

Abstract
Rickets is an important problem even in countries with adequate sun exposure. The causes of rickets/osteomalacia are varied and include nutritional deficiency, especially poor dietary intake of vitamin D and calcium. Non-nutritional causes include hypophosphatemic rickets primarily due to renal phosphate losses and rickets due to renal tubular acidosis. In addition, some varieties are due to inherited defects in vitamin D metabolism and are called vitamin D dependent rickets. This chapter highlights rickets/osteomalacia related to vitamin D deficiency or to inherited defects in vitamin D metabolism. Hypophosphatemic rickets and rickets due to renal tubular acidosis are discussed in other sections of the journal.
Keywords: Osteomalacia, rickets, Vitamin D, Vitamin D deficiency, Vitamin D dependent rickets
INTRODUCTION
Normal bone growth and mineralization depends on the availability of adequate calcium and phosphate. Deficient mineralization can result in rickets and/or osteomalacia. In rickets, there is deficient mineralization at the growth plate, whereas osteomalacia refers to impaired mineralization of the bone matrix. Rickets and osteomalacia usually occur together as long as the growth plates are open as in children; only osteomalacia occurs after the growth plates have fused as seen in adults.[1]
The mineralization defects can be classified as calcipenic (hypocalcemic) rickets caused by calcium deficiency and phosphopenic (hypophosphatemic) rickets caused by phosphate deficiency.[2] Vitamin D is a prohormone that is essential for normal absorption of calcium from the gut, and deficiency of vitamin D is usually more common than either isolated calcium or phosphorus deficiency and is the commonest cause of rickets/osteomalacia.
PATHOGENESIS
There are several types of cells constituting the bone. Osteoblasts are bone forming cells which lay down osteoid. Osteoclasts help in bone remodeling. Osteoid is subsequently mineralized by calcium salts. In rickets, the mineralization defect leads to the accumulation of osteoid in the bone tissue below the growth plate (metaphysis). This leads to weak bones and bowing over a period of time.
CAUSES OF RICKETS/OSTEOMALACIA
The causes of rickets include conditions that lead to hypocalcemia and/or hypophosphatemia, either isolated or secondary to vitamin D deficiency. To determine the optimal treatment, the cause needs to be identified.
Calcipenic rickets
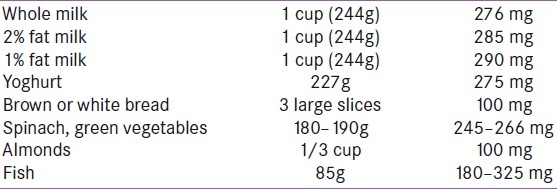
Calcipenic (hypocalcemic) rickets is characterized by deficiency of calcium or more commonly vitamin D. In Indian children living in the UK, diets are typically low in calcium and high in phytate. The calcium content of common foodstuffs is given in Table 1. Rickets can occur despite adequate vitamin D levels if the calcium intake is very low. This problem generally does not occur unless calcium intake is very low because vitamin D increases intestinal calcium absorption. Most children with calcium deficiency rickets have normal serum 25-hydroxyVitamin D [25(OH)D] and high serum 1,25-dihydroxyVitamin D [1,25(OH)2] concentrations, indicating adequate intake of vitamin D. These children may have an increased vitamin D requirement when measured by their response to vitamin D replacement. Thus, vitamin D requirements may be higher than expected in children who are calcium deficient.[3] In addition to this, low dietary calcium intake even without coexisting vitamin D deficiency increases serum 1,25(OH)2D concentrations, which in turn decrease the half-life of 25(OH)D, probably by increasing the catabolism of 25(OH)D. In the majority of South Asian children with rickets, 25(OH)D concentrations are also in the vitamin D deficient range.[4]
Table 1.
Calcium content of various foods
Phosphopenic rickets
Phosphopenic rickets is commonly caused by renal phosphate wasting. This may be isolated or part of a generalized renal tubular disorder such as Fanconi syndrome or Dent disease.[5] Isolated phosphate loss is seen in X-linked hypophosphatemic rickets (XLH), autosomal dominant hypophosphatemic rickets, tumor-induced osteomalacia, and hypophosphatemic rickets with hypercalciuria (autosomal recessive) disease. Nutritional deficiency is uncommon. These causes can be distinguished by measuring urinary amino acids, bicarbonate, glucose, calcium, and vitamin D concentrations.[6]
Another way to classify rickets is as nutritional and non-nutritional. Of the 126 children with non-nutritional (refractory) rickets, 64.3% had distal renal tubular acidosis (DRTA), 20.6% had renal failure,7.1% had proximal RTA, while 6.4% had hypophosphatemic rickets and 1.6% had vitamin D dependent rickets (VDDR).[6] Phosphopenic rickets is discussed in another chapter.
VITAMIN D AND RICKETS
Vitamin D is essential for skeletal health. It promotes differentiation of enterocytes and the intestinal absorption of calcium and phosphorus. This helps in bone mineralization. In conditions of hypocalcemia or hypophosphatemia, vitamin D stimulates bone resorption, thereby maintaining serum levels of calcium and phosphorus. vitamin D deficiency or resistance thus causes hypocalcemia and hypophosphatemia. Hypocalcemia stimulates the release of parathyroid hormone (PTH), which, through its actions on bone and kidney, partially corrects the hypocalcemia but enhances urinary phosphate excretion, leading to hypophosphatemia and osteomalacia. 25(OH)D also plays an important role in extraskeletal health. vitamin D deficiency may be associated with certain immunological conditions such as multiple sclerosis, type 1 diabetes, rheumatoid arthritis, inflammatory bowel disease, mood disorders, and cancers such as breast, prostate, and colon cancer. In adolescents, low serum vitamin D levels are associated with increased risk of hypertension, hyperglycemia, metabolic syndrome, and higher risk of upper respiratory infections. This chapter focuses on the skeletal manifestations of vitamin D deficiency.
VITAMIN D METABOLISM
Vitamin D, or calciferol, refers to a group of lipid-soluble compounds with a four-ringed cholesterol backbone. vitamin D exists as vitamin D2 or vitamin D3. Cholecalciferol, or vitamin D3, is formed by the action of ultraviolet-B (UV-B) radiation (wavelength 290–315 nm) on skin, which converts 7-dehydrocholesterol in epidermal keratinocytes and dermal fibroblasts to pre-vitamin D, which subsequently isomerizes to vitamin D3 in a nonenzymatic manner.[7,8] This is the form of vitamin D found in animal products (fortified milk, fatty fish, cod-liver oil, eggs) and some vitamin D supplements. Ergocalciferol, or vitamin D2, is formed when ergosterol in plants is exposed to sunlight. This is the form of vitamin D found in plant sources, fish, fortified milk, and in most vitamin D supplements [Table 2]. Some cereals and bread products are fortified with vitamin D. Less than 10% of vitamin D comes from natural dietary sources. Dietary vitamin D is fat soluble and is absorbed in the small intestine and incorporated into chylomicrons. Dietary vitamin D is carried to the liver, bound to vitamin D binding protein (DBP) in association with chylomicrons and lipoproteins, where it and endogenously synthesized vitamin D3 are metabolized.[9] vitamin D is then bound by the DBPs and transported via blood to the target organs. vitamin D is then hydroxylated in the liver to 25(OH)D (calcidiol). 25(OH)D, complexed with its plasma carrier, the DBP, is filtered through the glomerulus and reabsorbed in the proximal tubules by the endocytic receptor megalin.[10] Calcidiol is hydroxylated primarily in the kidney to 1,25(OH)2D (calcitriol), which is the most active form.[11] The plasma 1,25(OH)2D concentration depends on the availability of 25(OH)D and the activities of the renal enzymes 1-α-hydroxylase and 24-α-hydroxylase. The renal 1-α-hydroxylase enzyme is primarily regulated by PTH, serum calcium and phosphate concentrations, and fibroblast growth factor 23 (FGF23).[9] Increased PTH, calcitonin, and hypophosphatemia stimulate the enzyme and enhance 1,25(OH)2D production, while high calcium, hyperphosphatemia, and 1,25(OH)2D inhibit it.[12] 1,25(OH)2D inhibits the synthesis and secretion of PTH, providing negative feedback regulation of 1,25(OH)2D production. 1,25(OH)2D synthesis may also be modulated by vitamin D receptors on the cell surface; downregulation of these receptors may play an important role in regulating vitamin D activation.[13] FGF23, a newly described phosphaturic hormone, inhibits renal production of 1,25(OH)2D by inhibiting 1-α-hydroxylase in the renal proximal tubule and by simultaneously increasing the expression of 24-α-hydroxylase and production of 24,25(OH)2D (an inactive metabolite).[14] 1,25(OH)2D stimulates FGF23, creating a feedback loop. FGF23 decreases renal reabsorption of phosphate, and thereby counteracts the increased gastrointestinal phosphate reabsorption induced by 1,25(OH)2D, maintaining phosphate homeostasis.[15]
Table 2.
Vitamin D content of common foods

When hypocalcemia occurs, serum PTH concentration increases and enhances tubular reabsorption of calcium, as well as the activity of 1-α-hydroxylase in the kidney. This results in increased 1,25(OH)2D production, and in turn, intestinal calcium absorption. PTH also stimulates bone osteoclast activity to mobilize bone calcium stores, thereby increasing serum calcium. Both 1,25(OH)2D and 25(OH)D are degraded in part by being hydroxylated at the 24 position by a 24-hydroxylase. The activity of the 24-hydroxylase gene is increased by 1,25(OH)2D (which therefore promotes its own inactivation) and reduced by PTH (thereby allowing more active hormone to be formed).[9] Estrogen, placental growth hormone, and prolactin may also regulate vitamin D metabolism, playing a role during pregnancy to meet increased calcium demands. 1,25(OH)2D is also formed in some other tissues, but is used only within the tissues and not circulated. PTH- independent extrarenal production of 1,25(OH)2D from 25(OH)D is by activated macrophages in the lung and lymph nodes. The 1-α-hydroxylase enzyme is also expressed at other extrarenal sites, including the gastrointestinal tract, skin, vasculature, mammary epithelial cells, and in osteoblasts and osteoclasts.[16]
CAUSES OF VITAMIN D DEFICIENCY
Reduced cutaneous synthesis
Vitamin D deficiency can occur in people who live without sun exposure.
Sun exposure
Dietary vitamin D deficiency can also occur in children, with differences among ethnic groups depending on skin pigmentation and varied ingestion of supplements.[17] A Caucasian infant's vitamin D requirements are met by exposure to sunlight for 30 minutes per week, clothed only in a diaper, or for 2 hours per week fully clothed with no hat. Asians require approximately threefold longer periods of sunlight exposure because of the protective pigmentation in their skin and Africans need six times the same exposure.[18] Current American Academy of Pediatrics (AAP) recommendations include keeping infants <6 months old out of direct sunlight, selecting children's activities that minimize sunlight exposure, and using protective clothing and sunscreen. These recommendations are acceptable for light-skinned children in lower latitudes, particularly in the summer months. However, the effects of UV-B exposure on dark-skinned children who live in higher latitudes need to be explored. Above 37° north latitude, in the winter months, the number of UV-B photons reaching the earth's atmosphere is decreased by 80–100%, and as a consequence, little vitamin D3 is produced in the skin. Sunscreen absorbs UV-B and some UV-A light and prevents it from reaching and entering the skin. A sunscreen with a sun protection factor (SPF) of 8 can decrease vitamin D3 synthetic capacity by 95%, and SPF 15 can decrease it by 98%. For adequate vitamin D synthesis, exposure to the midday sun (between 1000 and 1500 hours) for 10–15 minutes in the spring, summer, and fall is considered sufficient for light-skinned people. After this extent of exposure, application of a sunscreen with an SPF of 15 is recommended to prevent the damaging effects of chronic excessive exposure to sunlight. Whole body clothing as noted in some cultures prevents adequate sun exposure, i.e. purdah system in Muslim community. Cloud cover, increasing water vapor, and industrial pollution can reduce the amount of UV-B that reaches the earth's surface, and industrial pollution has been associated with a greater prevalence of vitamin D deficiency rickets. This is because after prolonged UV-B radiation exposure, the vitamin D made in the skin is further degraded to the inactive vitamin D metabolites, tachysterol and lumisterol.
Cold climates
Vitamin D deficiency is also common at the end of the winter due to less sun exposure.[19] Vitamin D deficiency has been reported in dark-skinned immigrants from warm climates to cold climates. Asian Indian immigrants to the United States may have vitamin D deficiency, even with adequate sun exposure.[20]
Extensive burns
In patients with a history of extensive burn injuries, vitamin D synthesis in skin is below normal, even with sun exposure.[21]
Nutritional deficiency
Vitamin D deficiency can occur even with adequate sun exposure.[22] It can occur in people who consume foods that are not fortified with vitamin D or if there is intestinal malabsorption of vitamin D. There are few foods that naturally contain vitamin D and because most of these are meat or fish based, they may not be acceptable to cultures that favor a vegetarian diet. Currently, few foods are fortified with vitamin D. Routine vitamin D fortification should be considered for milk and other food products.
Elderly people
Cutaneous vitamin D production and vitamin D stores decline with age. vitamin D intake is often low in older subjects. Achlorhydria, which is common in the elderly, limits calcium absorption. Older persons, in addition, may also be confined indoors.[23]
Maternal vitamin D deficiency
Vitamin D is transferred from the mother to the fetus across the placenta, and reduced vitamin D stores in the mother are associated with lower vitamin D levels in the infant.[24] Low vitamin D levels during pregnancy have been associated with intrauterine growth retardation, premature labor, and hypertension, all of which increase the risk of low birth weight.
Prematurity
Vitamin D levels are low in premature infants, who have less time to accumulate vitamin D from the mother through transplacental transfer. The third trimester is a critical time for vitamin D transfer because this is when the fetal skeleton becomes calcified, requiring increased activation of 25(OH)D to 1,25(OH)2D in the maternal kidneys and placenta. vitamin D deficiency in the mother during this period can cause fetal vitamin D deficiency, and in severe cases, fetal rickets.[24,25] The premature infants are more likely to have enamel defects in both primary and permanent teeth because vitamin D sufficiency is necessary for normal fetal tooth development.
Exclusive breast feeding
The vitamin D content of breast milk is low (15–50 IU/L) even in a vitamin D replete mother.[26] Exclusively breastfed infants consuming an average of 750 mL of breast milk daily thus ingest only 10–40 IU/day of vitamin D. Most breastfed infants need to be exposed to sunlight for at least 30 minutes/ week while wearing only a diaper in order to maintain 25(OH)D levels at >20 ng/mL. vitamin D deficiency is uncommon in formula-fed infants because of the fortification of infant formulas (400 IU/L). However, it can still occur if the infant had low vitamin D stores at birth because of maternal vitamin D deficiency and if the vitamin D content of the formula is insufficient.[27]
Obesity
25(OH)D levels are low in obese individuals as vitamin D is sequestrated in fat. vitamin D requirements are thus higher in obese individuals.[28]
Hospitalized patients
Inadequate intake and lack of sun exposure cause vitamin D deficiency in this group of patients.[29]
Women treated for osteoporosis
Subclinical vitamin D deficiency is common in postmenopausal women on therapy for osteoporosis (bisphosphonates, raloxifene, calcitonin, or PTH).[30]
Renal diseases
Chronic renal disease
In patients with chronic kidney disease, calcitriol [1,25(OH)2D] production is low due to diminished glomerular filtration, loss of the 1-α-hydroxylase enzyme secondary to structural renal damage, and suppression of enzyme activity secondary to hyperphosphatemia. This results in hypocalcemia, hyperparathyroidism, and bone disease.[31] In addition to the deficiency of 1,25(OH)2D, recent studies have shown co-existence of 25(OH)D deficiency in pre-dialysis and dialysis patients, especially in female diabetics and patients on peritoneal dialysis.[32] Whether improving 25(OH) D concentrations benefits these patients is controversial, but Kidney Dialysis Outcome Initiative (KDOQI) guidelines have recommended supplementation.
Nephrotic syndrome
Most of the calcidiol in serum is bound to DBP. Patients with nephrotic syndrome lose DBP and may develop vitamin D deficiency.[33,34]
Distal renal tubular acidosis
Hypocalcemia can occur in dRTA, along with acidosis, which causes rickets. The presence of dRTA is suspected in conditions of hypokalemia, hyperchloremia, and nomal anion gap metabolic acidosis.[35] (RTA is discussed in another chapter.)
Gastrointestinal disease
Malabsorption associated with diseases of the small intestine, hepatobiliary tree with cholestatic liver disease, extrahepatic biliary obstruction, and diseases of pancreas may result in decreased absorption of vitamin D and/or depletion of endogenous 25(OH)D stores due to abnormal enterohepatic circulation. Malabsorption of vitamin D occurs as a consequence of steatorrhea, which disturbs fat emulsification and chylomicron-mediated absorption. Patients may have rickets or osteomalacia or only low bone density. Common examples are celiac disease, inflammatory bowel disease, food allergies, cholestasis, and exocrine pancreatic insufficiency (as in cystic fibrosis).[36]
Gastric bypass
Patients with short-limb bypass have secondary hyperparathyroidism (SHPT) in spite of normal 25(OH)D concentrations, due to calcium malabsorption. vitamin D deficiency also occurs in patients with partial or total gastrectomy for peptic ulcer disease or bariatric surgery due to the loss of gastrointestinal acidity, malfunction of the proximal small bowel which leads to vitamin D malabsorption, absence of adequate absorbing surface, or failure of intestinal mucosal cells to respond to vitamin D.[36,37]
Liver disease
Vitamin D is hydroxylated in the liver to produce calcidiol [25(OH)D]. Hence, patients with significant parenchymal or obstructive liver disease have reduced 25(OH)D. These patients manifest biochemical or histological evidence of osteomalacia only in the presence of concomitant nutritional deficiency or interruption of the enterohepatic circulation.[36,37]
Medications
Certain anticonvulsants and antiretroviral drugs used to treat HIV infection can precipitate vitamin D deficiency by enhancing catabolism of 25(OH)D and 1,25(OH)2D.[38] decreased circulating levels of calcidiol occur in patients on phenytoin, phenobarbitone, carbamazepine, isoniazid, rifampicin, and theophylline due to induction of P-450 enzyme activity, which metabolizes calcidiol to inactive vitamin D metabolites. Tenofovir can cause rickets. Abnormalities in calcium concentration are seen with medications used in the treatment of the complications of HIV, such as foscarnet, pentamidine, and recombinant growth hormone. vitamin D requirements are higher in patients on glucocorticoids because they inhibit intestinal vitamin D dependent calcium absorption. Ketoconazole and some other antifungal agents increase vitamin D requirements because they block 1-hydroxylation. Supplementation with vitamin D (400–4000 IU/day) may be needed for these patients.[39]
CLINICAL MANIFESTATIONS
The manifestations of rickets are initially seen at the distal forearm, knee, and costochondral junctions which are the sites of rapid bone growth. Sometimes, skeletal dysplasias may be mistaken for rickets as the clinical features of rickets and skeletal dysplasia with metaphyseal involvement can be similar; however, serum inorganic phosphorus and PTH concentrations usually are normal in children with skeletal dysplasia.
Skeletal findings
The skeletal changes are similar in calcipenic and phosphopenic rickets.[40] The anterior fontanelle closes by 18 months and posterior by 3 months normally. However, in rickets, there is a delay in the closure of the fontanelles. There is parietal and frontal bossing, craniotabes (soft skull bones) with ping pong bones in infants, enlargement of the costochondral junction of ribs (the “rachitic rosary”), Harrison sulcus due to the muscular pull of the diaphragm on the lower ribs, widening of the wrist and bowing of the distal radius and ulna, and progressive lateral bowing of the femur and tibia. Widening of ankle (double malleoli) can be seen. The age of the child and weight bearing determine the type and the site of deformities. In infants, the deformities are seen in forearm bones and the tibia. In toddlers, there is exaggeration of normal physiological bowing of the legs (genu varum). Older children have either genu valgum [Figure 1] or windswept deformity of the lower limbs (genu varum on one side and valgum on the other). There may be kyphosis or scoliosis. In adults, the bony deformities are unusual; however, in females there may be triradiate pelvis which makes normal vaginal delivery difficult.
Figure 1.
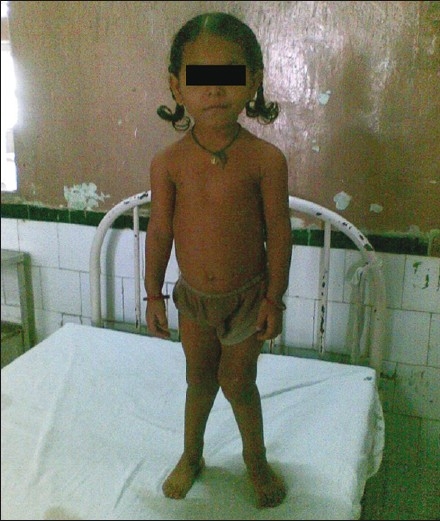
Bony deformities in rickets
Extraskeletal findings
The child may be asymptomatic or may present with pain, irritability, delay in motor milestones, and poor growth.[41] Visceroptosis leads to pot belly. Children may have waddling gait (antalgic gait). Presentation with hypocalcemic seizures is frequent in the first year of life.[41] Children with calcipenic rickets are prone to acquiring infectious diseases.[42] There is hypoplasia of dental enamel. Increased sweating is a common finding in young infants with calcipenic rickets and may be caused by bone pain. Phosphopenic rickets is characterized by predominantly lower limb deformities and dental abscesses. Children may present with irritability or paresthesias. In adolescents and adults, the growth plate is fused, and hence bony deformities are not seen. Thus, most adult patients may be asymptomatic. Sometimes, osteomalacia in adults starts insidiously as aches and pains in the lumbar (lower back) region and thighs, spreading later to the arms and ribs. The pain is symmetrical, non-radiating, and accompanied by tenderness in the involved bones. Proximal muscles are weak, and there is difficulty in climbing up stairs and getting up from a squatting position. Pathological fractures may develop due to weight bearing.
LABORATORY INVESTIGATIONS
The evaluation of a patient with clinical signs of rickets includes a dietary history, medication history, and history of liver or renal disease and malabsorption. The laboratory tests which aid in determining etiology include the following.
Radiographic findings
In children, the changes of rickets can be seen at the growth plate of rapidly growing bones. Thus, in the upper limbs, the changes are most prominent at the distal ulna, while in lower limbs the changes are most prominent at the metaphyses above and below the knees. There is widening of the epiphyseal plate due to unmineralized osteoid and loss of definition of the zone of provisional calcification at the epiphyseal/metaphyseal junction. There is cupping and splaying of the epiphyseal end of metaphysis with formation of cortical spurs and stippling. The appearance of the epiphyseal bone centers may be delayed, and these are small and osteopenic.[43] In children as well as adults, the shafts of the long bones are osteopenic and the cortices become thin. The trabecular pattern is fuzzy, coarse, and has a ground-glass appearance. Deformities of the shafts of the long bones are present and, in severe rickets, pathological fractures and looser zones (Milkman's fractures) may be noted.[44] Looser zones are pseudofractures, narrow radiolucent lines, 2–5 mm wide, with sclerotic borders, and are a characteristic radiologic finding in osteomalacia. They are bilateral and symmetric and lie perpendicular to the cortical margins of bones. They are found at the femoral neck, on the medial part of the femoral shaft, immediately under the lesser trochanter, or on the pubic and ischial rami. They also may occur in the ulna, scapula, clavicle, rib, and metatarsal bones. The term “Milkman syndrome” refers to the combination of multiple, bilateral, and symmetric pseudofractures in a patient with osteomalacia. Pseudofractures also can be seen as hot spots on bone scans. In the absence of looser zones, osteopenia may be the only finding and osteomalacia may be confused with osteoporosis. Looser zones have been postulated to represent either stress fractures that have been repaired by the laying down of inadequately mineralized osteoid or erosion of bone by arterial pulsations, since they often lie in apposition to arteries. There may be biconcave vertebral bodies (cod fish vertebrae). Skeletal changes due to longstanding SHPT are less frequent than the above abnormalities. They include subperiosteal resorption of the phalanges, bone cysts, and resorption of the distal ends of long bones such as the clavicle and humerus. More severe osteomalacia can lead to shortening and bowing of the tibia, pathologic fractures, and coxa profunda hip deformity.
Bone mineral density
Several studies have demonstrated markedly reduced spine, hip, and forearm bone density [as measured by dual-energy X-ray absorptiometry (DXA)] in patients with osteomalacia related to vitamin D deficiency. However, bone mineral density (BMD) is not required for the diagnosis of osteomalacia, and reduced BMD does not distinguish osteoporosis from osteomalacia. In contrast, BMD tends to be normal or increased (especially lumbar spine) in adults with XLH.
Bone biopsy
Bone biopsy with tetracycline labeling is the most accurate way to diagnose osteomalacia/rickets. However, it is infrequently performed because it is invasive and the diagnosis can usually be made from a combination of clinical and laboratory findings. The histomorphometric characteristics of osteomalacia include: Prolonged mineralization lag time, widened osteoid seams, and increased osteoid volume. All of these features are necessary for the diagnosis because other disorders may show one of these findings. Wide osteoid seams reflecting high turnover can be seen with hyperthyroidism, Paget's disease, and hyperparathyroidism. However, the mineral apposition rate is elevated in these disorders in contrast to osteomalacia.
Alkaline phosphatase (ALP) is an excellent marker of disease activity because it participates in the mineralization of bone and growth plate cartilage. ALP levels are usually 500 IU/L in neonates and 1000 IU/L in children up to 9 years of age and decrease after puberty. Serum ALP concentrations are elevated in both hypocalcemic and hypophosphatemic rickets.[45] ALP may be used to screen for rickets, with the caveat that rickets has sometimes been reported with normal ALP levels. When a high index of suspicion exists, a wrist or knee radiograph should be obtained.
Serum phosphorus concentrations usually are low in both calcipenic and phosphopenic rickets.[46]
The serum calcium concentration is normal in phosphopenic rickets. Serum calcium levels are usually low in calcipenic rickets, but may be normal in some stages of the disease due to a compensatory increase in PTH.[46]
The total reabsorption of phosphorus (TRP) and the maximal tubular reabsorption of phosphorus per glomerular filtration rate (TmP/GFR) usually are decreased in both types of rickets.
The serum concentration of PTH is elevated in calcipenic rickets. In contrast, with rare exception, PTH concentrations are normal in phosphopenic rickets. Hyperparathyroidism is associated with aminoaciduria which, therefore, is not seen in familial hypophosphatemia rickets (FHR). The diagnostic approach to suspected rickets is to use measurements of serum inorganic phosphorus (Pi) and PTH to distinguish calcipenic from phosphopenic rickets.[46]
Serum creatinine is high and GFR may be low in rickets associated with renal failure. Here, the phosphate level is high. Urine calcium levels are high in hypophosphatemic rickets with hypercalciuria and low in other varieties. Other lab investigations include urine pH, urine phosphorus, glucose, serum creatinine, liver enzymes, and arterial blood gas.[46]
For children with phosphopenic rickets, the causes can be distinguished by measuring urinary amino acids, bicarbonate, glucose, and calcium concentrations.
For children with calcipenic rickets, measurements of serum 25(OH)D help to distinguish rickets caused by vitamin D deficiency (the most common form) from other causes of calcipenic rickets. Serum concentration of 25(OH)D reflects the amount of vitamin D stored in the body and, consequently, is low in vitamin D deficiency, while it is normal or slightly increased in the other forms. 1,25(OH)2D can be low, normal, or increased in calcipenic rickets. 1,25(OH)2D levels initially increase in response to rising levels of PTH, but may subsequently decrease because its substrate, 25(OH)D, is limited. 1,25(OH)2D is increased in VDDR type II and hypophosphatemic rickets.
Vitamin D levels
The best way to assess vitamin D status is to measure 25(OH) D levels. As 25(OH)D levels fall, intestinal absorption of calcium falls, leading to a decrease in serum calcium. This causes a rise in serum PTH, which stimulates conversion of 25(OH)D to 1,25(OH)2D, and thereby maintains absorption of calcium.[47] Thus, optimal level of 25(OH) D is defined as the level which causes maximal suppression of PTH and maximum calcium absorption. The optimal 25(OH) D concentration may also be defined clinically, such as level needed for fracture reduction. There is no consensus on the optimal 25(OH) D concentration for skeletal health. The Institute of Medicine (IOM) supports 25(OH)D concentrations above 20 ng/mL. National Osteoporosis Foundation, International Osteoporosis Foundation, and American Geriatric Society suggest that a minimum level of 30 ng/mL is necessary in older adults to minimize the risk of falls and fracture. All agree that levels lower than 20 ng/mL are suboptimal for skeletal health.[48,49] The optimal serum 25(OH)D level for extraskeletal health is not established. A safe upper serum level has also not been determined. There is an increased risk for fractures and some cancers (e.g. pancreatic, prostate) and mortality with levels above 30–48 ng/mL.[50] The IOM systematic review favors maintaining the serum 25(OH)D concentration between 20 and 40 ng/mL, whereas others favor maintaining 25(OH)D levels between 30 and 50 ng/mL.
Currently accepted standards for defining vitamin D status in children and adolescents are: vitamin D sufficiency: 25(OH)D ≥ 20 ng/mL; vitamin D insufficiency: 25(OH)D between 15 and 20 ng/mL; and vitamin D deficiency: 25(OH)D ≤ 15 ng/mL. Severe deficiency is defined as a level <5 ng/mL. These cut-offs may need to be revised if future pediatric studies demonstrate efficacy of higher 25(OH)D level. Individuals with 25(OH)D levels of 100 ng/mL have been arbitrarily designated as having vitamin D excess and above 150 ng/mL is considered to be intoxication.
Vitamin D assays
The level of 25(OH)D is the best indicator of vitamin D status and stores. 25(OH)D is the main circulating form of vitamin D and has a half-life of 2–3 weeks. 1,25(OH)2D has a shorter half-life of about 4 hours, circulates in much lower concentrations than 25(OH)D, and is tightly regulated by PTH, calcium, and phosphate, such that it does not decrease significantly until vitamin D deficiency is severe. Also, its concentration correlates more with kidney function than with vitamin D deficiency. Hence, it is not useful for assessment of vitamin D stores. The vitamin D assay should measure both the D2 and D3 derivatives of 25(OH) D because both derivatives are biologically active after 1-α-hydroxylation. There are many commercially available 25(OH)D assays used for the determination of vitamin D status. These include chemical assays [high-pressure liquid chromatography (HPLC) and mass spectrometry (MS)] and binding assays – immunoassays [i.e. radioimmunoassay (RIA),[51] enzyme immunoassay (EIA), chemiluminescent immunoassay (CLIA)] and protein-binding assays [i.e. competitive protein-binding assays (CPBA) and automated chemiluminescence protein- binding assays (CLPBA)]. Variability among assays is an important problem. HPLC and liquid chromatography-mass spectroscopy (LC-MS) are the gold standards for vitamin D assay and give a higher vitamin D level. Immunoassays and protein-binding assays report total value. Chemical assays can report both vitamin D2 and d3.
PREVALENCE OF VITAMIN D DEFICIENCY
Overt vitamin D deficiency, characterized by hypocalcemia or hypophosphatemia and rickets or osteomalacia, is now uncommon in most developed countries. However, subclinical vitamin D deficiency is associated with osteoporosis, increased risk of falls, and possibly fractures. The prevalence of vitamin D deficiency varies with the definition used (<20 or <30 ng/mL). In the National Health and Nutrition Examination Survey (NHANES) 2000–2004, >30% of participants of age 12 years and older had 25(OH)D levels below 20 ng/mL.[52] In India, there is high prevalence of vitamin D deficiency, with a study reporting 90% prevalence in adults and 84% prevalence in pregnant women.[53] The prevalence of low vitamin D levels is increasing in the general population. Data from NHANES showed a decrease in mean 25(OH)D concentration from 30 to 24 ng/mL between 1988 and 2004. This may be due to assay changes, changes in milk intake, use of sun protection, and changes in body mass index (BMI).[54]
OPTIMAL INTAKE
Estimates of vitamin D requirements vary and depend in part on sun exposure and the standards used to define a deficient state. The IOM committee assumed minimal sun exposure when establishing the dietary reference intakes for vitamin D and identified a 25(OH)D level of at least 20 ng/mL as necessary to meet the needs of at least 97.5% of the population. The dietary reference intakes were based upon data examining the beneficial effects of calcium and vitamin D on skeletal health and not on extraskeletal health.
Adults
According to the recommendations given by the IOM in 2010, the Recommended dietary Allowance (RDA) of vitamin D for adults from 18 to 70 years is 600 IU.[55] Older persons (>71 years) confined indoors and other high-risk groups require higher intakes(RDA 800 IU).
Infants
The AAP and Lawson Wilkins Pediatric Endocrine Society recommend that all infants, including those who are exclusively breastfed, and older children and adolescents should have a minimum daily intake of 400 IU/day of vitamin D, which should be initiated within a few days after birth.[56] Most infant formulas contain at least 400 units/L of vitamin D, so formula-fed infants will also require supplementation to meet this goal unless they consume at least 1000 mL daily of formula milk. Another strategy to raise vitamin D levels in exclusively breastfed infants is to administer high doses of vitamin D (4000– 6400 IU/ day) to the lactating mother. This intervention sufficiently increases the vitamin D content of breast milk to allow sufficient vitamin D intake by the infant without causing hypervitaminosis d in the mother. More moderate vitamin D doses (e.g. maternal intake of 1500–2000 IU daily during pregnancy and lactation) are generally sufficient to maintain blood levels of vitamin D of >30 ng/mL in the mother and will improve the infant's vitamin D status at birth. However, these doses may not result in sufficient vitamin D in breast milk to meet the infant's needs, and supplementation may still be necessary for the infant.
Children
The intake of 200 IU daily was initially recommended to ensure serum 25(OH)D levels >11 ng/mL. This target serum level was later considered inadequate because these levels are not sufficient for preventing all cases of florid rickets. AAP then recommended vitamin D intake of at least 400 units/day for children who do not consume at least 1 L of vitamin D fortified milk daily.[56] The IOM guidelines issued in 2010 raised the RDA for vitamin D to 600 IU daily for healthy children aged 1–18 years.[57] The AAP has not as yet revised the previous guidelines. Children who are obese and those on anticonvulsants, glucocorticoids, and medications for HIV infection require higher doses.
Pregnant and lactating mothers
Intake of 600 IU is recommended. However, some studies suggest that higher intakes may be necessary to maintain normal levels of 25(OH)D during pregnancy and lactation.[57]
In 2010, the IOM defined the safe upper limit for vitamin D as 2500 IU/day for ages 1–3 years, 3000 IU/day for ages 4–8 years, and 4000 IU/day for ages 9–71+ years (including pregnant or lactating women).
SCREENING FOR VITAMIN D DEFICIENCY
It is not necessary to perform universal screening of serum 25(OH)D levels in the general population. In individuals who are in the high-risk groups described above, it is appropriate to measure serum 25(OH)D to supplement with the amount estimated to reach the target 25(OH)D level, and then to measure again 3–4 months later to verify that the target has been achieved.[56] Patients who present with nonspecific musculoskeletal pain should also be screened for vitamin D deficiency. Children with elevated levels of serum ALP (e.g. >500 IU/L in neonates or >1000 IU/L in children up to 9 years of age) should be screened.[58]
PREPARATIONS OF VITAMIN D
Multiple preparations of vitamin D are available. vitamin D, rather than its metabolites, is used as it is cheap and effective. The two common forms of vitamin D supplements are ergocalciferol and cholecalciferol. Some studies suggest that cholecalciferol (Vitamin D3) increases serum 25(OH) D more efficiently than does ergocalciferol (Vitamin D2).
Vitamin D3 is available in 400, 800, 1000, 2000, 5000, 10,000, and 60,000 unit capsules. It is available in some countries as an intramuscular injection (Arachital 6 lakhs units, which maintains vitamin D levels for 1 year). However, it can be extremely painful.
Vitamin D2 (ergocalciferol) is available for oral use in 400 and 50,000 unit capsules or in a liquid form (8000 units/mL).
Vitamin D metabolites
Vitamin D metabolites can be used to treat vitamin D deficiency, particularly when there is abnormal vitamin D metabolism (renal or liver disease). The recommended preparation and dose vary with the clinical condition.
Calcidiol: Calcidiol [25(OH)D] is available in capsules of 800 and 2000 IU. It does not require hepatic 25-hydroxylation, and is therefore most useful in patients with liver disease. The onset of action is more rapid and the half-life of 2–3 weeks is shorter than that of Vitamin D3 and similar to that of Vitamin D2. Vitamin D deficiency in patients with severe liver disease can be treated with calcidiol (2000–8000 IU/day). Calcitriol may be used in patients with severe liver disease who remain deficient after treatment with Vitamin D2 or Vitamin D3.
Calcitriol [1,25(OH)2D] is available in capsules of 0.25 and 0.5 mg and also as injection. It is most useful in those with decreased synthesis of calcitriol, as in chronic kidney disease or in type I and type II VDDR. Calcitriol has a rapid onset of action and its half-life is only 6 hours, is expensive, and does not build up vitamin D stores. Hence, calcitriol is not preferred for stoss therapy or for nutritional deficiency. It is associated with a high incidence of hypercalcemia, so the serum calcium should be monitored carefully. While using calcitriol as a supplement, 25(OH)D levels do not indicate clinical vitamin D status.
Doxercalciferol is used in the treatment of SHPT due to chronic kidney disease. It is the first pro-Vitamin D2 to be available in India as 0.5–2.5 mg capsules.
Dihydrotachysterol (DHT): DHT is a synthetic vitamin D analog activated in the liver, which does not require renal hydroxylation. dHT has a rapid onset of action (2 hours), a shorter half-life, and a greater effect on mineralization of bone salts than does vitamin D. dHT is available as tablets of 0.125, 0.2, and 0.5 mg. It is functionally equivalent to 1a-hydroxy vitamin D. It requires hepatic 25-hydroxylation prior to becoming therapeutically active. dHT can only treat hypocalcemia associated with vitamin D deficiency, but does not build up vitamin D stores. dHT can be used in the disorders for which calcitriol is used.
19-nor-1,25-dihydroxyvitamin D2 (paricalcitol), an analog of 1,25 dihydroxy ergocalciferol, the active form of vitamin D2, can be used in intravenous doses ranging from 0.04 to 0.24 μg/kg.
TREATMENT OF VITAMIN D DEFICIENCY
Adults
Dosing of vitamin D depends upon the nature and severity of the deficiency. In patients with normal absorptive capacity, for every 100 units of added vitamin D3, serum 25(OH)D concentrations increase by approximately 1.0 ng/mL. Multiple dosing regimens have been shown to treat vitamin D deficiency effectively. In a 2-month trial of oral vitamin D3 repletion in elderly women with hip fracture, the same cumulative dose given daily (1500 units), weekly (10,500 units), or monthly (45,000 units) resulted in similar increments in serum 25(OH)D levels. Although large annual doses of vitamin D3 increase serum 25(OH)D levels, they are not recommended as undesirable effects of increasing falls and fractures in older adults are seen. Various regimes which can be used are as follows:
High-risk individuals with serum 25(OH)D concentrations <20 ng/mL should be treated with 50,000 IU of vitamin D2 or d3 orally once per week for 6–8 weeks, followed by 800 units of vitamin D daily thereafter.
For high-risk individuals with serum 25(OH)D levels of 20–30 ng/mL, initial supplementation with 600–800 units of vitamin D3 daily may be sufficient to maintain levels in the target range.
In pregnant women, weekly doses of 50,000 units for 6–8 weeks are not recommended and 600–800 units of vitamin D3 daily are thought to be safer. Urinary calcium excretion increases in pregnancy, and it should be monitored when treating vitamin D deficiency, especially in women with a history of renal stones.
In malabsorption or patients with gastrectomy, generally high doses of vitamin D of 10,000–50,000 units daily are needed to correct the deficiency. Hydroxylated vitamin D metabolites are good options because they are more readily absorbed.
The above recommendations are in agreement with Endocrine Society practice guidelines on the treatment of vitamin D deficiency. In adults with vitamin D deficiency, however, the Endocrine Society guidelines suggest a maintenance dose of vitamin D2 or D3 (1500–2000 IU daily) to maintain a serum 25(OH)D concentration above 30 ng/mL.[59]
All patients should consume total calcium of at least 1000 mg (for ages 19–70 years) to 1200 mg (for women of ages 51 through 70 years and for all adults 71 years and older) per day. The upper level (UL) of intake for calcium in most adults is 2000–2500 mg daily. However, a higher calcium dose (up to 4 g/day) may be necessary in patients with malabsorption. Parenteral calcium as calcium gluconate (10–20 mg of elemental calcium per kg intravenously slowly over 5–10 minutes, usually given as 1–2 mL/kg of 10% calcium gluconate) becomes necessary in case of manifest tetany or convulsions. Repeat boluses may also be necessary. In addition, calcitriol may be necessary in doses of 20–100ng/ kg/day in two to three divided doses until calcium levels normalize. High doses of calcium are necessary early in the course of therapy, after which doses are reduced by half for the next 1–2 weeks. Once vitamin D supplementation has been reduced to 400 IU/day with normal PTH and 25(OH) D levels, calcium supplementation is usually not necessary.
Children
The most widely used treatment for vitamin D deficiency consists of vitamin D2 (ergocalciferol) or vitamin D3 (cholecalciferol). The dosing scheme recommended for treatment of vitamin D deficient rickets is 1000 IU daily for newborns <1 month, 1000–5000 IU daily for infants 1–12 months old, and 5000–10,000 IU daily for children 1 year and older. Treatment is continued until there is radiographic evidence of healing; subsequently, the dose of vitamin D is reduced to 400 IU daily. Calcium intake should be maintained at approximately 1000 mg/day (30–75 mg/ kg of elemental calcium per day in three divided doses) to avoid “hungry bone” syndrome (worsening hypocalcemia after the start of vitamin D therapy). This leads to resolution of the biochemical and radiological abnormalities within 3 months. Skeletal deformities regress completely after medical therapy. However, orthopedic intervention can be done if deformities do not improve even after radiologic appearance of the growth plates has normalized. An alternative protocol is “stoss therapy,” which consists of a high dose of oral vitamin D (600,000 IU) given on a single day, then maintained at 400–1000 IU of vitamin D per day, or 50,000 IU of vitamin D2 weekly for 8 weeks orally (teenagers) followed by 400 IU/day. This amount of vitamin D approximately corresponds to a 3-month course of 5000 IU/day and should be sufficient to induce healing within 3 months. High-dose vitamin D may need to be intermittently repeated (usually every 3 months) if poor compliance persists. Stoss therapy is useful when compliance is a problem. However, such high doses of vitamin D can lead to hypercalcemia. Doses of 150,000 or 300,000 IU are equally effective with lesser side effects.
ADVERSE EFFECTS OF VITAMIN D
Side effects of calcitriol therapy include hypercalcemia, hypercalciuria, nephrocalcinosis, and intraocular calcifications. There may be polyuria, pruritis, and azotemia. The first measurable consequences of vitamin D toxicity are hypercalciuria and hypercalcemia, which have been observed only at 25(OH)D levels above 88 ng/mL. Many patients take vitamin and mineral supplements that contain vitamin D without being aware of these. It is important to inquire about additional dietary supplements that patients may be taking before prescribing extra vitamin D.[60] Exposure to sunlight for extended periods of time does not normally cause vitamin D toxicity. Within about 20 minutes of UV exposure in light-skinned individuals, the concentrations of vitamin D precursors produced in the skin reach an equilibrium and any further vitamin D that is produced is degraded. According to some sources, endogenous production with full body exposure to sunlight is approximately 10,000 IU/day.
Some patients with vitamin D deficiency have coexisting primary hyperparathyroidism that is not recognized until vitamin D is repleted. vitamin D replacement in these individuals should be provided cautiously as hypercalcemia and hypercalciuria may develop. In contrast, in individuals with clinically significant vitamin D deficiency and SHPT, calcium concentrations are generally normal or at the lower end of normal (rarely below normal) and PTH concentrations are mildly elevated. The PTH level should return to normal upon vitamin D repletion. Urinary calcium is extremely low in patients with vitamin D deficiency and SHPT and takes months to normalize, whereas in primary hyperparathyroidism it normalizes very rapidly with vitamin D replacement.[61]
BENEFITS OF VITAMIN D REPLETION
Skeletal
The skeletal benefits of calcium and vitamin D supplementation have been demonstrated in prospective, randomized, placebo-controlled studies of calcium and vitamin D in community-dwelling older individuals. Lumbar spine and femoral neck bone density increased at annualized rates of 4–5% per year.[62] vitamin D supplementation may also contribute to a reduction in fracture risk due to improved muscle function and a reduction in the risk of falls.
Extraskeletal benefits
In addition to skeletal effects, vitamin D may have several other benefits, including beneficial effects on the immune and cardiovascular systems.
MONITORING
Healthy individuals on vitamin D supplementation do not require an initial or follow-up serum 25(OH)D measurement after starting supplementation, but in patients being treated for deficiency, close supervision is needed during treatment. It includes physical examination and biochemical evaluation every 2 weeks. The biochemical evaluation should include measurement of serum calcium, phosphorus, ALP, PTH levels, and urine calcium/creatinine ratio. Radiographs should be taken at 4 weeks. Response is deemed if radiographs show clear improvement and serum calcium and phosphorus levels have normalized, and ALP has started to decrease toward the reference range. The urinary calcium/creatinine ratio can still be at 0 or may have become measurable again at 4 weeks. Radiograph and biochemistry should be repeated after 3 months, when the growth plates should have regained a normal appearance and biochemistry should be normal and urine calcium should become detectable. Patients may be evaluated at 3-month intervals during maintenance therapy. Therefore, it is important to check the urinary calcium/creatinine ratio and kidney function (e.g. serum creatinine) at each visit. PTH should be assayed in 6 months. Hand radiographs, renal ultrasound, and ophthalmologic consultation (slit-lamp examination) should be performed once per year. 25(OH)D levels should be monitored yearly.
VITAMIN D DEPENDENT RICKETS
Vitamin D dependent rickets type I
VDDR type I (VDDR-I) is also known as pseudovitamin D-deficient rickets and was identified in 1961. VDDR-I is an autosomal recessive disorder. Patients with VDDR-I have inactivating mutations in the CYP27B1gene, located on 12 q13.3, which encodes the enzyme [25- (OH) D 1-α-hydroxylase or 1-α-hydroxylase], leading to 1-α-hydroxylase deficiency.[63] As a result, calcidiol is not hydroxylated to calcitriol and calcium is not absorbed normally. VDDR-I is characterized by early onset of skeletal disease (within the first year of life), severe hypocalcemia (sometimes with tetany), and moderate hypophosphatemia. Patients exhibit enamel hypoplasia, muscle weakness, hypotonia, motor retardation, and stunted growth. With progression, patients develop the classic radiographic signs of vitamin D deficiency rickets and bone biopsy evidence of osteomalacia. Biochemical evaluation shows hypocalcemia, increased PTH, and increase in urinary excretion of amino acids and phosphate. The characteristic biochemical findings of VDDR-I are normal serum levels of calcidiol and low levels of calcitriol. The clinical and biochemical evidence of rickets can be corrected with 1,25(OH)2D treatment. The dose depends upon the severity of disease and body weight. The initial dose for florid rickets is 1 mcg/day. Treatment is continued at this dose until bones are healed. Thereafter, the maintenance dose varies between 0.25 and 1 mcg/day, depending on the results of biochemical analyses. The aims of the treatment are to achieve normocalcemia, to maintain PTH levels within normal limits, and to avoid hypercalciuria.
Vitamin D dependent rickets type II
VDDR type II is also known as hereditary vitamin D resistant rickets (HVDRR). HVDRR is a very rare autosomal recessive form of rickets, with fewer than 50 known affected kindreds. It is associated with end-organ resistance to calcitriol, usually caused by mutations in the gene encoding the vitamin D receptor. The defect in the receptor interferes with the function of the hormone–receptor complex, thereby preventing calcitriol action.[64] The identified mutations or defects in the vitamin D receptor include: (a) failure of 1,25(OH)2D binding to available receptors, (b) reduction in 1,25(OH)2D receptor binding sites, (c) abnormal binding affinity of 1,25(OH)2D to receptor, (d) inadequate translocation of 1,25(OH)2D–receptor complex to the nucleus, and (e) diminished affinity of the 1,25(OH)2D–receptor complex for the dNA-binding domain, secondary to changes in the structure of receptor zinc-binding fingers.
The clinical spectrum of HVDRR varies with the type of mutation within the vitamin D receptor and residual vitamin D receptor activity. Affected children usually appear normal at birth, but develop rickets within the first 2 years of life. A unique feature of the syndrome is alopecia, which is seen in approximately two-thirds of cases and is a marker of disease severity. Alopecia results from the lack of vitamin D receptor action within keratinocytes. Additional ectodermal anomalies may also be seen, including multiple milia, epidermal cysts, and oligodontia. The treatment of HDVRR involves a therapeutic trial of calcitriol and calcium supplementation. The individual response varies with the severity of the receptor defect. Therapy is started at daily doses of 2 mcg of calcitriol and 1000 mg of calcium. However, administration of extremely high doses of calcitriol (up to 30–60 mcg/day) and calcium (up to 3 g/day) may be necessary. Long-term infusion of calcium into a central vein is an alternative for resistant patients and needs to be continued for many months. Oral calcium therapy may be sufficient once radiographic healing has been observed. During treatment, patients should initially be evaluated at least once per week. Serum calcium, phosphorus, ALP, creatinine, 1,25(OH)2D, PTH concentrations, and the urinary calcium/creatinine ratio should be measured. If the biochemical parameters do not respond, the dose of calcitriol should be gradually increased to reach serum concentrations of up to 100 times the normal mean. Failure of therapy should be considered if no biochemical response occurs after 3–5 months of treatment.
CONCLUSIONS
Vitamin D deficiency is an important cause of rickets/osteomalacia even in countries with adequate sun exposure. Screening for vitamin D deficiency is recommended in individuals at risk. Proper treatment corrects the disturbed bone metabolism and deformities. In addition, vitamin D has multiple extraskeletal benefits and treatment of vitamin D deficiency improves the quality of life. VDDR is another form of bone disease related to vitamin D, but unlike vitamin D deficient rickets it is hereditary. Though uncommon, recognition of VDDR is essential for treatment. The other important causes of rickets/osteomalacia are hypophosphatemic rickets and RTA which are discussed in other sections.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Pitt MJ. Rickets and osteomalacia are still around. Radiol Clin North Am. 1991;29:97–118. [PubMed] [Google Scholar]
- 2.Rauch F. The rachitic bone. Endocr Dev. 2003;6:69. doi: 10.1159/000072770. [DOI] [PubMed] [Google Scholar]
- 3.Ric John M. Pettifor vitamin D and/or calcium deficiency rickets in infants and children: Anglobal perspective. Indian J Med Res. 2008;127:245–9. [PubMed] [Google Scholar]
- 4.De Lucia MC, Mitnick ME. Carpenter Nutritional rickets with normal circulating 25-hydroxyvitamin D: A call for reexamining the role of dietary calcium intake in North American infants. J Clin Endocrinol Metab. 2003;88:3539. doi: 10.1210/jc.2002-021935. [DOI] [PubMed] [Google Scholar]
- 5.Sahay M, Vali SP, Ramesh VD. The Case. A child with metabolic acidosis and growth retardation. Kidney Int. 2009;75:1121–2. doi: 10.1038/ki.2009.25. [DOI] [PubMed] [Google Scholar]
- 6.Sahay M, Sahay R. Rickets in tropics. Int J Endocrinol Metab. 2010;23:1–5. doi: 10.1515/jpem.2010.098. [DOI] [PubMed] [Google Scholar]
- 7.Terushkin V, Bender A, Psaty EL, Engelsen O, Wang SQ, Halpern AC. Estimated equivalency of vitamin D production from natural sun exposure versus oral vitamin D supplementation across seasons at two US latitudes. J Am Acad Dermatol. 2010;62:929.e1. doi: 10.1016/j.jaad.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 8.Holick MF. vitamin D: A millenium perspective. J Cell Biochem. 2003;88:296. doi: 10.1002/jcb.10338. [DOI] [PubMed] [Google Scholar]
- 9.Christakos S, Ajibade dV, dhawan P, Fechner AJ, Mady LJ. vitamin D: Metabolism. Endocrinol Metab Clin North Am. 2010;39:243. doi: 10.1016/j.ecl.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nykjaer A, dragun d, Walther d, Vorum H, Jacobsen C, Herz J, et al. An endocytic pathway essential for renal uptake and activation of the steroid 25-(OH) vitamin D3. Cell. 1999;96:507. doi: 10.1016/s0092-8674(00)80655-8. [DOI] [PubMed] [Google Scholar]
- 11.Takeyama K, Kitanaka S, Sato T, Kobori M, Yanagisawa J, Kato S. 25-Hydroxyvitamin D3 1alpha-hydroxylase and vitamin D synthesis. Science. 1997;277:1827. doi: 10.1126/science.277.5333.1827. [DOI] [PubMed] [Google Scholar]
- 12.Portale AA, Halloran BP, Morris RC., Jr Physiologic regulation of the serum concentration of 1,25-dihydroxyvitamin D by phosphorus in normal men. J Clin Invest. 1989;83:1494. doi: 10.1172/JCI114043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iida K, Shinki T, Yamaguchi A, DeLuca HF, Kurokawa K, Suda T. A possible role of vitamin D receptors in regulating vitamin D activation in the kidney. Proc Natl Acad Sci U S A. 1995;92:6112. doi: 10.1073/pnas.92.13.6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prié D, Friedlander G. Reciprocal control of 1,25-dihydroxyvitamin D and FGF23 formation involving the FGF23/Klotho system. Clin J Am Soc Nephrol. 2010;5:1717. doi: 10.2215/CJN.02680310. [DOI] [PubMed] [Google Scholar]
- 15.Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol. 2006;17:1305–15. doi: 10.1681/ASN.2005111185. [DOI] [PubMed] [Google Scholar]
- 16.Bikle D. Nonclassic actions of vitamin D. J Clin Endocrinol Metab. 2009;94:26. doi: 10.1210/jc.2008-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haddad JG. vitamin D–solar rays, the Milky Way, or both? N Engl J Med. 1992;326:1213. doi: 10.1056/NEJM199204303261808. [DOI] [PubMed] [Google Scholar]
- 18.Binkley N, Novotny R, Krueger d, Kawahara T, Daida YG, Lensmeyer G, et al. Low vitamin D status despite abundant sun exposure. J Clin Endocrinol Metab. 2007;92:2130–5. doi: 10.1210/jc.2006-2250. [DOI] [PubMed] [Google Scholar]
- 19.Chatfield SM, Brand C, Ebeling PR, Russell dM. vitamin D deficiency in general medical inpatients in summer and winter. Intern Med J. 2007;37:377–82. doi: 10.1111/j.1445-5994.2007.01339.x. [DOI] [PubMed] [Google Scholar]
- 20.Awumey EM, Mitra DA, Hollis BW, Kumar R, Bell NH. vitamin D metabolism is altered in Asian Indians in the southern United States: A clinical research center study. J Clin Endocrinol Metab. 1998;83:169–73. doi: 10.1210/jcem.83.1.4514. [DOI] [PubMed] [Google Scholar]
- 21.Klein GL, Chen TC, Holick MF, Langman CB, Price H, Celis MM, et al. Synthesis of vitamin D in skin after burns. Lancet. 2004;363:291–2. doi: 10.1016/S0140-6736(03)15388-3. [DOI] [PubMed] [Google Scholar]
- 22.Carvalho NF, Kenney RD, Carrington PH, Hall DE. Severe nutritional deficiencies in toddlers resulting from health food milk alternatives. Pediatrics. 2001;107:E46. doi: 10.1542/peds.107.4.e46. [DOI] [PubMed] [Google Scholar]
- 23.Gloth FM, 3rd, Gundberg CM, Hollis BW, Haddad JG, Jr, Tobin JD. vitamin D deficiency in homebound elderly persons. JAMA. 1995;274:1683–6. doi: 10.1001/jama.1995.03530210037027. [DOI] [PubMed] [Google Scholar]
- 24.Hollis BW, Wagner CL. vitamin D requirements during lactation: High-dose maternal supplementation as therapy to prevent hypovitaminosis D for both the mother and the nursing infant. Am J Clin Nutr. 2004;80:1752S. doi: 10.1093/ajcn/80.6.1752S. [DOI] [PubMed] [Google Scholar]
- 25.Thandrayen K, Pettifor JM. Maternal vitamin D status: Implications for the development of infantile nutritional rickets. Endocrinol Metab Clin North Am. 2010;39:303. doi: 10.1016/j.ecl.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 26.Specker BL, Valanis B, Hertzberg V, Edwards N, Tsang RC. Sunshine exposure and serum 25-hydroxyvitamin D concentrations in exclusively breast-fed infants. J Pediatr. 1985;107:372. doi: 10.1016/s0022-3476(85)80509-6. [DOI] [PubMed] [Google Scholar]
- 27.Tsang RC, Zlotkin SH, Nichols BL, Hansen JW, editors. 2nd ed. Cincinnati, OH: Digital Education Publishing; 1997. Formula fed nutrition during infancy: Principles and Practice; p. 467. [Google Scholar]
- 28.Parikh SJ, Edelman M, Uwaifo GI, Freedman RJ, Semega-Janneh M, Reynolds J, et al. The relationship between obesity and serum 1, 25-dihydroxy vitamin D concentrations in healthy adults. J Clin Endocrinol Metab. 2004;89:1196–9. doi: 10.1210/jc.2003-031398. [DOI] [PubMed] [Google Scholar]
- 29.Thomas MK, Lloyd-Jones DM, Thadhani RI, Shaw AC, Deraska DJ, Kitch BT, et al. Hypovitaminosis d in medical inpatients. N Engl J Med. 1998;338:777–83. doi: 10.1056/NEJM199803193381201. [DOI] [PubMed] [Google Scholar]
- 30.Holick MF, Siris ES, Binkley N, Beard MK, Khan A, Katzer JT, et al. Prevalence of vitamin D inadequacy among postmenopausal North American women receiving osteoporosis therapy. J Clin Endocrinol Metab. 2005;90:3215–24. doi: 10.1210/jc.2004-2364. [DOI] [PubMed] [Google Scholar]
- 31.Elder GJ, Mackun K. 25-Hydroxyvitamin D deficiency and diabetes predict reduced BMD in patients with chronic kidney disease. J Bone Miner Res. 2006;21:1778. doi: 10.1359/jbmr.060803. [DOI] [PubMed] [Google Scholar]
- 32.Taskapan H, Ersoy FF, Passadakis PS, Tam P, Memmos dE, Katopodis KP, et al. Severe vitamin D deficiency in chronic renal failure patients on peritoneal dialysis. Clin Nephrol. 2006;66:247–55. doi: 10.5414/cnp66247. [DOI] [PubMed] [Google Scholar]
- 33.Vaziri ND. Endocrinological consequences of the nephrotic syndrome. Am J Nephrol. 1993;13:360. doi: 10.1159/000168650. [DOI] [PubMed] [Google Scholar]
- 34.Goldstein DA, Haldimann B, Sherman D, Norman AW, Massry SG. vitamin D metabolites and calcium metabolism in patients with nephrotic syndrome and normal renal function. J Clin Endocrinol Metab. 1981;52:116–21. doi: 10.1210/jcem-52-1-116. [DOI] [PubMed] [Google Scholar]
- 35.Oduwole AO, Giwa OS, Arogundade RA. Relationship between rickets and incomplete distal renal tubular acidosis in children. Ital J Pediatr. 2010;36:54. doi: 10.1186/1824-7288-36-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dibble JB, Sheridan P, Losowsky MS. A survey of vitamin D deficiency in gastrointestinal and liver disorders. Q J Med. 1984;53:119. [PubMed] [Google Scholar]
- 37.American Gastroenterological Association medical position statement: Guidelines on osteoporosis in gastrointestinal diseases. Gastroenterology. 2003;124:791. doi: 10.1053/gast.2003.50107. [DOI] [PubMed] [Google Scholar]
- 38.Sotaniemi EA, Hakkarainen HK, Puranen JA, Lahti RO. Radiologic bone changes and hypocalcemia with anticonvulsant therapy in epilepsy. Ann Intern Med. 1972;77:389. doi: 10.7326/0003-4819-77-3-389. [DOI] [PubMed] [Google Scholar]
- 39.Hahn TJ. Drug-induced disorders of vitamin D and mineral metabolism. Clin Endocrinol Metab. 1980;9:107. doi: 10.1016/s0300-595x(80)80023-5. [DOI] [PubMed] [Google Scholar]
- 40.Misra M, Pacaud D, Petryk A, Collett-Solberg PF, Kappy M. Drug and Therapeutics Committee of the Lawson Wilkins Pediatric Endocrine Society. vitamin D deficiency in children and its management: Review of current knowledge and recommendations. Pediatrics. 2008;122:398–417. doi: 10.1542/peds.2007-1894. [DOI] [PubMed] [Google Scholar]
- 41.Robinson PD, Högler W, Craig ME, Verge CF, Walker JL, Piper AC, et al. The re-emerging burden of rickets: A decade of experience from Sydney. Arch Dis Child. 2006;91:564–8. doi: 10.1136/adc.2004.069575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Najada AS, Habashneh MS, Khader M. The frequency of nutritional rickets among hospitalized infants and its relation to respiratory diseases. J Trop Pediatr. 2004;50:364. doi: 10.1093/tropej/50.6.364. [DOI] [PubMed] [Google Scholar]
- 43.Oestreich AE. The acrophysis: A unifying concept for understanding enchondral bone growth and its disorders. II. Abnormal growth. Skeletal Radiol. 2004;33:119. doi: 10.1007/s00256-003-0735-9. [DOI] [PubMed] [Google Scholar]
- 44.Chapman T, Sugar N, Done S, Marasigan J, Wambold N, Feldman K. Fractures in infants and toddlers with rickets. Pediatr Radiol. 2010;40:1184–9. doi: 10.1007/s00247-009-1470-8. [DOI] [PubMed] [Google Scholar]
- 45.Whyte MP. Physiological role of alkaline phosphatase explored in hypophosphatasia. Ann N Y Acad Sci. 2010;1192:190–200. doi: 10.1111/j.1749-6632.2010.05387.x. [DOI] [PubMed] [Google Scholar]
- 46.Baroncelli GI, Bertelloni S, Ceccarelli C, Amato V, Saggese G. Bone turnover in children with vitamin D deficiency rickets before and during treatment. Acta Paediatr. 2000;89:513. doi: 10.1080/080352500750027763. [DOI] [PubMed] [Google Scholar]
- 47.Durazo-Arvizu RA, Dawson-Hughes B, Sempos CT, Yetley EA, Looker AC, Cao G, et al. Three-phase model harmonizes estimates of the maximal suppression of parathyroid hormone by 25-hydroxyvitamin D in persons 65 years of age and older. J Nutr. 2010;140:595–9. doi: 10.3945/jn.109.116681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dawson-Hughes B, Heaney RP, Holick MF, Lips P, Meunier PJ, Vieth R. Estimates of optimal vitamin D status. Osteoporos Int. 2005;16:713–6. doi: 10.1007/s00198-005-1867-7. [DOI] [PubMed] [Google Scholar]
- 49.Vieth R. What is the optimal vitamin D status for health? Prog Biophys Mol Biol. 2006;92:26. doi: 10.1016/j.pbiomolbio.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 50.Sanders KM, Stuart AL, Williamson EJ, Simpson JA, Kotowicz MA, Young D, et al. Annual high-dose oral vitamin D and falls and fractures in older women: A randomized controlled trial. JAMA. 2010;303:1815–22. doi: 10.1001/jama.2010.594. [DOI] [PubMed] [Google Scholar]
- 51.Binkley N, Krueger D, Cowgill CS, Plum L, Lake E, Hansen KE, et al. Assay variation confounds the diagnosis of hypovitaminosis D: A call for standardization. J Clin Endocrinol Metab. 2004;89:3152. doi: 10.1210/jc.2003-031979. [DOI] [PubMed] [Google Scholar]
- 52.Yetley EA. Assessing the vitamin D status of the US population. Am J Clin Nutr. 2008;88:558S. doi: 10.1093/ajcn/88.2.558S. [DOI] [PubMed] [Google Scholar]
- 53.Goswami R, Mishra SK, Kochupillai N. Prevalence and potential significance of vitamin D deficiency in Asian Indians. Indian J Med Res. 2008;127:229–38. [PubMed] [Google Scholar]
- 54.Looker AC, Pfeiffer CM, Lacher DA, Schleicher RL, Picciano MF, Yetley EA. Serum 25-hydroxyvitamin D status of the US population: 1988-1994 compared with 2000-2004. Am J Clin Nutr. 2008;88:1519. doi: 10.3945/ajcn.2008.26182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ross AC, Manson JE, Abrams SA, Aloia JF, Brannon PM, Clinton SK, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: What clinicians need to know. J Clin Endocrinol Metab. 2011;96:53–8. doi: 10.1210/jc.2010-2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Misra M, Pacaud D, Petryk A, Collett-Solberg PF, Kappy M. drug and Therapeutics Committee of the Lawson Wilkins Pediatric Endocrine Society. vitamin D deficiency in children and its management: Review of current knowledge and recommendations. Pediatrics. 2008;122:398–417. doi: 10.1542/peds.2007-1894. [DOI] [PubMed] [Google Scholar]
- 57.Washington, DC: National Academy Press; 2010. [Last Accessed on 2010 Dec 14]. Institute of Medicine, Food and Nutrition Board. dietary reference intakes for calcium and vitamin D. Available from: http://books.nap.edu/openbook.php?record_id = 13050 . [Google Scholar]
- 58.Wharton B, Bishop N. Rickets. Lancet. 2003;362:1389. doi: 10.1016/S0140-6736(03)14636-3. [DOI] [PubMed] [Google Scholar]
- 59.Holick MF, Binkley NC, Bischoff-Ferrari HA, Gordon CM, Hanley dA, Heaney RP, et al. Evaluation, treatment, and prevention of vitamin D deficiency: An Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96:1911–30. doi: 10.1210/jc.2011-0385. [DOI] [PubMed] [Google Scholar]
- 60.Vieth R. vitamin D supplementation, 25-hydroxyvitamin D concentrations, and safety. Am J Clin Nutr. 1999;69:842–56. doi: 10.1093/ajcn/69.5.842. [DOI] [PubMed] [Google Scholar]
- 61. [Last Accessed on 2010 dec 01]. Available from: http://www.iom.edu/Reports/2010/Dietary-Reference-Intakes-for-Calcium-and-Vitamin-D/Report-Brief.aspx .
- 62.Adams JS, Kantorovich V, Wu C, Javanbakht M, Hollis BW. Resolution of vitamin D insufficiency in osteopenic patients results in rapid recovery of bone mineral density. J Clin Endocrinol Metab. 1999;84:2729. doi: 10.1210/jcem.84.8.5899. [DOI] [PubMed] [Google Scholar]
- 63.Kim CJ, Kaplan LE, Perwad F, Huang N, Sharma A, Choi Y, et al. vitamin D 1alpha-hydroxylase gene mutations in patients with 1alpha-hydroxylase deficiency. J Clin Endocrinol Metab. 2007;92:3177–82. doi: 10.1210/jc.2006-2664. [DOI] [PubMed] [Google Scholar]
- 64.Brooks MH, Bell NH, Love L, Stern PH, Orfei E, Queener SF, et al. Vitamin-D-dependent rickets type II.Resistance of target organs to 1,25-dihydroxyvitamin D. N Engl J Med. 1978;298:996–9. doi: 10.1056/NEJM197805042981804. [DOI] [PubMed] [Google Scholar]