. doi: 10.1083/jcb.148.4.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burden SJ. Building the vertebrate neuromuscular synapse. J Neurobiol. 2002;53:501–511. doi: 10.1002/neu.10137. [DOI] [PubMed] [Google Scholar]
- Burden SJ, Sargent PB, McMahan UJ. Acetylcholine receptors in regenerating muscle accumulate at original synaptic sites in the absence of the nerve. J Cell Biol. 1979;82:412–425. doi: 10.1083/jcb.82.2.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartaud A, Strochlic L, Guerra M, Blanchard B, Lambergeon M, Krejci E, Cartaud J, Legay C. MuSK is required for anchoring acetylcholinesterase at the neuromuscular junction. J Cell Biol. 2004;165:505–515. doi: 10.1083/jcb.200307164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casar JC, McKechnie BA, Fallon JR, Young MF, Brandan E. Transient up-regulation of biglycan during skeletal muscle regeneration: delayed fiber growth along with decorin increase in biglycan-deficient mice. Dev Biol. 2004;268:358–371. doi: 10.1016/j.ydbio.2003.12.025. [DOI] [PubMed] [Google Scholar]
- Chen F, Liu Y, Sugiura Y, Allen PD, Gregg RG, Lin W. Neuromuscular synaptic patterning requires the function of skeletal muscle dihydropyridine receptors. Nat Neurosci. 2011;14:570–577. doi: 10.1038/nn.2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggan JS, Grutzendler J, Bishop DL, Cook MR, Gan W, Heym J, Lichtman JW. Age-associated synapse elimination in mouse parasympathetic ganglia. J Neurobiol. 2004;60:214–226. doi: 10.1002/neu.20022. [DOI] [PubMed] [Google Scholar]
- Fallon JR, Nitkin RM, Reist NE, Wallace BG, McMahan UJ. Acetylcholine receptor-aggregating factor is similar to molecules concentrated at neuromuscular junctions. Nature. 1985;315:571–574. doi: 10.1038/315571a0. [DOI] [PubMed] [Google Scholar]
- Feng G, Krejci E, Molgo J, Cunningham JM, Massoulié J, Sanes JR. Genetic analysis of collagen Q: Roles in acetylcholinesterase and butyrylcholinesterase assembly and in synaptic structure and function. J Cell Biol. 1999;144:1349–1360. doi: 10.1083/jcb.144.6.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferns MJ, Campanelli JT, Hoch W, Scheller RH, Hall Z. The ability of agrin to cluster AChRs depends on alternative splicing and on cell surface proteoglycans. Neuron. 1993;11:491–502. doi: 10.1016/0896-6273(93)90153-i. [DOI] [PubMed] [Google Scholar]
- Fox MA, Sanes JR, Borza DB, Eswarakumar VP, Fässler R, Hudson BG, John SW, Ninomiya Y, Pedchenko V, Pfaff SL, Rheault MN, Sado Y, Segal Y, Werle MJ, Umemori H. Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell. 2007;129:179–193. doi: 10.1016/j.cell.2007.02.035. [DOI] [PubMed] [Google Scholar]
- Ganju P, Walls E, Brennan J, Reith AD. Cloning and developmental expression of Nsk2, a novel receptor tyrosine kinase implicated in skeletal myogenesis. Oncogene. 1995;11:281–290. [PubMed] [Google Scholar]
- Glass DJ, DeChiara TM, Bowen DC, Stitt TN, Radziejewski C, Bruno J-A, Ryan TE, Gies DR, Shah S, Burden SJ, DiStefano PS, Valenzuela DM, Yancopoulos GD. The receptor tyrosine kinase, MuSK, is required for neuromuscular junction formation in vivo, and is a component of a receptor complex for agrin. Soc Neurosci Abstr. 1996a;22:1476. [Google Scholar]
- Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattsson K, Burden SJ, DiStefano PS, Valenzuela DM, DeChiara TM, Yancopoulos GD. Agrin acts via a MuSK receptor complex. Cell. 1996b;85:513–523. doi: 10.1016/s0092-8674(00)81252-0. [DOI] [PubMed] [Google Scholar]
- Godfrey EW, Nitkin RM, Wallace BG, Rubin LL, McMahan UJ. Components of Torpedo electric organ and muscle that cause aggregation of acetylcholine receptors on cultured muscle cells. J Cell Biol. 1984;99:615–627. doi: 10.1083/jcb.99.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon H, Hall ZW. Glycosaminoglycan variants in the C2 muscle cell line. Dev Biol. 1989;135:1–11. doi: 10.1016/0012-1606(89)90152-8. [DOI] [PubMed] [Google Scholar]
- Grady RM, Akaaboune M, Cohen AL, Maimone MM, Lichtman JW, Sanes JR. Tyrosine-phosphorylated and nonphosphorylated isoforms of alpha-dystrobrevin: roles in skeletal muscle and its neuromuscular and myotendinous junctions. J Cell Biol. 2003;160:741–752. doi: 10.1083/jcb.200209045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ZW. Multiple forms of acetylcholinesterase and their distribution in endplate and non-endplate regions of rat diaphragm muscle. J Neurobiol. 1973;4:343–361. doi: 10.1002/neu.480040404. [DOI] [PubMed] [Google Scholar]
- Herbst R, Burden SJ. The juxtamembrane region of MuSK has a critical role in agrin-mediated signaling. EMBO J. 2000;19:67–77. doi: 10.1093/emboj/19.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt KH, Campbell KP. Assembly of the sarcoglycan complex. Insights for muscular dystrophy. J Biol Chem. 1998;273:34667–34670. doi: 10.1074/jbc.273.52.34667. [DOI] [PubMed] [Google Scholar]
- Hopf C, Hoch W. Tyrosine phosphorylation of the muscle-specific kinase is exclusively induced by acetylcholine receptor-aggregating agrin fragments. Eur J Biochem. 1998;253:382–389. doi: 10.1046/j.1432-1327.1998.2530382.x. [DOI] [PubMed] [Google Scholar]
- Hrabovska A, Bernard V, Krejci E. A novel system for the efficient generation of antibodies following immunization of unique knockout mouse strains. PLoS One. 2010;5:e12892. doi: 10.1371/journal.pone.0012892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing L, Lefebvre JL, Gordon LR, Granato M. Wnt signals organize synaptic prepattern and axon guidance through the zebrafish unplugged/MuSK receptor. Neuron. 2009;61:721–733. doi: 10.1016/j.neuron.2008.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, Dustin ML, Burden SJ. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell. 2008;135:334–342. doi: 10.1016/j.cell.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong XC, Barzaghi P, Ruegg MA. Inhibition of synapse assembly in mammalian muscle in vivo by RNA interference. EMBO Rep. 2004;5:183–188. doi: 10.1038/sj.embor.7400065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan P, Hocking AM, Scholtz JM, Pace CN, Holik KK, McQuillan DJ. Distinct secondary structures of the leucine-rich repeat proteoglycans decorin and biglycan. Glycosylation-dependent conformational stability. J Biol Chem. 1999;274:10945–10950. doi: 10.1074/jbc.274.16.10945. [DOI] [PubMed] [Google Scholar]
- Kummer TT, Misgeld T, Sanes JR. Assembly of the postsynaptic membrane at the neuromuscular junction: paradigm lost. Curr Opin Neurobiol. 2006;16:74–82. doi: 10.1016/j.conb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Lechner BE, Lim JH, Mercado ML, Fallon JR. Developmental regulation of biglycan expression in muscle and tendon. Muscle Nerve. 2006;34:347–355. doi: 10.1002/mus.20596. [DOI] [PubMed] [Google Scholar]
- Legay C. Why so many forms of acetylcholinesterase? Microsc Res Tech. 2000;49:56–72. doi: 10.1002/(SICI)1097-0029(20000401)49:1<56::AID-JEMT7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Lichtman JW, Magrassi L, Purves D. Visualization of neuromuscular junctions over periods of several months in living mice. J Neurosci. 1987;7:1215–1222. doi: 10.1523/JNEUROSCI.07-04-01215.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lwebuga-Mukasa JS, Lappi S, Taylor P. Molecular forms of acetylcholinesterase from Torpedo californica: their relationship to synaptic membranes. Biochemistry. 1976;15:1425–1434. doi: 10.1021/bi00652a012. [DOI] [PubMed] [Google Scholar]
- Marques MJ, Conchello JA, Lichtman JW. From plaque to pretzel: fold formation and acetylcholine receptor loss at the developing neuromuscular junction. J Neurosci. 2000;20:3663–3675. doi: 10.1523/JNEUROSCI.20-10-03663.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall LM, Sanes JR, McMahan UJ. Reinnervation of original synaptic sites on muscle fiber basement membrane after disruption of the muscle cells. Proc Natl Acad Sci U S A. 1977;74:3073–3077. doi: 10.1073/pnas.74.7.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masiakowski P, Yancopoulos GD. The Wnt receptor CRD domain is also found in MuSK and related orphan receptor tyrosine kinases [letter] Curr Biol. 1998;8:R407. doi: 10.1016/s0960-9822(98)70263-5. [DOI] [PubMed] [Google Scholar]
- McEwan PA, Scott PG, Bishop PN, Bella J. Structural correlations in the family of small leucine-rich repeat proteins and proteoglycans. J Struct Biol. 2006;155:294–305. doi: 10.1016/j.jsb.2006.01.016. [DOI] [PubMed] [Google Scholar]
- McMahan UJ, Sanes JR, Marshall LM. Cholinesterase is associated with the basal lamina at the neuromuscular junction. Nature. 1978;271:172–174. doi: 10.1038/271172a0. [DOI] [PubMed] [Google Scholar]
- Megeath LJ, Fallon JR. Intracellular calcium regulates agrin-induced acetylcholine receptor clustering. J Neurosci. 1998;18:672–678. doi: 10.1523/JNEUROSCI.18-02-00672.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado ML, Amenta AR, Hagiwara H, Rafii MS, Lechner BE, Owens RT, McQuillan DJ, Froehner SC, Fallon JR. Biglycan regulates the expression and sarcolemmal localization of dystrobrevin, syntrophin, and nNOS. FASEB J. 2006;20:1724–1726. doi: 10.1096/fj.05-5124fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JE, Beauchamp JR, Pagel CN, Peckham M, Ataliotis P, Jat PS, Noble MD, Farmer K, Partridge TA. Myogenic cell lines derived from transgenic mice carrying a thermolabile T antigen: a model system for the derivation of tissue-specific and mutation-specific cell lines. Developmental Biology. 1994;162:486–498. doi: 10.1006/dbio.1994.1103. [DOI] [PubMed] [Google Scholar]
- Nitkin RM, Smith MA, Magill C, Fallon JR, Yao YM, Wallace BG, McMahan UJ. Identification of agrin, a synaptic organizing protein from Torpedo electric organ. J Cell Biol. 1987;105:2471–2478. doi: 10.1083/jcb.105.6.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa E, Mizuno Y, Hagiwara Y, Sasaoka T, Yoshida M. Molecular and cell biology of the sarcoglycan complex. Muscle Nerve. 2005;32:563–576. doi: 10.1002/mus.20349. [DOI] [PubMed] [Google Scholar]
- Peng HB, Xie H, Rossi SG, Rotundo RL. Acetylcholinesterase clustering at the neuromuscular junction involves perlecan and dystroglycan. J Cell Biol. 1999;145:911–921. doi: 10.1083/jcb.145.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punga AR, Lin S, Oliveri F, Meinen S, Rüegg MA. Muscle-selective synaptic disassembly and reorganization in MuSK antibody positive MG mice. Exp Neurol. 2011;230:207–217. doi: 10.1016/j.expneurol.2011.04.018. [DOI] [PubMed] [Google Scholar]
- Rafii MS, Hagiwara H, Mercado ML, Seo NS, Xu T, Dugan T, Owens RT, Hook M, McQuillan DJ, Young MF, Fallon JR. Biglycan binds to alpha- and gamma-sarcoglycan and regulates their expression during development. J Cell Physiol. 2006;209:439–447. doi: 10.1002/jcp.20740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi SG, Rotundo RL. Localization of “non-extractable” acetylcholinesterase to the vertebrate neuromuscular junction. J Biol Chem. 1993;268:19152–19159. [PubMed] [Google Scholar]
- Rotundo RL. Expression and localization of acetylcholinesterase at the neuromuscular junction. J Neurocytol. 2003;32:743–766. doi: 10.1023/B:NEUR.0000020621.58197.d4. [DOI] [PubMed] [Google Scholar]
- Ryall JG, Schertzer JD, Lynch GS. Cellular and molecular mechanisms underlying age-related skeletal muscle wasting and weakness. Biogerontology. 2008;9:213–228. doi: 10.1007/s10522-008-9131-0. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- Smith CL, Mittaud P, Prescott ED, Fuhrer C, Burden SJ. Src, Fyn, and Yes are not required for neuromuscular synapse formation but are necessary for stabilization of agrin-induced clusters of acetylcholine receptors. J Neurosci. 2001;21:3151–3160. doi: 10.1523/JNEUROSCI.21-09-03151.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanco AM, Werle MJ. Agrin and acetylcholine receptors are removed from abandoned synaptic sites at reinnervated frog neuromuscular junctions. J Neurobiol. 1997;33:999–1018. [PubMed] [Google Scholar]
- Vincent A, McConville J, Farrugia ME, Newsom-Davis J. Seronegative myasthenia gravis. Semin Neurol. 2004;24:125–133. doi: 10.1055/s-2004-829589. [DOI] [PubMed] [Google Scholar]
- Watty A, Neubauer G, Dreger M, Zimmer M, Wilm M, Burden SJ. The in vitro and in vivo phosphotyrosine map of activated MuSK. Proc Natl Acad Sci U S A. 2000;97:4585–4590. doi: 10.1073/pnas.080061997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherbee SD, Anderson KV, Niswander LA. LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development. 2006;133:4993–5000. doi: 10.1242/dev.02696. [DOI] [PubMed] [Google Scholar]
- Williams AH, Valdez G, Moresi V, Qi X, McAnally J, Elliott JL, Bassel-Duby R, Sanes JR, Olson EN. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science. 2009;326:1549–1554. doi: 10.1126/science.1181046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzemann V. Development of the neuromuscular junction. Cell Tissue Res. 2006;326:263–271. doi: 10.1007/s00441-006-0237-x. [DOI] [PubMed] [Google Scholar]
- Wu H, Xiong WC, Mei L. To build a synapse: signaling pathways in neuromuscular junction assembly. Development. 2010;137:1017–1033. doi: 10.1242/dev.038711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Bianco P, Fisher LW, Longenecker G, Smith E, Goldstein S, Bonadio J, Boskey A, Heegaard AM, Sommer B, Satomura K, Dominguez P, Zhao C, Kulkarni AB, Robey PG, Young MF. Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice. Nat Genet. 1998;20:78–82. doi: 10.1038/1746. [DOI] [PubMed] [Google Scholar]
- Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L. LRP4 serves as a coreceptor of agrin. Neuron. 2008;60:285–297. doi: 10.1016/j.neuron.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Glass DJ, Yancopoulos GD, Sanes JR. Distinct domains of MuSK mediate its abilities to induce and to associate with postsynaptic specializations. J Cell Biol. 1999;146:1133–1146. doi: 10.1083/jcb.146.5.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]