

Abstract
Nineteen Wnt ligands and 10 Frizzled (Fz) receptors mediate multiple distinct cellular events during neuronal development. However, their precise roles in cell-type specification and organogenesis are poorly delineated because of overlapping functions and expression profiles. Here, we have explored the role of two closely related Frizzled receptors, Fz5 and Fz8, in mouse retinal development. We previously showed that Fz5−/− mice exhibit mild coloboma and microphthalmia at ∼50% penetrance. Fz8 expression overlaps with Fz5 in the neural retina and optic fissure/disc. Mice lacking Fz8 show minimal eye and retinal defects. The embryos lacking both Fz5 and Fz8 die early in development, but a majority of triallelic Fz5−/−;Fz8+/− mutants survive until birth. The triallelic mutant develops severe retinal coloboma and microphthalmia with full penetrance. At the cellular level, impaired neurogenesis is indicated by increased early-born retinal neurons that result from accelerated cell cycle exit of progenitors. Deficiency of apical retinal neuroepithelium is indicated by altered localization of apical junction markers, such as atypical protein kinase C, RhoA and β-catenin. Hes1 expression, which is critical for retinal progenitor expansion, is down-regulated in the triallelic mutant mouse. Furthermore, blocking Frizzled receptors in cultured retinal explants led to basally shifted divisions of retinal progenitors. Together, our studies suggest a dose-dependent regulation of signaling by Fz5 and Fz8 in optic fissure/disc formation and progenitor expansion.
INTRODUCTION
How diverse neurons are produced in precise numbers during development is poorly understood. Stringent control mechanisms appear to dictate the production of neuronal diversity and eliminate unwanted cells by apoptosis (1–3). Control of cell cycle length and time of exit are expected to modulate the nature and extent of the neural progenitors. Intrinsic genetic programs and extracellular signaling cues contribute to the expansion of neural progenitor pool (4–6). During retinal development, six major types of neurons and one type of glia are produced in a conserved and overlapping order (7,8). Transcriptional factors provide key intrinsic control, qualitatively directing cell fate specification and differentiation. For example, Pax6 is required for the multipotent state of the retinal progenitors (9); Math5, Brn3 proteins and Islet-1 control ganglion cell production (10–12); and OTX2, RORβ, NRL and CRX control rod and cone photoreceptor differentiation (13). The major signaling pathways, including Notch, Hedgehog and Wnt, mediate the responses to extrinsic cues and modulate the plasticity of specific pools or proportion of retinal progenitors (14), thereby exerting a spatiotemporal regulation by defining the number of each cell type produced along the window of retinal neurogenesis.
Signaling pathways coordinate the spatiotemporal regulation of neurogenesis by integrating microenvironment to the intrinsic genetic program. Unlike transcription regulatory factors, the impact of extrinsic cues on retinal progenitor fate is permissive and quantitative rather than instructive. As a result, imbalance in signaling dose can lead to disproportionate production of cell types, consequently affecting retinal size, architecture and/or function. For instance, disruption of the Notch pathway by targeted deletion of Notch1 receptor, its mediator Rbpj or its effector Hes-1 can cause microphthalmia and increased production of early-born cones and/or ganglion cells at the expense of retinal progenitors (15–17). Conditional deletion of Shh signaling leads to overproduction of retinal ganglion cells as a result of precocious cell cycle exit of the retinal progenitors (18).
The signaling dosage for retinal neurogenesis is coordinated, at least in part, by the polarized neuroepithelium, providing an environment for fine-tuning the neuroblast fate decision(s). This seems to be a dogma for the whole central nervous system neurogenesis where, for example, in zebrafish neural tube, the progenitors produced apical daughters (based on cleavage plane) biased to make neurons (19). Cell division and fate choice coincide with an apical–basal gradient of Notch activity in the retinal neuroepithelium (20). Insufficient interkinetic nuclear movement of neuroblasts causes early cell cycle exit due to reduced exposure to high dose of Notch at the apical retina (20,21). Similarly, ganglion cell-derived Shh (18) is present at a high dosage at the basal retina and contributes to the maintenance of neural progenitors. Interestingly, Notch and Shh pathways share a downstream target, Hes1, during retinal neurogenesis (22); this observation has led to the hypothesis that different signaling pathways might converge and coordinate the signaling dosage for progenitor proliferation/expansion.
Multiple components of the Wnt pathway are expressed in the early embryonic mouse retina (23), indicating their role in retinogenesis. Notably, Wnt2b is critical for peripheral retinal fate in the chick (24,25), and Wnt3a promotes adult retinal neurogenesis upon injury in the mouse (26). Canonical Wnt signaling appears to be active only in the ciliary marginal zone of developing mouse retina, as revealed by Tcf-β-gal reporter analysis and loss- or gain-of-function studies of β-catenin (23,27–29). Additionally, conditional ablation of β-catenin, a downstream target of the canonical Wnt pathway, in the developing retina affects retinal lamination but not neurogenesis (25), while inactivation of β-catenin in retinal pigment epithelium (RPE) cells causing transformation of RPE to neural retina (30). Finally, in Xenopus, the Frizzled 5-mediated canonical pathway acts through Sox2 to regulate neuronal potential of the progenitor (31), although this does not apply to mouse knockout models (28,32).
To further explore the role of Wnt–Frizzled signaling in retinal neurogenesis and its contribution to retinal diseases, we have focused on the major Wnt receptors, Frizzled (Fz), which localize to several regions of the CNS and mediate both canonical and non-canonical Wnt pathways (33). The major cellular processes modulated by Fz receptors include planar cell polarity (PCP) in the inner-ear sensory neurons (34), neural tube closure (34), axon pathfinding (35–37), retinal vasculature development and regression (32,38,39), retinal optic fissure closure (32) and survival of thalamic neurons (40).
Previously, we and others have shown that Fz5 was expressed in developing mouse retina (27,28,32), brain ventricles and thalamic nuclei. Mutation in Fz5 in the mouse caused retina vitreous persistent fetal vasculature (PFV) (28,32), microphthalmia, retinal coloboma (32) and missing thalamic parafascicular nucleus (40). As Fz8 and Fz5 have the highest similarity and are activated by Wnt9b (40,41), we examined whether Fz8 and Fz5 receptors function in a dose-dependent manner during retinal neurogenesis. Here, we demonstrate that Fz8 and Fz5 together maintain the organization of retinal neural epithelia, thereby regulating the retinal progenitor pool and neurogenesis.
RESULTS
Fz8 expression in developing mouse retina
To investigate the Fz8 expression, we used a knock-in β-gal reporter at the endogenous locus in Fz8+/lacZ mice (42) that show no distinct phenotype compared with the wild-type (WT) mice. Fz8 is expressed in the forebrain and edge of the eye field during early embryogenesis (Fig. 1A–D). At embryonic day 13.5 (E13.5), strong staining was observed in optic disc and retinal proliferating marginal zone (PMZ), while weaker signal was detected in neural retina (Supplementary Material, Fig. S1). At E14.5 and E16.5, in bisected whole-mount retina, Fz8 expression was observed in optic disc, PMZ and neural retina with a preference to the ventral (Fig. 1E–G). In 4-week-old retina, Fz8 is expressed in all retina layers with less staining in the outer nuclear layer (ONL) (Fig. 1H). Immunofluorescence labeling of dissociated cells from adult Fz8+/− retina (Fig. 1I) revealed Fz8 expression in ganglion cells (marked by Brn3a and NFL), Müller glia (marked by Sox9) and Calbindin-positive cells. Fz8 expression in optic disc region and neural retina overlaps with the Fz5 during the early development (Supplementary Material, Fig. S1A–C) (27,32). Fz8 expression in Müller glia in adult retina also overlaps with Fz5, and its expression in Calbindin-positive cells potentially overlaps with Fz5 expression in amacrine cells (compare Fig. 1I and Supplementary Material, Fig. S1D and E).
Figure 1.
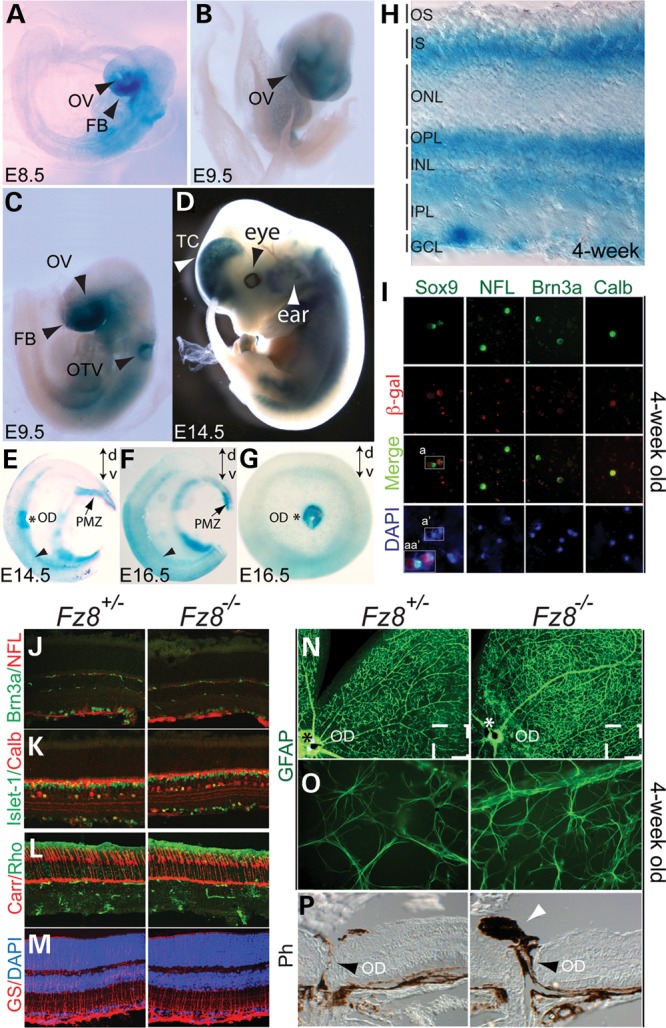
Fz8 retinal expression during mouse development and phenotypic analysis of Fz8 mutant retina. (A–H) Fz8 expression was visualized by a β-gal reporter from its knock-in endogenous locus. (A–C) Fz8 was expressed in the areas of embryonic forebrain (FB), optic vesicle (OV) and otic vesicle (OTV) at early stages of embryonic day 8.5 (E8.5) and E9.5. (D) Fz8 expression was continuously seen in forebrain ear and eye regions at E14.5. (E and F) Bisected X-gal stained whole-mount retinas at E14.5 (E) and E16.5 (F) showing Fz8 enhanced expression in optic disc (OD, asterisk), PMZ (arrows) and ventral retina (arrowhead). Dorsal–ventral orientation (d-v) is indicated by up-down arrows in each panel. (G) A top view of an X-gal-stained E16.5 retina showing a preferential Fz8 expression in optic disc and ventral retina. (H) A 4-week-old X-gal-stained retinal section shows that Fz8 was expressed through all retinal layers. OS, outer segment; IS, inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; IPL, inner plexiform layer; GCL, ganglion cell layer. (I) Co-immunostaining using anti-β-gal antibody (red) and other retinal cell markers (green) shows that Fz8 is expressed in Müller cell, ganglion cell (RGC) and Calbidin-positive cells (likely amacrine cells). Merged images show co-labeling of β-gal with Sox9 for Müller cell, NFL and Brn3a for ganglion cell, and Calbindin (Calb) positive cells. DAPI staining (blue) shows number of cells in the pictured field. Boxed areas indicate that same cells are being labeled by Sox9 and β-gal. a, double-labeled cells with Sox9 and β-gal; a′, DAPI-stained cell nuclei; and aa″, zooming in on merged channels of Sox9, β-gal and DAPI in a and a′ areas. (J–M), normal development of retinal cell types in Fz8 mutant retinas: no significant differences in immunostained Fz8 heterozygous and mutant retinas for Brn3a- and NFL-positive RGCs (J, green and red are for RGCs and RGC axons, respectively), Islet-1-positive RGCs, ACs and bipolar cells (K, green), calbindin (Calb)-positive ACs and HCs (K, red), rhodopsin-labeled rods (L, green), cone arrestin (Carr)-labeled cones (L, red) and glutamate synthetase (GS)-labeled Müller glia. Non-specific staining (green signals in IPL and OPL) in (J) and (L) is from anti-mouse Ig secondary antibody. (N–P) Increased GFAP staining of astrocytes dendrites in the mutant retina, and mild optic disc defects. (N) Low magnification showing the GFAP stained whole-mount retinas showing enhanced GFAP staining. (O) Zoom-in areas roughly correspond to the boxed areas in (N), showing individually stained astrocytes dendrites. (P) A frequent observation (6/11 mutant mice) of optic disc defects associated with the enhanced GFAP staining, featured by PFV (white arrowhead). The optic disc (OD, black arrowhead) does not seem to close properly in the mutant mice.
Ocular phenotype in the Fz8−/− mice
We then examined the retinal phenotypes of Fz8−/− mice. The major cell types such as cones, rods, ganglion cells, amacrine cells and bipolar cells are developed correctly in the Fz8−/− retina (Fig. 1J–M). However, a fraction of the mutant retina showed a modest increase in glial fibrillary acidic protein staining of astrocytes (Fig. 1N and O) with an ectopic pigment stalk sitting on the optic nerve head (Fig. 1P), which has been well described as PFV (32) and resembles the human eye disease called persistent hyperplastic primary vitreous (28). The phenotype, although weaker and with incomplete penetrance (6 out of 11 mice examined), was broadly similar to the optic disc defects in the Fz5−/− mutant retina (28,32).
Severe retinal coloboma, microphthalmia, astrocytic gliosis and axon sprouting in Fz5 and Fz8 compound mutant retina
The phenotypic similarity of Fz5 and Fz8 mutants in optic disc development led us to hypothesize that the two receptors work together during retinal neurogenesis since their expression also overlaps in the neural retina. We therefore generated Fz5 and Fz8 compound mutant mice. Mice carrying double null alleles of Fz5 and Fz8 died before E13.5 likely due to systemic angiogenesis defects, making it difficult to analyze the retinal phenotype. However, adding one Fz8 WT allele to the double null (Fz5−/−;Fz8+/−) allowed these mice to survive until E17.5 or birth. Occasional pups survived to 1 month but had significantly reduced body weight and more severe eye defects (Fig. 2, and data not shown). In contrast, Fz5+/−;Fz8+/− mice did not show apparent abnormalities and are therefore indistinguishable from the WT. Henceforth, Fz5−/−;Fz8+/− and Fz5+/−;Fz8+/− mice are referred as compound mutant/mutant and WT/control, respectively. We observed that optic fissure failed to close in all Fz5−/−;Fz8+/− retinas at all stages examined. Retinal coloboma was readily visible at E15.5 (Fig. 2A), with a lack of ventral retinal tissue in the young adult (Fig. 2F). Pax2 expression in the mutant retina expanded laterally (Fig. 2B and C). Overgrown embryonic fetal vasculature packed behind the embryonic lens (Fig. 2D and E). Increased neural fiber thickness and abnormal axon routing were also consistently observed by staining of neural filament using anti-NFL-70 (Fig. 2D and E).
Figure 2.

Severe retinal coloboma, microphthalmia, retinal gliosis and axon sprouting in the Fz5−/−;Fz8+/− mutant retina. (A) Lacking ventral retinal tissue, severe retinal coloboma and microphthalmia at E15.5 in the Fz5−/−;Fz8+/− compound mutant retina (arrowheads). (B) Staining of Pax2 at optic fissure is more restricted in the WT (arrowheads, left panel) than in the mutant (arrowheads, right panel). Asterisks indicate the optic discs. (C) Merged images of Pax2 staining with DAPI indicate the retinal structure at optic disc. (D and E) Widened optic fissure with massive RGC axons routing out the mutant retina at E17.5. Misrouting of axons often happens (white arrow, right panel) with a consistently observed thickened retinal neural fiber layer and malformation of retinal fetal vasculature (dashed lines enclosure). (E) DAPI staining of the same sections in (D) showing the retinal structure of optic disc area (asterisk) at E17.5. Dashed lines indicate the embryonic vasculature tissues. (F) A brightfield (BF) picture of 4-week-old mutant eyeball showing the server ventral retinal coloboma (arrowhead). The pupil-iris boundary is demarcated by dashed lines. (G) Phase contrast image of a 4-week-old retinal section showing severely deformed mutant retina and intraocular pigmented PFV (arrowhead). (H) Boxed area in G showing overwhelming GFAP-stained astroglia lining the surface of unclosed optic fissure. (I) NFL-stained axon sprouting in the 4-week-old compound mutant retina in both GCL and OPL (arrows). (J) Images from (I) merged with DAPI showing inner retina thickness (white brackets, INL and IPL) is significantly reduced in the mutant compared with the WT. Mutant ONL thickness is similar to the WT at this stage (yellow brackets). (K) Abnormal glial activation in the mutant retina. GFAP-labeled astrocytes sparsely distribute below the GCL in the WT retina, while they spread along the neural fiber layer in the mutant retina (arrows) and go up to through the mutant retina. (L) DAPI-stained retinal nuclear layers merged with GFAP staining shown in (K).
Occasionally, compound mutant survived to 4 weeks of age developed severe microphthalmia and retinal coloboma (Fig. 2F). PFV was also evident (Fig. 2G) with highly pigmented mesenchymal cells intermingled with GFAP-stained astrocytes (data not shown). Astrocytic gliosis is apparent along the surface of open optic fissure by GFAP staining (Fig. 2H). Axonal bundles sprout in both neural fiber and outer plexiform layer (OPL), as revealed by NFL staining (Fig. 2I and J). The axonal sprouting phenotype was also observed in Fz5−/− single mutant, which occurred later at 6 months of age and was relatively milder (Supplementary Material, Fig. S2). Despite the whole retina being thinner, INL was more affected compared with the ONL (Fig. 2J and L). Müller glia and/or astrocytes were activated in vast regions of the retina with thickened fiber reaching through the INL (Fig. 2K and L).
The more severe and complete penetrance of ocular defects in Fz5−/−;Fz8+/− mice stands in contrast to those of either Fz8−/− or Fz5−/− alone, suggesting that Fz5 and Fz8 function together dose-dependently during retinal development. We have also examined the Fz5+/−;Fz8−/− triallelic combination and found the retina developed normally in this mutant. Therefore, Fz5 provides more genetic load than Fz8 during retinal development.
Increased early-born retinal neurons and accelerated neurogenesis in Fz5 and Fz8 compound mutant retina
Despite the smaller eye size, the ganglion cell layer labeled by Brn3a is thicker in the compound mutant retina at E15.5 (Fig. 3A and B). Similarly, Islet-1 labeled early-born neurons including ganglion cell and amacrine cells were also increased when examined at E17.5 (Fig. 3C–E). We therefore hypothesized that early-born neurons were generated over a broader window of time during early neurogenesis in the compound mutant retina. To examine this possibility, we re-evaluated neurogenesis in the E13.5 mutant retina. Indeed, we observed advanced and broad areas of neurogenesis occurring as early as E13.5 in the mutant retina using Tuj1 as a marker of postmitotic neuronal precursors (Fig. 3F and H). Segmentation of the retina showed that the advancing area (AA) for retinal neurogenesis is more expanded in the mutants (Fig. 3G and I), suggesting a faster spreading of retinal neurogenesis. Quantitative analysis revealed a 6–10% increase in the neurogenesis area of the mutant retina.
Figure 3.

Increased early-born neuronal production and neurogenesis in the Fz5−/−;Fz8+/− mutant retina. (A and B) Two consecutive Brn3a-stained retinal sections at E15.5 showing increased RGC production in the mutant retina [brackets, sectioning orientation is drawn in (J), line through the embryonic eye]. (C–E) Increased Islet-1-labeled early-born neuronal precursors in the Fz5−/−;Fz8+/− retina at E17.5. (C) Islet-1-stained INBL (under dashed lines) of the mutant retina is significantly thicker in the mutant compared with the WT. Correspondingly, the newly born neuronal precursors in outer neuroblast layer (ONBL, above dashed lines) are also significantly increased [boxed area, also in (D)]. (D) A close view of the retina in the boxed regions in (C) showing the increased Islet-1 cells in both INBL and ONBL. Arrowheads point some individual Islet-1-positive neurons in ONBL. (E) Quantification of Islet-1-positive cells in the ONBL: (a) index values from Islet-1 cells in ONBL divided by the counting area; (b) area ratio of INBL/NBL (total neuroblast layer). Students’ t-test was used to obtain P-values from the comparison of paired groups of sections between WT and mutant. (F–I) Quantitative analysis of neurogenesis area in WT and mutant retina at E13.5 based on Tuj1 staining. (F and H) Two representative E13.5 retinal sections at consecutive levels of both WT and mutant [sectioning orientation shown in (J)] stained with Tuj1 showing advanced neurogenesis in the mutant retina (AA, advancing area; and CA, central area). AA of the mutant retina expanded more than the WT. (G and I) Pie chart expression of retinal neurogenesis in comparison of the WT and the mutant based on Tuj1 staining. Retina sections were segmented using inner retina surface curvature as reference. The segmentation points (G, drawn in green) are the angular vertex of the smallest angle formed by the crossing of lines tangential to the inner retinal surface, where the biggest curvature occurs. A segmentation line was then drawn from a segmental point parallel to the direction of a nearest Tuj1-positive neuroblast. The line is roughly vertical to the tangential outer retinal surface (short green lines). The segmental lines are roughly the boundary between the central (CA) and advancing differentiating areas (AA). The outer boundary of the AA area is defined by Tuj1-postive INBL peripheral boundary (red lines in G). A line is drawn following INBL peripheral boundary parallel to the nearest Tuj1-positive neuroblast as AA border. Area counting was conducted using MetaMorph software. Student's t-test was used to obtain P-values. There are ∼7% more Tuj1-positive area in the mutant retina in the sampled region of the E13.5 retina. (J) Schematic description of sectioning and sampling of retinal tissues. Total 9 (n= 3×3) sections from three animals were subjected to Tuj1 staining and thereafter statistical analysis.
Normal mitotic division rate, increased BrdU-retained early-born neurons and cell death
The accelerated neurogenesis indicates cell cycle defects of the proliferating progenitors. One possibility of the cell cycle defects is gross reduction in proliferation of the progenitors. This should be reflected by expression of proliferating antigens, for example, Ki67. However, no obvious changes were detected in either Ki67 expression territory or intensity at E13.5 (Fig. 4A) and E17.5 (Fig. 4F–H), indicating a grossly normal proliferation in the mutant retina. We then examined whether the mitotic rate has slowed down in the mutant retina because of the microphthalmia phenotype. Quantitative analysis of phospho-histone 3 (pH3)-labeled dividing cells (Fig. 4B and C) on retinal apical surface and single pulse of 1 h BrdU labeled proliferating cells (data not shown) showed no significant difference between WT and mutant retinas. Another possibility was that although progenitor cells divided at a normal rate in the mutant apical retina, more daughter cells exited cell cycle by asymmetrical divisions. To test this possibility, we delivered a single pulse of BrdU at E13 and monitored BrdU retention after 60 h, at which time the progenitors would have gone through three cell cycles. Bright and round BrdU+ cells are predicted to be newly post-mitotic with higher labeling since BrdU was not diluted in these cells. We observed an increase in BrdU-positive cells in both inner neuroblast layer (INBL) and apical outer neuroblast layer (ONBL) in all sections at E15.5 (Fig. 4D and E). The BrdU-positive cells in apical ONBL likely corresponded to early-born cones, although this was not tested directly. Similar result of BrdU retention was also seen after 16 h pulse (Supplementary Material, Fig. S3A and B). We then immunostained the retina for cyclin D kinase inhibitor p27kip1, which promotes the exit of retinal progenitors from cell cycle (43). We observed a stronger staining of p27Kip1 at the apical mutant retina, and a reduction in INBL staining (Supplementary Material, Fig. S4A), suggesting a role for p27Kip1 in maintaining cell morphology and in cell migration (44,45). Interestingly, total p27Kip1 protein was slightly but consistently reduced, whereas cyclin D1 remained unchanged (Supplementary Material, Fig. S4B). Together, our data suggest that accelerated neurogenesis is likely due to an early retreat of progenitors from the cell cycle, probably caused by increased asymmetrical divisions rather than the rate of cell division.
Figure 4.

Increased BrdU-labeled postmitotic neuronal precursors, and cell death during early retinogenesis of Fz5−/−;Fz8+/− mutant retina. (A) Ki67 expression in proliferating retinal progenitors of mutant retina is grossly normal at E13.5. (B) pH3-labeled dividing cells at the apical surface of the E15.5 WT and mutant retina. Enlarged views of cornered areas in (B) are shown below with arrowheads indicating dividing cells. (C) Schematic pH3-positive cells' counting and analysis. The pH3-positive cells are normalized to the circumferential length (arbitrary length unit detected by the MetaMorph imaging software). No statistical difference is detected between the WT and the mutant retina [P= 0.46, sectioning and sampling is described in (J)]. (D) BrdU was injected at E13 and analyzed 60 h later, which is around E15.5, by IHC. Bright BrdU+ cells were considered newly post-mitotic since they likely exit cell cycle soon after the BrdU injection. BrdU incorporation in proliferating cells would get diluted within few cell cycles. Increased BrdU retention in apical cells (arrowheads), presumably early-born cones, in ONBL neuroblast and INBL cells indicate an early cell cycle exit of mutant retinal progenitors. BrdU labeling below INBL is retinal embryonic vasculature cells (asterisk). (E) Quantitative analysis of BrdU retention in both INBL and apical retina (AR). In these areas, only cells that retained nuclear BrdU-labeling (arrowheads), which could be easily defined by shape and labeling strength were counted as the post-mitotic cells at the moment BrdU is pulsed. BrdU retention in ONBL in the mutant retina, although qualitatively more, is more diffused, which probably reflect many progenitors went through additional rounds of cell cycles causing more dilution of BrdU. Therefore, these cells were excluded from counting. A significant increase in BrdU-positive cells was detected in both INBL (P< 0.001) and AR (P< 0.01) by Student’s t-test in the mutant retina. (F–H) Grossly normal expression of Ki67 at E17.5 (green) in the mutant retina with an increased cell death primarily in the INBL (red, cleaved form of caspase-3). (F) Dorsal half a retinal section from the central region; (G) ventral half of a retinal section from the central region; (H) boxed area in (G) showing apoptotic cells in the INBL. There are barely detectable apoptotic cells in the INBL in the WT retina at this age (0.75 cells /section), but more in the mutants (26.7 cells/section). Retinal sections from three independent animals of each genotype were counted for caspase-3-positive cells. Note the strong yellow staining on each section adjacent to nerve fiber layer is non-specific staining.
We also detected increased cell death in the INBL at E17.5 (Fig. 4F–H). The increased cell death appears to be a secondary event in post-mitotic cells since Fz5 is not expressed in the differentiated neurons in INBL, and Fz8 alone does not cause retinal neuronal cell death.
Deficiency of retinal apical junctions and down-regulation of RhoA and β-catenin
We hypothesized that early cell cycle exit of the mutant progenitors might result from defects in apical-basal polarity of the neural epithelia. Consistent with this hypothesis, Frizzled receptors are shown to be central players of the PCP pathway that is involved in apical–basal polarity of the embryonic epiblasts during gastrulation (46,47). We first examined retinal apical neuroepithelium by staining for β-catenin, an important apical junction marker that is critical for retinal lamination and its loss causes disorganized retinal neural epithelium (25). We detected a significant reduction in β-catenin on apical surface of the mutant retina at E15.5 (Fig. 5A). F-actin that is enriched at the apical surface was also reduced at apical junctions (Fig. 5B). Upstream of actin assembly, RhoA-GTPase, an important player in the PCP pathway and enriched in apical junction, was also mislocalized (Fig. 5C). The localizations of other junction proteins, atypical protein kinase C and N-cadherin (Fig. 5D and E), were also compromised at the apical junctions, indicating perturbation of retinal apical-basal polarity. Interestingly, at the total protein level, we observed that both RhoA and β-catenin were down-regulated significantly in the mutant retina by western blots (Fig. 5F and G). We also examined the expression of laminin, an ECM protein that is affected by PCP pathways (46). Laminin deposition was reduced and shifted more to the periphery in the mutant retina at E13.5 (Supplementary Material, Fig. S5). Together, these results show a deficiency in apical junctions of retinal neuroepithelium, which might lead to aberrant neurogenesis.
Figure 5.
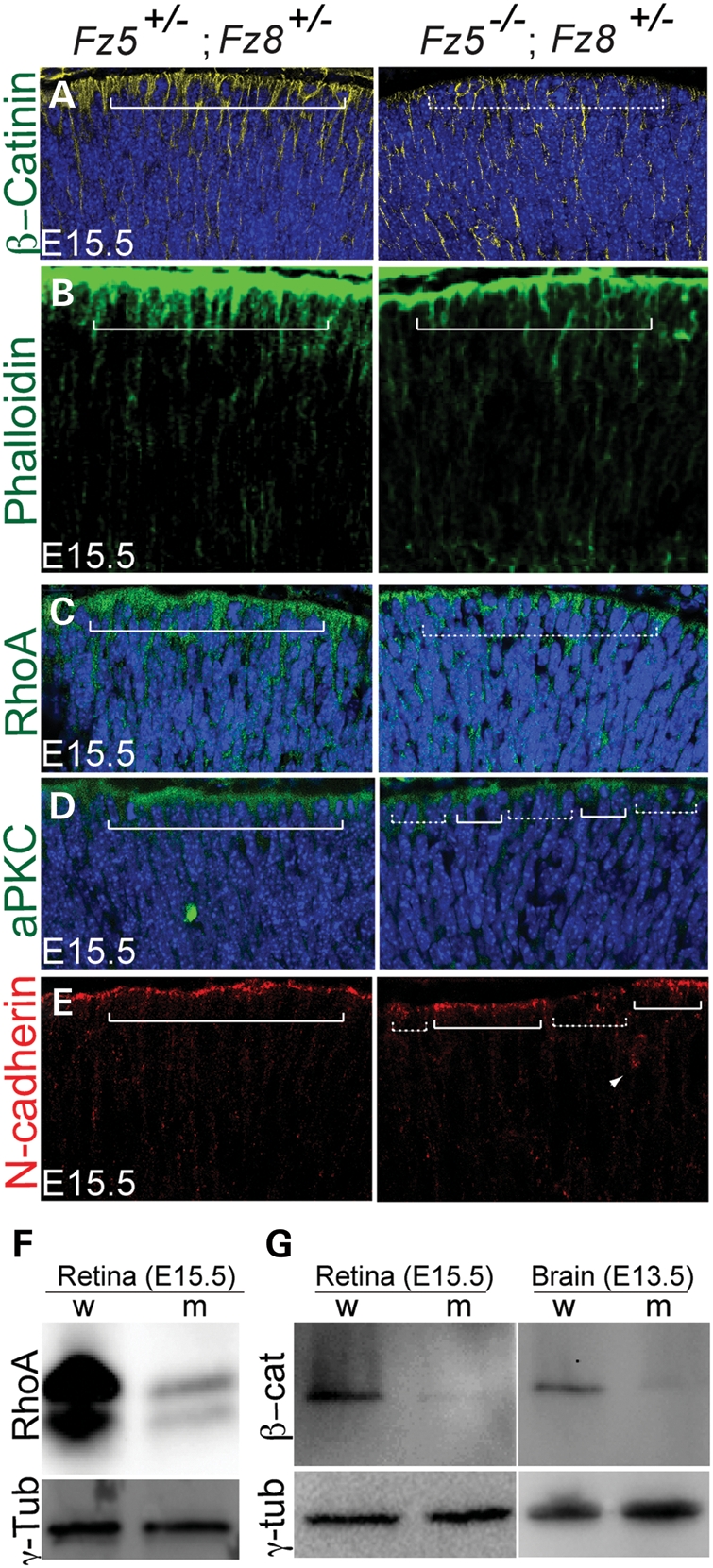
Retinal apical junction is perturbed in developing the Fz5−/−;Fz8+/− mutant retina. (A) Mislocalization of β-catenin in the mutant retina. β-Catenin localization is enriched on the apical retina (left panel, solid bracket), but became more homogenous in the mutant retina. The DAPI-stained apical nuclei in the WT are well topped by β-catenin-enriched apical cytoplasm, while they are seemingly more naked in the mutant (dashed bracket). (B) In the WT E15.5 retina, phalloidin-stained F-actins are enriched on the apical retinal surface (bracket, left panel), while in the mutant retina less staining is seen (right panel). (C) Reduction and discontinuity of RhoA enrichment on apical surface of the Fz5−/−;Fz8+/− mutant retina (dashed bracket). (D) Discontinuity of aPKC on the apical junction of the mutant retina (compare regions above solid and dashed brackets in the mutant retina). (E) Discontinuity of N-cadherin staining at the mutant retinal apical junctions. Ectopic N-cadherin staining is also sometimes seen in the subapical area in the mutant retina (arrow in right panel), but not in the WT. (F and G) Western blots using whole retina and brain tissue extracts of E15.5 embryos showing significant down-regulation of RhoA and β-catenin at protein level. w, wild-type; m, mutant. Retinal sections were cut through the central–ventral area horizontally, where the mutant retinal phenotype is readily obvious under the microscope. Each panel represents five projected images at 1 μm step. All mutant retinal sections are shown in right panels.
Cell-autonomous retraction of neuroblast apical processes and ectopic progenitor divisions upon blocking Frizzled receptors in vitro
To further elucidate the effect of apical junction deficiency, we generated mosaic Frizzled mutant neuroblast cells by injection of 4-hydroxytamoxifen (4HT) to control Cre-ER activity in excising the Fz5ckoAP allele (32). The apical processes of many mutant neuroblasts retracted from retinal surface (Fig. 6A and B). In concordance, we also observed that γ-tubulin-labeled centrioles in retinal progenitors of the compound mutant retina delocalized from the retinal apical surface at E13.5 and E15.5 (Fig. 6C and D). Thus, deficiency at apical junctions might cause the retraction of apical processes of retinal neuroblasts. Our data also indicated that Frizzled receptors function primarily in a cell-autonomous manner in the neural retina.
Figure 6.

Cell-autonomous neuroblast retraction and ectopic retinal progenitor divisions upon blocking Frizzled receptors in vitro. (A and B) Mosaic mutant analysis shows single or clustered mutant neuroblasts labeled by alkaline phosphatase (AP) reporter. (A) An overview of AP-labeled isolated or clustered neuroblasts in the E13 retina by pharmacological delivery of 4HT (see Materials and Methods). Left panel, WT neuroblasts; right panel, mutant neuroblast. Boxed regions are shown in (B). Le, lens. (B) Boxed areas in (A) show the detached neuroblasts from apical surface of the mutant retina (brackets, right panel). (C and D) γ-Tubulin-labeled centrosomes on the apical surface of E13.5 and E15.5 retinas. In the Fz5−/−;Fz8+/− retina (right panels), more centrioles were delocalized to the sub-apical area (short lines), probably because neuroblasts defects were occurring at the apical ends. (E and F) Representative retinal explants' sections stained for γ-tubulin and pH3. In control (E, F, left panels), most of the dividing cells (pH3 positive) are restricted to the apical surface, while they are ectopic and basally shifted in Fz8Ig-CRD-treated retinal explants (right panels). γ-Tubulin staining (red) lined up the apical surface of both control and treated retinal explants. (Ga) Increased total pH3 dividing cells in Fz8Ig-CRD-treated retinas. pH3-positive cells are counted as an average of nine sections from three retina explants (three sections from each retina) Students’ t-test obtained a P-value of 0.042. (b) Percentage of apical-localized pH3 cells calculated from (a). About 68% of pH3-positive cells localized apically in Fz8Ig-CRD-treated explants, while in the controls, this number is 93% (P< 0.01).
The apical niche of the neuroepithelium is where the progenitors renew and generate neurons. In many cases, disruption of apical-basal polarity can result in ectopic progenitor divisions; however, this is barely observed in the Fz5−/−; Fz8+/− retina. Nevertheless, we detected ectopic progenitors' divisions along the retinal neuroepithelium by blocking the Frizzled receptor function in cultured retinal explants, using a secreted form of the Fz8 cystine-rich domain (Fz8Ig-CRD) (Fig. 6E and F). Fz8 CRD is highly conserved among Frizzled members (40) and widely inhibits Frizzled receptor-mediated canonical Wnt pathway. Fz8Ig-CRD was added to cultured E13.5 retinal explants. After 42 h incubation, >30% of the dividing progenitors in treated retinas divided ectopically or shifted basally along the retinal epithelium (Fig. 6E–G); however, γ-tubulin-marked neuroblast apical centrioles were roughly in continuum (Fig. 6E and F), indicating the ectopic divisions are not artifacts of distortion or folding of the cultured retinal explants. Interestingly, a significant increase in the dividing rate of the progenitors (labeled by pH3) was also observed (Fig. 6E–G), which has not been seen in the Fz5−/−;Fz8+/− retina. One interpretation for these outcomes is that the remained 50% of the Fz8 dose in the triallelic mutant retina can be completely blocked by applying Fz8Ig-CRD to the retinal explants in the in vitro culture system; alternatively, other Frizzled receptors which may compensate for the Fz5/8 function loss in the compound mutant retina could also be further blocked in vitro. In any case, more severe retinal neurogenesis defects were caused.
Altered gene expression of signaling pathways required for neurogenesis in the mutant retina
To address more directly the cause of earlier cell cycle exit in the mutant retina, we evaluated Notch and Shh signaling pathways by in situ hybridization and quantitative PCR (q-PCR). While Notch1 receptor expression appeared unaffected in the mutant retina (Fig. 7A and C, Supplementary Material, Table S1, P= 0.18), its downstream target, Hes1, was reduced significantly in most area of the retina at E13.5 (Fig. 7B and C, Supplementary Material, Table S1, P= 0.0067). Hes5 expression seems slightly increased on the other hand in the mutant retina, but with a P-value of 0.14 (Supplementary Material, Table S1). Since it has been shown that Hes1 is also a major target of Shh in the retina independent of Notch signaling (22), we examined expression of Shh downstream effectors Gli2 and Gli3. Although Gli2 is required for the Hes1 activation in the developing retina (22), it remains unchanged in the mutant by q-PCR assay, so does Gli3. Therefore, the compromised Hes1 expression in the Fz5 and Fz8 triallelic mutant retina is likely mediated through Notch signaling.
Figure 7.

Altered gene expression of signaling pathways. (A and B) In situ hybridization at E13.5 shows that although Notch1 expression is essentially unaffected (A), its downstream effector, Hes1, is grossly reduced through the mutant retina (B). The dashed lines demarcated the boundary between INBL, ONBL and PFV tissue. (C) q-PCR detection of gene expression involving Notch and Shh signaling and apical-basal polarity. Consistent with in situ hybridization, Hes1 expression is downregulated in the mutant retina ∼3.5-fold (P< 0.01). There is a moderate upregulation of RhoA expression (P= 0.02) and unchanged β-catenin expression in the mutant retina. The expression of Shh downstream effectors, Gli2 and Gli3, was not altered. Y-axis: fold change of gene expression in the mutant retina with respect to control. Positive values indicate upregulation, while negative values indicate downregulation. Value of either ‘−1’ or ‘1’ indicates no changes. There are no values between −1 and 1 since the expression ratios <1 were treated with negative reciprocal, and plotted along the downregulation direction. (D) A schematic model for Fz5 and Fz8 functions during retinal neurogenesis. During the retinal neurogenesis, Fz8 and Fz5 are critical for neuroblast apical junction maintenance through a set of apical complex proteins, which include RhoA and β-catenin. This is important for the neuroblast polarity and organization. In the Fz5−/−;Fz8+/− mutant retina, the retinal apical deficiency subsequently affect Notch and/or Shh signaling components, of which Hes-1 is an example. This in turn pushes more progenitors to exit cell cycle. In such, the size of the proliferating progenitor pool in the mutant retina becomes smaller (see schematic pie charts), which is likely the cause of microphthalmia and retinal coloboma.
Interestingly, we found an upregulated RhoA (P= 0.02) and unchanged β-catenin expression in contrast to the down-regulation of both proteins detected by western blot in Figure 5. This seemingly paradox could be interpreted as possible alternations of protein stability of RohA and β-catenin in the mutant retina, which could be regulated by both canonical and/or non-canonical Wnt–Frizzled pathways.
DISCUSSION
Here, we demonstrate that Wnt receptors, Fz5 and Fz8, are critical for modulating progenitor cell cycle exit in the mammalian retina, thereby contributing to retinal progenitor expansion. We also show that Frizzled receptors play a role in maintaining retinal neuroblast apical junctions, which in turn may affect reception of Notch signaling along the retinal epithelium. Our data provide insight for the relationship of Frizzled receptors mediated retinal neural epithelial integrity with signaling pathways critical for retinal neurogenesis. We propose a working model for Frizzled receptors' function in retinal neuroblasts based on our findings (shown in Fig. 7D).
Fz5 and Fz8 coordinate optic fissure/disc formation and axon integrity in a dose-dependent manner
The complexity of Wnt–Frizzled signaling is compounded by the existence of multiple members in both Wnt and Frizzled families (33). For example, Fz3 and Fz6 function redundantly in the inner ear, yet have distinct roles in the brain and hair follicles (34,36). In our studies, mutations in Fz5 or Fz8 cause varying degrees of optic fissure/disc abnormalities. The two genes do not fully compensate for each other in the retina, as mutation in each of them causes phenotypes. However, mutation in both genes causes more severe retinal phenotypes with full penetrance, indicating Fz5 and Fz8 function in the dose-dependent manner in the retina. In the Fz5+/−; Fz8−/− triallelic combination, retinal development proceeds as in Fz8−/− mutant. Thus, the ocular defect is most pronounced in the Fz5−/−;Fz8+/− mutant followed by the Fz5−/− single mutant, whereas in the Fz5+/−;Fz8−/− compound and the Fz8−/− single mutant the retinal defect is minimal. These phenotypic variations suggest that Fz5 and Fz8 receptors are signaling similar cellular events in the developing retina, with Fz5 being the dominant contributor by far. As such, the variable severity in ocular defects would simply reflect the varying levels of combined functional Fz5/Fz8 dosage among these mutants.
The dosage effect is also reflected by the aberrant axons' sprouting in the Fz5−/−;Fz8+/− mutant retina, which is clearly evident in 4-week-old mice yet absent in the Fz5−/− mutant mice until 6 months of age. Additionally, broadly blocking Frizzled receptors in vitro to furtherer reduce Frizzled receptors dosage produced a qualitatively similar, but even more severe defect in retinal progenitors mitotic divisions, further support this notion.
Role of Frizzled receptors in maintaining retinal progenitor pools and implications for microphthalmia and coloboma diseases
We propose that microphthalmia, coloboma and excessive production of early neurons in the Fz5−/−;Fz8+/− embryonic retina are due to altered homeostasis of retinal progenitor expansion. Similar phenotypes of abnormal CNS neurogenesis are caused by mutations in Shh and Notch signaling pathways. We saw no significant changes in the mitotic division rate in the Fz5−/−;Fz8+/− mutant retinal progenitors. Rather, we identified a moderate acceleration in cell cycle exit of retinal progenitors. Three lines of evidence support this conclusion. First, Brn3a-labeled RGCs and Islet-1-labeled postmitotic neurons are increased in embryonic mutant retinas. Secondly, Tuj1-labeled early postmitotic neurons increased and expanded more peripherally in the E13.5 mutant retina. And finally, BrdU pulse at E13 showed a significant increase in postmitotic labeling in the mutant retina. The increased apoptosis in INBL of the Fz5−/−;Fz8+/− retina is likely to be secondary to the prematurity of excessive early-born neurons populated in the INBL. This is because Fz5 is not expressed in INBL and Fz8 mutation alone does not confer the early cell death. Therefore, accelerated cell cycle exit leading to the depletion of retinal progenitors, in combination with the INBL apoptosis, appear to be the major cause of microphthalmia, coloboma. Whether the accelerated cell cycle exit is because of a moderate shift from symmetric proliferative to asymmetric neuronal divisions (one progenitor + one neuron) requires further investigation.
A more direct cause of accelerated neurogenesis of RGC could be explained by gross reduction in Hes1 expression in the mutant retina. Hes1 is an important target of both Notch and Shh signaling pathways (22). Shh is also critical for optic fissure/disc formation (48) (49) and astrocytes development (50), RGC production (18) (51) and retinal lamination (52). Hes1 mutant also show a retinal morphogenesis defects caused wide open of the ventral retinal at E15.5 and over production of RGCs (52) (17). The reduction in Hes1 in the retina and optic fissure caused by loss of Fz5 and Fz8 suggests that cross-talks between Frizzled and other signaling pathways are required for in retinal neurogenesis and structural development. Furthermore, the enhancement of Fz8, Fz5 and Hes1 (data not shown) expression in optic fissure along with the severer optic fissure phenotype implies that a higher dosage Frizzled signaling is required to maintain this structure.
Role of Fz8 and Fz5 in retinal neuroblast apical junctions
The apical–basal polarity of the embryonic retina is crucial for the maintenance of the retinal progenitor homeostasis and differentiation. Several events take place asymmetrically along the apical-basal axis: (i) mitotic divisions occurring on the apical surface of the retinal neuroblasts (53); (ii) Notch signaling gradient along the apical to basal axis of the neuroblast being critical for the maintenance of retinal progenitors' properties (20); and (iii) an efficient interkinetic nuclear migration along the apical–basal axis driving the maturation of the neuroblast(s) for cell division (21,54–56). Little is known about signaling pathways participating in establishment of retinal neuroblast apical junctions and neurogenesis niche at the apical surface. Non-canonical Wnt–Frizzled signaling is reportedly involved in multiple aspects of cell or tissue polarity (57), where the cytoskeleton reassembly is an important downstream event (46,47,58). We observed that two PCP targets, F-actin and RhoA, which normally are enriched at the apical adherens junctions, were reduced in the mutant retina. Partial loss of β-catenin, aPKCλ and N-cadherin staining from the apical domain in the mutant is also indicative of diminished apical junctional complexes. Furthermore, by western blotting, both β-catenin and RhoA levels are much lower in the mutant. However, we detected increased expression of RhoA but no change in β-catenin RNA by q-PCR analysis, suggesting that reduced β-catenin and RhoA protein levels likely result from post-transcriptional mechanisms. For instance, reduction in β-catenin could be explained by insufficiency of Frizzled signaling, which then leads to an overly active β-catenin destruction complex. While canonical Wnt/ β-catenin signaling does not appear to regulate TCF family of transcription factors during retinal neurogenesis, there are no data indicating the initial step of canonical signaling, namely the stabilization of β-catenin is not operative in the developing retina.
The disturbance of retinal neuroblast apical processes is likely to be a cell-autonomous event as suggested by mosaic analysis. The retraction of processes from the apical surface in single mutant neuroblast may directly lead to apical junction defects when all neuroblasts along the retinal neurogenesis window are considered.
Interactions among signaling pathways during retinal neurogenesis and optic fissure development
Attenuated Hes1 expression indicated that the Notch signaling was compromised probably due to insufficient cell–cell interaction of the neuroblasts resulting from their apical deficiency. In this regard, Notch signaling belongs to juxtaposed signaling category, requiring direct cell–cell contact. Even though Notch receptors and ligands remain largely intact, Notch signaling still could be disturbed by compromised cell–cell contact, eventually causing reduced Hes1 expression. Meanwhile, Shh signaling may not be compromised since Shh is a diffusible morphogen less relying on cell–cell contacts. In accordance, we did not detect altered expression of Gli2, which is required for Shh-mediated Hes1 activation (22). In addition, Hes1 seems to suppress RGC fate only (17). Therefore, the increased apical cone-like cells in the Frizzled mutant retina might be contributed by other Notch signaling members, for example Hes5 or Rpbj (17), mutations in which also give cone phenotypes. Whether Frizzled receptors receive Wnts signaling in the retina or use other ligands remains to be explored.
Taken together, we propose a model in which Fz8 and Fz5 are required for establishing and/or maintaining retinal neuroblast apical junctions, which are critical for the communication of signaling pathways. In the Fz5−/−;Fz8+/− mutant retina, disturbance of retinal apical junctions leads to compromised Notch signaling in the defective neuroblasts, biasing retinal progenitors toward premature cell cycle exit, which in turn causes disproportion of retinal progenitor pool size. Alternatively, the perturbed retinal apical junction may also affect the efficiency of interkinetic nuclear migration of the neuroblasts, thereby altering their exposure to the signaling cues in the developing retina. In any event, impaired neurogenesis will result in microphthalmia and/or coloboma.
MATERIALS AND METHODS
Generation of Fz5 and Fz8 compound alleles
All procedures involving the use of mice were approved by Animal Care and Use Committee of the National Eye Institute. Fz5 straight and conditional knockout mice and Rosa26-CreER: Fz5ckoAP/+ mice have been described previously (40). Briefly, Fz5 straight knockout (Fz8lacZ/lacZ) mice are lethal at around E9.5 due to placenta defects; hence, Sox2-Cre [not expressed in placenta; (59)] is combined with the Fz5ckoAP/+ allele to produce null mutants. Fz8 knockout was created by replacing its coding region with a β-gal reporter [generously provided by Jeremy Nathans; (42)]. Multiple intercrosses were used to produce experimental parental lines, designated as Sox2-Cre;Fz5lacZ/+:Fz8lacZ/lacZ, Rosa26-CreER; Fz5lacZ/+ and Fz5ckoAP/ckoAP:Fz8lacZ/+ based on the names of transgenes, endogenous alleles and knock-in reporters. None of the lines individually showed phenotypes pertaining to our analysis described in the results. Crossing the two parental lines generated mice with a range of genotypes, from which Sox2-Cre;Fz5lacZ/AP:Fz8lacZ/+, Sox2-Cre;Fz5AP/+:Fz8lacZ/+, Rosa26-CreER; Fz5ckoAP/+;Fz8lacZ/+ and CreER; Fz5lacZ/AP:Fz8lacZ/+ mice were used for experiments. Sox2-Cre;Fz5lacZ/AP:Fz8lacZ/lacZ mice, which are equivalent to the double knockout of Fz5 and Fz8, had paler and smaller embryonic size and died before E13.5. Genotyping methods have been described earlier (40).
Mosaic mutant analysis of retinal neuroblasts
Mosaic mutational analysis was performed as described previously (32). Briefly, 4HT was introduced by intraperitoneal injection into pregnant mice carrying Rosa26-CreER; Fz5lacZ/ckoAP;Fz8lacZ/+ and Rosa26-CreER; Fz5ckoAP/+;Fz8lacZ/+ pups at 5 µg/g body weight at E10. Embryos were harvested at E13 and subjected to alkaline phosphatase staining (32).
Histology and imaging
Procedures for preparation of flat mount retina, light and electron microscopy sections have been described previously (32). Standard protocols were used for X-gal and X-phos staining, in situ hybridization and immunohistochemistry (IHC) (32). Antibodies and dilutions used in this study are: rabbit anti-Sox9, 1:500 (Chemicon AB5535); rabbit ant-NFL, 1:500 (Chemicon MAB5294); mouse anti-Brn3a, 1:2000 (Santa Cruz, sc-8429); rabbit anti-calbindin, 1:1000 (Calbiochem, PC253L); rat anti-β-gal, 1:1000 (a gift from Tom Glaser); mouse anti-GFAP, 1:500 (Millipore, MAB3402); mouse anti-Islet-1, 1:2000 (DSHB, 40.3A4); rabbit anti-phospho-Histone H3, 1:150 (Cell Signaling, 9701); mouse anti-Tuj1, 1:1000 (Covance, MMS-435P); mouse anti-Ki67, 1:200 (abcam, ab8191); rabbit anti-Caspase-3, 1:200 (Cell Signaling, 9661); rabbit anti-laminin, 1:50 (Sigma-Aldrich, L9393); Phalloidin-Alexa 594, 1:200 (Invitrogen); mouse anti-γ-tubulin, 1:500 (Sigma-Aldrich, T6557); rabbit cone arrestin, 1:500 (Millipore, ab15282); mouse rhodopsin 1D4, 1:200 (ab5417); mouse Glutamine synthetase, 1:200 (laboratories, 610518); rat anti-BrdU, 1:200 (ab6326); anti-PKCλ 1:300 (cell signaling, 9378); anti-N-cadherin, 1:200 (Invitrogen, 33-3900); anti-RhoA, 1:1000, sigma (SAB1400017); anti- β-catenin (610154, BD transduction laboratories); mouse anti-p27Kip1, 1:1000 (BD transduction, 610241); and mouse anti-Ccnd1, 1:250 (Santa Cruz, SC 450). Fluorescent images were collected by Olympus epifluorescence microscope, Leica SP2 or Olympus FV1000 confocal microscope.
Retinal tissue extraction and western blot
The embryonic retina was dissected and extracted using the radio immunoprecipitation assay lysis buffer, following vortex for 2 min. Tissue lysates were then centrifuged at 14 000g for 10 min, and the supernatant used for sodium dodecyl sulfate polyacrylamide gel electrophoresis. Immunoblots were probed with anti-RhoA, β-catenin, Ccnd1 and p27Kip1 (antibody resources are listed as above for IHC).
Retinal cell dissociation and immunofluorescence studies
Adult retinas were dissected, cut into pieces and digested in 15 ml conical tubes containing papain solution at 8°C for 30 min. The samples were then shifted to 28°C for another 10 min incubation with intermittent shaking. Digested tissues were mixed using the Pasteur pipette and centrifuged at 150g at 4°C for 5 min. After resuspension in anti-papain solution and another spin, cell pellet was resuspended in DMEM/F12 medium with 10% serum. Cells were then spread on L-lysine coated slides and incubated for 2 h at 37°C. After rinsing several times with phosphate buffered saline or DMEM/F12 medium, cells were fixed and stored in methanol at −80°C for immunofluorescence labeling, as described for IHC using tissue sections.
BrdU pulse assay
BrdU (50 μg/g body weight) was injected in E13-timed pregnant mice intraperitoneally. Mice were sacrificed after 16 or 60 h of BrdU injection. Retina sections were subjected to IHC. BrdU-positive cells are then counted separately in ONBL and INBL. For 16 h BrdU incorporation, cell counting is implemented by using the ImageJ software to set signal threshold for difficulty of getting single cell resolution. For 60 h BrdU incorporation study, BrdU-positive cells were counted in INBL and apical surface of the ONBL.
Cell counting
A group of three sections from each of the Islet-1, pH3 or BrdU-labeled embryonic retina were collected from the central area. Three retinal sections each from three independent animals were subjected to cell counting for each marker. The number of Islet-1-positive cells in ONBL was normalized to the counting area × 1000 using Meta imaging (series 7.5) software. Islet-1-positive cells in INBL are very dense, thus counting was performed by staining area and normalized to total neuroblast layer (NBL) area. Data are presented as percentage. The number of pH3-positive cells was normalized to the retinal apical circumferential length, which is measured by MetaMorph software and given arbitrary unit value. For 16 h BrdU single pulse, BrdU signal (intensity) in ONBL was detected by NIH ImageJ by setting threshold, while in INBL was counted by cells. The use of the unconventional counting method for ONBL was because BrdU labeling after 16 h was very intense in ONBL; single cell morphology was barely distinguishable and, therefore, software was applied. While in INBL, most of labeled cells are post-mitotic and well separated, therefore, suitable for counting by cells. Students' t-test was used to obtain P-values in paired groups of sections.
Production of Fz8Ig-CRD conditional medium and retinal explants culture
HEK293 cells were transfected with Fz8Ig-CRD plasmid, in which Fz8 extracellular CRD was fused with human immunoglobin G (IgG) heavy chain, or with IgG heavy chain gene alone (as control), driven by a CMV promoter in a pRK5 plasmid backbone (40). The secretion of Fz8IgCRD was tested by the immunoblot analysis of the culture medium (data not shown).
Retinas were dissected from E13.5 mice, cultured in a drop of Fz8IgCRD conditioned or DMEM/F12 medium on a Nucleopore filter (Whatman) floated in wells filled with cultured medium of a six-well dish. After 42 h incubation, retinas were fixed and sectioned for the IHC analysis. pH3-positive cells were counted from three retinas (three sections/retinas). Statistical analysis was conducted using Microsoft Excel.
RNA preparation and quantitative PCR analysis
Total RNA was prepared from two to three retinas using TriPure Isolation Reagent (Roche) and RNeasy Mini Kit (Qiagen). Three independent samples of total RNA from WT and mutant mice were used for the synthesis of first strand of cDNA using SuperScript® First-Strand synthesis system (Invitrogen). RT–qPCR was then performed using SYBR® Green-Based Detection of gene expression (Applied Biosystem, 7900HT). Data analysis employed the Livak ΔΔCt method, in which gene expression fold change is calculated as 2−ΔΔCt. Change ratios of <1 were treated with negative reciprocals for the presentation purpose.
FUNDING
This research was supported by intramural program of the National Eye Institute.
Supplementary Material
ACKNOWLEDGEMENTS
We are indebted to Jeremy Nathans for generously providing Fz8 knockout mice prior to publication, Tom Glaser for anti-β-gal antibody and Matthew Brooks for qPCR analysis. We thank Jeremy Nathans, Yanshu Wang, Amir Rattner, Sheldon Miller, Thad Whitaker, Yide Mi, Norimoto Gotoh, Jacob Nellissery, Linn Gieser and Oleg Bulgakov for advice and assistance. The technical support of NEI imaging, transgenic and mouse facilities is gratefully acknowledged.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Cowan W.M., Fawcett J.W., O'Leary D.D., Stanfield B.B. Regressive events in neurogenesis. Science. 1984;225:1258–1265. doi: 10.1126/science.6474175. [DOI] [PubMed] [Google Scholar]
- 2.Burek M.J., Oppenheim R.W. Programmed cell death in the developing nervous system. Brain Pathol. 1996;6:427–446. doi: 10.1111/j.1750-3639.1996.tb00874.x. [DOI] [PubMed] [Google Scholar]
- 3.Nijhawan D., Honarpour N., Wang X. Apoptosis in neural development and disease. Annu. Rev. Neurosci. 2000;23:73–87. doi: 10.1146/annurev.neuro.23.1.73. [DOI] [PubMed] [Google Scholar]
- 4.McConnell S.K. Constructing the cerebral cortex: neurogenesis and fate determination. Neuron. 1995;15:761–768. doi: 10.1016/0896-6273(95)90168-x. [DOI] [PubMed] [Google Scholar]
- 5.Cayouette M., Poggi L., Harris W.A. Lineage in the vertebrate retina. Trends Neurosci. 2006;29:563–570. doi: 10.1016/j.tins.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 6.Yoon K., Gaiano N. Notch signaling in the mammalian central nervous system: insights from mouse mutants. Nat. Neurosci. 2005;8:709–715. doi: 10.1038/nn1475. [DOI] [PubMed] [Google Scholar]
- 7.Young R.W. Cell differentiation in the retina of the mouse. Anat. Rec. 1985;212:199–205. doi: 10.1002/ar.1092120215. [DOI] [PubMed] [Google Scholar]
- 8.Livesey F.J., Cepko C.L. Vertebrate neural cell-fate determination: lessons from the retina. Nat. Rev. Neurosci. 2001;2:109–118. doi: 10.1038/35053522. [DOI] [PubMed] [Google Scholar]
- 9.Marquardt T., Ashery-Padan R., Andrejewski N., Scardigli R., Guillemot F., Gruss P. Pax6 is required for the multipotent state of retinal progenitor cells. Cell. 2001;105:43–55. doi: 10.1016/s0092-8674(01)00295-1. [DOI] [PubMed] [Google Scholar]
- 10.Brown N.L., Patel S., Brzezinski J., Glaser T. Math5 is required for retinal ganglion cell and optic nerve formation. Development. 2001;128:2497–2508. doi: 10.1242/dev.128.13.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elshatory Y., Everhart D., Deng M., Xie X., Barlow R.B., Gan L. Islet-1 controls the differentiation of retinal bipolar and cholinergic amacrine cells. J. Neurosci. 2007;27:12707–12720. doi: 10.1523/JNEUROSCI.3951-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Badea T.C., Cahill H., Ecker J., Hattar S., Nathans J. Distinct roles of transcription factors brn3a and brn3b in controlling the development, morphology, and function of retinal ganglion cells. Neuron. 2009;61:852–864. doi: 10.1016/j.neuron.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swaroop A., Kim D., Forrest D. Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat. Rev. Neurosci. 2010;11:563–576. doi: 10.1038/nrn2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agathocleous M., Harris W.A. From progenitors to differentiated cells in the vertebrate retina. Annu. Rev. Cell Dev. Biol. 2009;25:45–69. doi: 10.1146/annurev.cellbio.042308.113259. [DOI] [PubMed] [Google Scholar]
- 15.Jadhav A.P., Mason H.A., Cepko C.L. Notch 1 inhibits photoreceptor production in the developing mammalian retina. Development. 2006;133:913–923. doi: 10.1242/dev.02245. [DOI] [PubMed] [Google Scholar]
- 16.Yaron O., Farhy C., Marquardt T., Applebury M., Ashery-Padan R. Notch1 functions to suppress cone-photoreceptor fate specification in the developing mouse retina. Development. 2006;133:1367–1378. doi: 10.1242/dev.02311. [DOI] [PubMed] [Google Scholar]
- 17.Riesenberg A.N., Liu Z., Kopan R., Brown N.L. Rbpj cell autonomous regulation of retinal ganglion cell and cone photoreceptor fates in the mouse retina. J. Neurosci. 2009;29:12865–12877. doi: 10.1523/JNEUROSCI.3382-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y., Dakubo G.D., Thurig S., Mazerolle C.J., Wallace V.A. Retinal ganglion cell-derived sonic hedgehog locally controls proliferation and the timing of RGC development in the embryonic mouse retina. Development. 2005;132:5103–5113. doi: 10.1242/dev.02096. [DOI] [PubMed] [Google Scholar]
- 19.Alexandre P., Reugels A.M., Barker D., Blanc E., Clarke J.D. Neurons derive from the more apical daughter in asymmetric divisions in the zebrafish neural tube. Nat. Neurosci. 13:673–679. doi: 10.1038/nn.2547. [DOI] [PubMed] [Google Scholar]
- 20.Del Bene F., Wehman A.M., Link B.A., Baier H. Regulation of neurogenesis by interkinetic nuclear migration through an apical-basal notch gradient. Cell. 2008;134:1055–1065. doi: 10.1016/j.cell.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baye L.M., Link B.A. Nuclear migration during retinal development. Brain Res. 2008;1192:29–36. doi: 10.1016/j.brainres.2007.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wall D.S., Mears A.J., McNeill B., Mazerolle C., Thurig S., Wang Y., Kageyama R., Wallace V.A. Progenitor cell proliferation in the retina is dependent on Notch-independent Sonic hedgehog/Hes1 activity. J. Cell Biol. 2009;184:101–112. doi: 10.1083/jcb.200805155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu H., Mohamed O., Dufort D., Wallace V.A. Characterization of Wnt signaling components and activation of the Wnt canonical pathway in the murine retina. Dev. Dyn. 2003;227:323–334. doi: 10.1002/dvdy.10315. [DOI] [PubMed] [Google Scholar]
- 24.Kubo F., Takeichi M., Nakagawa S. Wnt2b inhibits differentiation of retinal progenitor cells in the absence of Notch activity by downregulating the expression of proneural genes. Development. 2005;132:2759–2770. doi: 10.1242/dev.01856. [DOI] [PubMed] [Google Scholar]
- 25.Fu X., Sun H., Klein W.H., Mu X. Beta-catenin is essential for lamination but not neurogenesis in mouse retinal development. Dev. Biol. 2006;299:424–437. doi: 10.1016/j.ydbio.2006.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osakada F., Ooto S., Akagi T., Mandai M., Akaike A., Takahashi M. Wnt signaling promotes regeneration in the retina of adult mammals. J. Neurosci. 2007;27:4210–4219. doi: 10.1523/JNEUROSCI.4193-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burns C.J., Zhang J., Brown E.C., Van Bibber A.M., Van Es J., Clevers H., Ishikawa T.O., Taketo M.M., Vetter M.L., Fuhrmann S. Investigation of Frizzled-5 during embryonic neural development in mouse. Dev. Dyn. 2008;237:1614–1626. doi: 10.1002/dvdy.21565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J., Fuhrmann S., Vetter M.L. A nonautonomous role for retinal frizzled-5 in regulating hyaloid vitreous vasculature development. Invest. Ophthalmol. Vis. Sci. 2008;49:5561–5567. doi: 10.1167/iovs.08-2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu H., Xu S., Wang Y., Mazerolle C., Thurig S., Coles B.L., Ren J.C., Taketo M.M., van der Kooy D., Wallace V.A. Ciliary margin transdifferentiation from neural retina is controlled by canonical Wnt signaling. Dev. Biol. 2007;308:54–67. doi: 10.1016/j.ydbio.2007.04.052. [DOI] [PubMed] [Google Scholar]
- 30.Seitz R., Hackl S., Seibuchner T., Tamm E.R., Ohlmann A. Norrin mediates neuroprotective effects on retinal ganglion cells via activation of the Wnt/beta-catenin signaling pathway and the induction of neuroprotective growth factors in Muller cells. J. Neurosci. 2010;30:5998–6010. doi: 10.1523/JNEUROSCI.0730-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Raay T.J., Moore K.B., Iordanova I., Steele M., Jamrich M., Harris W.A., Vetter M.L. Frizzled 5 signaling governs the neural potential of progenitors in the developing Xenopus retina. Neuron. 2005;46:23–36. doi: 10.1016/j.neuron.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 32.Liu C., Nathans J. An essential role for frizzled 5 in mammalian ocular development. Development. 2008;135:3567–3576. doi: 10.1242/dev.028076. [DOI] [PubMed] [Google Scholar]
- 33.Gordon M.D., Nusse R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 2006;281:22429–22433. doi: 10.1074/jbc.R600015200. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y., Guo N., Nathans J. The role of Frizzled3 and Frizzled6 in neural tube closure and in the planar polarity of inner-ear sensory hair cells. J. Neurosci. 2006;26:2147–2156. doi: 10.1523/JNEUROSCI.4698-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lyuksyutova A.I., Lu C.C., Milanesio N., King L.A., Guo N., Wang Y., Nathans J., Tessier-Lavigne M., Zou Y. Anterior-posterior guidance of commissural axons by Wnt-frizzled signaling. Science. 2003;302:1984–1988. doi: 10.1126/science.1089610. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y., Zhang J., Mori S., Nathans J. Axonal growth and guidance defects in Frizzled3 knock-out mice: a comparison of diffusion tensor magnetic resonance imaging, neurofilament staining, and genetically directed cell labeling. J. Neurosci. 2006;26:355–364. doi: 10.1523/JNEUROSCI.3221-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y., Thekdi N., Smallwood P.M., Macke J.P., Nathans J. Frizzled-3 is required for the development of major fiber tracts in the rostral CNS. J. Neurosci. 2002;22:8563–8573. doi: 10.1523/JNEUROSCI.22-19-08563.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu Q., Wang Y., Dabdoub A., Smallwood P.M., Williams J., Woods C., Kelley M.W., Jiang L., Tasman W., Zhang K., et al. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116:883–895. doi: 10.1016/s0092-8674(04)00216-8. [DOI] [PubMed] [Google Scholar]
- 39.Ye X., Wang Y., Cahill H., Yu M., Badea T.C., Smallwood P.M., Peachey N.S., Nathans J. Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell. 2009;139:285–298. doi: 10.1016/j.cell.2009.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu C., Wang Y., Smallwood P.M., Nathans J. An essential role for Frizzled5 in neuronal survival in the parafascicular nucleus of the thalamus. J. Neurosci. 2008;28:5641–5653. doi: 10.1523/JNEUROSCI.1056-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smallwood P.M., Williams J., Xu Q., Leahy D.J., Nathans J. Mutational analysis of Norrin-Frizzled4 recognition. J. Biol. Chem. 2007;282:4057–4068. doi: 10.1074/jbc.M609618200. [DOI] [PubMed] [Google Scholar]
- 42.Ye X., Wang Y., Rattner A., Nathans J. Genetic mosaic analysis reveals a major role for frizzled 4 and frizzled 8 in controlling ureteric growth in the developing kidney. Development. 2011;138:1161–1172. doi: 10.1242/dev.057620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dyer M.A., Cepko C.L. p27Kip1 and p57Kip2 regulate proliferation in distinct retinal progenitor cell populations. J. Neurosci. 2001;21:4259–4271. doi: 10.1523/JNEUROSCI.21-12-04259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Belletti B., Pellizzari I., Berton S., Fabris L., Wolf K., Lovat F., Schiappacassi M., D'Andrea S., Nicoloso M.S., Lovisa S., et al. p27kip1 controls cell morphology and motility by regulating microtubule-dependent lipid raft recycling. Mol. Cell Biol. 2010;30:2229–2240. doi: 10.1128/MCB.00723-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Besson A., Gurian-West M., Schmidt A., Hall A., Roberts J.M. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004;18:862–876. doi: 10.1101/gad.1185504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Veeman M.T., Nakatani Y., Hendrickson C., Ericson V., Lin C., Smith W.C. Chongmague reveals an essential role for laminin-mediated boundary formation in chordate convergence and extension movements. Development. 2008;135:33–41. doi: 10.1242/dev.010892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tao H., Suzuki M., Kiyonari H., Abe T., Sasaoka T., Ueno N. Mouse prickle1, the homolog of a PCP gene, is essential for epiblast apical-basal polarity. Proc. Natl Acad. Sci. USA. 2009;106:14426–14431. doi: 10.1073/pnas.0901332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morcillo J., Martinez-Morales J.R., Trousse F., Fermin Y., Sowden J.C., Bovolenta P. Proper patterning of the optic fissure requires the sequential activity of BMP7 and SHH. Development. 2006;133:3179–3190. doi: 10.1242/dev.02493. [DOI] [PubMed] [Google Scholar]
- 49.Dakubo G.D., Wang Y.P., Mazerolle C., Campsall K., McMahon A.P., Wallace V.A. Retinal ganglion cell-derived sonic hedgehog signaling is required for optic disc and stalk neuroepithelial cell development. Development. 2003;130:2967–2980. doi: 10.1242/dev.00515. [DOI] [PubMed] [Google Scholar]
- 50.Wallace V.A., Raff M.C. A role for Sonic hedgehog in axon-to-astrocyte signalling in the rodent optic nerve. Development. 1999;126:2901–2909. doi: 10.1242/dev.126.13.2901. [DOI] [PubMed] [Google Scholar]
- 51.Zhang X.M., Yang X.J. Regulation of retinal ganglion cell production by Sonic hedgehog. Development. 2001;128:943–957. doi: 10.1242/dev.128.6.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tomita K., Ishibashi M., Nakahara K., Ang S.L., Nakanishi S., Guillemot F., Kageyama R. Mammalian hairy and Enhancer of split homolog 1 regulates differentiation of retinal neurons and is essential for eye morphogenesis. Neuron. 1996;16:723–734. doi: 10.1016/s0896-6273(00)80093-8. [DOI] [PubMed] [Google Scholar]
- 53.Malicki J. Cell fate decisions and patterning in the vertebrate retina: the importance of timing, asymmetry, polarity and waves. Curr. Opin. Neurobiol. 2004;14:15–21. doi: 10.1016/j.conb.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 54.Baye L.M., Link B.A. Interkinetic nuclear migration and the selection of neurogenic cell divisions during vertebrate retinogenesis. J. Neurosci. 2007;27:10143–10152. doi: 10.1523/JNEUROSCI.2754-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Norden C., Young S., Link B.A., Harris W.A. Actomyosin is the main driver of interkinetic nuclear migration in the retina. Cell. 2009;138:1195–1208. doi: 10.1016/j.cell.2009.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schenk J., Wilsch-Brauninger M., Calegari F., Huttner W.B. Myosin II is required for interkinetic nuclear migration of neural progenitors. Proc. Natl Acad. Sci. USA. 2009;106:16487–16492. doi: 10.1073/pnas.0908928106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lai S.L., Chien A.J., Moon R.T. Wnt/Fz signaling and the cytoskeleton: potential roles in tumorigenesis. Cell Res. 2009;19:532–545. doi: 10.1038/cr.2009.41. [DOI] [PubMed] [Google Scholar]
- 58.Goto T., Davidson L., Asashima M., Keller R. Planar cell polarity genes regulate polarized extracellular matrix deposition during frog gastrulation. Curr. Biol. 2005;15:787–793. doi: 10.1016/j.cub.2005.03.040. [DOI] [PubMed] [Google Scholar]
- 59.Hayashi S., Lewis P., Pevny L., McMahon A.P. Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech. Dev. 2002;119(Suppl. 1):S97–S101. doi: 10.1016/s0925-4773(03)00099-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.