

Abstract
Mutations of glutamate dehydrogenase cause the hyperinsulinism/hyperammonemia syndrome by desensitizing glutamate dehydrogenase to allosteric inhibition by GTP. Normal allosteric activation of glutamate dehydrogenase by leucine is thus uninhibited, leading us to propose that children with hyperinsulinism/hyperammonemia syndrome will have exaggerated acute insulin responses to leucine in the postabsorptive state. As hyperglycemia increases β-cell GTP, we also postulated that high glucose concentrations would extinguish abnormal responsiveness to leucine in hyperinsulinism/hyperammonemia syndrome patients. After an overnight fast, seven hyperinsulinism/hyperammonemia syndrome patients (aged 9 months to 29 yr) had acute insulin responses to leucine performed using an iv bolus of l-leucine (15 mg/kg) administered over 1 min and plasma insulin measurements obtained at −10, −5, 0, 1, 3, and 5 min. The acute insulin response to leucine was defined as the mean increase in insulin from baseline at 1 and 3 min after an iv leucine bolus. The hyperinsulinism/hyperammonemia syndrome group had excessively increased insulin responses to leucine (mean ± SEM, 73 ± 21 μIU/ml) compared with the control children and adults (n = 17) who had no response to leucine (1.9 ± 2.7 μU/ml; P < 0.05). Four hyperinsulinism/hyperammonemia syndrome patients then had acute insulin responses to leucine repeated at hyperglycemia (blood glucose, 150–180 mg/dl). High blood glucose suppressed their abnormal baseline acute insulin responses to leucine of 180, 98, 47, and 28 μU/ml to 73, 0, 6, and 19 μU/ml, respectively. This suppression suggests that protein-induced hypoglycemia in hyperinsulinism/hyperammonemia syndrome patients may be prevented by carbohydrate loading before protein consumption.
Recently, we and others described an unusual form of congenital hyperinsulinism, the hyperinsulinism/hyperammonemia syndrome (HI/HA), which is associated with symptomatic hypoglycemia and persistent, asymptomatic hyperammonemia (1, 2). HI/HA is caused by dominantly expressed regulatory mutations of glutamate dehydrogenase (GDH) (3), a mitochondrial matrix enzyme involved in the pathway of leucine-stimulated insulin secretion (4-8). GDH from HI/HA patients has impaired sensitivity to its allosteric inhibitor, GTP, thus causing excessive enzyme activity (3, 9). This finding of GDH mutations in children with HI/HA suggests that strict regulation of GDH is necessary to maintain normal control of insulin secretion. Gao et al. demonstrated that glucose suppresses leucine-stimulated insulin secretion in isolated pancreatic rat islets (10), presumably because the increased energy state of the β-cell that accompanies increased glucose metabolism elevates GTP concentrations, thus inhibiting GDH activity.
As leucine stimulates insulin release by allosterically activating GDH (11-15), the possibility of mutant GDH playing a role in children who were previously described as having leucine-sensitive hypoglycemia seemed plausible. We postulated that because of impaired inhibitory control of GDH, HI/HA patients would hyperrespond to leucine-stimulated insulin secretion. We also hypothesized that elevated plasma glucose would reduce insulin responses to leucine in HI/HA patients. To test these hypotheses, the present studies examined the acute insulin responses to iv bolus injection of leucine (leu-AIR) in HI/HA patients and in children with other forms of congenital hyperinsulinism.
Experimental Subjects
The clinical characteristics of seven patients with HI/HA who were studied are summarized in Table 1. These patients were from six unrelated families. Patients 2 and 5 had de novo mutations, and patients 1, 3, and 6 had familial GDH mutations. Patient 3 was the daughter of patient 6. A parent of patient 4 had a presumed germline mutation because patient 4 had an affected sister (not studied), but neither parent carried the mutation in peripheral blood leukocytes. All of the HI/HA patients had persistently elevated plasma ammonium concentrations (ranging from 40–164 μmol/liter). All were successfully treated with diazoxide to control hypoglycemia. The clinical features of patients 1, 2, and 4 (9) and patients 3, 5, and 6 (16) have been described previously.
TABLE 1.
Patients with HI/HA syndrome
| Patient no. | Gender | Age (yr) | GLUD1 mutation | Plasma ammonium (normal, 9–33 μmol/liter) |
|---|---|---|---|---|
| 1 | F | 1 | Ser448Pro | 100 |
| 2 | F | 3 | Lys450Glu | 80–90 |
| 3 | F | 3 | Arg269His | 40–70 |
| 4 | M | 10 | His454Tyr | 128 |
| 5 | F | 15 | Arg269His | 75–92 |
| 6 | M | 29 | Arg269His | 164 |
| 7 | F | 11 | Pending | 50–80 |
The leu-AIRs in the HI/HA patients were compared with those in a control group consisting of 11 adults and 6 children. The normal adults, aged 19–48 yr, were all healthy; none was obese or had a history of hypoglycemia or diabetes mellitus. The control children, aged 2 months to 5 yr, were undergoing evaluation for suspected hypoglycemia. Four had ketotic hypoglycemia, and 2 were infants who were shown to have normal fasting homeostasis after resolution of transient neonatal hyperinsulinism.
The leu-AIRs were also studied in two disease control groups with other types of hyperinsulinism. One group consisted of seven children, aged 9 months to 13 yr, with hyperinsulinism typical of diazoxide-unresponsive potassium channel (KATP) defects. Six of the seven had known mutations of the KATP channel sulfonylurea receptor (SUR1). Although five had previously undergone partial pancreatectomies, all seven continued to have hyperinsulinemic hypoglycemia. The second disease control group consisted of eight children, aged 1 wk to 15 yr, with nonhyperammonemic hyperinsulinism. In contrast to children with the KATP hyperinsulinism phenotype, these eight children were all diazoxide responsive. Three had transient neonatal hyperinsulinism associated with maternal hypertension or prematurity. The remaining five had persistent congenital diazoxide-responsive hyperinsulinism (CDR), for which the responsible genetic defects have yet to be identified.
Materials and Methods
Leu-AIR tests were performed after a 6- to 12-h fast by administering a 1% solution of l-leucine (15 mg/kg, iv) over 1–2 min. Blood samples for insulin and glucose measurements were obtained from a separate vein at −10, −5, 0, 1, 3, 5, 10, 20, 30, 40, and 60 min relative to the leucine infusion. In small infants, sampling frequency was reduced to −5, 0, 1, 3, and 5 min. The leu-AIR was defined as the mean increase in insulin at 1 and 3 min from the baseline value.
For patients treated with diazoxide, this drug was withheld for at least 5 d. Octreotide or glucagon medications were withdrawn at least 24 h before the study. Dextrose was infused iv as necessary to maintain a plasma glucose range of 60–90 mg/dl before the leucine test.
In four HI/HA patients the leu-AIR test was repeated during hyperglycemia. After the leu-AIR at normal concentrations of blood glucose was completed, 10% dextrose was infused over 30–60 min to raise the blood glucose to 150–180 mg/dl. Once the blood glucose plateaued for 20 min at this blood glucose range, the leu-AIR was repeated. Patient 7 had these leu-AIRs performed separately on 2 consecutive d. In addition, a child with diazoxide-sensitive HI (CDR-3), her affected father, and three normal controls (aged 5–38 yr) underwent leu-AIR testing at hyperglycemia.
Whole blood glucose was measured using a Hemocue glucose analyzer (Hemocue, Inc., Mission Viejo, CA). Plasma insulin concentrations were determined by an ELISA method (ALPCO, Inc., Windham, NH). The lower detection limit of this assay was 3 μU/ml. Plasma insulin concentrations in adult controls were quantified by RIA (Linco Research, Inc., St. Charles, MO); the lower limit of detection was 2.5 μU/ml. Plasma leucine was measured on an amino acid analyzer (Beckman Coulter, Inc., Palo Alto, CA).
An alternate Welch t test was used for statistical analysis (Instat for Macintosh, version 2.00, GraphPad Software, Inc., San Diego, CA).
The research was approved by The Children’s Hospital of Philadelphia institutional review board. Written informed consent was obtained from all subjects or from the parents of children under age 18 yr. Assent was obtained from older children.
Results
Figure 1 illustrates a leu-AIR test in an HI/HA child. After leucine infusion, the plasma leucine concentration increased 10-fold and then returned nearly to baseline within 30 min. Similar changes were found in the four normal adult controls whose leucine concentrations were measured (mean peak plasma leucine, 750 μmol/liter, range, 555-1030 μmol/liter). Plasma insulin concentrations rose 10-fold to a peak of 122 μU/ml in the HI/HA patient. This transient rise in plasma insulin after leucine did not cause hypoglycemia in this patient.
Fig. 1.
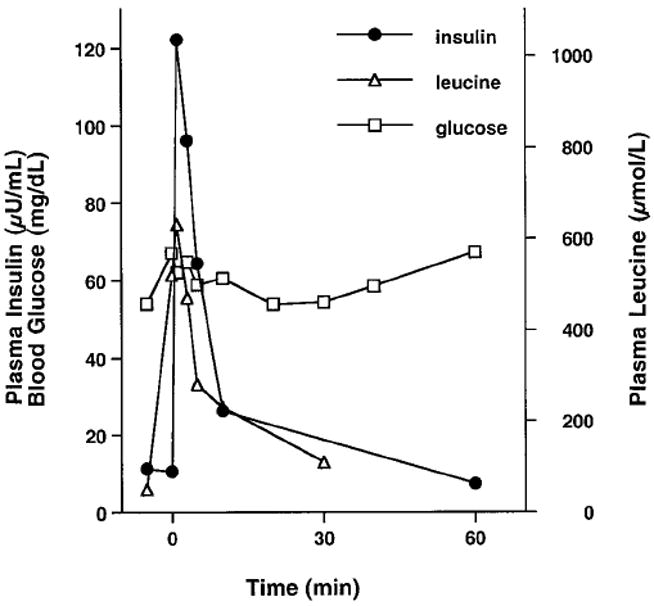
Acute insulin response to stimulation with bolus iv leucine in HI/HA patient 2.
Table 2 summarizes the results of the leu-AIR tests in the seven HI/HA patients. All seven had brisk insulin responses, with leu-AIRs ranging from 16–180 μU/ml. The hyperresponsiveness to leucine stimulation found in HI/HA patients was not affected by age. In contrast, normal controls did not respond to leucine. Responses in reference control children and adults were similar (leu-AIR, 3.1 ± 0.9 and 1.2 ± 0.8 μU/ml, respectively). Baseline blood glucose was somewhat lower in HI/HA patients than in the control groups, but baseline insulin concentrations were similar. The mean leu-AIR in HI/HA children was significantly greater than that in the control group (P < 0.05).
TABLE 2.
Acute insulin responses to stimulation with bolus iv leucine (Leu-AIR) in patients with HI/HA syndrome
| Patient no. | Baseline blood glucose (mg/dl) | Baseline insulin (μU/ml) | Leu-AIR (μU/ml) |
|---|---|---|---|
| 1 | 65 | 13 | 29 |
| 2 | 60 | 11 | 98 |
| 3 | 80 | 7.7 | 72 |
| 4 | 65 | 6.0 | 84 |
| 5 | 67 | 3.0 | 16 |
| 6 | 68 | 20 | 28 |
| 7 | 69 | 20 | 180 |
| Mean ± SEM | 68 ± 6a | 12 ± 3 | 73 ± 21b |
| Controls (n = 17) | 79 ± 2 | 8 ± 5 | 1.9 ± 2.7 |
| Range | −3.4 to 6.5 |
P < 0.01 vs. controls.
P < 0.05 vs. controls.
Table 3 shows the leu-AIRs in children with the KATP channel hyperinsulinism phenotype and in children with diazoxide-responsive hyperinsulinism not associated with hyperammonemia. Children with KATP hyperinsulinism had a mean leu-AIR significantly lower than that of the HI/HA group (P < 0.05). Only two of the seven KATP hyperinsulinism children had leu-AIRs (9 and 13 μU/ml) above the range of the normal control groups. None fell within the range of the HI/HA group. Previous pancreatectomy did not correlate to lack of response to leucine; both KATP hyperinsulinism responders had previously undergone pancreatectomy, whereas the two children who had not undergone pancreatectomy, one of whom was a sibling of a responder, did not respond to leucine (1.7 and 2.5 μU/ml). Four of the five children with CDR without hyperammonemia had abnormal leu-AIRs that ranged from 9–85 μU/ml. One child with a leu-AIR of 45 μU/ml had a maternal aunt and uncle who had previously been reported as having leucine-sensitive hypoglycemia (17). One child with transient neonatal HI had a leu-AIR of 13 μU/ml.
TABLE 3.
Leu-AIR Tests in patients with non-HI/HA forms of hyperinsulinism
| Patient | Plasma ammonium (μmol/liter) | Baseline blood glucose (mg/dl) | Baseline insulin (μU/ml) | Leu-AIR (μU/ml) |
|---|---|---|---|---|
| KATP hyperinsulinism (mean ± SEM; n = 7) | 77 ± 4 | 8.1 ± 1.9 | 4.7 ± 1.9a | |
| Range | 0–13 | |||
| Other forms of hyperinsulinism | ||||
| Transient neonatal (TN) | ||||
| TN-1, 1 wk | 19–41 | 84 | 13 | 13 |
| TN-2, 4 wk | 40 | 109 | 3.0 | 2.5 |
| TN-3, 6 wk | 54 | 82 | 4.9 | 4.8 |
| Congenital, diazoxide-responsive | ||||
| CDR-1, 15 yr | 25–37 | 75 | 7.8 | 2.1 |
| CDR-2, 14 months | 27 | 94 | 9.2 | 9.0 |
| CDR-3, 19 months | 22–39 | 67 | 2.1 | 19 |
| CDR-4, 21 months | 21–33 | 68 | 2.9 | 45 |
| CDR-5, 29 months | 8–24 | 64 | 6.2 | 85 |
P < 0.05 vs. HI/HA patients.
Ten minutes after leucine infusion, HI/HA cases 1 and 6 had blood glucose concentrations of 46 and 42 mg/dl. These cases were two of the earliest studied, and both responded promptly to treatment with dextrose infusions. In the other five HI/HA patients, blood glucose concentrations remained above 54 mg/dl. None of the subjects in the normal or disease control groups became hypoglycemic after administration of the leucine bolus.
To test the effect of glucose on leucine-stimulated insulin secretion, we repeated leu-AIRs in four HI/HA patients. As shown in Fig. 2, progressively smaller insulin responses to leucine occurred with increasing blood glucose in HI/HA patient 6. Insulin responsiveness to leucine stimulation was also suppressed by hyperglycemia in the three other HI/HA patients studied (Table 4). In patients 2 and 6, suppression was complete, whereas in patients 5 and 7, suppression of leu-AIR was only partial. In patient 7 the leu-AIR test at hyperglycemia was performed on a separate day, indicating that glucose suppression of leucine-stimulated insulin secretion is not due to down-regulation by the previous leucine stimulus. In addition, patient CDR-3 and her affected father both had potentiation rather than suppression of their leu-AIR tests during hyperglycemia. Hyperglycemia had no effect on leu-AIR tests in the three normal controls.
Fig. 2.
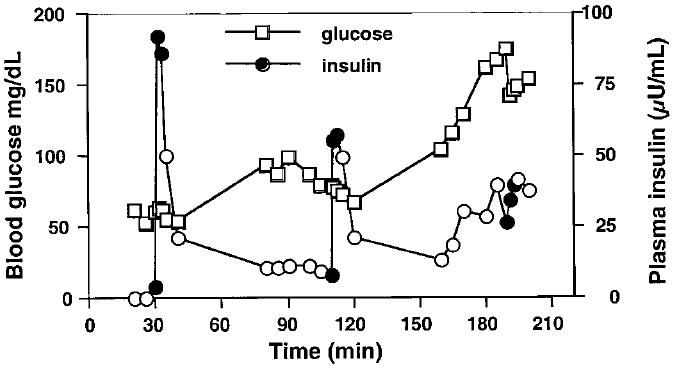
Effect of glucose on leucine-stimulated insulin secretion in HI/HA patient 6. Blood glucose (□) was maintained at 50–65, 70–80, and 160–180 mg/dl with iv dextrose. Leucine was administered at 30, 110, and 190 min. ●, The 0, 1, and 3 min insulin measurements that define the leu-AIR; ○, other insulins.
TABLE 4.
Effect of hyperglycemia on Leu-AIR in HI/HA patients
| Patient no. | Baseline blood glucose (mg/dl) | Baseline Leu-AIR (μU/ml) | Hyperglycemic blood glucose (mg/dl) | Hyperglycemic Leu-AIR (μU/ml) |
|---|---|---|---|---|
| HI/HA | ||||
| 2 | 61 | 98 | 161 | 0 |
| 5 | 67 | 28 | 170 | 19 |
| 6 | 62 | 47 | 160 | 6 |
| 7 | 69 | 180 | 150 | 73 |
| Congenital, diazoxide-responsive HI | ||||
| CDR-3 | 67 | 16 | 150 | 30 |
| CDR-3 father | ||||
| Test 1 | 90 | 19 | ||
| Test 2 | 96 | 0 | 170 | 45 |
| Normals | ||||
| 38 yr | 81 | 6.5 | 165 | 11 |
| 33 yr | 76 | 9 | 180 | −15 |
| 5 yr | 79 | 0 | 145 | 2 |
Discussion
The results of this study indicate that in patients with HI/HA due to mutations of GDH, insulin secretion is hyperresponsive to leucine stimulation. Because of impaired allosteric inhibition of GDH by GTP in these patients, leucine stimulation of glutamate oxidation by GDH is accentuated and leads to exaggerated insulin secretion. This hyperresponsiveness to acute iv leucine injection is consistent with the increased insulin secretion that was reported in a child with the hyperinsulinism/hyperammonemia syndrome after an oral leucine load (1). This patient was subsequently shown to have an S448P GDH mutation (3). We have also documented that patients with HI/HA are susceptible to protein-induced hypoglycemia (18) consistent with the present observation of leucine sensitivity.
The finding of hyperresponsiveness to leucine in children with HI/HA suggests that some patients previously described as having idiopathic leucine-sensitive hypoglycemia of infancy had regulatory mutations of GDH. Numerous cases of leucine-sensitive hypoglycemia have been reported in infants and children since its first description in 1955 by Cochrane et al. (17, 19-21). Since the recognition of hyperinsulinism as the basis for hypoglycemia in children with nesidioblastosis or idiopathic hypoglycemia, children with hyperinsulinism have been assumed to be leucine sensitive. The present results, however, indicate that children with the autosomal recessive KATP channel form of hyperinsulinism are not hyperresponsive to leucine. Even the children without pancreatectomies did not hyperrespond to leucine, indicating that this lack of responsiveness cannot be attributed to pancreatectomy causing insufficient β-cell mass. The absence of leucine responsiveness suggests that low leucine or protein-restricted diets may not be effective in the severe form of hyperinsulinism due to KATP channel mutations.
The results of the present study demonstrate that other forms of hyperinsulinism that have not yet been characterized have leucine sensitivity comparable to that of HI/HA. These forms resemble HI/HA in being diazoxide responsive, suggesting at least partially functional KATP channels, but do not have elevated concentrations of plasma ammonium, suggesting that they involve sites other than GDH. Possible defects in these children could include disorders of KATP channel-independent pathways of insulin secretion that respond to leucine or increased ATP generation through enhanced oxidation of leucine via α-ketoisocaproate. Hypersensitivity of the KATP channel to inhibition by increases in the ATP/ADP ratio is a third possibility, as inhibition of the channel with tolbutamide can induce leucine sensitivity in normal adults (22).
The leu-AIR test used in the present study is both simpler and safer than the oral and iv leucine tolerance tests that have been used previously. The oral leucine tolerance test originally described by Cochrane et al. (19) required the development of hypoglycemia within 1 h for interpretation. Subsequently, an iv variant of the leucine tolerance test was developed using a 75 mg/kg dose infused over 30 min. With this test as well, a positive response required the development of hypoglycemia within 30–60 min (21). Neither the oral nor the iv leucine tolerance test specifically examined insulin responses, and only one series of insulin responses to oral leucine in children with various forms of hypoglycemia has been reported (21). An additional major problem with the oral and iv leucine tolerance tests has been difficulty in obtaining reliable results because of unstable baseline blood glucose concentrations in many children with hyperinsulinism. Most importantly, both tests are hazardous because of the likelihood of provoking severe hypoglycemia in sensitive individuals. In contrast, the leu-AIR test specifically examines insulin responses over a short interval of less than 5 min and can be performed using dextrose infusions to maintain normal blood glucose concentrations. Although two of our initial HI/HA patients developed mild hypoglycemia 10–15 min after leucine infusion, frequent monitoring and infusion of dextrose permitted hypoglycemia to be avoided in subsequent studies. As the changes in plasma insulin and leucine are transient, the leu-AIR test can be performed serially with acute insulin response tests to other secretogogues, such as calcium and tolbutamide, to examine multiple pathways of insulin regulation (23).
Glucose suppression of leucine sensitivity in HI/HA patients suggests that the β-cell phosphate potential plays a critical role in modulating GDH-mediated insulin release in humans and is similar to observations in isolated islets from laboratory animals. The suppression of the leu-AIR by glucose was not an artifact of repetitive stimulation, as it has been shown that repetitive stimulation does not alter the AIRs to glucose (24). In addition, glucose suppression of leucine sensitivity was demonstrated in one HI/HA patient in whom testing was performed on separate days. Glucose suppression of leucine sensitivity may be clinically important, as carbohydrate priming may reduce the risk of protein-induced hypoglycemia, a prominent feature of HI/HA (18).
The results of the present study clearly establish that leucine-sensitive hypoglycemia is a specific component of the HI/HA syndrome and that GDH plays a key role in modulating amino acid-stimulated insulin secretion. Although other specific forms of hyperinsulinism may be associated with leucine hypersensitivity, at least some of the previously reported cases of leucine-sensitive hypoglycemia probably had the HI/HA syndrome due to regulatory mutations of GDH.
Acknowledgments
We thank the patients, the General Clinical Research Center staff, and The Children’s Hospital of Philadelphia nurses without whom this work would not have been possible. We are indebted to Dr. Lester Baker for his contributions to the field of hypoglycemia and his mentorship.
This work was supported in part by NIH Grants T32-DK-07314 (to A.K., R.F., and D.N.) and M01-RR-00240, R01–53012, and R01-DK-56268 (to C.A.S.), Lawson Wilkins Pediatric Endocrine Society Research Fellowships (to A.K.), fellowship grants from Pharmacia & Upjohn, Inc., and Eli Lilly & Co. (to A.K.), and the American Diabetes Association (to C.A.S.).
Abbreviations
- CDR
Congenital diazoxide-responsive hyperinsulinism
- GDH
glutamate dehydrogenase
- HI/HA
hyperinsulinism/hyperammonemia syndrome
- KATP
ATP-sensitive potassium channel
- leu-AIR
acute insulin response to leucine
- SUR1
sulfonylurea receptor
References
- 1.Zammarchi E, Filippi L, Novembre E, Donati MA. Biochemical evaluation of a patient with a familial form of leucine-sensitive hypoglycemia and concomitant hyperammonemia. Metabolism. 1996;45:957–960. doi: 10.1016/s0026-0495(96)90262-0. [DOI] [PubMed] [Google Scholar]
- 2.Weinzimer SA, Stanley CA, Berry GT, Yudkoff M, Tuchman M, Thornton PS. A syndrome of congenital hyperinsulinism and hyperammonemia. J Pediatr. 1997;130:661–664. doi: 10.1016/s0022-3476(97)70256-7. [DOI] [PubMed] [Google Scholar]
- 3.Stanley CA, Lieu YK, Hsu BYL, et al. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med. 1998;338:1352–1357. doi: 10.1056/NEJM199805073381904. [DOI] [PubMed] [Google Scholar]
- 4.Colman RF. Glutamate dehydrogenase (bovine liver) In: Kuby SA, editor. A study of enzymes. New York: CRC Press; 1991. pp. 173–192. [Google Scholar]
- 5.Fahien LA, Kmiotek E. Regulation of glutamate dehydrogenase by palmitoylcoenzyme A. Arch Biochem Biophys. 1981;212:247–253. doi: 10.1016/0003-9861(81)90364-7. [DOI] [PubMed] [Google Scholar]
- 6.Zaleski J, Wilson DF, Erecinska M. Glutamine metabolism in rat hepatocytes. Stimulation by a nonmetabolizable analog of leucine. J Biol Chem. 1986;261:14082–14090. [PubMed] [Google Scholar]
- 7.Zaleski J, Wilson DF, Erecinska M. β-2-Aminobicyclo-(2.2.1)-heptane-2-carboxylic acid. A new activator of glutaminase in intact rat liver mitochondria. J Biol Chem. 1986;61:14082–14090. [PubMed] [Google Scholar]
- 8.Sener A, Malaisse WJ. l-Leucine and a nonmetabolized analogue activate pancreatic islet glutamate dehydrogenase. Nature. 1980;288:187–189. doi: 10.1038/288187a0. [DOI] [PubMed] [Google Scholar]
- 9.Stanley CA, Fang J, Kutyna K, Hsu BYL, et al. Molecular basis and characterization of the hyperinsulinism/hyperammonemia syndrome: predominance of mutations in exons 11 and 12 of the glutamate dehydrogenase gene. Diabetes. 2000;49:667–673. doi: 10.2337/diabetes.49.4.667. [DOI] [PubMed] [Google Scholar]
- 10.Gao Z, Li G, Najafi H, Wolf BA, Matschinsky FM. Glucose regulation of glutaminolysis and its role in insulin secretion. Diabetes. 1999;48:1535–1547. doi: 10.2337/diabetes.48.8.1535. [DOI] [PubMed] [Google Scholar]
- 11.Bryla J, Michalik M, Nelson J, Erecinska M. Regulation of the glutamate dehydrogenase activity in rat islets of Langerhans and its consequence on insulin release. Metabolism. 1994;43:1187–1195. doi: 10.1016/0026-0495(94)90064-7. [DOI] [PubMed] [Google Scholar]
- 12.Sener A, Malaisse-Lagae F, Malaisse WJ. Stimulation of pancreatic islet metabolism and insulin release by a nonmetabolizable amino acid. Proc Natl Acad Sci USA. 1981;78:5460–5464. doi: 10.1073/pnas.78.9.5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gylfe E. Comparison of the effects of leucines, non-metabolizable leucine analogues and other insulin secretogogues on the activity of glutamate dehydrogenase. Acta Diabet Lat. 1976;13:20–24. doi: 10.1007/BF02591577. [DOI] [PubMed] [Google Scholar]
- 14.Fahien LA, MacDonald MJ, Kmiotek EH, Mertz RJ, Fahien CM. Regulation of insulin release by factors that also modify glutamate dehydrogenase. J Biol Chem. 1988;263:13610–13614. [PubMed] [Google Scholar]
- 15.Malaisse WJ. Insulin biosynthesis and secretion in vitro. In: Alberti KGMM, DeFronzo RA, Keen H, Zimmet P, editors. International textbook of diabetes mellitus. Chichester: Wiley & Sons; 1992. pp. 261–283. [Google Scholar]
- 16.MacMullen C, Fang J, Hsu BY, et al. Hyperinsulinism/hyperammonemia syndrome in children with regulatory mutations in the inhibitory binding domain of glutamate dehydrogenase. J Clin Endocrinol Metab. 2001;86:1782–1787. doi: 10.1210/jcem.86.4.7414. [DOI] [PubMed] [Google Scholar]
- 17.DiGeorge AM, Auerbach VH. Leucine induced hypoglycemia: a review and speculations. Am J Med Sci. 1960;99:792–801. [Google Scholar]
- 18.Hsu B, Kelly A, Thornton PS, et al. Protein-sensitive and fasting hypoglycemia in children with the hyperinsulinism/hyperammonemia syndrome. J Pediatr. 2001;138:383–389. doi: 10.1067/mpd.2001.111818. [DOI] [PubMed] [Google Scholar]
- 19.Cochrane WA, Payne WW, Simpkiss MJ, Woolf LI. Familial hypoglycemia precipitated by amino acids. J Clin Invest. 1955;35:411–422. doi: 10.1172/JCI103292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mabry CC, DiGeorge AM, Auerbach VH. Leucine-induced hypoglycemia: clinical observations and diagnostic considerations. J Pediatr. 1960;57:526–538. doi: 10.1016/s0022-3476(60)80080-7. [DOI] [PubMed] [Google Scholar]
- 21.Grant DB. Serum insulin changes following administration of l-leucine to children. Arch Dis Child. 1967;43:69–72. doi: 10.1136/adc.43.227.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fajans SS, Floyd FC, Knopf RF, et al. A difference in the mechanism by which leucine and other amino acids induce insulin release. J Clin Endocrinol Metab. 1967;27:1600–1606. doi: 10.1210/jcem-27-11-1600. [DOI] [PubMed] [Google Scholar]
- 23.Ferry RJ, Jr, Kelly A, Grimberg A, et al. Calcium-stimulated insulin secretion in diffuse and focal forms of congenital hyperinsulinism. J Pediatr. 2000;137:239–246. doi: 10.1067/mpd.2000.107386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koncz L, Soeldner JS, Otto H, et al. Insulin secretory dynamics after two consecutive intravenous stimulations with glucose and/or tolbutamide. Metabolism. 1979;28:1183–1197. doi: 10.1016/0026-0495(79)90130-6. [DOI] [PubMed] [Google Scholar]