

Abstract
The enteric nervous system (ENS) comprises a complex neuronal network that regulates peristalsis of the gut wall and secretions into the lumen. The ENS is formed from a multipotent progenitor cell population called the neural crest, which is derived from the neuroepithelium. Neural crest cells (NCCs) migrate over incredible distances to colonize the entire length of the gut and during their migration they must survive, proliferate and ultimately differentiate. The absence of an ENS from variable lengths of the colon results in Hirschsprung's disease (HSCR) or colonic aganglionosis. Mutations in about 12 different genes have been identified in HSCR patients but the complex pattern of inheritance and variable penetrance suggests that additional genes or modifiers must be involved in the etiology and pathogenesis of this disease. We discovered that Tcof1 haploinsufficiency in mice models many of the early features of HSCR. Neuroepithelial apoptosis diminished the size of the neural stem cell pool resulting in reduced NCC numbers and their delayed migration along the gut from E10.5 to E14.5. Surprisingly however, we observe continued and complete colonization of the entire colon throughout E14.5–E18.5, a period in which the gut is considered to be non- or less-permissive to NCC. Thus, we reveal for the first time that reduced NCC progenitor numbers and delayed migration do not unequivocally equate with a predisposition for the pathogenesis of HSCR. In fact, these deficiencies can be overcome by balancing NCC intrinsic processes of proliferation and differentiation with extrinsic influences of the gut microenvironment.
INTRODUCTION
A normal functioning bowel requires the presence of a complete enteric nervous system (ENS) throughout its entire length. The mammalian ENS is derived from a migratory progenitor cell population called the neural crest (1,2). More specifically, neural crest cells (NCCs) within the vagal region of the neural tube (adjacent to somites 1–7) of embryonic day (E) 9.0 embryos, delaminate and travel ventrally through the embryo reaching the foregut by E9.5. During the next 5 days of embryogenesis, vagal NCC advance throughout the entire extent of the bowel (3–6) and coalesce into discrete ganglia that comprise the myenteric and submucosal plexi (7). The absence of ganglia from variable portions of the colon is a characteristic feature of Hirschsprung's disease (HSCR), a common human disease that affects 1 : 5000 live births (8).
Insights into the etiology and pathogenesis of HSCR have been obtained from analyses of NCC development in genetically mutant mice and in neural tube ablation/grafting experiments performed in avian embryos (8–14). These experiments suggest that normal ENS formation depends upon a critical balance between NCC survival, proliferation, differentiation and migration during all stages of ENS development. Tight control of these processes ensures that a sufficient progenitor cell pool enters the foregut at the correct time and furthermore that the correct balance of NCC proliferation and differentiation is maintained as these cells migrate along the gut. This preserves a critical number of dividing cells, which together with specific cell–cell interactions established at the NCC migration wavefront facilitates their advancement along the entire length of the gut.
The NCC micro-environment plays a critical role in regulating the extent of ENS formation through its influence on NCC number and their colonization of the gut. Glial cell-derived neurotrophic factor (GDNF) is a ligand for the receptor tyrosine kinase (RET), and modulating the level of this mesenchymal factor in vivo alters NCC survival, proliferation, migration and differentiation along the gut (15–21). Extracellular matrix (ECM) components such as tenascin-C and fibronectin that are present within the cecum and proximal colon may also influence NCC migration and development (22). Increased laminin is detected in the colon of Endothelin3 (Edn3ls/ls)-deficient mice that exhibits perturbed endothelin receptor B (Ednrb) signaling. Consequently, this promotes neuronal differentiation at the expense of NCC migration (23,24) resulting in incomplete colonization of the colon (25). Furthermore, age-dependent changes in the ECM have been postulated to inhibit the migration of Ednrb−/− NCC along the colon beyond E14.5 (26). Consistent with this idea, NCC invasion of the colon has been shown to decrease with increasing age (25). These data suggest that there is a limited temporal window available for NCC colonization of the colon after which the environment becomes either non-permissive or less-permissive.
To date, over a dozen HSCR disease-associated genes have been identified, with RET being the most significant as it accounts for 50% of familial and about 20% of sporadic cases (27). Mutations have also been described in the RET ligands, GDNF and neurturin, in the co-receptor GFRα1, as well as in endothelin-signaling genes, NRG1, KBP, L1-CAM and the transcription factors SOX10, ZEB2 and PHOX2B (9,12,13,28–30). However, a large number of HSCR cases are currently genetically undetermined. Therefore, additional genes or modifiers must be involved in the complex pattern of inheritance and variable penetrance observed in HSCR. To this end, we have identified Tcof1, which encodes a putative nucleolar protein known as Treacle, as an important regulator of vagal NCC development and ENS formation. Tcof1 loss-of-function results in a deficiency of vagal NCC and their delayed colonization of the gut during early embryogenesis, which mimics the early stages of HSCR. Surprisingly however, complete ENS formation is achieved by E18.5. Consequently, we discovered that precise regulation of progenitor pool proliferation enables NCC colonization of the entire colon, beyond stages that are typically considered to be less or non-permissive. Thus, complete ENS formation depends upon a critical balance between intrinsic and extrinsic signals that regulate the survival, proliferation, migration and differentiation of vagal NCC.
RESULTS
Tcof1+/− mice model features of HSCR
NCCs play essential roles in the development of craniofacial structures, the outflow tract of the heart and other systems such as the ENS. We have previously demonstrated that Tcof1/TCOF1 plays an important role in neuroepithelial cell and NCC development with respect to craniofacial development and pathogenesis of Treacher Collins syndrome (31,32). Tcof1 is widely expressed during embryogenesis (31,33) and Tcof1−/− embryos die between implantation and gastrulation which demonstrates that Tcof1 plays important functions in many cell types. Haploinsufficiency of Tcof1/Treacle however, primarily affects neuroepithelial cell and NCC development. One logical explanation for these haploinsufficient effects is that Tcof1/Treacle has been shown both in vivo and in vitro to play a critical role in regulating ribosome biogenesis and cell proliferation. Thus, during early embryogenesis, the neuroepithelium proliferates rapidly while at the same time generating an entirely new cell population; the neural crest, which makes it particularly sensitive to the loss of Tcof1/Treacle. Congenital defects that arise through deficiencies in NCC are collectively termed neurocristopathies and often multiple parts of the body are simultaneously affected. Therefore, we explored the requirement for Tcof1 in the vagal NCC population that forms the ENS. To determine the extent of colonization of the gut by NCC in E10.5–E14.5 embryos, we whole-mount immunostained wild-type (Tcof1+/+) and Tcof1+/− guts with TuJ1, the neuronal antibody specific to βIII-tubulin which labels cell bodies and nerve fibers (Fig. 1). While Tcof1+/− embryos are slightly smaller than their wild-type counterparts, we observed no developmental delay. For these experiments, we size-matched the guts in order to ensure that any changes we observed in NCC reflected true ENS defects rather than effects caused by any size difference between embryos. We also did not observe any differences in the length of the gut tube. At E10.5, NC-derived cells expressing TuJ1 were detected within the esophagus, stomach and along most of the length of the small intestine (SI) in Tcof1+/+ embryos. In contrast, the migration wavefront was delayed in all of the Tcof1+/− guts examined, such that NC-derived cells had advanced to a maximum of only half the SI length. Furthermore, TuJ1+ neuronal cell bodies in the SI of Tcof1+/− embryos were typically organized in thin chains with extensive fasciculation of the nerve fibers, which is distinct from the widespread dispersed colonization observed in wild-type embryos (Fig. 1 and Supplementary Material, Fig. S1).
Figure 1.
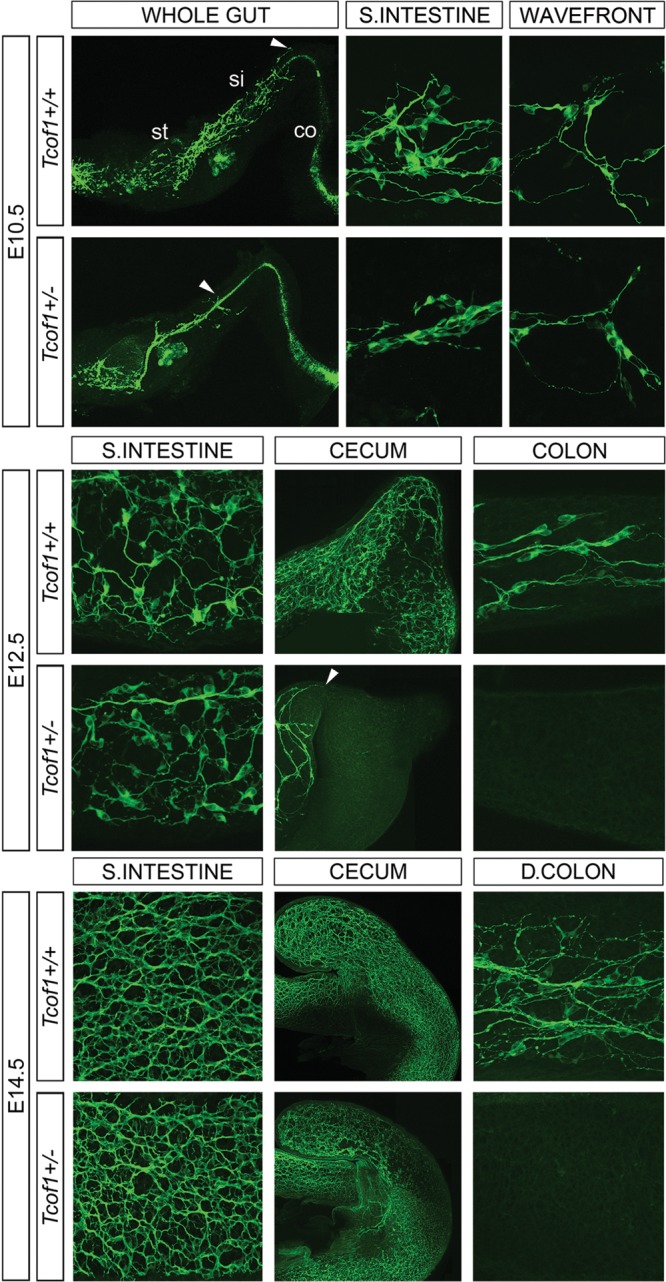
Delayed NCC migration in E10.5, E12.5 and E14.5 Tcof1+/− guts. TuJ1 immunostaining of E10.5 whole guts shows that NC-derived cells have colonized ≈50% of the SI in Tcof1+/− guts in comparison with wild-type guts where the migration wavefront is at the end of the SI (arrowhead). The cells appear to have migrated collectively in a thick chain in the SI in Tcof1+/− guts rather than extending evenly across the entire wall of the gut in Tcof1+/+. At E12.5, the migration wavefront is in the colon in Tcof1+/+ guts but has only reached the cecum in Tcof1+/− guts (arrowhead). There appears to be reduced or delayed NCC differentiation in Tcof1+/− guts at this stage. By E14.5, the characteristic network of NC-derived cells is present along the entire length of the gut in Tcof1+/+ mice, while <50% of Tcof1+/− guts contain aganglionic regions. A similar network is present in the SI in both genotypes. c, colon; D, distal; si, small intestine; st, stomach.
By E12.5, NC-derived cells had continued to travel along the gut wall in Tcof1+/+ guts such that ∼30% of the colon length exhibited TuJ1+ staining (Fig. 1 and Supplementary Material, Fig. S1). Moreover, NC-derived cells were arranged into a dense network along the SI and throughout the cecum (Fig. 1). In contrast, TuJ1+ cells were typically restricted to the SI and concomitantly were absent from the cecum in 67% of Tcof1+/− embryos analyzed (n= 15; Supplementary Material, Fig. S1). Cell bodies were more apparent within the SI of Tcof1+/− embryos with less fasciculation of nerve fibers suggesting either reduced or delayed neuronal differentiation.
As development proceeded, NCC continued to invade the mesenchyme of the gut wall of both Tcof1+/+ and Tcof1+/− embryos. At E14.5, the entire colon length contained TuJ1+ cells in Tcof1+/+ embryos (n= 18; Fig. 1 and Supplementary Material, Fig. S1), and neuronal cell bodies and axons were organized into a defined network in the SI (Fig. 1). All Tcof1+/− embryos examined contained an equivalent network within the SI, despite the fact that only about 50% of guts were completely colonized by NC-derived cells (n= 23; Fig. 1 and Supplementary Material, Fig. S1). The considerable delay in the colonization of the gut by NCC in E10.5–E14.5 in Tcof1+/− embryos is very similar to that described in animal models of HSCR (34).
Neural tube apoptosis reduces the progenitor cell pool in Tcof1+/− embryos
The delayed and reduced colonization of the gut wall by NCC in Tcof1+/− embryos could arise as a consequence of deficiencies in progenitor cell proliferation and survival. To test this hypothesis, we co-immunostained cryosections of E9.5–E10.5 Tcof1+/+ and Tcof1+/− embryos at the vagal neural tube level (somites 1–7) with p75 to identify NCC and either terminal dUTP nick end labeling (TUNEL) to mark apoptotic cells or phosphoHistone H3 (pHH3) to determine their mitotic index. These experiments revealed that the neural tube (NT) in Tcof1+/− embryos is visibly smaller and narrower than in Tcof1+/+ embryos, however, no developmental delay was noted (Fig. 2A). Consistent with this, we observed extensive TUNEL staining in the NT of Tcof1+/− embryos compared with wild-type (n= 4 and 3, respectively; Fig. 2A). Counting p75+ cells within these sections revealed a 40% reduction in the total numbers of NCC in Tcof1+/− embryos when compared with controls (Fig. 2B). However, in contrast there was no difference in the small fraction of TUNEL-positive NCC that had migrated from the NT into the foregut (Fig. 2B). Similarly, there was no significant difference in the mitotic index of NCC that had migrated towards and into the foregut (n= 3; Fig. 2B). Collectively, these results show that the vagal NCC progenitor pool is considerably diminished in Tcof1+/− embryos as a consequence of neural stem cell apoptosis within the NT.
Figure 2.
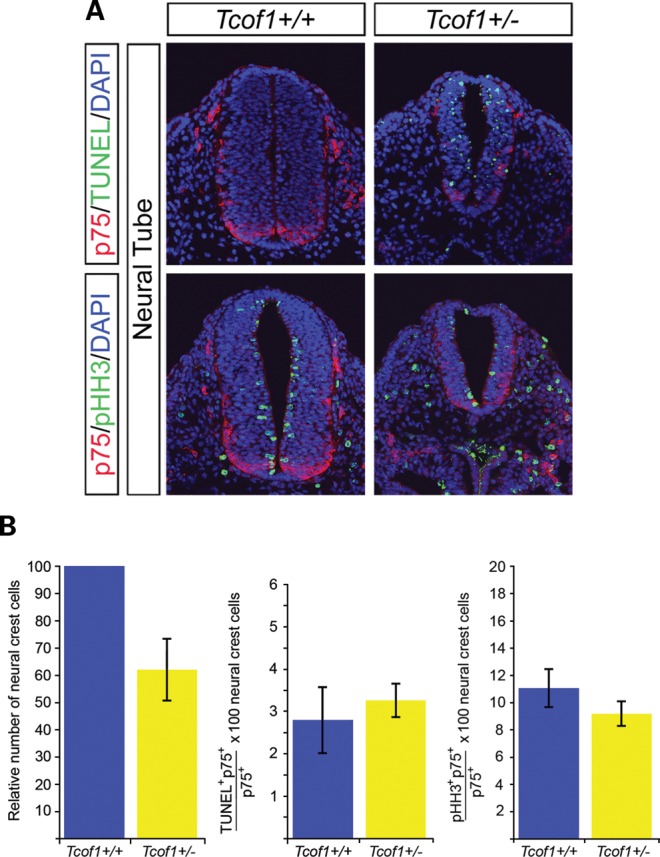
Neural tube apoptosis reduces the NCC numbers that migrate towards and into the foregut in Tcof1+/− mice. (A) Examination of apoptosis and proliferation was performed using immunostaining of cryosections of Tcof1+/+ and Tcof1+/− embryos with the NCC marker p75 and either TUNEL or pHH3, respectively. DAPI stained all nuclei. TUNEL staining was observed in the neural tube of Tcof1+/− embryos which are also smaller than their wild-type counterparts. No apparent proliferative differences were observed between genotypes. (B) Histograms showing that NCC numbers were reduced by 40% in Tcof1+/− embryos in comparison with Tcof1+/+. Similar percentages of apoptotic or proliferating NCC were found between genotypes.
NCC are less committed in Tcof1+/– embryos
To investigate whether Tcof1 haploinsufficiency alters the specification of the NCC that migrate towards and into the foregut, we co-immunostained cryosections of E10 Tcof1+/+ and Tcof1+/− embryos at the vagal neural tube level (somites 1–7) with the ENS progenitor marker Sox10 and the neuronal commitment marker, RET (Supplementary Material, Fig. S2A). These experiments revealed a significant reduction in the proportion of RET+ neural crest cells (RET+Sox10+/Sox10+) within the foregut of Tcof1+/− compared with Tcof1+/+ embryos (70 ± 9.8% versus 91 ± 2.5%, P= 0.002, n= 3; Supplementary Material, Fig. S2B). Thus, more multipotent progenitor cells enter the foregut in Tcof1+/− embryos compared with wild-type animals.
Continued NCC migration in Tcof1+/− guts beyond E14.5
To ascertain whether the loss of Tcof1 resulted in terminal colonic aganglionosis later in development, guts were dissected from E18.5 Tcof1+/+ and Tcof1+/− embryos and TuJ1-immunolabeled. NC-derived cells were present throughout the entire length of the colon in all of the wild-type and mutant embryos examined (Fig. 3). This was a surprising and unexpected finding as the colon is thought to become non-permissive or less-permissive to NCC colonization after E14.5. However, despite the fact that we observe a considerable delay in NCC colonization of the gut in Tcof1+/− embryos at E14.5, NCC are still capable of completing colonization of the colon by E18.5. This challenges the dogma in the field that there is a limited temporal window in which NCC can colonize the full extent of the colon, the failure of which manifests as HSCR.
Figure 3.
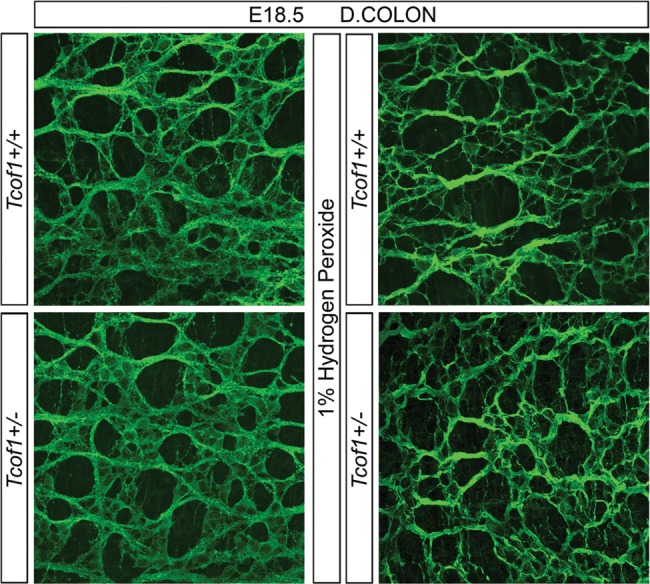
A complete ENS network is seen in Tcof1+/− guts at E18.5. TuJ1 immunostaining of whole guts at E18.5 shows the ENS network within the distal colon of E18.5 Tcof1+/+ and Tcof1+/− untreated and H2O2-treated guts. D, distal.
Increased NCC proliferation and reduced neuronal differentiation enable the continued advance of NCC along the gut wall
To determine the mechanism by which NCC in Tcof1+/− embryos are capable of continued migration along the gut tube beyond stages previously thought to be non-permissive, we analyzed NCC proliferation and neuronal differentiation at E11.5 when NCC were colonizing the cecum and at E13.5 during their advance along the colon. The proportion of proliferating NCC at E11.5 was scored in Tcof1+/+ and Tcof1+/− size-matched whole guts co-immunostained with p75 and either pHH3 or an antibody to detect BrdU incorporation (DNA synthesis label). The proportion of dividing NCC was quantified at both the migration wavefront and along the SI (Fig. 4A and B). At this stage, an equivalent proportion of dividing NCC was observed at both the wavefront and along the SI using both pHH3 and BrdU labeling in wild-type guts (11.6 ± 2% versus 11.5 ± 2% for pHH3 and 50.8 ± 2% versus 53 ± 2% for BrdU, respectively). However, we observed a significant increase in the percentage of proliferating NCC at the migration wavefront in Tcof1+/− guts using both pHH3 (16.9 ± 2% versus 11.6 ± 2%, P= 0.002, n= 4; Fig. 4A) and BrdU labeling (58.5 ± 4 versus 50.8 ± 2, P= 0.001, n= 4 and 7, respectively; Fig. 4B). In contrast, no difference in NCC proliferation was observed along the length of the SI (Fig. 4A and B). Examination of neuronal differentiation was also performed at E11.5 via co-immunostaining Tcof1+/+ and Tcof1+/− guts with p75 and TuJ1. Again, we noted a similar percentage of NCC-expressing TuJ1 at both the migration wavefront and along the SI in wild-type guts (19.9 ± 3% versus 18.6 ± 4%). Although, a significant reduction in the extent of neuronal differentiation both at the migration wavefront (5.8 ± 3% versus 19.9 ± 3%, n= 5 and 7, respectively; Fig. 4C) and along the length of the SI (8.3 ± 2% versus 18.6 ± 4%; Fig. 4C) was counted in Tcof1+/− compared with Tcof1+/+ embryos. The reduced neuronal differentiation detected with TuJ1 was also reflected in a smaller fraction of p75+ cells expressing RET scored in cryosections of the gut at this same stage (Supplementary Material, Fig. S2B). This indicates that NCC at the wavefront in Tcof1+/− embryos exhibit increased proliferation at the expense of differentiation and this may account for their capacity to colonize the entire extent of the colon.
Figure 4.

Increased proliferation at the NCC migration wavefront and reduced neuronal differentiation along the gut at E11.5 in Tcof1+/− embryos. Immunostaining of E11.5 whole guts with p75 (red) and either pHH3 (green) or BrdU (green) revealed no difference in proliferation between genotypes along the intestines. Dividing cells can be identified by the presence of green staining in the nucleus. However, a significant increase in NCC proliferation at the migration wavefront was counted in Tcof1+/− embryos compared with wild-type. Reduced neuronal differentiation was scored throughout the intestines in Tcof1+/− compared with Tcof1+/+ guts immunostained with p75 (red) and TuJ1 (green). *P < 0.05 (Student's t-test).
Unlike what we observed above, by E13.5, the proportion of dividing NCC was significantly increased at the migration wavefront compared with the SI in wild-type guts (43 ± 2% versus 22 ± 2%, P= 4.9×10−6). In addition to this, and consistent with what we had seen earlier, NCC proliferation was increased at the migration wavefront in the colon of E13.5 Tcof1+/− embryos compared with control Tcof1+/+ embryos (50 ± 5% versus 43 ± 2%, P= 0.04, n= 4; Supplementary Material, Fig. S3). In contrast, equivalent proportions of proliferating NCC were observed along the SI in comparisons between wild-type and mutant embryos (28 ± 5 versus 22 ± 2, P= 0.07; Supplementary Material, Fig. S3). Analysis of differentiation at this stage using co-immunostaining of whole guts with p75 and the neuronal marker, Hu showed a significant decrease in the percentage of NCC labeled with Hu at the migration wavefront compared with the SI in wild-type guts (12.9 ± 2% versus 18.5 ± 2%, P= 0.01). When we compared the neuronal differentiation at the migration wavefront between size-matched mutant and wild-type guts, we found a reduction in the percentage of NCC labeled with Hu in Tcof1+/− when compared with Tcof1+/+ guts (7.5 ± 1% versus 12.9 ± 2%, P= 0.003, n= 4; Supplementary Material, Fig. S3). In contrast, similar levels of neuronal differentiation were detected along the length of the SI between animals (15.8 ± 1 versus 18.5 ± 2, P= 0.09; Supplementary Material, Fig. S3) reflecting the comparable TuJ1 staining densities observed at E14.5 (Fig. 1). Therefore, the balance of increased NCC proliferation at the migration wavefront both at E11.5 and E13.5, together with reduced neuronal differentiation throughout the gut at E11.5 and at the wavefront at E13.5 in Tcof1+/− embryos, ensures that despite the initial reduction in the size of the progenitor pool that migrates into the foregut, a sufficient number of proliferating NCC are maintained that are capable of colonizing the gut wall at later developmental stages.
Manipulating the progenitor cell pool size in Tcof1+/− embryos
Tcof1 haploinsufficiency alone appears insufficient to cause aganglionosis. However, the considerable initial retardation in the extent of migration and colonization of the gastrointestinal tract in Tcof1+/− embryos is similar to that observed in mouse models of HSCR (34). Furthermore, diminishment of NCCs via neural tube ablation experiments in avians results in the absence of the ENS in the hindgut, mimicking the HSCR phenotype in humans (2,35–37). Moreover, analyses in avian embryos have suggested that only a minimal number of NCC is required to successfully complete colonization of the entire gut (35). Therefore, we hypothesized that Tcof1/Treacle may be a modifier of or be sensitive to conditions that promote the pathogenesis of colonic aganglionosis. It is therefore formally possible that the colon is successfully colonized by E18.5 in Tcof1+/− mice because the 40% reduction of NCC numbers in these mice does not reduce the progenitor cell pool below a critical minimal level or ‘tipping point’ (Fig. 2B). In order to reduce the NCC numbers further in Tcof1+/− mice, and test the hypothesis that a minimal number of NCC are required for complete colonization of the gut, we exposed Tcof1+/− embryos to oxidative stress. NCC cultures have been reported to be devoid of significant superoxide dismutase activity (38), such that exogenous oxidation preferentially affects NCC survival and proliferation (39). Therefore, we injected pregnant mice with either phosphate-buffered saline (PBS) or the oxidant, hydrogen peroxide (H2O2), beginning at E7, prior to vagal NCC formation and delamination. Subsequently, guts were harvested from treated embryos at E11.5 and the extent of ENS formation was examined. Analysis of Tcof1+/− guts using p75 and TuJ1 revealed that 67% of those harvested from H2O2-injected mice (n= 8/12; Fig. 5) exhibited a migration delay when compared with only 47% of Tcof1+/− guts treated with PBS (n= 9/19; Fig. 5). Thus, exogenous oxidative stress enhanced the frequency of retarded NCC migration and perturbed ENS formation. In addition, there was a statistical difference in the location of the caudal-most NCC in Tcof1+/− guts from mice treated with H2O2 when compared with PBS (P= 0.005), with the NCC migration wavefront being more severely retarded in guts of H2O2-treated Tcof1+/− embryos (Fig. 5).
Figure 5.

Oxidation treatment reduces the NCC migration in Tcof1+/− guts. Injection of pregnant females with 1% H2O2 prior to NCC delamination results in reduced NCC colonization of the gut in Tcof1+/− mice compared with wild-type and PBS-injected Tcof1+/− embryos. Immunostaining of E11.5 Tcof1+/+ and Tcof1+/− whole guts with p75 (red) and TuJ1 (green) showed a migration delay in 47% of Tcof1+/− injected with PBS which increased to ≈70% in H2O2-treated embryos. The percentage of SI and colon length colonized by NCC is represented by arrows. The number of guts with NCC at this position is above the arrow. (+/+), Tcof1+/+; (+/−), Tcof1+/−.
Since H2O2 exacerbated the retardation of NCC colonization of the gut in Tcof1+/− embryos when compared with PBS injected or untreated Tcof1+/− mice, we examined whether this effect was due to a further reduction in the size of the progenitor cell pool in these mice. Embryos from H2O2-injected mice were harvested at E9.5–E10 and cryosections of the vagal neural tube level (somites 1–7) were co-immunostained with p75 and either TUNEL or pHH3 as described above (Supplementary Material, Fig. S4A). Quantification of p75+ cells revealed about a 60% reduction in the NCC progenitor cell pool in Tcof1+/− embryos treated with H2O2 compared with wild-type littermates (Supplementary Material, Fig. S4B). This is reflected in the smaller number of NCC around the foregut (Supplementary Material, Fig. S4A). TUNEL staining was again apparent within the NT of only Tcof1+/− embryos (Supplementary Material, Fig. S4A) and analysis of the NCC that had migrated towards and into the foregut revealed no difference in the proportion of TUNEL+ NCC in comparisons between H2O2-treated wild-type and Tcof1+/− embryos (4.2 ± 0.9% versus 4.2 ± 0.5%, n= 4 and 5, respectively; Supplementary Material, Fig. S4B). However, there was a statistically significant increase in the number of apoptotic NCC in H2O2-treated Tcof1+/− embryos when compared with untreated Tcof1+/− embryos (4.2 ± 0.5% compared with 3.3 ± 0.3%, P= 0.04; compare Supplementary Material, Fig. S4B with Fig. 2B). Although a similar NCC mitotic index was observed in H2O2-treated wild-type and Tcof1+/− embryos, this was significantly reduced in comparison with untreated Tcof1+/+ (3.7 ± 1% versus 11 ± 1.4%, P= 0.0005; compare Supplementary Material, Fig. S4B with Fig. 2B) and untreated Tcof1+/− (4.3 ± 1% versus 9.3 ± 0.9%, P= 0.002; compare Supplementary Material, Fig. S4B with Fig. 2B) embryos. Thus, exogenous oxidative stress compromises survival in Tcof1+/− and proliferation in both wild-type and Tcof1+/− embryos; however, Tcof1+/− embryos are more sensitive to oxidative stress as they exhibit an exacerbated 60% reduction in NCC required to colonize the gut and form the ENS.
Complete ENS formation in Tcof1+/− despite severe reduction in NCC numbers
The extensive reduction in initial NCC numbers in H2O2-treated Tcof1+/− embryos due to increased apoptosis and reduced proliferation en route to the foregut is associated with significantly retarded colonization of the intestine at E11.5. However, TuJ1 staining of guts in E18.5 Tcof1+/− and wild-type mice treated with H2O2 showed a similarly complete ENS network throughout the entire length of the colon (n= 10; Fig. 3). Interestingly, progenitor cell pool size reductions of the magnitude observed in H2O2-treated Tcof1+/− mice have previously been reported in other mouse mutants to result in almost complete intestinal aganglionosis (40). However, despite a nearly two-thirds reduction of NCC in H2O2-treated Tcof1+/− embryos, these residual cells are still capable of completing colonization of the entire colon and forming a complete ENS during embryonic development.
Increased NCC proliferation and reduced neuronal migration in H2O2-injected Tcof1+/− embryos maintain a sufficient progenitor cell pool
To understand why the gut is completely colonized at E18.5 in H2O2-injected Tcof1+/− embryos, we analyzed NCC proliferation and neuronal differentiation at E11.5 using p75 and either BrdU or TuJ1. A similar effect on NCC proliferation was seen in these guts as detected previously in untreated embryos where there was a greater percentage of BrdU+ NCC only at the migration wavefront in Tcof1+/− embryos compared with guts in H2O2-treated Tcof1+/+ embryos (58.6 ± 7% versus 47 ± 5%, P= 0.05). No difference in proliferation was observed along the SI (47 ± 8% versus 47 ± 3%). However, neuronal differentiation was significantly reduced in guts from H2O2-treated Tcof1+/− embryos when compared with controls both at the migration wavefront (5.5 ± 1% versus 16 ± 0.5, P= 2×10−6) and along the length of the SI (6 ± 1% versus 15.7 ± 4%, P= 0.002). Thus, despite the dramatic effects of H2O2 on the NCC progenitor pool and exacerbated retardation of gut colonization in Tcof1+/− embryos, increased NCC proliferation at the migration wavefront and an overall reduction in neuronal differentiation facilitate the eventual—albeit delayed—formation of a complete ENS.
Changes in ECM molecules in Tcof1+/− guts
Since temporal changes in the gut microenvironment may affect NCC migration along the colon (25,26), we examined the expression of the cell adhesion molecules, laminin and β1 integrin in the colon of E13 and E14 Tcof1+/+ and Tcof1+/− embryos. Laminin was detected in the basal laminae around the epithelium, around blood vessels and in the serosal layer along the length of the colon and the intensity of staining appeared to be stronger and more diffuse in the serosal layer of Tcof1+/− embryos when compared with Tcof1+/+ controls (Fig. 6). At E13, β1 integrin expression was observed across the colon with higher levels around blood vessels. Subsequently, the expression of β1 integrin increases with developmental age such that the staining is greater at E14, especially in the terminal colon (Fig. 6). Initially at E13.5, less β1 integrin expression was detected in the proximal colon of Tcof1+/− embryos when compared with Tcof1+/+ embryos (38 ± 2 versus 51 ± 1 mean pixels/measured area; Fig. 6). However, by E14.5, expression in the proximal colon was noticeably higher in the Tcof1+/− embryos when compared with wild-type. In contrast, β1 integrin levels in the distal colon were comparable between mutants and controls (89 ± 2 versus 71 ± 2 mean pixels/area measured, Fig. 6). Thus, we observed an increase in laminin expression throughout the colon during development in Tcof1+/− embryos compared with wild-type. However, the change in β1 integrin expression between the genotypes was restricted to the proximal colon, where it had previously been shown to be required to modulate the effects of tenascin-C and fibronectin (22). Although the activity of laminin and β1 integrin dynamically changes in the gut throughout embryogenesis and there are spatially quantitative differences between wild-type and Tcof1+/− individuals, neither molecule is absent. Thus, the extracellular matrix molecules appear not to play a critical role in regulating NCC colonization of the colon in Tcof1+/− embryos. Rather, Tcof1+/− appears to be an important regulator of vagal NCC number and ENS formation via its effects on vagal NCC formation, survival, proliferation and differentiation.
Figure 6.
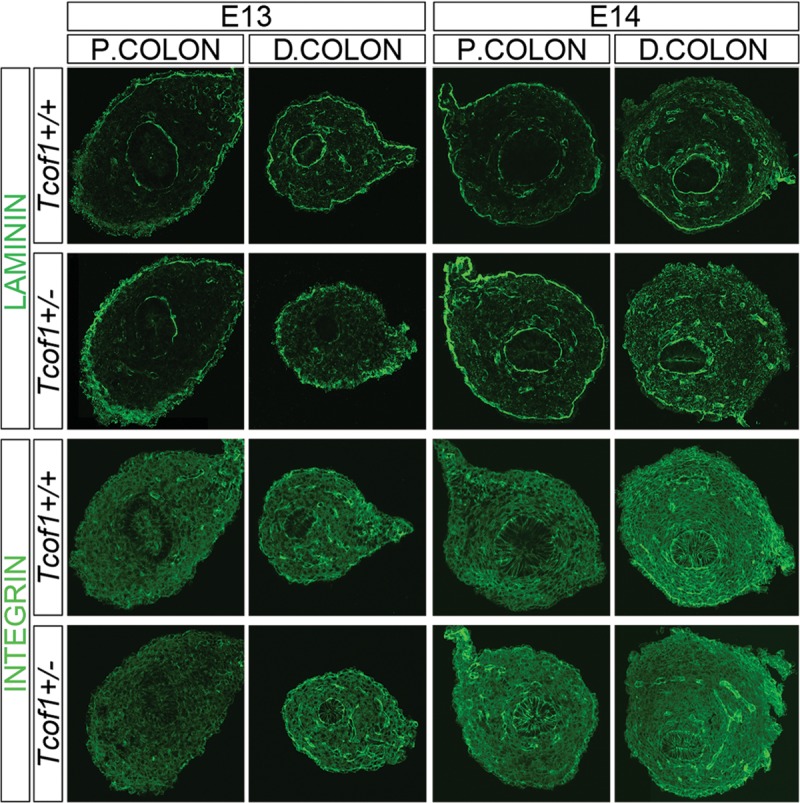
Increased laminin and reduced β1 integrin expression in Tcof1+/− guts compared with Tcof1+/+. Immunostaining of E13 and E14 colon cryosections with laminin and β1 integrin antibodies. Greater levels and more diffuse laminin staining in Tcof1+/− colon compared with wild-type at both stages. The expression of β1 integrin increased with developmental age in both genotypes; however, β1 integrin was initially reduced in the proximal colon at E13, but later at E14, was increased in this region in Tcof1+/− colon in comparison with Tcof1+/+. P, proximal; D, distal.
DISCUSSION
In this study, we have identified Tcof1 as a novel key regulator of vagal NCC development. Tcof1+/− mice exhibit many of the early developmental features of HSCR. We show that neuroepithelial apoptosis reduces the progenitor cell pool in Tcof1+/− mice resulting in delayed NCC migration along the gut from E10.5 to E14.5. This phenotype is very similar to that previously characterized in other animal models of HSCR. However, we report for the first time in mammalian ENS development, increased proliferation at the NCC migratory wavefront which enables the maintenance of a sufficient progenitor cell pool capable of continued advance along the colon and beyond stages at which the gut microenvironment is considered to be non-permissive or less-permissive to NCC.
Unlike many animal models of HSCR, where ENS progenitor cell numbers are reduced by apoptosis during their migration into the foregut (40–43), loss of Tcof1 causes NT apoptosis thereby preventing the formation and delamination of sufficient numbers of NCC. A similar mechanism has been shown to underlie the cardiac phenotype observed in Pax3−/− mice (44–46) and it may also contribute to the almost complete aganglionosis observed in these mice as well (47). HSCR is a multigenic disorder, however, mutations in known HSCR-associated genes account for <50% of reported cases. Furthermore, not only are the genetics of HSCR complex but they are also not strictly Mendelian. In addition, incomplete penetrance, inter- and intrafamilial variation are commonly observed as part of the condition. Thus, there is an increasing body of evidence that points towards interactions between known HSCR susceptibility genes and modifier genes. Interestingly, both Tcof1 and Pax3 loss-of-function induces p53-dependent cell death (32,44). Therefore, it will be interesting in the future to test whether Tcof1 might act as a modifier of colonic aganglionosis in the manifestation of HSCR through a synergistic interaction with Pax3.
In an effort to determine whether complete ENS formation observed within Tcof1+/− embryos was due to the fact that the progenitor cell pool had not been sufficiently reduced below a critical threshold level or ‘tipping’ point, we treated mice with the oxidizing agent H2O2. Oxidation has previously been shown to induce apoptosis in migrating NCC, thereby reducing the total cardiac NCC pool (39). We observed equivalent effects in our studies as evidenced by a significant increase in TUNEL+ NCC migrating towards and into the foregut of Tcof1+/− embryos treated with H2O2 compared with untreated mice. However, unlike the previous report (39), no significant apoptotic effect was apparent in wild-type embryos. In contrast, however, we did observe reduced proliferation of p75+ cells migrating towards and into the foregut within these embryos. The defects in cardiac NCC development elicited by oxidation were attributed to decreased Pax3 expression (39), however, we observed no change in Pax3 levels in Tcof1+/− embryos. Nonetheless, the sensitivity of Tcof1+/− embryos to exogenous oxidation and the similar apoptotic effects of oxidation on cardiac and vagal NCC in Pax3+/− and Tcof1+/− embryos heighten the potential for synergistic interactions in ENS formation and the pathogenesis of HSCR.
Complete ENS formation along the entire length of the gut requires a critical number of NCC and proliferation is a crucial regulator of this process in both avians and mice (15,35,48–50). Differential proliferation has been reported at the migration wavefront of cranial and enteric NCC in avian embryos (49–51). However, this may be species-specific, as we and others have previously documented no significant differences in NCC proliferation rates in different gut regions of E11.5 and E12.5 wild-type mammalian embryos (52,53). It is also possible that the differences observed between the mouse and avian gut data could arise from technical differences in the way that the experiments were performed. The avian gut analysis was performed on explant cultures which do not grow significantly during the period of investigation, while mouse data were collected from guts that were dissected from embryos that were growing during the normal process of development.
We did, however, discover inverse differential proliferation and differentiation between the migration wavefront in the colon and along the SI at E13.5, such that the proliferation was increased, while the differentiation was decreased at the migration wavefront compared with the SI. This might be reasonably expected as the SI is completely colonized at this stage with neuronal differentiation well underway, while NCC are still migrating along the colon. Despite the initial reduction in NCC numbers that enter Tcof1+/− foreguts and the additional dramatic effect of oxidation on this population, increased NCC proliferation at the migration wavefront maintains a sufficient progenitor cell pool enabling the eventual and complete colonization of the gut in untreated and H2O2-treated Tcof1+/− embryos.
The capability of NCC to completely colonize the gut at late developmental stages in Tcof1+/− embryos is drastically different from the results observed when the initial population of progenitor cells is reduced within avian embryos (2,35,54). In these experiments, NCC were delayed throughout all the stages of development examined. They were halted specifically within the duodenum, equivalent to the anterior extent of the Nerve of Remak resulting in almost complete intestinal aganglionosis (2,35,54).
NCC proliferation and differentiation need to be tightly coordinated during development to ensure a complete and fully functioning ENS is established along the entire gut wall. The consequence of a failure to maintain this critical balance is evident in the many animal models of HSCR (8,13,42). Therefore, delayed specification of the NCC in Tcof1+/− embryos as evidenced via reduced numbers of RET+ progenitor cells located around the foregut together with diminished neuronal differentiation of NCC within the gut of Tcof1+/− embryos collectively contributed to the maintenance of a sufficient progenitor cell pool capable of continued advancement along the colon at later developmental stages.
To date, only two other mutants, Gdnf +/− and L1cam null mice, have been reported to exhibit early NCC migration delays, yet still complete formation of a proper ENS (15,43,55,56). However, the migration delay in these mice is not as extensive as that observed in Tcof1+/− embryos as NCC have colonized the cecum by E12.5 in Gdnf and L1cam mutants and this is not the case in Tcof1+/− embryos. The early ENS phenotype in Tcof1+/− embryos is in fact more similar to that described for endothelin signaling loss-of-function mice (34,57–59), where NCC in about 70% of cases examined at E12.5 have not entered the colon. It has been proposed that the colonic microenvironment becomes non-permissive or less-permissive to NCC migration with developmental age since restricted migration of NCC into aganglionic colon was seen at E14.5 when compared with E11.5 (25). This change has been attributed to a temporal increase in laminin expression during normal embryonic development (26). Enhanced laminin expression was also detected in Edn3 mutants when compared with wild-type guts (23,24) and it has been proposed that laminin may restrict NCC migration with increasing neuronal differentiation. The expression of β1 integrin also needs to be regulated in order to ensure complete gut colonization as it is required to modulate the effects of high levels of tenascin-C and fibronectin within the cecum and proximal colon that may inhibit NCC invasion (22). We observed spatiotemporal changes and differences in laminin and β1 integrin expression in the gastrointestinal tract of Tcof1+/− embryos when compared with wild-type, however, colonization of the colon well after E14.5 in Tcof1+/− embryos challenges the notion that the environment of the gut becomes non-permissive to migration at any particular developmental stage. Our results therefore highlight the importance of coordinately regulating NCC proliferation with differentiation as modulating this balance can overcome dramatic deficiencies in the initial vagal NCC progenitor pool.
Taken together, our data show that Tcof1+/− mice model many of the features of HSCR since apoptosis within the NT reduces the pool of NCC progenitor cells that migrate towards and into the gut in Tcof1+/− embryos causing an early developmental ENS defect. However, we demonstrate for the first time that a sufficient pool of dividing progenitors cells can be maintained within the gut through increased NCC proliferation specifically at the migration wavefront in combination with reduced neuronal differentiation. Together with an appropriate colonic microenvironment, this enables the formation of a complete ENS beyond developmental stages at which the colon was considered to be non-permissive or less-permissive to NCC. Therefore, we show that normal ENS formation can be achieved by balancing NCC intrinsic processes with those of the gut microenvironment. This demonstrates the important role played by Tcof1/Treacle in vagal NCC development and sets the stage for future investigations of Tcof1 as a novel modifier of colonic aganglionosis in the pathogenesis of HSCR.
MATERIALS AND METHODS
Animals
All animal protocols were approved by the Institutional Animal Care and Use Committee of Stowers Institute for Medical Research. Genetic analysis was performed as detailed previously (33); however, briefly, the severity and penetrance of characteristic Treacher Collins syndrome facial defects in Tcof1 heterozygous mice are dependent upon the genetic background. Therefore, consistent with previous studies (31,32,60), we maintained the Tcof1+/− line on a pure DBA background, and then outcrossed to C57BL/6 in order to generate embryos with the characteristic features of Treacher Collins syndrome. Importantly, although the embryos analyzed were of mixed DBA × C57BL/6 background, the characteristic mutant phenotype was consistently reproducible with minimal inter-embryo variability at each developmental stage and as previously reported, all neonates displayed gasping behavior and abdominal distension and died within 24h of birth.
Immunohistochemistry
Whole-gut immunolabeling was performed after dissection from the embryo. Tissues were fixed for 2h at room temperature (RT) in 4% paraformaldehyde in PBS before being rinsed several times with PBS. They were incubated in blocking solution (10% heat-inactivated sheep serum in PBS + 0.1% Triton X-100) for 2h at RT. Primary antibodies (see table) in blocking solution were added overnight at 4°C. The following day, they were rinsed in PBS three times for 5 min and then for 1h at RT prior to the addition of appropriate secondary antibodies (1:500 Alexa, Invitrogen; see Supplementary Material, Table S1) diluted in blocking solution for 4h at RT. Guts were rinsed in PBS, mounted in Vectashield with DAPI (Vector Laboratories) and analyzed using an LSM5 PASCAL confocal microscope (Carl Zeiss). Composite images were compiled using Adobe Photoshop software, brightness and contrast may have been modified.
For immunolabeling 10 μm cryosections of whole embryos or dissected guts, the slides were placed into blocking solution for 30min before being immunostained for 2h at RT with the primary antibodies (see Supplementary Material, Table S1). After PBS washing, secondary antibodies in blocking solution were added for 2h at RT before rinsing in PBS and mounting in Vectashield with DAPI (Vector Laboratories). Immunohistochemistry with the Sox10 antibody was as described except that the signal was amplified using DSB-XTM biotin donkey anti-goat IgG (1:100, Invitrogen) and Streptavidin Alexa 568 (1:300, Invitrogen). Apoptosis was detected using the in situ Cell Death Detection Kit Fluorescein (Roche) after the immunostaining according to the manufacturer’s instructions. Mean pixel intensity values were calculated using Image J software.
BrdU incorporation
BrdU (Sigma) was injected intraperitoneally (IP) (1 μl/g of animal weight of a 100 mg/ml stock solution) into pregnant mice. Embryos were harvested 45min later, guts were dissected and immunostained with p75 as described above. They were then post-fixed in 4% PFA for 10min and treated with 2 m HCl at 37°C for 30min prior to the incubation with the BrdU antibody.
Hydrogen peroxide treatment
IP injection of 1% hydrogen peroxide solution was given to pregnant mice (10 μl/g of animal weight) once at E7, twice at E7.5 with a 6h time interval and then once at E8.5. Females were sacrificed at E10, E11.5 and E18.5 to determine any effects of the treatment upon NCC death, proliferation and colonization of the gut.
NCC death and proliferation analysis of sections
Apoptosis within the NCC that had migrated towards the foregut was determined on sections of embryos from E9.5 to E10.5. TUNEL/p75 double-positive cells were scored in 15 cryosections of the embryo at the vagal neural tube level (somites 1–7) and represented as a percentage of total number of p75+ cells. The mitotic index was defined in a similar manner using pHH3+/p75+ cells shown as a fraction of the total p75+ cells. Data are mean ± standard deviation (SD). Statistical analysis was carried out using an unpaired t-test and we considered P-values >0.05 not significant.
NCC proliferation and neuronal differentiation analysis in whole guts
The extent of proliferation was determined at the migration wavefront of NCC by counting double-positive pHH3/p75 or BrdU/p75 of the first 50 p75+ cells from the analysis of 1.5 µm optical sections collected using a 63X lens using a LSM5 PASCAL confocal microscope. Cell proliferation along the SI was obtained by examining a minimum of four regions along the entire length. Neuronal differentiation at the NCC migration wavefront and along the SI was measured at E11.5 and E13.5 as described above except that TuJ1+/p75+ or Hu+/p75+ cells were identified. Data are mean ± SD. We considered P-values >0.05 not significant.
SUPPLEMENTARY MATERIAL
FUNDING
Research in the Trainor Laboratory is supported by the Stowers Institute for Medical Research, March of Dimes (no. 6FY05-82) and National Institute of Dental and Craniofacial Research (RO1 DE 016082-01). Research in the Dixon laboratory is supported by the National Institutes of Health (P50 DE 016215) and the Medical Research Council, UK (G81/535).
Supplementary Material
ACKNOWLEDGEMENTS
The authors are greatly appreciative of all members of the Trainor Laboratory for their comments and suggestions during the generation of this work. We thank Dr Miles Epstein for the kind gift of the Hu antibody; Melissa Childers for her expertise with the maintenance of mutant mouse lines; Teri Johnson, Sharon Beckham, Nancy Thomas, Nannette Marsh and Karen Smith for histological assistance and advice. We would also like to thank Richard Alexander for training on Image J software
Conflict of Interest Statement: None declared.
REFERENCES
- 1.Le Douarin N.M., Teillet M.A. The migration of neural crest cells to the wall of the digestive tract in avian embryo. J. Embryol. Exp. Morphol. 1973;30:31–48. [PubMed] [Google Scholar]
- 2.Yntema C.L., Hammond W.S. The origin of intrinsic ganglia of trunk viscera from vagal neural crest in the chick embryo. J. Comp. Neurol. 1954;101:515–541. doi: 10.1002/cne.901010212. doi:10.1002/cne.901010212. [DOI] [PubMed] [Google Scholar]
- 3.Durbec P.L., Larsson-Blomberg L.B., Schuchardt A., Costantini F., Pachnis V. Common origin and developmental dependence on c-ret of subsets of enteric and sympathetic neuroblasts. Development. 1996;122:349–358. doi: 10.1242/dev.122.1.349. [DOI] [PubMed] [Google Scholar]
- 4.Kapur R.P., Yost C., Palmiter R.D. A transgenic model for studying development of the enteric nervous system in normal and aganglionic mice. Development. 1992;116:167–175. doi: 10.1242/dev.116.Supplement.167. [DOI] [PubMed] [Google Scholar]
- 5.Natarajan D., Marcos-Gutierrez C., Pachnis V., de Graaff E. Requirement of signalling by receptor tyrosine kinase RET for the directed migration of enteric nervous system progenitor cells during mammalian embryogenesis. Development. 2002;129:5151–5160. doi: 10.1242/dev.129.22.5151. [DOI] [PubMed] [Google Scholar]
- 6.Young H.M., Hearn C.J., Ciampoli D., Southwell B.R., Brunet J.F., Newgreen D.F. A single rostrocaudal colonization of the rodent intestine by enteric neuron precursors is revealed by the expression of Phox2b, Ret, and p75 and by explants grown under the kidney capsule or in organ culture. Dev. Biol. 1998;202:67–84. doi: 10.1006/dbio.1998.8987. doi:10.1006/dbio.1998.8987. [DOI] [PubMed] [Google Scholar]
- 7.Hirose T., O'Brien D.A., Jetten A.M. RTR: a new member of the nuclear receptor superfamily that is highly expressed in murine testis. Gene. 1995;152:247–251. doi: 10.1016/0378-1119(94)00656-d. doi:10.1016/0378-1119(94)00656-D. [DOI] [PubMed] [Google Scholar]
- 8.Vuoriluoto K., Haugen H., Kiviluoto S., Mpindi J.P., Nevo J., Gjerdrum C., Tiron C., Lorens J.B., Ivaska J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene. 2011;30:1436–1448. doi: 10.1038/onc.2010.509. doi:10.1038/onc.2010.509. [DOI] [PubMed] [Google Scholar]
- 9.Amiel J., Sproat-Emison E., Garcia-Barcelo M., Lantieri F., Burzynski G., Borrego S., Pelet A., Arnold S., Miao X., Griseri P., et al. Hirschsprung disease, associated syndromes and genetics: a review. J. Med. Genet. 2008;45:1–14. doi: 10.1136/jmg.2007.053959. doi:10.1136/jmg.2007.053959. [DOI] [PubMed] [Google Scholar]
- 10.Anderson R.B., Newgreen D.F., Young H.M. Neural crest and the development of the enteric nervous system. Adv. Exp. Med. Biol. 2006;589:181–196. doi: 10.1007/978-0-387-46954-6_11. doi:10.1007/978-0-387-46954-6_11. [DOI] [PubMed] [Google Scholar]
- 11.Shilatifard A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr. Opin. Cell. Biol. 2008;20:341–348. doi: 10.1016/j.ceb.2008.03.019. doi:10.1016/j.ceb.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gershon M.D. Genes and lineages in the formation of the enteric nervous system. Curr. Opin. Neurobiol. 1997;7:101–109. doi: 10.1016/s0959-4388(97)80127-4. doi:10.1016/S0959-4388(97)80127-4. [DOI] [PubMed] [Google Scholar]
- 13.Laranjeira C., Pachnis V. Enteric nervous system development: recent progress and future challenges. Auton. Neurosci. 2009;151:61–69. doi: 10.1016/j.autneu.2009.09.001. doi:10.1016/j.autneu.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 14.Zhang D., Brinas I.M., Binder B.J., Landman K.A., Newgreen D.F. Neural crest regionalisation for enteric nervous system formation: implications for Hirschsprung's disease and stem cell therapy. Dev. Biol. 2010;339:280–294. doi: 10.1016/j.ydbio.2009.12.014. doi:10.1016/j.ydbio.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 15.Flynn B., Bergner A.J., Turner K.N., Young H.M., Anderson R.B. Effect of Gdnf haploinsufficiency on rate of migration and number of enteric neural crest-derived cells. Dev. Dyn. 2007;236:134–141. doi: 10.1002/dvdy.21013. doi:10.1002/dvdy.21013. [DOI] [PubMed] [Google Scholar]
- 16.Gianino S., Grider J.R., Cresswell J., Enomoto H., Heuckeroth R.O. GDNF availability determines enteric neuron number by controlling precursor proliferation. Development. 2003;130:2187–2198. doi: 10.1242/dev.00433. doi:10.1242/dev.00433. [DOI] [PubMed] [Google Scholar]
- 17.Moore M.W., Klein R.D., Farinas I., Sauer H., Armanini M., Phillips H., Reichardt L.F., Ryan A.M., Carver-Moore K., Rosenthal A. Renal and neuronal abnormalities in mice lacking GDNF. Nature. 1996;382:76–79. doi: 10.1038/382076a0. doi:10.1038/382076a0. [DOI] [PubMed] [Google Scholar]
- 18.Mwizerwa O., Das P., Nagy N., Akbareian S.E., Mably J.D., Goldstein A.M. Gdnf is mitogenic, neurotrophic, and chemoattractive to enteric neural crest cells in the embryonic colon. Dev. Dyn. 2011;240:1402–1411. doi: 10.1002/dvdy.22630. doi:10.1002/dvdy.22630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pichel J.G., Shen L., Sheng H.Z., Granholm A.C., Drago J., Grinberg A., Lee E.J., Huang S.P., Saarma M., Hoffer B.J., et al. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382:73–76. doi: 10.1038/382073a0. doi:10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- 20.Sanchez M.P., Silos-Santiago I., Frisen J., He B., Lira S.A., Barbacid M. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature. 1996;382:70–73. doi: 10.1038/382070a0. doi:10.1038/382070a0. [DOI] [PubMed] [Google Scholar]
- 21.Shen L., Pichel J.G., Mayeli T., Sariola H., Lu B., Westphal H. Gdnf haploinsufficiency causes Hirschsprung-like intestinal obstruction and early-onset lethality in mice. Am. J. Hum. Genet. 2002;70:435–447. doi: 10.1086/338712. doi:10.1086/338712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Breau M.A., Dahmani A., Broders-Bondon F., Thiery J.P., Dufour S. Beta1 integrins are required for the invasion of the caecum and proximal hindgut by enteric neural crest cells. Development. 2009;136:2791–2801. doi: 10.1242/dev.031419. doi:10.1242/dev.031419. [DOI] [PubMed] [Google Scholar]
- 23.Jacobs-Cohen R.J., Payette R.F., Gershon M.D., Rothman T.P. Inability of neural crest cells to colonize the presumptive aganglionic bowel of ls/ls mutant mice: requirement for a permissive microenvironment. J. Comp. Neurol. 1987;255:425–438. doi: 10.1002/cne.902550309. doi:10.1002/cne.902550309. [DOI] [PubMed] [Google Scholar]
- 24.Wu J.J., Chen J.X., Rothman T.P., Gershon M.D. Inhibition of in vitro enteric neuronal development by endothelin-3: mediation by endothelin B receptors. Development. 1999;126:1161–1173. doi: 10.1242/dev.126.6.1161. [DOI] [PubMed] [Google Scholar]
- 25.Hotta R., Anderson R.B., Kobayashi K., Newgreen D.F., Young H.M. Effects of tissue age, presence of neurones and endothelin-3 on the ability of enteric neurone precursors to colonize recipient gut: implications for cell-based therapies. Neurogastroenterol. Motil. 2010;22:331–e386. doi: 10.1111/j.1365-2982.2009.01411.x. doi:10.1111/j.1365-2982.2009.01411.x. [DOI] [PubMed] [Google Scholar]
- 26.Druckenbrod N.R., Epstein M.L. Age-dependent changes in the gut environment restrict the invasion of the hindgut by enteric neural progenitors. Development. 2009;136:3195–3203. doi: 10.1242/dev.031302. doi:10.1242/dev.031302. [DOI] [PubMed] [Google Scholar]
- 27.Parisi M.A., Kapur R.P. Genetics of Hirschsprung disease. Curr. Opin. Pediatr. 2000;12:610–617. doi: 10.1097/00008480-200012000-00017. doi:10.1097/00008480-200012000-00017. [DOI] [PubMed] [Google Scholar]
- 28.Garcia-Barcelo M.M., Tang C.S., Ngan E.S., Lui V.C., Chen Y., So M.T., Leon T.Y., Miao X.P., Shum C.K., Liu F.Q., et al. Genome-wide association study identifies NRG1 as a susceptibility locus for Hirschsprung's disease. Proc. Natl Acad. Sci. USA. 2009;106:2694–2699. doi: 10.1073/pnas.0809630105. doi:10.1073/pnas.0809630105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heanue T.A., Pachnis V. Enteric nervous system development and Hirschsprung's disease: advances in genetic and stem cell studies. Nat. Rev. Neurosci. 2007;8:466–479. doi: 10.1038/nrn2137. doi:10.1038/nrn2137. [DOI] [PubMed] [Google Scholar]
- 30.Batlle E., Sancho E., Franci C., Dominguez D., Monfar M., Baulida J., Garcia De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell. Biol. 2000;2:84–89. doi: 10.1038/35000034. doi:10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 31.Dixon J., Jones N.C., Sandell L.L., Jayasinghe S.M., Crane J., Rey J.P., Dixon M.J., Trainor P.A. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc. Natl Acad. Sci. USA. 2006;103:13403–13408. doi: 10.1073/pnas.0603730103. doi:10.1073/pnas.0603730103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones N.C., Lynn M.L., Gaudenz K., Sakai D., Aoto K., Rey J.P., Glynn E.F., Ellington L., Du C., Dixon J., et al. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat. Med. 2008;14:125–133. doi: 10.1038/nm1725. doi:10.1038/nm1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dixon J., Brakebusch C., Fassler R., Dixon M.J. Increased levels of apoptosis in the prefusion neural folds underlie the craniofacial disorder, Treacher Collins syndrome. Hum. Mol. Genet. 2000;9:1473–1480. doi: 10.1093/hmg/9.10.1473. doi:10.1093/hmg/9.10.1473. [DOI] [PubMed] [Google Scholar]
- 34.Barlow A., de Graaff E., Pachnis V. Enteric nervous system progenitors are coordinately controlled by the G protein-coupled receptor EDNRB and the receptor tyrosine kinase RET. Neuron. 2003;40:905–916. doi: 10.1016/s0896-6273(03)00730-x. doi:10.1016/S0896-6273(03)00730-X. [DOI] [PubMed] [Google Scholar]
- 35.Barlow A.J., Wallace A.S., Thapar N., Burns A.J. Critical numbers of neural crest cells are required in the pathways from the neural tube to the foregut to ensure complete enteric nervous system formation. Development. 2008;135:1681–1691. doi: 10.1242/dev.017418. doi:10.1242/dev.017418. [DOI] [PubMed] [Google Scholar]
- 36.Aihara Y., Hayashi Y., Hirata M., Ariki N., Shibata S., Nagoshi N., Nakanishi M., Ohnuma K., Warashina M., Michiue T., et al. Induction of neural crest cells from mouse embryonic stem cells in a serum-free monolayer culture. Int. J. Dev. Biol. 2010;54:1287–1294. doi: 10.1387/ijdb.103173ya. doi:10.1387/ijdb.103173ya. [DOI] [PubMed] [Google Scholar]
- 37.Maurer J., Fuchs S., Jager R., Kurz B., Sommer L., Schorle H. Establishment and controlled differentiation of neural crest stem cell lines using conditional transgenesis. Differentiation. 2007;75:580–591. doi: 10.1111/j.1432-0436.2007.00164.x. doi:10.1111/j.1432-0436.2007.00164.x. [DOI] [PubMed] [Google Scholar]
- 38.Davis W.L., Crawford L.A., Cooper O.J., Farmer G.R., Thomas D.L., Freeman B.L. Ethanol induces the generation of reactive free radicals by neural crest cells in vitro. J. Craniofac. Genet. Dev. Biol. 1990;10:277–293. [PubMed] [Google Scholar]
- 39.Morgan S.C., Relaix F., Sandell L.L., Loeken M.R. Oxidative stress during diabetic pregnancy disrupts cardiac neural crest migration and causes outflow tract defects. Birth Defects Res. A. Clin. Mol. Teratol. 2008;82:453–463. doi: 10.1002/bdra.20457. doi:10.1002/bdra.20457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stanchina L., Baral V., Robert F., Pingault V., Lemort N., Pachnis V., Goossens M., Bondurand N. Interactions between Sox10, Edn3 and Ednrb during enteric nervous system and melanocyte development. Dev. Biol. 2006;295:232–249. doi: 10.1016/j.ydbio.2006.03.031. doi:10.1016/j.ydbio.2006.03.031. [DOI] [PubMed] [Google Scholar]
- 41.Maka M., Stolt C.C., Wegner M. Identification of Sox8 as a modifier gene in a mouse model of Hirschsprung disease reveals underlying molecular defect. Dev. Biol. 2005;277:155–169. doi: 10.1016/j.ydbio.2004.09.014. doi:10.1016/j.ydbio.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 42.Stanchina L., Van de Putte T., Goossens M., Huylebroeck D., Bondurand N. Genetic interaction between Sox10 and Zfhx1b during enteric nervous system development. Dev. Biol. 2010;341:416–428. doi: 10.1016/j.ydbio.2010.02.036. doi:10.1016/j.ydbio.2010.02.036. [DOI] [PubMed] [Google Scholar]
- 43.Wallace A.S., Schmidt C., Schachner M., Wegner M., Anderson R.B. L1cam acts as a modifier gene during enteric nervous system development. Neurobiol. Dis. 2010;40:622–633. doi: 10.1016/j.nbd.2010.08.006. doi:10.1016/j.nbd.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 44.Pani L., Horal M., Loeken M.R. Rescue of neural tube defects in Pax-3-deficient embryos by p53 loss of function: implications for Pax-3- dependent development and tumorigenesis. Genes. Dev. 2002;16:676–680. doi: 10.1101/gad.969302. doi:10.1101/gad.969302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phelan S.A., Ito M., Loeken M.R. Neural tube defects in embryos of diabetic mice: role of the Pax-3 gene and apoptosis. Diabetes. 1997;46:1189–1197. doi: 10.2337/diab.46.7.1189. doi:10.2337/diabetes.46.7.1189. [DOI] [PubMed] [Google Scholar]
- 46.Epstein J.A. Pax3 and vertebrate development. Methods Mol. Biol. 2000;137:459–470. doi: 10.1385/1-59259-066-7:459. [DOI] [PubMed] [Google Scholar]
- 47.Lang D., Chen F., Milewski R., Li J., Lu M.M., Epstein J.A. Pax3 is required for enteric ganglia formation and functions with Sox10 to modulate expression of c-ret. J. Clin. Invest. 2000;106:963–971. doi: 10.1172/JCI10828. doi:10.1172/JCI10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bondurand N., Natarajan D., Barlow A., Thapar N., Pachnis V. Maintenance of mammalian enteric nervous system progenitors by SOX10 and endothelin 3 signalling. Development. 2006;133:2075–2086. doi: 10.1242/dev.02375. doi:10.1242/dev.02375. [DOI] [PubMed] [Google Scholar]
- 49.Simpson M.J., Landman K.A., Hughes B.D., Newgree D.F. Looking inside an invasion wave of cells using continuum models: proliferation is the key. J. Theor. Biol. 2006;243:343–360. doi: 10.1016/j.jtbi.2006.06.021. doi:10.1016/j.jtbi.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 50.Simpson M.J., Zhang D.C., Mariani M., Landman K.A., Newgreen D.F. Cell proliferation drives neural crest cell invasion of the intestine. Dev. Biol. 2007;302:553–568. doi: 10.1016/j.ydbio.2006.10.017. doi:10.1016/j.ydbio.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 51.Kulesa P.M., Teddy J.M., Stark D.A., Smith S.E., McLennan R. Neural crest invasion is a spatially-ordered progression into the head with higher cell proliferation at the migratory front as revealed by the photoactivatable protein, KikGR. Dev. Biol. 2008;316:275–287. doi: 10.1016/j.ydbio.2008.01.029. doi:10.1016/j.ydbio.2008.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Young H.M., Turner K.N., Bergner A.J. The location and phenotype of proliferating neural-crest-derived cells in the developing mouse gut. Cell Tissue Res. 2005;320:1–9. doi: 10.1007/s00441-004-1057-5. doi:10.1007/s00441-004-1057-5. [DOI] [PubMed] [Google Scholar]
- 53.Walters L.C., Cantrell V.A., Weller K.P., Mosher J.T., Southard-Smith E.M. Genetic background impacts developmental potential of enteric neural crest-derived progenitors in the Sox10Dom model of Hirschsprung disease. Hum. Mol. Genet. 2010;19:4353–4372. doi: 10.1093/hmg/ddq357. doi:10.1093/hmg/ddq357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peters-van der Sanden M.J., Kirby M.L., Gittenberger-de Groot A., Tibboel D., Mulder M.P., Meijers C. Ablation of various regions within the avian vagal neural crest has differential effects on ganglion formation in the fore-, mid- and hindgut. Dev. Dyn. 1993;196:183–194. doi: 10.1002/aja.1001960305. doi:10.1002/aja.1001960305. [DOI] [PubMed] [Google Scholar]
- 55.Wallace A.S., Tan M.X., Schachner M., Anderson R.B. L1cam acts as a modifier gene for members of the endothelin signalling pathway during enteric nervous system development. Neurogastroenterol. Motil. 2011;11:e510–e522. doi: 10.1111/j.1365-2982.2011.01692.x. doi:10.1016/S0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- 56.Anderson R.B., Turner K.N., Nikonenko A.G., Hemperly J., Schachner M., Young H.M. The cell adhesion molecule l1 is required for chain migration of neural crest cells in the developing mouse gut. Gastroenterology. 2006;130:1221–1232. doi: 10.1053/j.gastro.2006.01.002. doi:10.1053/j.gastro.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 57.Baynash A.G., Hosoda K., Giaid A., Richardson J.A., Emoto N., Hammer R.E., Yanagisawa M. Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell. 1994;79:1277–1285. doi: 10.1016/0092-8674(94)90018-3. doi:10.1016/0092-8674(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 58.Hosoda K., Hammer R.E., Richardson J.A., Baynash A.G., Cheung J.C., Giaid A., Yanagisawa M. Targeted and natural (piebald-lethal) mutations of endothelin-B receptor gene produce megacolon associated with spotted coat color in mice. Cell. 1994;79:1267–1276. doi: 10.1016/0092-8674(94)90017-5. doi:10.1016/0092-8674(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 59.Druckenbrod N.R., Powers P.A., Bartley C.R., Walker J.W., Epstein M.L. Targeting of endothelin receptor-B to the neural crest. Genesis. 2008;46:396–400. doi: 10.1002/dvg.20415. doi:10.1002/dvg.20415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dixon J., Dixon M.J. Genetic background has a major effect on the penetrance and severity of craniofacial defects in mice heterozygous for the gene encoding the nucleolar protein Treacle. Dev. Dyn. 2004;229:907–914. doi: 10.1002/dvdy.20004. doi:10.1002/dvdy.20004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.