

Abstract
Cobalamins are stored in high concentrations in the human liver and thus are available to participate in the regulation of hepatotropic virus functions. We show that cyanocobalamin (vitamin B12) inhibited the HCV internal ribosome entry site (IRES)-dependent translation of a reporter gene in vitro in a dose-dependent manner without significantly affecting the cap-dependent mechanism. Vitamin B12 failed to inhibit translation by IRES elements from encephalomyocarditis virus (EMCV) or classical swine fever virus (CSFV). We also demonstrate a relationship between the total cobalamin concentration in human sera and HCV viral load (a measure of viral replication in the host). The mean viral load was two orders of magnitude greater when the serum cobalamin concentration was above 200 pM (P < 0.003), suggesting that the total cobalamin concentration in an HCV-infected liver is biologically significant in HCV replication.
The hepatitis C virus (HCV) is an important human pathogen, infecting about 1% of the global population. Approximately 30% of chronically infected carriers develop serious liver disease, making it the single leading indicator for liver transplantation (1). HCV is believed to be noncytopathic, and hepatocellular injury likely results from an abortive immune response to virus-infected cells. During HCV infection, HCV RNA replicates by using a virally encoded RNA-dependent RNA polymerase without a DNA intermediate (2). Because the virion presumably contains only the positive-strand genomic RNA and the virally encoded structural proteins, translation of the HCV genome to produce the viral proteins required for replication is an early obligatory step in the replication cycle. Translation initiates in the 5′ untranslated region (UTR) and proceeds 5′ to 3′ along the positive strand genomic RNA, whereas transcription to synthesize the negative-strand RNA is thought to initiate in the 3′ UTR of the positive strand and proceeds from 3′ to 5′. It is unlikely that the transcription complex (replicase) of a positive-strand RNA virus can compete with actively translating ribosomal subunits for the same RNA template (3–5). Therefore, HCV should down-regulate its own translation to achieve initiation of transcription. Such regulatory mechanisms probably affect initiation, rather than elongation, thus allowing the translation machinery to clear the template (4). The virus can use a virally encoded protein or cellular factor as a regulatory agent, targeting the HCV internal ribosome entry site (IRES) (6). It is reasonable to suggest that a highly tissue-specific virus like HCV may evolve to use a compound in which that tissue is rich for such a regulatory purpose.
HCV initiates translation by a mechanism that is distinct from the usual eukaryotic cap-dependent mechanism (7). Antiviral drug design targeting this mechanism has been proposed (8, 9), because it may yield a therapy with high viral specificity and low host cellular toxicity. The putative regulatory mechanism outlined above suggests that partial inhibition of HCV translation may enhance HCV transcription by increasing the proportion of positive strands available to the HCV replicase (4). Indeed, the RNA-dependent RNA polymerase is not required stoichiometrically, and once a critical concentration of the viral proteins has been established in the early stages of infection, the requirement for transcription should become paramount. A partial inhibition of translation could create a mixed population of RNA available for either translation or transcription, and produce an optimal balance between these two viral functions.
The HCV IRES (Fig. 1) initiates translation by binding to the ribosomal complex and efficiently presenting it to the start codon without scanning (10). Because it is able to initiate translation without the usual complement of eukaryotic initiation factors (eIFs), the HCV translation mechanism has been described as prokaryotic-like (11). Poliovirus, which also utilizes a cap-independent IRES translation mechanism, down-regulates the host cellular cap-dependent mechanism without affecting its own IRES-dependent mechanism, demonstrating that the IRES mechanism can be functionally distinguished (12).
Figure 1.

The proposed secondary structure of the genotype 1b HCV IRES, adapted from Honda et al. (6). The authentic initiation codon is boxed. (Inset) Comparison between HCV and classical swine fever virus (CSFV) IRES structures in domains IIIe, IIIf, and IV.
A critical element in the molecular scaffolding of the HCV IRES is an unusual RNA pseudoknot structure near the authentic start codon (13) (Fig. 1, domain IIIf). Spedding and Draper (14) have demonstrated that an RNA pseudoknot in the α operon of Escherichia coli is used to regulate translation. Initiation of translation is down-regulated by a feedback mechanism, whereby the S4 translational repressor protein encoded in the α operon binds to and stabilizes the RNA structure immediately downstream from the start codon. The initiation complex becomes trapped on the Shine–Dalgarno sequence. We (data not shown) and others (15–17) have observed that the HCV core protein can inhibit the HCV IRES activity in vitro, possibly by using a similar mechanism. The E. coli pseudoknot lies primarily upstream from the start codon, but incorporates a sequence within the open reading frame. The complete HCV IRES also requires part of the polyprotein coding region for full IRES activity (18, 19). We speculate that this coding sequence may be involved in a complex RNA structure, and the stability of this structure affects the efficiency of HCV IRES-dependent translation initiation. Increased stability of an RNA hairpin in domain IV of the HCV IRES has been shown to sharply reduce HCV IRES efficiency (6). Similar to the role proposed for the RNA structure in the α operon of E. coli, the RNA pseudoknot in domain IIIe and the stable RNA hairpin in domain IV of the HCV IRES, which includes the authentic start codon, are thought to be structural elements required for the recognition and binding of the incoming ribosomal subunits (9, 11). After initiation of translation, the base-pairing in the coding region must be destroyed to elongate the nascent protein chain. Based upon these striking similarities, we suggest that compounds that bind to and stabilize the RNA structure surrounding the authentic HCV start codon will inhibit HCV IRES-dependent initiation of translation.
Specific binding interactions between RNA and small molecules or proteins are often involved in the regulation of posttranscriptional gene expression (20). The tuberactinomycin class drug viomycin provides a precedent for the regulation of biological function mediated by recognition and binding of tertiary RNA structures. This cyclic peptide binds RNA pseudoknots strongly in vitro, and all of its known biological targets contain RNA pseudoknots (21). Viomycin inhibits normal eukaryotic cap-dependent translation by binding to a pseudoknot in the ribosomal RNA. When added to an in vitro translation reaction, viomycin very strongly inhibited HCV IRES-dependent translation as well as cap-dependent translation (data not shown). Because viomycin acts on the ribosomal RNA, an inhibitor specific to HCV IRES-dependent translation was required to validate our hypothesis.
The binding of cyanocobalamin (vitamin B12) to RNA aptamers selected from a random RNA library represents one of the strongest binding affinities yet reported for a small molecule to RNA (Kd = 88 nM) (22). Because the criterion for selection of the aptamer was binding affinity and unusual ionic conditions were required, this may be regarded as a lower limit Kd for vitamin B12 bound to RNA. The selected RNA aptamers potentially formed RNA pseudoknots, and subsequent binding studies and structural analysis showed that cyanocobalamin did specifically bind to these RNA structures (23, 24).
Because cobalamins, the class of compounds to which vitamin B12 belongs, are found most abundantly in the human liver (25), we suggest that B12 may bind to a similar structure in the HCV genome and may serve as a natural inhibitor of viral translation, thus affecting viral replication. If this suggestion is true, a relationship between cobalamin concentration in the liver and viral replication is expected in HCV carriers. The aims of this research were to demonstrate and characterize the inhibition of HCV IRES-dependent translation by vitamin B12 in vitro, and to provide evidence for a biological relevance of this observation by demonstrating a clinical relationship between HCV replication and B12 in vivo.
Materials and Methods
Plasmids and Reagents.
Vitamin B12 was purchased (Sigma) as reagent grade and was not further purified. Solutions containing vitamin B12 were shielded from UV radiation throughout the experiments. The plasmids described in Fig. 2a contained the elements of the 5′ UTR from HCV (341 nucleotides), classical swine fever virus (CSFV, 373 nucleotides), and encephalomyocarditis virus (EMCV, 586 nucleotides) cloned into pBluescript Ks and were generous gifts from Rene Rijnbrand (Univ. of Texas Medical Branch, Galveston). The T368-CAT plasmid (Fig. 2b) was constructed by cloning the HCV IRES element from the Australian HCV isolate (26) (pSTDL, genotype 1b), which includes the entire 5′ UTR (368 nucleotides) plus the first 27 nucleotides of the polyprotein coding region, into pGEMT-Easy (Promega) upstream from the chloramphenicol acetyltransferase (CAT) reporter gene. RNA representing the first 368 nucleotides of the HCV genome was transcribed from the cDNA of the Australian HCV isolate, pSTDL (26).
Figure 2.

Plasmids used for in vitro translation inhibition assays. (a) The bicistronic constructs under the control of a T7 transcriptional promoter contained the Renilla luciferase (R-Luc) reporter gene, a viral IRES element, and the chloramphenicol acetyltransferase (CAT) reporter gene. The viral sequences incorporated in these constructs consisted of the 5′ UTRs of EMCV (586 nucleotides), CSFV (373 nucleotides), and HCV (341 nucleotides), respectively. (b) The monocistronic construct (T368-CAT) contained the HCV IRES sequence, consisting of the entire 5′ UTR (341 nucleotides) and 27 nucleotides of the polyprotein coding sequence, and the CAT reporter gene cloned into the pGEMT-Easy (Promega) expression vector.
In Vitro Translation Assays.
mRNA was transcribed in vitro (Ribomax, Promega) from the linearized plasmids and then translated in vitro in a separate reaction (rabbit reticulocyte lysate systems, Promega) by following the protocols supplied by the manufacturer. In vitro transcribed mRNA lacking an IRES structure was capped by incorporating m7G(5′)ppp(5′)G RNA capping analog (Life Technologies) in the transcription reaction mixture. The observed inhibition by vitamin B12 in the in vitro translation reaction was optimized by using 80 ng of mRNA in a 25-μl reaction mixture and by incubating the mRNA with vitamin B12 at room temperature for 10 min before addition to the rabbit reticulocyte lysate mixture. When RNA transcribed from the monocistronic plasmid (Fig. 2b) was translated, the control RNA (5′-cap-modified firefly luciferase RNA) supplied with the in vitro translation system was incorporated as an internal positive control. In the HCV IRES competition experiments, the in vitro translation reactions were performed as described above except that a large excess (4 μg) of HCV RNA representing the first 368 nucleotides of the Australian HCV isolate (26) was incorporated into the translation mixture. The products from the translation reactions were resolved by SDS/PAGE, visualized with a PhosphorImager (Molecular Dynamics), and quantified by using the ImageQuant software package (Molecular Dynamics). To allow for any nonspecific effects, the IRES efficiency for each concentration of vitamin B12 was defined as the fraction of total translation products that were produced by the HCV IRES-dependent mechanism (IRES efficiency = [CAT]/([CAT] + [Luc])). These data were plotted and fitted to a rectangular hyperbolic binding function by using the KaleidaGraph software package (Synergy Software, Reading, PA).
HCV Viral Load and Total Serum Cobalamin.
Fifty samples of stored sera from HCV carriers were tested for HCV viral load and total cobalamin level. These carriers were unstratified and chosen randomly from a pool of HCV carriers representing all clinical stages of disease state. The HCV viral load (number of copies of HCV RNA per milliliter of serum) was estimated by using the Roche Amplicon HCV monitor. The virus RNA was extracted from serum samples and the reverse transcription (RT)-PCR was performed according to the supplier's instructions. Total serum cobalamin concentrations were measured by RIA (Abbott Laboratories). These data were plotted by using the KaleidaGraph software package and were statistically evaluated by using the Mann–Whitney U test—a two-group ordinal comparison to assess the probability that two groups come from a single ordering.
Results and Discussion
Selective Inhibition of HCV IRES-Dependent Translation.
Initially, two separate mRNAs, one containing the HCV IRES upstream from the CAT reporter gene (Fig. 2b) and the other containing the 5′-cap-modified firefly luciferase reporter gene, were added to the same in vitro translation experiment, with and without vitamin B12. Vitamin B12 selectively inhibited HCV IRES-dependent translation without substantially affecting cap-dependent translation (Fig. 3). Nonspecific binding of vitamin B12 to the RNA or an interaction between vitamin B12 and the translational machinery would be expected to inhibit the translation of both reporter genes.
Figure 3.

Selective inhibition of the HCV IRES in vitro. Lane 1 shows the expression of both the HCV IRES-dependent CAT reporter gene and the cap-dependent firefly (FF)-Luc reporter gene in the lysate in the absence of added vitamin B12. Lane 2 shows the selective inhibition when the mRNA was preincubated with vitamin B12 (2.5 mM final concentration on the lysate) before translation. The unidentified minor bands in the gel represent peptide fragments that are characteristic products of the FF-Luc control RNA.
Functional Determination of the Dissociation Constant, Kd.
The inhibition observed in Fig. 3 was concentration dependent, and the data fit a rectangular hyperbolic binding curve (Fig. 4, r = 0.98). In some experiments we have observed a complete inhibition of HCV IRES-dependent translation, but we reproducibly observed a maximum inhibition of ≈60%. The determined Kd of 170 μM is much higher than the Kd reported for the binding of vitamin B12 to the selected RNA aptamer (88 nM) (22). Such a high Kd (Kd ≈ 10−2 M) has been reported (20) for small molecule RNA ligands that are significant for substrate–ligand recognition. We were unable to directly measure a specific dissociation constant for cyanocobalamin to the HCV IRES or fragments thereof by using frontal affinity chromatography, nitrocellulose filter binding, or biosensor assays (data not shown). The weak binding responsible for the observed inhibition may be obscured by a background of nonspecific binding to the negatively charged linear polymeric acceptor (27). Thus, a functional assay may be the only useful method to characterize this binding event. A normal healthy liver, which stores the body's reservoir of cobalamins (analogues of vitamin B12), contains ≈555 ng of total cobalamin per gram of wet tissue (25), suggesting that the unusually high functional Kd may represent a biologically relevant concentration of cobalamin in the tissue. Because these data were collected from multiple independent experiments, using both monocistronic and bicistronic constructs, the curve fit demonstrates the reproducibility of the inhibition. The data are consistent with the inhibition arising from a single binding event, although other models are possible.
Figure 4.

Vitamin B12 concentration dependence of the observed HCV IRES-dependent translational inhibition. The HCV IRES efficiency was plotted as a function of final vitamin B12 concentration from a bicistronic mRNA (Fig. 2a, ▴) or from a monocistronic mRNA (Fig. 2b, ●). The IRES efficiency, defined as [CAT]/([CAT] + [Luc]), was calculated for each reaction and normalized to the reaction with no added vitamin B12. The curve fit a regular hyperbolic binding function (r = 0.98). The minimum IRES efficiency was ≈0.40, suggesting a maximum of 60% inhibition relative to the uninhibited control.
Specificity of Inhibition for the HCV IRES.
Bicistronic constructs (Fig. 2a) were used to demonstrate the specificity for the HCV IRES structure. The use of bicistronic constructs is a standard method to compare the efficiencies of different IRES elements (28). Constructs containing the IRES elements from HCV, CSFV, and EMCV were used. CSFV, in the pestivirus genus of the family Flaviviridae, uses an IRES that is similar to the HCV IRES in both structure and mechanism (11, 29). The IRES of EMCV, which belongs to the cardiovirus genus of the family Picornaviridae, differs significantly from the HCV IRES in structure and mechanism (30). Fig. 5 shows that vitamin B12 selectively inhibited HCV IRES-dependent translation without substantially affecting the cap-dependent cistron in the same RNA, consistent with the data in Fig. 3. The EMCV IRES was expected to provide a negative control, on the basis of our hypothetical mechanism. Indeed, vitamin B12 had no significant effect on the efficiency of EMCV IRES-dependent translation. Because the sequences of the reporter genes were identical in all of the bicistronic constructs, it is unlikely that the observed inhibition of the HCV IRES resulted from an interaction between vitamin B12 and the RNA encoding the CAT or luciferase reporter proteins.
Figure 5.

Specificity for the HCV IRES. Lanes 1–3 show the in vitro translation products from bicistronic mRNA containing EMCV, CSFV, and HCV IRES elements, respectively. Lanes 4–6 show the in vitro translation products formed when vitamin B12 was incorporated in the reaction mixture to a final concentration of 3 mM. The faster-migrating species is the HCV IRES-dependent CAT protein and the slower-migrating species is the cap-dependent R-Luc protein. The IRES efficiency for each reaction, defined as in Fig. 4 but not normalized, is listed below the corresponding lane. Lane 6 shows the characteristic 60% inhibition relative to lane 3.
Surprisingly, vitamin B12 also failed to inhibit translation initiated by the structurally similar CSFV IRES. Because the pestivirus IRES element contained a pseudoknot of similar structure and in approximately the same position relative to the start codon (29), we considered it likely that vitamin B12 would inhibit CSFV IRES-dependent translation. This observation suggests an unusual specificity for the HCV IRES. There are two important structural differences between the HCV and the CSFV IRES elements. First, the CSFV IRES contains six unpaired residues immediately preceding the obligatory base-paired sequence of the pseudoknot that are not contained in the HCV pseudoknot. This unpaired sequence makes the CSFV pseudoknot considerably more flexible. Since the HCV pseudoknot is a necessary structural element whose primary sequence is not essential for IRES function (13), the rigidity of this structural region may be an important element for specific recognition. Second, the CSFV IRES has not been reported to contain the structural element present in domain IV of the HCV IRES (Fig. 1). This domain may play a critical role in the down-regulation of HCV translation, since the HCV IRES efficiency is strongly affected by small changes in the thermodynamic stability of this RNA structure in this region (6).
Trapping Assays.
RNA representing the complete HCV IRES (368 nucleotides), but lacking a reporter gene, was added to the standard in vitro translation assay. In the presence of an excess of both vitamin B12 and the HCV IRES RNA, both cap-dependent and HCV IRES-dependent translation mechanisms were inhibited (Fig. 6). This nonspecific inhibition was strongly dependent upon the concentration of added HCV IRES RNA, and was not observed when similar concentrations of nonspecific RNA or fragments of the HCV IRES lacking domain IV were used (S.S.T., E.J.G., and W.B.L., unpublished results). Because added HCV IRES RNA in the absence of vitamin B12 had no significant effect on translation, it is unlikely that the observed inhibition in the presence of vitamin B12 arose from a posttranscriptional gene silencing mechanism (31) or from the previously observed activation of RNase L by double-stranded RNA in rabbit reticulocyte lysate (32). These data suggest that the vitamin B12-HCV IRES complex may have sequestered one or more required translational factors. By analogy to E. coli (14), we suggest that the ribosomal complex was trapped on the loading site by a vitamin B12-stabilized downstream RNA structure. A detailed biophysical characterization of the vitamin B12 trapping effect on the HCV IRES RNA-ribosome complex in support of this interpretation will be presented elsewhere (S.S.T., E.J.G., and W.B.L., unpublished work).
Figure 6.

Trapping by the HCV IRES–vitamin B12 complex. (a)
Nonspecific inhibition of translation was observed when the full-length
HCV IRES RNA (368 nucleotides) was added to a standard translation
assay in the presence of 250 μM vitamin B12 (lane 4). This inhibition
was not observed when either the HCV IRES RNA or vitamin B12 was
omitted from the reaction (lanes 2 and 3, respectively). The gel was
deliberately overloaded to convincingly illustrate the absence of
protein in lane 4. (b) The suggested mechanism of the
observed translational inactivation. Vitamin B12 binds specifically to
the HCV IRES, trapping the preinitiation complex [minimally the 40S
ribosomal subunit and the eIF2/Met-tRNA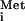 /GTP
ternary complex (11)] on the ribosomal subunit loading site.
/GTP
ternary complex (11)] on the ribosomal subunit loading site.
Vitamin B12 and HCV Pathology in Humans.
The above in vitro results suggest that vitamin B12 may have the capacity to modulate the replication of HCV in the host by inhibiting HCV IRES-dependent translation. However, there is no reliable cell culture system or small animal model available to evaluate this possibility. Because cobalamins are stored in high concentrations in the liver, we wished to determine whether any functional relationship existed in vivo between HCV RNA and total cobalamin concentration in the serum of human HCV carriers. The concentration of viral RNA in the serum is a good estimate of HCV replication, because the half-life of the virion in the host is estimated to be only a few hours (33). We conducted a retrospective study of stored sera from a random unstratified sample of 50 HCV carriers. We determined the HCV viral loads (copies of HCV RNA per ml of serum) and total cobalamin concentrations for each of these samples. Fig. 7 is a plot of the log of the HCV viral load as a function of total serum cobalamin concentration. These data adopted a sigmoidal shape that resembled a dose–response curve, with the transition at ≈200 pM total cobalamin. The mean values for the maximum and minimum viral load were separated by approximately two orders of magnitude, and the cobalamin transition range was less than 100 pM. If cobalamin concentration in the liver influences HCV replication, this narrow transition region may suggest an unusually tight regulation. The magnitudes of the viral load above and below 200 pM total serum cobalamin are statistically distinct (Mann–Whitney U test, P < 0.003). Because the normal range of total serum cobalamin in humans is ≈200 and 800 pM, this plot suggests that HCV carriers with unusually low total serum cobalamin concentration exhibit an unusually low rate of HCV replication. These data demonstrate a relationship between HCV replication and endogenous cobalamin concentration. To our knowledge, no correlation has been demonstrated between HCV viral load and any other measurable parameter (34). In our study, neither HCV viral load nor total serum cobalamin concentration correlated with alanine aminotransferase (ALT) levels (data not shown), in agreement with a recent report (35).
Figure 7.

Relationship between HCV viral load and total serum cobalamin concentration. The HCV viral load was plotted against the total serum cobalamin concentration. The mean of the data below 200 pM differed significantly from the mean of the data above 200 pM (Mann–Whitney U test, P < 0.003).
Conclusions
In this study we have demonstrated that vitamin B12 inhibits HCV IRES-dependent initiation of translation in vitro. Furthermore, this inhibition is selective for the HCV IRES in the presence of cap-dependent RNA, and it is specific for the HCV IRES relative to other viral IRES elements. Although other possibilities cannot be excluded on the basis of these data, the mechanism of this inhibition likely involves a direct interaction between vitamin B12 and the HCV IRES RNA. Preincubation of the HCV IRES-containing mRNA with vitamin B12 before translation enhanced the observed inhibition, supporting this interpretation. It is unlikely that vitamin B12 inhibits by binding to the ribosomal RNA, as has been demonstrated for viomycin inhibition (21), because such inhibition would be nonspecific, affecting both the cap-dependent and the HCV IRES-dependent mechanisms. Similarly, a mechanism whereby vitamin B12 sequesters an HCV IRES-specific eIF is unlikely. Indeed, HCV IRES-dependent translation requires fewer eIFs than eukaryotic cap-dependent translation (11), although a putative HCV IRES-specific eIF has been recently reported (36). The nonspecific inhibition of translation by a putative HCV IRES RNA–vitamin B12 complex, but not by the individual components of this complex (Fig. 6), also supports a direct vitamin B12–RNA interaction. Experiments to directly observe and quantify the putative direct interaction between vitamin B12 and the HCV IRES RNA have proven surprisingly difficult, probably as a result of the high Kd observed in our functional assay and the potential for nonspecific binding to the charged phosphate backbone. Such a high Kd suggests that the putative mechanism of inhibition has a low thermodynamic requirement. This is consistent with inhibition of initiation (6) and is not consistent with simple competitive inhibition. The ribosomal subunits are known to bind to a structural element in the HCV IRES (domain IIIe, Fig. 1), and, once bound, they do not scan (7). The ribosomal complex thus formed will have rigorous spatial requirements with respect to the start codon. Efficient initiation, for example, requires that the authentic start codon is located within seven nucleotides of its wild-type position (37). Minimal vitamin B12 stabilization of an RNA structure that prevents the start codon from engaging with the ribosomal complex is consistent with the observations reported here.
Our retrospective study in human HCV carriers demonstrated a biological relationship between the total serum cobalamin concentration and HCV viral load. In the plot in Fig. 7, viral load was lower in HCV carriers who exhibited cobalamin concentrations near the lower end of the normal human serum concentration range. From these data we theorize that HCV may have evolved to use the high concentration of cobalamins in hepatocytes to strike a balance between translation and transcription that results in optimal levels of virus replication. In cobalamin deficiency, HCV may be unable to replicate efficiently because the replicase cannot compete effectively with translating ribosomal complexes for the same RNA template. At high cobalamin concentrations, reduced viral translation may result in concentrations of viral proteins that are inadequate for replication and packaging. Although the mechanism is as yet unclear, these observations suggest a relationship between the pathogenesis of HCV and the total amount of cobalamin in the infected tissue.
Do cobalamins play a natural role in the regulation of the HCV replication cycle? We have shown that vitamin B12 can specifically inhibit HCV IRES-dependent translation in vitro, albeit at unusually high concentrations. We have also demonstrated an in vivo relationship between total serum cobalamin and HCV replication in human carriers. But are these observations related? While it is reasonable to suggest that a hepatotropic virus may evolve to use materials in which the liver is rich for viral regulation, clearly, an acceptable cell culture system is required to adequately answer that question. There is no reliable cell culture system or small animal model available for the study of HCV replication. Subgenomic constructs that incorporate the HCV IRES and reporter genes are used to study HCV IRES-dependent translation in cell culture. We transfected BT7-H cells with the plasmids described in Fig. 2, and the HCV IRES efficiency was monitored as a function of vitamin B12 concentration in the medium (data not shown). We observed an inconsistent 0- to 5-fold reduction in HCV IRES-dependent CAT expression in the presence of vitamin B12 in the medium. Intracellular cobalamin concentrations, however, are difficult to control in cell culture. In normal concentrations, cobalamins cannot gain entry to the cell without the cobalamin transport protein, transcobalamin II (TCII) (38). Indeed, one reason for the addition of serum to cell culture media is to provide the required TCII for cobalamin transport into the cell (39). As added complications, the cobalamin–TCII complex is required for healthy cell proliferation (40), and internalization of this complex depends on the cellular expression of the TCII receptor (38). A cobalamin-deficient cell culture system was only recently reported (41).
Once elucidated, the mechanism of inhibition of translation by vitamin B12 may be useful in engineering antiviral agents that show increased potency while retaining the high level of specificity for HCV.
Acknowledgments
We thank Dr. R. Rijnbrand for his generous gift of the bicistronic plasmids. We are grateful to Roche Molecular Systems for the kind gift of the HCV monitor kits. This research was supported by the National Health and Medical Research Council of Australia. This is publication 130 from the Sir Albert Sakzewski Virus Research Centre. The Hepatitis Research Unit within the Sir Albert Sakzewski Virus Research Centre is a member of the Australian Centre for Viral Hepatitis Inc.
Abbreviations
- HCV
hepatitis C virus
- UTR
untranslated region
- IRES
internal ribosome entry site
- eIF
eukaryotic initiation factor
- CAT
chloramphenicol acetyltransferase
- CSFV
classical swine fever virus
- EMCV
encephalomyocarditis virus
References
- 1.Cuthbert J A. Clin Microbiol Rev. 1994;7:505–532. doi: 10.1128/cmr.7.4.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choo Q L, Kuo G, Weiner A J, Overby L R, Bradley D W, Houghton M. Science. 1989;244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 3.Wang S, Guo L, Allen E, Miller W A. RNA. 1999;5:728–738. doi: 10.1017/s1355838299981979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barton D J, Morasco B J, Flanegan J B. J Virol. 1999;73:10104–10112. doi: 10.1128/jvi.73.12.10104-10112.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gamarnik A V, Andino R. Genes Dev. 1998;12:2293–2304. doi: 10.1101/gad.12.15.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Honda M, Brown E A, Lemon S M. RNA. 1996;2:955–968. [PMC free article] [PubMed] [Google Scholar]
- 7.Rijnbrand R C, Lemon S M. Curr Top Microbiol Immunol. 2000;242:85–116. doi: 10.1007/978-3-642-59605-6_5. [DOI] [PubMed] [Google Scholar]
- 8.Zhao W D, Wimmer E, Lahser F C. J Virol. 1999;73:1546–1554. doi: 10.1128/jvi.73.2.1546-1554.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hellen C U, Pestova T V. J Viral Hepat. 1999;6:79–87. doi: 10.1046/j.1365-2893.1999.00150.x. [DOI] [PubMed] [Google Scholar]
- 10.Reynolds J E, Kaminski A, Carroll A R, Clarke B E, Rowlands D J, Jackson R J. RNA. 1996;2:867–878. [PMC free article] [PubMed] [Google Scholar]
- 11.Pestova T V, Shatsky I N, Fletcher S P, Jackson R J, Hellen C U. Genes Dev. 1998;12:67–83. doi: 10.1101/gad.12.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kleijn M, Vrins C L, Voorma H O, Thomas A A. Virology. 1996;217:486–494. doi: 10.1006/viro.1996.0143. [DOI] [PubMed] [Google Scholar]
- 13.Wang C, Le S Y, Ali N, Siddiqui A. RNA. 1995;1:526–537. [PMC free article] [PubMed] [Google Scholar]
- 14.Spedding G, Draper D E. Proc Natl Acad Sci USA. 1993;90:4399–4403. doi: 10.1073/pnas.90.10.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shimoike T, Tani H, Aizaki H, Suzuki T, Matsuura Y, Miyamura T. In: Sixth International Symposium on Hepatitis C and Related Viruses: Molecular Virology and Pathogenesis. Liang T J, Hoofnagle J H, editors. Bethesda, MD: National Institutes of Health; 1999. p. 146. [Google Scholar]
- 16.Shimoike T, Mimori S, Tani H, Matsuura Y, Miyamura T. J Virol. 1999;73:9718–9725. doi: 10.1128/jvi.73.12.9718-9725.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hwang L H, Hsieh C L, Chung Y L, Chen D S. In: Sixth International Symposium on Hepatitis C and Related Viruses: Molecular Virology and Pathogenesis. Liang T J, Hoofnagle J H, editors. Bethesda, MD: National Institutes of Health; 1999. p. 139. [Google Scholar]
- 18.Reynolds J E, Kaminski A, Kettinen H J, Grace K, Clarke B E, Carroll A R, Rowlands D J, Jackson R J. EMBO J. 1995;14:6010–6020. doi: 10.1002/j.1460-2075.1995.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hwang L H, Hsieh C L, Yen A, Chung Y L, Chen D S. Biochem Biophys Res Commun. 1998;252:455–460. doi: 10.1006/bbrc.1998.9673. [DOI] [PubMed] [Google Scholar]
- 20.Draper D E. Annu Rev Biochem. 1995;64:593–620. doi: 10.1146/annurev.bi.64.070195.003113. [DOI] [PubMed] [Google Scholar]
- 21.Wallis M G, Streicher B, Wank H, von Ahsen U, Clodi E, Wallace S T, Famulok M, Schroeder R. Chem Biol. 1997;4:357–366. doi: 10.1016/s1074-5521(97)90126-5. [DOI] [PubMed] [Google Scholar]
- 22.Lorsch J R, Szostak J W. Biochemistry. 1994;33:973–982. doi: 10.1021/bi00170a016. [DOI] [PubMed] [Google Scholar]
- 23.Sussman D, Nix J C, Wilson C. Nat Struct Biol. 2000;7:53–57. doi: 10.1038/71253. [DOI] [PubMed] [Google Scholar]
- 24.Sussman D, Greensides D, Reilly K, Wilson C. Acta Crystallogr D Biol Crystallogr. 1999;55:326–328. doi: 10.1107/S0907444998004922. [DOI] [PubMed] [Google Scholar]
- 25.Beck W S. In: B12. Dolphin D, editor. Vol. 2. New York: Wiley; 1982. pp. 1–30. [Google Scholar]
- 26.Takyar S T, Li D, Wang Y, Trowbridge R, Gowans E J. Hepatology. 2000;32:382–387. doi: 10.1053/jhep.2000.9094. [DOI] [PubMed] [Google Scholar]
- 27.Winzor D J, Sawyer W H. Quantitative Characterization of Ligand Binding. New York: Wiley–Liss; 1995. pp. 128–146. [Google Scholar]
- 28.Buratti E, Gerotto M, Pontisso P, Alberti A, Tisminetzky S G, Baralle F E. FEBS Lett. 1997;411:275–280. doi: 10.1016/s0014-5793(97)00715-1. [DOI] [PubMed] [Google Scholar]
- 29.Sizova D V, Kolupaeva V G, Pestova T V, Shatsky I N, Hellen C U. J Virol. 1998;72:4775–4782. doi: 10.1128/jvi.72.6.4775-4782.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hellen C U, Wimmer E. Curr Top Microbiol Immunol. 1995;203:31–63. doi: 10.1007/978-3-642-79663-0_2. [DOI] [PubMed] [Google Scholar]
- 31.Fire A. Trends Genet. 1999;15:358–363. doi: 10.1016/s0168-9525(99)01818-1. [DOI] [PubMed] [Google Scholar]
- 32.Williams B R, Gilbert C S, Kerr I M. Nucleic Acids Res. 1979;6:1335–1350. doi: 10.1093/nar/6.4.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neumann A U, Lam N P, Dahari H, Gretch D R, Wiley T E, Layden T J, Perelson A S. Science. 1998;282:103–107. doi: 10.1126/science.282.5386.103. [DOI] [PubMed] [Google Scholar]
- 34.Puoti C, Stati T, Magrini A. Liver. 1999;19:104–109. doi: 10.1111/j.1478-3231.1999.tb00018.x. [DOI] [PubMed] [Google Scholar]
- 35.Fanning L, Kenny E, Sheehan M, Cannon B, Whelton M, O'Connell J, Collins J K, Shanahan F. Hepatology. 1999;29:904–907. doi: 10.1002/hep.510290310. [DOI] [PubMed] [Google Scholar]
- 36.Kruger M, Beger C, Li Q X, Welch P J, Tritz R, Leavitt M, Barber J R, Wong-Staal F. Proc Natl Acad Sci USA. 2000;97:8566–8571. doi: 10.1073/pnas.97.15.8566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rijnbrand R C, Abbink T E, Haasnoot P C, Spaan W J, Bredenbeek P J. Virology. 1996;226:47–56. doi: 10.1006/viro.1996.0626. [DOI] [PubMed] [Google Scholar]
- 38.Seetharam B. Annu Rev Nutr. 1999;19:173–195. doi: 10.1146/annurev.nutr.19.1.173. [DOI] [PubMed] [Google Scholar]
- 39.Baker H, DeAngelis B, Frank O. Experientia. 1988;44:1007–1010. doi: 10.1007/BF01939904. [DOI] [PubMed] [Google Scholar]
- 40.McLean G R, Pathare P M, Wilbur D S, Morgan A C, Woodhouse C S, Schrader J W, Ziltener H J. Cancer Res. 1997;57:4015–4022. [PubMed] [Google Scholar]
- 41.McLean G R, Williams M J, Woodhouse C S, Ziltener H J. Leuk Lymphoma. 1998;30:101–109. doi: 10.3109/10428199809050933. [DOI] [PubMed] [Google Scholar]