

Abstract
Parkinson's disease (PD) is characterized by movement disorders, including bradykinesia. Analysis of inherited, juvenile PD, identified several genes linked via a common pathway to mitochondrial dysfunction. In this study, we demonstrate that the larva of the Drosophila parkin mutant faithfully models the locomotory and metabolic defects of PD and is an excellent system for investigating their inter-relationship. parkin larvae displayed a marked bradykinesia that was caused by a reduction in both the frequency of peristalsis and speed of muscle contractions. Rescue experiments confirmed that this phenotype was due to a defect in the nervous system and not in the muscle. Furthermore, recordings of motoneuron activity in parkin larvae revealed reduced bursting and a striking reduction in evoked and miniature excitatory junction potentials, suggesting a neuronal deficit. This was supported by our observations in parkin larvae that the resting potential was depolarized, oxygen consumption and ATP concentration were drastically reduced while lactate was increased. These findings suggest that neuronal mitochondrial respiration is severely compromised and there is a compensatory switch to glycolysis for energy production.
parkin mutants also possessed overgrown neuromuscular synapses, indicative of oxidative stress, which could be rescued by overexpression of parkin or scavengers of reactive oxygen species (ROS). Surprisingly, scavengers of ROS did not rescue the resting membrane potential and locomotory phenotypes. We therefore propose that mitochondrial dysfunction in parkin mutants induces Parkinsonian bradykinesia via a neuronal energy deficit and resulting synaptic failure, rather than as a consequence of downstream oxidative stress.
INTRODUCTION
The recognition of Mendelian inheritance in some forms of Parkinson's disease (PD) has revolutionized our understanding of the causation of this disease (1). Mutations in PD-related genes (e.g. parkin, α-synuclein, PINK1, LRRK2) have now been replicated in a range of model organisms (2). Drosophila models share many features with the human disease, including the loss of dopaminergic neurons in the central nervous system (CNS) and reduced locomotion. Other similarities are evident at the cellular level, particularly mitochondrial malfunction and disorganization (3–5). The advanced fly genetic toolbox (6) has also facilitated deep insights into the cellular and metabolic pathways in which the PD-related gene products participate. A key result has been the identification of a single cellular pathway focussing on 4E-BP, to which several genes (parkin, PINK1 and LRRK2) contribute (7), suggesting the potential for novel drug therapies across several genotypes.
Apart from dopaminergic cell death, few Drosophila studies have analysed neuronal phenotypes, focusing instead on spermatozoa, eye or muscle. Our first focus, therefore, is what are the neurophysiological (rather than muscle) defects that lead to reduced locomotion? For this, we turned our attention to the larval (juvenile) system, as it helps us understand the initial stages of the neurodegenerative process. The larval neuromuscular junction is one of the best-characterized synapses (8). It is essential for normal movement. Here, we describe our finding that parkin mutant larvae have defective locomotion, with a bradykinesia-like phenotype. This is accompanied by reduced resting and synaptic potentials. The inability to maintain the normal muscle resting potential suggested that the mitochondria could not produce enough ATP: indeed, we find that parkin larvae have reduced ATP and oxygen consumption. Furthermore, they have elevated lactate, indicating that glycolysis may be elevated in response to the ATP deficit.
Our second question is how does oxidative stress relate to mitochondrial dysfunction? Fly models of PD are highly sensitive to oxidative stress, for example to paraquat (9,10), and are rescued by overexpression of genes that reduce reactive oxygen species (ROS) (11–14). It has recently been recognized that oxidative stress induces autophagy and synaptic overgrowth (15,16). We find that parkin mutants indeed have synaptic overgrowth, and this is rescued by over-expression of ROS scavengers as well as by parkin. However, as we find that ROS scavengers do not rescue the locomotion or electrophysiological defects, we are genetically separating the fundamental parkin deficit of energy production from its downstream oxidative stress consequences.
RESULTS
parkin larvae move slowly
The tracks of parkin mutant larvae crawling across an agar plate are shorter for animals migrating for a set interval of time (2 min) than wild-type as the parkin larvae move more slowly (Fig. 1A). Their mean speed is reduced to 62% of the wild-type. The parkin heterozygote crawls at the same speed as the wild-type, as would be expected from this recessive mutation. Using the GAL4/UAS system, we tested for rescue by global expression of wild-type parkin in the mutant background and found the velocity to be fully restored. As parkin mutants have both neuronal (11) and muscle (17) phenotypes, we then tested for tissue-specific rescue using neuronal (using elav GAL4) and muscle (G14 GAL4) drivers. The neuronal driver rescued locomotion to wild-type levels, but no significant rescue was achieved with the muscle driver (Fig. 1B). We confirmed the data from Figure 1 using two additional wild-types, a second heterozygote and two other parkin mutant combinations, with both the park25 homozygote null and parkz3678 hypomorph crawling more slowly than the other genotypes (CS, w−, CS/park25, nested ANOVA, parkin versus wild-types; F1,113 = 34, P< 0.001). Further confirmation of the role of neuronal parkin in locomotion was provided by using the same drivers to test for rescue in the park25 homozygote background. The global (Act5C GAL4) or neuronal drivers gave partial rescue (71% and 77%, respectively, nested ANOVA F77,1 = 7.4, P= 0.008) but no rescue was seen using the muscle driver (mean larval speed 93% of the park25 homozygote, ANOVA, Bonferroni post hoc test, P= 1.0).
Figure 1.

Larval locomotion is reduced in parkin mutants. (A) Tracks of parkin transheterozygotes crawling for 2 min are shorter than those of wild-type. (B) Quantification shows that the mean velocity of the parkin mutant is 62% of wild-type (Bonferroni post hoc comparison: P= 0.003). A significant rescue is seen with the expression of wild-type parkin in the transheterozygote background using the global or neuronal drivers but not with the muscle driver (Bonferroni post hoc tests: P = 0.009, 0.003, 0.253, respectively). At least 10 larvae of each genotype, total 130. Genotypes—wild-type: CS/w−; heterozygote: CS/parkz3678; parkin: park25/parkz3678. Drivers—global: Act5C; neuronal: elav3EI; muscle: G14. All rescue experiments are in park25/parkz3678 background.
We assayed the strength and frequency of peristaltic contractions directly, using a larval length assay, in which a restrained larva pulls on a flexible beam (Fig. 2A). Wild-type larvae pulled regularly on the beam, while the parkin knockouts had a reduced frequency of contractions (32% of wild-type; Fig. 2B and D). However, the peak–peak change in length did not differ between the wild-type and parkin null (Fig. 2C). This suggests that the reduced velocity of the larvae was due to less frequent steps, rather than a reduced step size. A particular feature of the parkin larval contractions was the slow rate at which the larva shortens, akin to bradykinesia (Fig. 2B). We also tested for rescue by neuronal or muscle driver. Only the neuronal driver restored the contraction frequency to wild-type levels (Fig. 2D).
Figure 2.

Locomotor contractions are less frequent in parkin larvae. (A) A larval extensometer, constructed from a flexible photobeam and quadrant photodiode. (B) Peristaltic waves recorded by the extensometer show that contractions are fewer in parkin knockouts than in wild-type, but the peak–peak excursion is not affected. (C) The mean force is the same in wild-type and parkin knockout (ANOVA: F1,27 = 2.8, P= 0.10). (D) The frequency of peristaltic contractions is reduced in the parkin knockout (ANOVA, Bonferroni post hoc test, P< 0.001), and completely rescued by neuronal expression of wild-type parkin in nerve (Bonferroni post hoc test versus parkin P= 0.014; versus wild-type P= 0.56). Expression in muscle is ineffective (Bonferroni P= 0.38). Genotypes and number of larvae—wild-type: CS 13; parkin: park25/park25 15; drivers: neuronal: elav3EI 10; muscle: G14 12. Rescues are in park25/park25 background.
The peristaltic longitudinal muscles are innervated by segmental nerves arising from the ventral nerve cord. To examine the neural input to the muscles, we severed all the segmental nerves and used a suction electrode on a segmental nerve to record the efferent activity of the motoneurons. The wild-type nerve recordings showed regular bursts of action potentials, along with occasional periods of tonic activity (Fig. 3). In the parkin null, the frequency of bursts was significantly reduced (to 44% of wild-type; Fig. 3B).
Figure 3.

Central pattern generator activity is reduced in parkin knockout. (A) The frequency of bursts in the motoneurons is reduced in the parkin knockout, with the wild-type showing a regular rhythm (and occasional long bursts), while the parkin mutant is much quieter. (B) The reduction (mean: 56%) is significant (ANOVA F1,9 = 6.1, P= 0.039). Genotypes—wild-type: CS; parkin: park25/park25.
parkin larvae have reduced resting and synaptic membrane potentials
The muscles used in locomotion include the longitudinal body wall muscles 6 and 7. Since their neuromuscular junction is easily accessible, it has been extensively studied, and is now a model synapse (8,18). These muscles are innervated by two motoneurons, so that intracellular recordings from them usually show two sizes of synaptic potentials (19). These are called the Is and Ib excitatory junction potentials (EJPs), though the motoneurons can also be recruited together to produce a composite EJP. Both types of EJPs were present in both the parkin muscles (Fig. 4A), but the parkin mutant showed significant differences from wild-type.
Figure 4.

Resting and synaptic potentials are reduced in parkin mutants. (A) Intracellular recordings from larval muscles show the more positive resting potential in the parkin larvae along with smaller EJPs, than in the control, wild-type larva. Both traces show two sizes of spontaneous EJPs: the large ones are called type Is, and the small ones type Ib; additional compound EJPs were also frequently recorded. (B) The mean muscle resting membrane potential (RMP) in the parkin mutant is 10.7 ± 1.7 mV more positive than the wild-type control (Bonferroni post hoc estimate, P < 0.001). Expressing wild-type parkin in this transheterozygote background using a neuronal driver did not rescue the RMP deficit (Bonferroni P= 1.0), but using a muscle-specific driver a significant rescue was seen (Bonferroni post hoc estimate, 6.2 ± 1.5, P = 0.001). (C) EJPs (type Is) are smaller in parkin mutants than in controls. (i) For each genotype, EJPs are smaller at more positive RMPs, but at any given RMP, EJPs from control muscles are bigger than those from parkin mutants as indicated by the regression lines. (ii) We tested for rescue as follows: we calculated the parkin regression line to provide the expected size of the EJP in each preparation, and calculated the difference of the observed EJP from this expected value. Expressing parkin neuronally in this mutant background partially rescued the EJPs (t13 = 2.6, P = 0.023), but no rescue was seen with muscle expression (t12 = 0.46, P = 0.65). As a control, we checked that the wild-type EJPs are significantly bigger than expected from the parkin regression line (one-sample t-test: t10 = 6.8, P < 0.001). Data from the same set of muscle recordings as (B); 63 EJPs at least 11 in each genotype. Genotypes—wild-type: CS/w−; parkin: park25/parkz3678. Drivers—neuronal; elav3EI; muscle—G14. Rescues are all in park25/parkz3678 background.
First, the resting potential of muscles 6/7 was much more positive in the parkin larvae than in the controls (Fig. 4A–C): the wild-type resting membrane potential (RMP) is −63.6 ± 1.7 mV (mean ± SE) and the mean parkin −52.9 ± 1.0 mV. We confirmed these data in an independent sample of 292 muscles from two wild-type controls (CS, CS/w−) and two parkin genotypes (park25/park25, park25/parkZ3678), where the mean difference in RMP was 12.3 mV (nested ANOVA F291,1 = 168, P< 0.001).
Secondly, the size of the EJP was reduced in the parkin mutants. The parkin knockout might be expected to show smaller synaptic potentials in any case, because the membrane potential of the parkin knockout is more positive, and therefore closer to the synaptic reversal potential. However, in recordings made at the same RMP, the parkin larvae still had smaller EJPs than the wild-type control. This is shown in Figure 4Ci, where the size of the EJP is plotted as a function of the RMP. The regression line for the wild-type data lies above the regression line for the parkin mutants.
We took a sub-sample of our electrophysiological data, using larvae raised on the same batch of food, and tested for rescue of the resting and synaptic potentials by expressing wild-type parkin in nerve or muscle in the transheterozygote background (Fig. 4B, Cii). Muscle expression of parkin in the transheterozygote background rescued the muscle RMP by 6.2 ± 1.5 mV (60% of the deficit), but not the synaptic potential, although these data come from the same larvae. On the other hand, the neuronal expression of wild-type parkin did not affect the RMP, but did rescue the size of the synaptic potential by 2.0 ± 0.8 mV. (Here we note that the choice of G14 and elav3E1 drivers clearly separated the neuronal and muscle phenotypes.)
The larval neuromuscular junction also shows miniature EJPs (mEJPs): using data from preparations with matched RMPs, the mEJPs from parkin transheterozygotes were smaller than those from wild-type larvae (modal amplitude: 0.5 and 0.7 mV, respectively; Fig. 5).
Figure 5.

Miniature excitatory junction potentials (mEJPs) are smaller in parkin mutants than in wild-type larvae. (A) Traces from parkin and wild-type larvae. (B) Histograms of the amplitude of 550 mEJPs taken from 13 muscles, matched for resting membrane potential (RMP), show that peak amplitude is ∼2 mV smaller in the parkin larvae (Kolmogorov–Smirnov test statistic 3.5, P < 0.001). RMP—wild-type: −63.8 ± 1.1, n = 7; parkin: −63.0 ± 0.85, n= 6. Genotypes—wild-type: CS/w−; parkin: park25/parkz3678.
parkin larvae show compromised respiration
The failure to maintain the RMP along with data from adults showing defective mitochondrial fission/fusion (17,20) suggested that the rate at which parkin larvae generate ATP by aerobic respiration might be reduced. In order to address this we used two separate technologies—capillary respirometer or an oxygen electrode. The capillary respirometer allows us to follow gas exchange in air, while the oxygen electrode, using larvae immersed in HL3 solution, allows us to examine the impact of respiratory poisons on oxygen consumption. We found with both techniques that the parkin larvae had 70% of the oxygen consumption of the wild-type controls (Fig. 6Ai,ii). parkin larvae were also more sensitive to metabolic poisons: 6 mm cyanide blocked parkin oxygen consumption completely, but reduced only wild-type respiration by 40%, while 6 mm iodoacetate blocked 62% of wild-type respiration, and 87% of parkin oxygen consumption (Fig. 6Aii). The ATP levels of the parkin knockout larvae were reduced to 14% of the wild-type value (Fig. 6B) and the lactate concentration was nearly doubled (+80%, Fig. 6C). We also obtained similar data in the park25/parkZ3678 transheterozygotes (mean increase in lactate over wild-type, 88%).
Figure 6.

parkin larvae have reduced aerobic metabolism and compensate with increased glycolysis. (A) Reduction in oxygen consumption. (i) Measurement with a capillary respirometer shows that the oxygen consumption is 70% of the parkin larvae than in the wild-type larvae (ANOVA F17,1 = 22.8, P< 0.001). (ii) Measurement with an oxygen electrode confirms that the oxygen consumption of parkin is 70% of wild-type (ANOVA F23,1 = 78, P< 0.001). Addition of 6 mm cyanide or iodoacetate reduces respiration, having a bigger impact on the parkin than the wild-type larvae. Data normalized to wild-type as 100%. (B) The ATP concentration is lower in parkin than in the wild-type larvae (ANOVA F15,1 = 104, P< 0.001). (C) The lactate concentration is higher in the parkin than in the wild-type controls (ANOVA F23,1 = 16, P= 0.001). Data normalized as moles lactate/gram of protein, with wild-type as 100%. Genotypes—wild-type: CS; parkin: park25/park25.
ROS scavengers rescue overgrown synapses, but not membrane potential or locomotor defects in parkin larvae
Oxidative stress is frequently associated with PD (in both the human disease and model organisms). Since oxidative stress leads to synaptic overgrowth (16), we counted the synaptic boutons on muscle 6/7 of the third instar parkin larvae, as this provides a convenient index of the synapse size. The sample images from the neuromuscular junctions of parkin transheterozygotes showed more boutons than those from micrographs of wild-type (CS) larvae (Fig. 7A). The average data for boutons/muscle surface area showed that Drosophila larvae that are null for parkin have ∼50% more synaptic boutons than the wild-type controls and heterozygote (Fig. 7B). We confirmed this increase by examining two other wild-type strains (w−, yw), a second heterozygote and three other parkin mutant combinations: the mean increase was 51 ± 4% (nested ANOVA F1,371 = 73, P< 0.001). The raw bouton counts from parkin mutants were also greater than wild-type (mean increase 18%, nested ANOVA, F1,143 = 37, P < 0.001). To confirm that the increase in boutons is due to the parkin knockout, the wild-type parkin was expressed in the parkin background, giving a complete rescue of the synaptic overgrowth phenotype (Fig. 7B). Both neuronal and muscle drivers also gave complete rescue of the synaptic overgrowth (Fig. 7B). We confirmed this data in the transheterozygote background, where we find that both neuronal and muscle expression of parkin rescue the synaptic overgrowth (ANOVA, Bonferroni post hoc tests, P< 0.001, N = 213).
Figure 7.
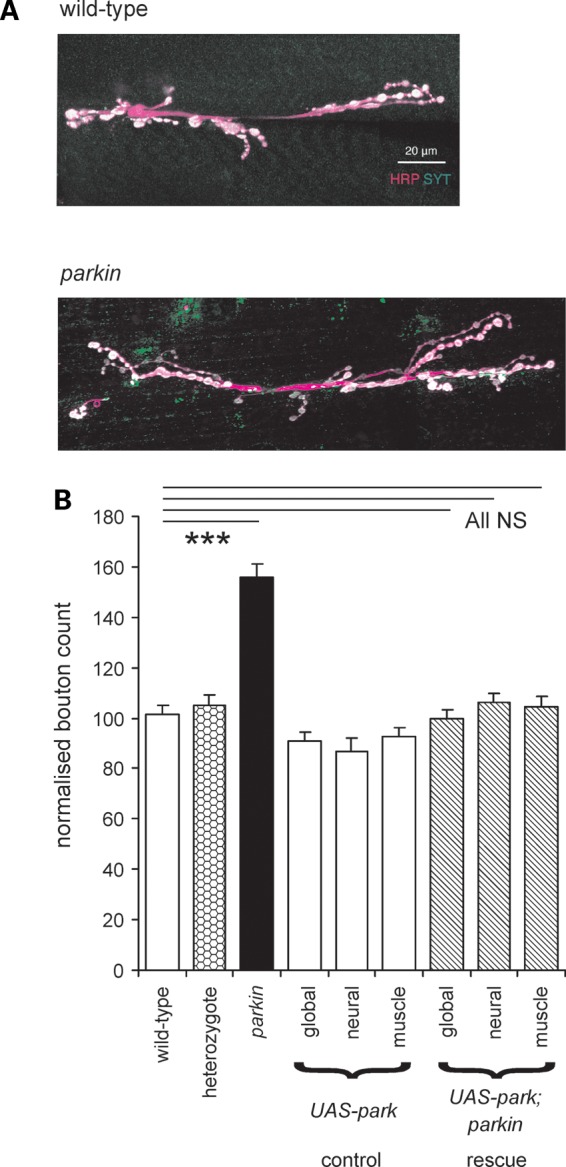
Overgrown synapses are characteristic of parkin mutants. (A) There are more boutons (white) in the parkin neuromuscular junction than in the control wild-type. Images from confocal stacks of synaptic terminals on muscles 6 and 7 stained with horseradish peroxidase antibody (HRP, magenta) show the neuronal membrane and with synaptotagmin (SYT, green) antibody to show the terminal boutons. (B) Quantification of the bouton count at 372 synapses confirms that parkin mutants have excess boutons (+53%, Bonferroni P < 0.001). Expression of wild-type parkin by the global, neuronal or muscle drivers achieves substantial rescue of the synaptic overgrowth (down to 3% above the control values, Bonferroni comparison with wild-type each = 1.0). There is no significant difference between the synaptic overgrowth in the three rescues (ANOVA, F2,78 = 0.81, P < 0.445). At least 16 neuromuscular junctions for each genotype, totally 223. Data normalized as ratio of boutons/muscle area, with wild-type as 100%. Genotypes—wild-type: CS; parkin: park25/park25. Drivers—global: Act5C; neuronal; elav3EI; muscle: G14. Rescues are in park25/park25 background.
Having demonstrated synaptic overgrowth, we wanted to examine the role of oxidative stress in the parkin phenotype. Overexpression of genes that scavenge free radicals (superoxide dismutase, catalase) significantly rescued the overgrowth phenotype, down from 53% to 20% more than the wild-type (Fig. 8A). In contrast to these data, we found no rescue of either the muscle RMP or the larval locomotory defect with expression of any of these scavengers (Fig. 8B and C).
Figure 8.

In the parkin null background, overexpression of oxidative stress transgenes rescues synaptic overgrowth but not muscle RMP or larval locomotion. (A) The normalized bouton count is reduced from the parkin mutant peak (∼+50% of the CS wild-type) to +20% by overexpression of catalase or superoxide dismutase (Sod1 or Sod2) with a global driver. The effect of the oxidative stress transgenes is significant (Bonferroni comparison with parkin P< 0.001 for each transgene). At least 24 neuromuscular junctions for each genotype, totally 224. (B) Oxidative stress gene overexpression did not rescue the muscle RMP (comparison with parkin mutants, Bonferroni tests all P= 1.0). At least 18 muscles for each genotype, 213 in total. (C) The oxidative stress gene overexpression did not rescue larval locomotion (comparison with parkin mutants Bonferroni tests all P= 1.0). At least 12 larvae for each genotype, total 171. Genotypes—(A and C) wild-type: CS; parkin: park25/park25 outcrossed to the Act5C driver used to express each transgene in park25/park25 background. (B) Wild-type CS/w−; parkin; parkZ3678/park25, transgene expression driven by the Act5C driver.
DISCUSSION
We have found that the parkin gene knockout and hypomorph have reduced locomotion due to slower movement resembling bradykinesia, reduced synaptic potentials, decreased quantal size, more positive RMP, lower oxygen consumption, reduced ATP and increased lactate, along with overgrown synapses. Our results provide a key description of the inter-related actions of a PD-related gene on the electrophysiology, anatomy and respiration of the most genetically tractable model organism, open to cellular and molecular analysis.
Although several laboratories have generated parkin knockouts and hypomorphs (9,17,21,22), only adult phenotypes were reported previously. This is the first report of larval defects associated with mutations in this early-onset gene. Indeed, one group specifically mentioned that they detected no larval phenotype (9). Unlike these groups, we obtain few adult park25 or parkZ3678 homozygotes, so we suspect that the severity of parkin phenotypes is affected by differences in food, possibly trace minerals (23,24) that modulate oxidative stress.
Our key observation is that the neuronal parkin knockout produces a bradykinesia-like reduction in locomotion. Others have shown loss of mobility in geotaxic assays of adult flies following manipulation of PD-related genes in muscles (11,25). We have taken this further by showing that the larval deficit is neuronal, not muscular as slower peristalsis results from reduced activity in the motoneurons. We show this by two ways: genetically and physiologically. Genetically, we show that (in the early stages of the degeneration) the nervous system is the main tissue affected by parkin as we can rescue the locomotory and bradykinesia phenotypes by neuronal expression of wild-type parkin. Physiologically, we show that both synaptic transmission and central pattern generator (CPG) activity are reduced. The reduction in CPG activity may result from reduced neurotransmission. The CPG may also be slowed by pumping deficits, as the low levels of ATP will increase the time taken to correct the ionic imbalance that builds up in neurons during each peristaltic wave (26). In addition, locomotion is also modulated by aminergic (dopamine, serotonin etc) neurons that depend on the Drosophila vesicular aminergic transporter (DVMAT). Mutations in this gene severely slow crawling (27). Since adult data show an interaction of DVMAT with parkin (21), so too, may larval bradykinesia result from parkin-induced disruption of aminergic transmission.
The smaller EJPs and mEJPs in the parkin knockout indicate a reduction in synaptic transmission. This is most likely to result from reduced glutamate release—perhaps, as a result of problems with its transport into vesicles in an ATP-dependent manner. The recent suggestion that parkin may affect transmission through its interaction with endocytic proteins, e.g. endophilin-A (28) is unlikely to explain our data: although parkin is known to function as a ubiquitin E3 ligase, failure of parkin to ubiquitinate and so degrade endophilin is unlikely to reduce synaptic transmission. A third possibility is that the motoneuron, like the muscle, has a more positive resting potential and this affects the dynamics of glutamate release. Finally, we note that the neuronal rescue did not fully restore the size of the EJP, so that a reduction in muscle impedance or post-synaptic ionic dynamics may also contribute to the reduction in synaptic transmission. Although the original descriptions of the phenotype of parkin null mice were divergent (29), more recent evidence indicates that these mice do have reduced dopamine release at striatal synapses (30). At the fly neuromuscular junction, manipulations of another PD-related gene, LRRK2, and its Drosophila homologue dLRRK, show complex effects on synaptic transmission. These include a reduction in synaptic transmitter release in the dLRRK knockout (31). However, the other effects of LRRK2 and parkin manipulations are dissimilar, indicating that these genes may not always be acting through a common cellular mechanism.
The more positive RMP in our parkin larvae is most likely to be due to a reduction in ATP synthesis, which requires oxygen. We have demonstrated this using two techniques to record O2 consumption and by measuring ATP levels directly. A similar reduction in ATP production (by 58%) is reported in human fibroblasts from patients with parkin mutations (32). Low ATP will lead to reduced pumping of ions across the membrane and so a more positive RMP (although this may be counteracted by increased opening of ATP-sensitive potassium channels (33)). Reports of Drosophila mutants with a more positive membrane potential are rare, but in one, the phosphoglycerate kinase mutant, nubian, this is due to reduced ATP levels (34). In the parkin knockout, we predict that neurons (as well as muscles) will also have a more positive membrane potential because the mutation is expressed globally.
The reduction of oxygen consumption indicates severe mitochondrial dysfunction; not surprising in the light of the reports of deficient mitochondrial fission/fusion and mitochondrial swelling in a range of fly models of PD (3,17,20,35). The parkin larvae respond to this by increasing glycolysis, as indicated by the increase in lactate levels. Increased lactate has been reported in some PD patients (36), but not in others (37). This discrepancy might be due to a general increase in lactate (and changes in anaerobic metabolism) with age (38), and so our data, along with improvements in genotyping and other molecular techniques may make the role of glycolytic biomarkers in the progression of PD worth revisiting.
Mitochondrial dysfunction will also induce oxidative stress. A key response to oxidative stress is the induction of autophagy (15), which causes increased growth of neuronal terminals (16). As with our parkin, so also manipulations of a second PD-related gene, dLRRK cause overgrowth of the larval neuromuscular junction (31). We note that, in both transgenics, the overgrowth phenotype is effectively rescued by global, neuronal or muscle expression of wild-type protein. This shows a difference from our physiological and locomotor phenotypes, which were rescued by neuronal—but not muscle—expression of parkin.
We find excellent rescue of the neuronal overgrowth by relieving oxidative stress. Increased expression of cytosolic (catalase, superoxide dismutase 1) or mitochondrial (superoxide dismutase 2) enzymes that scavenge ROS are equally effective. Similarly, adult fly models of PD are rescued by overexpression of genes that reduce ROS, e.g. glutathione S-transferase or thioredoxin (11–14).
However, over-expressing oxidative stress transgenes rescues only the neuronal overgrowth; the RMP and locomotor phenotype are not affected. (This is a major contrast to parkin expression, which rescues each of the oxidative stress, RMP and locomotor phenotypes.) Thus, expression of the ROS scavengers exposes fundamental physiological deficits, including the failure to maintain the RMP and the failure of the neuronal network of the CPG. In the same way, antioxidants like vitamin E have powerful cellular actions, but are not generally effective in the treatment of PD patients (39,40).
In conclusion, we find that parkin mutants recapitulate the bradykinesia symptoms of PD, and we suggest that this is due to an energy deficit rather than oxidative stress. parkin-Dependent mitochondrial dysfunction reduces ATP levels and the ability of neurons to maintain an RMP. Oxidative stress has received considerable attention as a target for treating ageing and neurodegenerative disorders; however, this study suggests that energy metabolism is a more important target for the treatment of PD.
MATERIALS AND METHODS
Flies
Flies were routinely raised at 25°C. Wild-type flies (Canton-S, CS and w−, w1118) were obtained from our laboratory stocks, while the parkin25(17), parkinZ3678 (11) and UAS-parkinC2 (wild-type Drosophila parkin (11)) lines were a gift from Alex Whitworth & Leo Pallanck. The G14 GAL4 line (from Akinao Nose, Tokyo), UAS stocks and Actin5C and elav3E1 GAL4 lines (from the Bloomington Stock centre) were recombined with the parkin lines as required. All experiments were performed on the third instar larvae, selected as they crawled on the side of the vial or over the surface of the food.
In all experiments, we find significant differences between the Canton-S (CS) wild-type and park25/park25 null. When we compare the wild-type outcross, CS/w− and the park25/parkZ3678 transheterozygote, we observe the same effects. All the parkin rescue experiments were performed both in the null background and the transheterozygote background; we find no differences due to background.
Larval locomotion
Larval locomotion was recorded using our standard procedure (41). Larval progress across a horizontal agar-filled Petri dish was recorded on to a computer disk for 2 min at a frame rate of 12 frames/min. Positions were determined using the MTrack2 plugin (42) (see also online http://valelab.ucsf.edu/~nico/IJplugins/MTrack2.html) to ImageJ (http://rsbweb.nih.gov/ij/).
Larval length assay
Larval length assay was performed as described recently (41). A larva pulls on a flexible plastic optical pipe, through which the beam of a red LED is transmitted onto a photodetector. The four optosensors in the detector allow the linear movement of the beam to be computed, with the signal recorded on computer disk at 0.1 Hz.
Saline solution
All experiments were performed in HL3 solution (43). Composition in millimolars—sodium chloride, 70; potassium chloride, 5; calcium chloride, 1; sodium hydrogen carbonate, 10; BES (N,N-bis(2-hydroxyethyl)-2-aminoethanesulphonic acid), 5; trehalose, 5; sucrose, 115; pH 7.5.
Segmental nerve recordings
Segmental nerve recordings were made by dissecting open the larva in HL3 saline, and removing its gut, fat body and reproductive organs. The segmental nerves were all severed and the CNS end of one drawn up into a glass suction pipette. The signal was amplified 1000 times and recorded on disk at 10 kHz.
Intracellular recording from the longitudinal muscles
Intracellular recording from the longitudinal muscles 6/7 was made with sharp glass micropipettes, with tip resistance 10–15MΩ. A high-resistance input 50× amplifier was used, and data stored on computer disk at 5 kHz. All data were recorded in HL3 saline. Spontaneous EJPs were recorded from preparations in which the CNS was left in place with intact innervation and the size of the large (Is) EJPs measured; mEJPs were analysed in preparations from which the CNS had been removed. Data from larvae with the RMP more positive than −40 mV were discarded.
Oxygen consumption
Oxygen consumption was recorded with two techniques. Firstly, using a capillary respirometer three larvae were placed in an unfilamented borosilicate glass capillary (internal diameter 1.16 mm, length 150 mm; Clark Electromedical, Pangbourne, UK) and one end sealed with cotton wool/nail varnish. Carbon dioxide was absorbed by ∼2 mg sodium hydroxide, and the volume change monitored by the movement of a paraffin oil droplet. The respiratory quotient was determined from larvae tested with and without the hydroxide to absorb carbon dioxide. No movement of the paraffin droplet was seen in replicates without larvae. Experiments were all performed at 21°C. Secondly, using a Clark oxygen electrode, in which larvae to a mass of 50 mg (∼30–40 larvae) were placed in the recording chamber filled with 3 ml HL3 saline.
ATP and lactate were measured in larval haemolymph using kits from proteinkinase.de (Kassel, Germany) and Eton Biosciences Inc. (San Diego, USA), respectively, following the manufacturers' protocols. Bradford assays (44) were used to measure the protein concentrations in the samples.
Synaptic boutons
Synaptic boutons were counted as described previously (16), using a polyclonal synaptotagmin antibody to mark synaptic-release sites and horseradish peroxidase antiserum to mark the nerve membrane.
Statistics
Statistics were evaluated in SPSS. In experiments with multiple genotypes, ANOVA followed by post hoc tests were used to compare two genotypes and nested ANOVA to compare groups of related genotypes (e.g. all parkin lines with all wild-type lines). Where Bonferroni probabilities are reported, at least the same probability level was seen in the Tukey HSD test. In the figures, NS denotes not significant and *P < 0.05, **P < 0.01, ***P < 0.001.
FUNDING
This work was supported by the Parkinson's Disease Society (now Parkinson's UK, Grant K-0804), BBSRC (for studentship to A.V.), The UK Medical Research Council project grant G0400580 (S.T.S.) and The Wellcome Trust, and Nuffield Foundation for funding. We are grateful to John Sparrow and Sangeeta Chawla for their comments on the manuscript. Funding to pay the Open Access publication charges for this article was provided by the Wellcome Trust.
ACKNOWLEDGEMENTS
We thank Alex Whitworth (University of Sheffield) for fly stocks, Harry Whitwell for assistance with experiments.
Conflict of Interest statement: None declared.
REFERENCES
- 1.Hardy J. Genetic analysis of pathways to Parkinson disease. Neuron. 2010;68:201–206. doi: 10.1016/j.neuron.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dawson T.M., Ko H.S., Dawson V.L. Genetic animal models of Parkinson's disease. Neuron. 2010;66:646–661. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whitworth A.J. Drosophila models of Parkinson's disease. Adv. Genet. 2011;73:1–50. doi: 10.1016/B978-0-12-380860-8.00001-X. [DOI] [PubMed] [Google Scholar]
- 4.Lu B. Recent advances in using Drosophila to model neurodegenerative diseases. Apoptosis. 2009;14:1008–1020. doi: 10.1007/s10495-009-0347-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Botella J.A., Bayersdorfer F., Gmeiner F., Schneuwly S. Modelling Parkinson's disease in Drosophila. NeuroMol. Med. 2009;11:268–280. doi: 10.1007/s12017-009-8098-6. [DOI] [PubMed] [Google Scholar]
- 6.Venken K., Simpson J., Bellen H. Genetic manipulation of genes and cells in the nervous system of the fruit fly. Neuron. 2011;72:202–230. doi: 10.1016/j.neuron.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tain L.S., Mortiboys H., Tao R.N., Ziviani E., Bandmann O., Whitworth A.J. Rapamycin activation of 4E-BP prevents Parkinsonian dopaminergic neuron loss. Nat. Neurosci. 2009;12:1129–1135. doi: 10.1038/nn.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Budnik V., Ruiz-Canada C. The fly neuromuscular junction: structure and function. 2nd edn. San Diego, CA, USA: Academic Press; 2006. [Google Scholar]
- 9.Pesah Y., Pham T., Burgess H., Middlebrooks B., Verstreken P., Zhou Y., Harding M., Bellen H., Mardon G. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development. 2004;131:2183–2194. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- 10.Meulener M., Whitworth A.J., Armstrong-Gold C.E., Rizzu P., Heutink P., Wes P.D., Pallanck L.J., Bonini N.M. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson's disease. Curr. Biol. 2005;15:1572–1577. doi: 10.1016/j.cub.2005.07.064. [DOI] [PubMed] [Google Scholar]
- 11.Whitworth A.J., Theodore D.A., Greene J.C., Benes H., Wes P.D., Pallanck L.J. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson's disease. Proc. Natl Acad. Sci. USA. 2005;102:8024–8029. doi: 10.1073/pnas.0501078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Im J.Y., Lee K.W., Junn E., Mouradian M.M. DJ-1 protects against oxidative damage by regulating the thioredoxin/ASK1 complex. Neurosci. Res. 2010;67:203–208. doi: 10.1016/j.neures.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Umeda-Kameyama Y., Tsuda M., Ohkura C., Matsuo T., Namba Y., Ohuchi Y., Aigaki T. Thioredoxin suppresses Parkin-associated endothelin receptor-like receptor-induced neurotoxicity and extends longevity in Drosophila. J. Biol. Chem. 2007;282:11180–11187. doi: 10.1074/jbc.M700937200. [DOI] [PubMed] [Google Scholar]
- 14.Botella J.A., Bayersdorfer F., Schneuwly S. Superoxide dismutase overexpression protects dopaminergic neurons in a Drosophila model of Parkinson's disease. Neurobiol. Dis. 2008;30:65–73. doi: 10.1016/j.nbd.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 15.Shen W., Ganetzky B. Autophagy promotes synapse development in Drosophila. J. Cell. Biol. 2009;187:71–79. doi: 10.1083/jcb.200907109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milton V.J., Jarrett H.E., Gowers K., Chalak S., Briggs L., Robinson I.M., Sweeney S.T. Oxidative stress induces overgrowth of the Drosophila neuromuscular junction. Proc. Natl Acad. Sci. USA. 2011;108:17521–17526. doi: 10.1073/pnas.1014511108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greene J.C., Whitworth A.J., Kuo I., Andrews L.A., Feany M.B., Pallanck L.J. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl Acad. Sci. USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bellen H.J., Tong C., Tsuda H. 100 years of Drosophila research and its impact on vertebrate neuroscience: a history lesson for the future. Nat. Rev. Neurosci. 2010;11:514–522. doi: 10.1038/nrn2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurdyak P., Atwood H.L., Stewart B.A., Wu C.F. Differential physiology and morphology of motor axons to ventral longitudinal muscles in larval Drosophila. J. Comp. Neurol. 1994;350:463–472. doi: 10.1002/cne.903500310. [DOI] [PubMed] [Google Scholar]
- 20.Poole A.C., Thomas R.E., Andrews L.A., McBride H.M., Whitworth A.J., Pallanck L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl Acad. Sci. USA. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sang T.K., Chang H.Y., Lawless G.M., Ratnaparkhi A., Mee L., Ackerson L.C., Maidment N.T., Krantz D.E., Jackson G.R. A Drosophila model of mutant human Parkin-induced toxicity demonstrates selective loss of dopaminergic neurons and dependence on cellular dopamine. J. Neurosci. 2007;27:981–992. doi: 10.1523/JNEUROSCI.4810-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cha G.H., Kim S., Park J., Lee E., Kim M., Lee S.B., Kim J.M., Chung J., Cho K.S. Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. Proc. Natl Acad. Sci. USA. 2005;102:10345–10350. doi: 10.1073/pnas.0500346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saini N., Schaffner W. Zinc supplement greatly improves the condition of parkin mutant Drosophila. Biol. Chem. 2010;391:513–518. doi: 10.1515/BC.2010.052. [DOI] [PubMed] [Google Scholar]
- 24.Saini N., Oelhafen S., Hua H., Georgiev O., Schaffner W., Bueler H. Extended lifespan of Drosophila parkin mutants through sequestration of redox-active metals enhancement of anti-oxidative pathways. Neurobiol. Dis. 2010;40:82–92. doi: 10.1016/j.nbd.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 25.Feany M.B., Bender W.W. A Drosophila model of Parkinson's disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- 26.Pulver S.R., Griffith L.C. Spike integration and cellular memory in a rhythmic network from Na+/K+ pump current dynamics. Nat. Neurosci. 2010;13:53–59. doi: 10.1038/nn.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simon A.F., Daniels R., Romero-Calderon R., Grygoruk A., Chang H.Y., Najibi R., Shamouelian D., Salazar E.D., Solomon M., Ackerson L.C., et al. Drosophila vesicular monoamine transporter mutants can adapt to reduced or eliminated vesicular stores of dopamine and serotonin. Genetics. 2009;181:525–541. doi: 10.1534/genetics.108.094110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trempe J.F., Chen C.X.Q., Grenier K., Camacho E.M., Kozlov G., McPherson P.S., Gehring K., Fon E.A. SH3 domains from a subset of BAR proteins define a Ubl-binding domain and implicate Parkin in synaptic ubiquitination. Mol. Cell. 2009;36:1034–1047. doi: 10.1016/j.molcel.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 29.Perez F.A., Palmiter R.D. Parkin-deficient mice are not a robust model of parkinsonism. Proc. Natl Acad. Sci. USA. 2005;102:2174. doi: 10.1073/pnas.0409598102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kitada T., Pisani A., Karouani M., Haburcak M., Martella G., Tscherter A., Platania P., Wu B., Pothos E.N., Shen J. Impaired dopamine release and synaptic plasticity in the striatum of Parkin–/– mice. J. Neurochem. 2009;110:613–621. doi: 10.1111/j.1471-4159.2009.06152.x. [DOI] [PubMed] [Google Scholar]
- 31.Lee S., Liu H.P., Lin W.Y., Guo H., Lu B. LRRK2 kinase regulates synaptic morphology through distinct substrates at the presynaptic and postsynaptic compartments of the Drosophila neuromuscular junction. J. Neurosci. 2010;30:16959–16969. doi: 10.1523/JNEUROSCI.1807-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mortiboys H., Thomas K.J., Koopman W.J.H., Klaffke S., bou-Sleiman P., Olpin S., Wood N.W., Willems P.H.G.M., Smeitink J.A.M., Cookson M.R. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann. Neurol. 2008;64:555–565. doi: 10.1002/ana.21492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liss B., Haeckel O., Wildmann J., Miki T., Seino S., Roeper J. K-ATP channels promote the differential degeneration of dopaminergic midbrain neurons. Nat. Neurosci. 2005;8:1742–1751. doi: 10.1038/nn1570. [DOI] [PubMed] [Google Scholar]
- 34.Wang P., Saraswati S., Guan Z., Watkins C.J., Wurtman R.J., Littleton J.T. A Drosophila temperature-sensitive seizure mutant in phosphoglycerate kinase disrupts ATP generation and alters synaptic function. J. Neurosci. 2004;24:4518. doi: 10.1523/JNEUROSCI.0542-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ziviani E., Tao R.N., Whitworth A.J. Drosophila Parkin requires PINK1 for mitochondrial translocation and ubiquitinates Mitofusin. Proc. Natl Acad. Sci. USA. 2010;107:5018. doi: 10.1073/pnas.0913485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bowen B.C., Block R.E., Sanchez-Ramos J., Pattany P.M., Lampman D.A., Murdoch J.B., Quencer R.M. Proton MR spectroscopy of the brain in 14 patients with Parkinson disease. Am. J. Neuroradiol. 1995;16:61–68. [PMC free article] [PubMed] [Google Scholar]
- 37.Yamamoto M., Ujike H., Wada K., Tsuji T. Cerebrospinal fluid lactate and pyruvate concentrations in patients with Parkinson's disease and mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) J. Neurol. Neurosurg. Psych. 1997;62:290. doi: 10.1136/jnnp.62.3.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ross J.M., Oberg J., Brene S., Coppotelli G., Terzioglu M., Pernold K., Goiny M., Sitnikov R., Kehr J., Trifunovic A., et al. High brain lactate is a hallmark of aging and caused by a shift in the lactate dehydrogenase A/B ratio. Proc. Natl Acad. Sci. USA. 2010;107:20087–20092. doi: 10.1073/pnas.1008189107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parkinson Study Group. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson's disease. N. Engl. J. Med. 1993;328:176–183. doi: 10.1056/NEJM199301213280305. [DOI] [PubMed] [Google Scholar]
- 40.Löhle M., Reichmann H. Clinical neuroprotection in Parkinson's disease: still waiting for the breakthrough. J. Neurol. Sci. 2010;289:104–114. doi: 10.1016/j.jns.2009.08.025. [DOI] [PubMed] [Google Scholar]
- 41.Hill J.A. The roles of tyramine and octopamine in Drosophila melanogaster. 2008 PhD Thesis, University of York. [Google Scholar]
- 42.Klopfenstein D.R., Vale R.D. The lipid binding pleckstrin homology domain in UNC-104 kinesin is necessary for synaptic vesicle transport in Caenorhabditis elegans. Mol. Biol. Cell. 2004;15:3729–3739. doi: 10.1091/mbc.E04-04-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stewart B.A., Atwood H.L., Renger J.J., Wang J., Wu C.F. Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J. Comp. physiol. A. 1994;175:179–191. doi: 10.1007/BF00215114. [DOI] [PubMed] [Google Scholar]
- 44.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]