

Abstract
Background
Polymorphic tandem repeat typing is a new generic technology which has been proved to be very efficient for bacterial pathogens such as B. anthracis, M. tuberculosis, P. aeruginosa, L. pneumophila, Y. pestis. The previously developed tandem repeats database takes advantage of the release of genome sequence data for a growing number of bacteria to facilitate the identification of tandem repeats. The development of an assay then requires the evaluation of tandem repeat polymorphism on well-selected sets of isolates. In the case of major human pathogens, such as S. aureus, more than one strain is being sequenced, so that tandem repeats most likely to be polymorphic can now be selected in silico based on genome sequence comparison.
Results
In addition to the previously described general Tandem Repeats Database, we have developed a tool to automatically identify tandem repeats of a different length in the genome sequence of two (or more) closely related bacterial strains. Genome comparisons are pre-computed. The results of the comparisons are parsed in a database, which can be conveniently queried over the internet according to criteria of practical value, including repeat unit length, predicted size difference, etc. Comparisons are available for 16 bacterial species, and the orthopox viruses, including the variola virus and three of its close neighbors.
Conclusions
We are presenting an internet-based resource to help develop and perform tandem repeats based bacterial strain typing. The tools accessible at http://minisatellites.u-psud.fr now comprise four parts. The Tandem Repeats Database enables the identification of tandem repeats across entire genomes. The Strain Comparison Page identifies tandem repeats differing between different genome sequences from the same species. The "Blast in the Tandem Repeats Database" facilitates the search for a known tandem repeat and the prediction of amplification product sizes. The "Bacterial Genotyping Page" is a service for strain identification at the subspecies level.
Background
Molecular epidemiology, the integration of molecular typing and conventional epidemiological studies, is likely to add significant value to analyses of infections caused by pathogenic bacteria (see [1] for review). Multilocus Sequence Typing (MLST) for instance is now a major reference method for the molecular epidemiology of Neisseria meningitidis and other human pathogens [2]. In this kind of assay, a set of typically 7 genes is partially sequenced, and the resulting data is converted into sequence types, which can be easily stored in databases, and compared to others. However a number of significant pathogens, including M. tuberculosis [3], B. anthracis and Y. pestis [4] are not amenable to this approach, because of the recent emergence of these pathogens and the resulting rarity of sequence variations. In these pathogens, tandem repeats (TRs) are a source of very informative markers for strain genotyping [5-10]. Tandem repeats in pathogenic bacteria were initially identified within genes associated with bacterial virulence [11,12]. In other instances, the contribution of tandem repeats to genome polymorphism was established after extensive searches based for instance on AFLP (amplified fragment length polymorphism) profiling. This is well illustrated by B. anthracis, in which polymorphic bands in AFLP patterns [13] were subsequently demonstrated by sequencing to be due to tandem repeat variations [14]. Eventually, some of these tandem repeats have been shown to directly contribute to phenotypic variations of the B. anthracis exosporium which makes the outer layer of the spores [15]. The frequent observation that tandem repeat-containing genes are often associated with outer membrane proteins suggests that such genes help bacteria adapt to their environment, and may be to some extent mutation hotspots as a result of positive selection.
Polymorphic tandem repeats (VNTRs, for Variable Number of Tandem Repeats), once identified, provide convenient tools requiring ordinary molecular biology equipment and the data can be easily exchanged and compared. The resulting assay, called MLVA (for multiple locus VNTR analysis) can even be automated [16]. We have developed tools which facilitate the bioinformatics step of genome analysis required to start a project. A previously described Tandem Repeats Database enables the identification of tandem repeats across entire genomes [9,10,17-19]. It has been constantly updated, with now more than a hundred bacterial genomes available, compared to 35 at the onset of the database. We present here a new and major development of this resource which takes advantage of the fact that more than two different strains from the same species have now been sequenced at least for a number of major human pathogens. As a result, the tools accessible over the Internet at http://minisatellites.u-psud.fr now comprise four complementary parts. The newly added resource, the Strain Comparison Page, takes advantage of the availability of genome sequences from more than one strain from a growing number of species to directly identify tandem repeats differing between the sequenced strains. This is of interest because the vast majority of tandem repeats is often not polymorphic [19]. The "Blast in the Tandem Repeats Database" page facilitates the search for a known tandem repeat, the prediction of PCR amplification products size, and the verification of primer specificity. Once an MLVA assay has been set up, and carefully validated by typing collections of isolates, it is relatively easy to construct databases of genotypes to be used locally or which can be queried across the Internet. The "Bacterial Genotyping Page" illustrates a freely accessible, fast and easy to use internet-based service for strain comparisons, in which a user can compare a genotype produced for one of his isolates to the existing data.
Construction and content
The Tandem Repeats Database main page
Tandem repeats were identified from finished microbial genome sequences (as listed by the Genome OnLine Database [20]) using the tandem repeats finder (TRF) software [21,22] with the following options: alignment parameters, "2,3,5" (these parameters are the less stringent ones), minimum alignment score to report repeat, 50 (this score allows to detect short structures), maximum period size, 500 base-pairs. When the program reported redundant (overlapping) repeats, the redundancy was eliminated as described in [23], before import in the database. The database uses Microsoft Access 2000 and the querying process uses Active Server Pages (ASP, Microsoft) with Perlscripts or VBscripts. Perl was obtained from the ActiveState Programmer Network [24]. The database is hosted on a server running under Windows 2000 server (Microsoft). The tandem repeats database main page is described in more detail in [9].
The Strain Comparison page
Sequence comparisons used BLAST [25]. The BLAST software was obtained from the NCBI FTP site [26]. The flanking sequences of TRs from one strain were compared to the whole sequence of the other strain (and reciprocally, to avoid missing some tandem repeats that would not appear in the tandem repeats database for one strain because they were not detected by the Tandem Repeats Finder [21] -for instance because there is only one copy of the repeated unit in the considered strain). The resulting list of matching tandem repeats was then imported in the database, where it can be queried. The comparison of more than two strains was made possible through a supplemental step before import in the database: the synthesis of several 2-strains comparisons, of the same "reference" strain against each of the others (matching between TRs of the different strains was deduced from the positions on the reference strain).
The Blast page
The Blast Page allows users to run BLAST [25] in the tandem repeats and flanking sequences from the database via Perlscripts. The Blast outputs are linked to the database, in order to easily obtain the description of identified tandem repeats.
The Bacterial Genotyping page
The web-page site performing identifications was developed using the BNserver application (version 3.0, Applied-Maths, Belgium) and ASP (Microsoft) using Perlscript. The typing results (gel images and resulting data) were managed using the Bionumerics software package as described in [10]. The output of a query is a list of strains and genotypes from the database together with similarity scores.
Utility
The procedure to find polymorphic tandem repeats (TRs) for use in strain typing
Figure 1 shows the steps leading from a genome sequence to the exploitation of polymorphic tandem repeats for bacterial strain genotyping. Although Tandem Repeats are easily identified using the Tandem Repeats Database, TR polymorphism must be evaluated by typing across a set of relevant strains. If the sequences of several strains of the species of interest are available, the Strain Comparison Page can be used to directly identify tandem repeats predicted to be polymorphic in size between the two (or more) sequenced strains. However, it is important to keep in mind that the tandem repeats predicted as being polymorphic will depend on the sequenced strains and well-planned surveys of isolates will still be necessary. The available tools do not replace this validation step, as the value of each marker must be carefully established on an appropriate set of isolates. The definition of an appropriate set of isolates depends upon the question which is being addressed, i.e. large scale or local epidemiology. The Blast Page has been implemented in the tandem repeats database in order to easily determine the size of the expected PCR amplification products. The database is also manually updated to contain PCR conditions as well as polymorphism index, and links to the original reports [27] (input from users is welcome). Eventually, when an MLVA assay has been fully developed and validated, typing data can be made accessible so that individual queries can be run. The Bacterial Genotyping Page illustrates how this could work. The genotyping data for a strain can be entered and submitted via this page. The output is the description of the closest strains. The data which has been submitted is not incorporated in the database itself, since this would require stringent data validation steps. In the following sections, we are presenting the web-based resources associated with this procedure.
Figure 1.
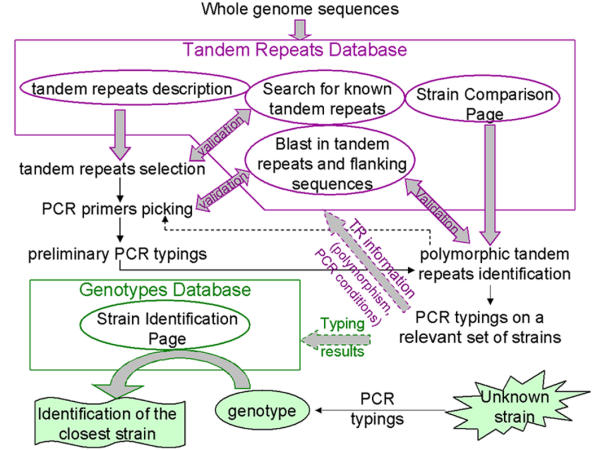
The procedure to find polymorphic tandem repeats for use in strain typing. The steps leading from the release of a complete (or incomplete) genome sequence to the validation of new polymorphic markers are described. The purpose of the web-based tools developed is to facilitate the bioinformatics data-management steps.
The "Strain Comparison" pages
The strain comparison pages are available via [28]. The comparison of two strains is based on a pre-computed BLAST [25] analysis of the flanking sequences of tandem repeats from one strain against the other, and vice-versa. Figure 2 summarizes the results of this first step for 23 comparisons. Three indexes are scored (see figure legend): (1) the "mean %identity" between the flanking sequences is a measure of single nucleotide polymorphism (SNPs) frequency (not insertions-deletions), (2) the proportion (%) of flanking sequences that matched the flanking sequence of its homologue in the other strain on more than half of the 500 bp assayed here – i.e. that were not rearranged, by insertion of mobile elements for instance -, (3) the proportion (%) of tandem repeats that were found to be of a different length between the two strains being compared. In addition, the positions of matching tandem repeats in the two genomes is plotted to reveal large-scale genome rearrangements. A number of situations are observed: for instance Yersinia pestis orientalis strain CO-92 [29], and medievalis strain KIM5 P12 [30] show a very high "mean %identity" (99.96 %), in agreement with the recent emergence of Yersinia pestis [4]. In spite of this, the two strains differ by a high number of large rearrangements (as seen on the plot), which reflects the high genome plasticity observed in this species [31], together with a relatively high rate of polymorphic tandem repeats (8.47%). In contrast, Listeria monocytogenes strain EGD-e and Listeria innocua strain Clip 11262 have a lower homology (90.19%) and only 3.99% of polymorphic tandem repeats in spite of the evolutionary distance (see Figure 2).
Figure 2.
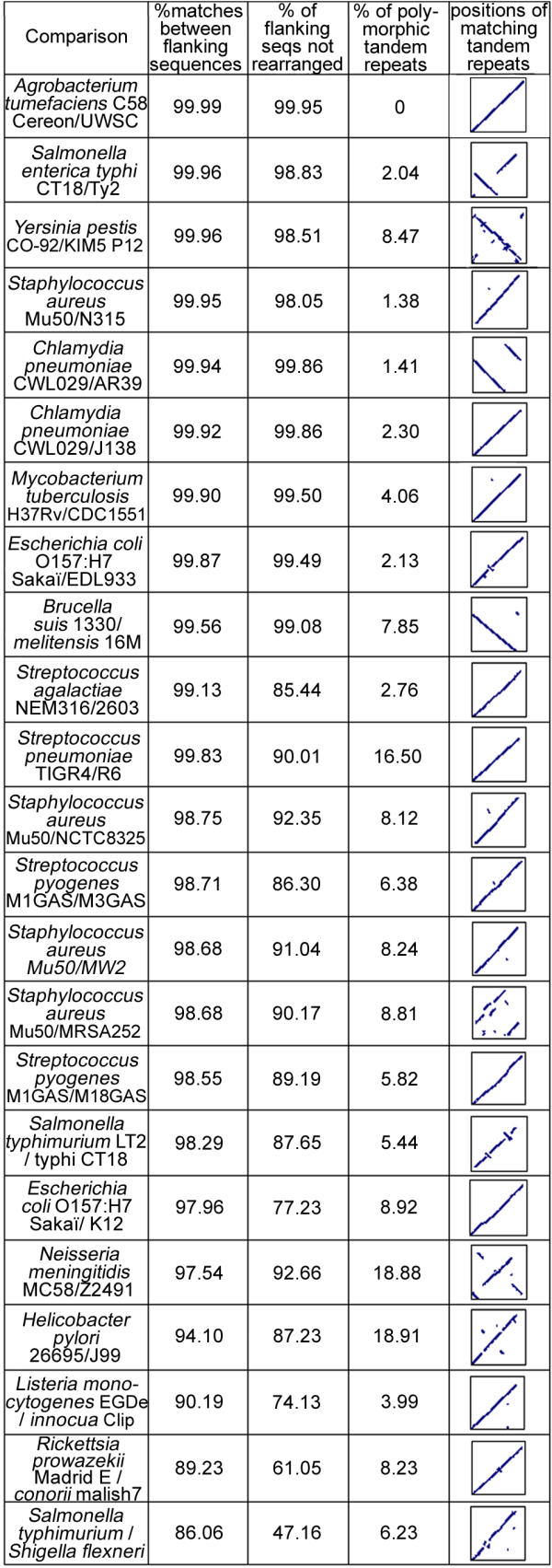
Comparison of strains using different indexes. The four columns correspond to (from left to right): (1) mean %identity provided by BLAST when the match occurred on more than half the length of the 500 bp of submitted flanking sequence ; (2) proportion (%) of flanking sequences that matched on more than half their length between the two strains ; (3) proportion (%) of tandem repeats of a different size in the two strains ; and (4) plot of the positions of homologous tandem repeat loci in the two genomes which indirectly reflects large scale genome rearrangements. Species are listed according to the first index (mean %identity)
The strain comparison page allows queries in the tandem repeats database according to the tandem repeat length difference between the two strains compared, and also to other tandem repeats characteristics (unit length, copy number, etc...). Figure 3 illustrates a query done for Mycobacterium tuberculosis strains H37Rv and CDC1551 [32]: the query "length difference ≥ 5 bp" identifies 58 tandem repeats (8 are shown on Figure 3). This prediction has been tested for the 30 loci amenable to PCR analysis and polymorphism has been confirmed in all cases [10].
Figure 3.
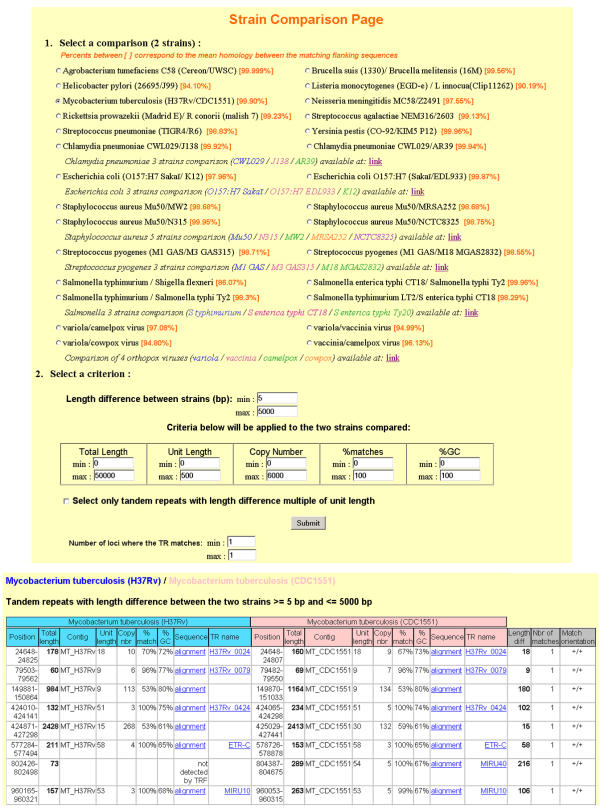
Example of a query in the Strain Comparison Page. On the top, the query page shows the 28 comparisons currently available (others will be added as new genome sequences are finished and released). Bottom, the result of a query performed for Mycobacterium tuberculosis strains H37Rv and CDC1551 is summarized.
When more than two strains have been sequenced, a synthesis of the results of several 2-strains comparisons is also available. Figure 4 illustrates a query made for Escherichia coli strains O157:H7 Sakaï, O157:H7 EDL933, K12, and UPEC-CFT073 [33-35]: 87 tandem repeats were found with 2 to 4 alleles among the 4 strains (18 of which are listed in Figure 4).
Figure 4.

Example of a query in the Strain Comparison Page for more than two strains. Top, the query page shows the 6 comparisons currently available (others will be added as new genome sequences are finished and released). Bottom, the result of a query performed for Escherichia coli strains O157:H7 Sakaï, O157:H7 EDL933, K12 and UPEC-CFT073 is summarized. In several loci, the size of the repeat is listed differently for the different strains, which is due to different detections by the Tandem Repeats Finder, usually as a result of internal variations within the tandem array. Total length is calculated from positions of matching flanking sequences in the different strains, and does not necessarily correspond to the length of the tandem repeat detected by TRF in the locus. "Number of alleles" refers to the number of predicted sizes differing by at least 5 bp among the strains compared.
The "Blast in the Tandem Repeats Database" page
To facilitate the identification of already studied tandem repeats, we implemented BLAST [25] against the tandem repeats from the database, i.e. the tandem repeats themselves and their flanking sequences. The Blast page is available at [36]. All bacteria can be queried at once, which allows the identification of tandem repeats families, conserved in several bacterial species. Another page is dedicated to the Blast of PCR primers and provides the size of the PCR products in all the species/strains where the primers match. Figure 5 shows the results of searching the PCR primer pair from tandem repeat H37Rv_0024_18 bp [10] in all bacteria: as expected, the PCR primer pair matches Mycobacterium tuberculosis strains H37Rv and CDC1551, providing different PCR product lengths.
Figure 5.

Example of a query in the "Blast of PCR primers" page, providing the length of the PCR products in the strains/species where the primer pair matches, and links to the corresponding tandem repeats descriptions.
The Bacterial Genotyping page
The Bacterial Genotyping page [37] provides one illustration on how tandem repeat typing data can be made available via internet to allow external users to query genotyping data (Bacillus anthracis, Yersinia pestis, Mycobacterium tuberculosis, Pseudomonas aeruginosa for the moment) and compare a new strain to existing data as previously described in [10]. For each locus, allele sizes can be selected among a list of possibilities (observed sizes). The results of the query indicate a similarity score and include links to the complete data recorded for each strain listed. This page is just meant as an illustration and prototype. MLVA reference data could also be made available for downloading as tabular data files, or can be copied from published datasets, which can then be complemented by in-house data, and analyzed by the appropriate clustering software.
Discussion
Bacterial genomes evolution
As shown by the indexes from Figure 2, there are different ways to represent the divergence/similarity between two strains. They are not correlated, suggesting independent evolution processes. First, the "mean %identity" between two genomes reflects point mutations, and is an indicator of the time passed since the two strains diverged. For instance, Yersinia pestis is known to be of recent emergence [4] and shows a high "mean %identity" between strains CO-92 (orientalis) and KIM5 P12 (medievalis). In contrast, and as shown by the dot plot, large genome rearrangements occurred in this genome, which is representative of a high genome plasticity [31]. The index "% of flanking sequences not rearranged" is an indicator of small-scale genome rearrangements, such as the insertions of mobile elements. This index is low for genomes rich in mobile elements, like Streptococcus agalactiae, in which such elements significantly contribute to strain diversity [38]. Finally, the index "% of polymorphic tandem repeats" between two strains represents the tandem repeats evolution rate. For the moment, the mechanisms of bacterial VNTRs mutations have not been precisely investigated, but it seems likely to be independent of the other processes mentioned, as there are no correlations between the indexes. Figure 2 provides clues to assess which typing method(s) will be efficient in the different species. For instance, the two bacterial species Salmonella typhimurium strain LT2 [39] and Shigella flexneri strain 2a301 [40] share only 86.06% of sequence identity, clearly making the identification of matching tandem repeats between the two species difficult and of low significance. MLVA analysis appears to be of highest interest for the subspecies typing of highly monomorphic species including Yersinia pestis, Bacillus anthracis, Mycobacterium tuberculosis and Brucella [9,10,41].
Strain comparison efficiency
The sequencing of more than one strain for some bacterial species allows direct identification of polymorphic tandem repeats, assuming that no sequencing errors occurred. Earlier investigations provide good reasons to believe that tandem repeats in the size range considered here (a few hundred base-pairs) are correctly sequenced, and consequently, that the strain comparison data is reliable. As a negative control, the comparison of two independent sequences from the same strain of Agrobacterium tumefaciens strain (C58), one from Cereon genomics [42] and the other from Washington University [43], shows that no length polymorphism is detected among tandem repeats (Figure 2) between the two independent sequences. As a positive control, the tandem repeats predicted to be polymorphic by genome sequence comparison between the two strains of M. tuberculosis have indeed been proved polymorphic by PCR typing of isolates [10].
Selection based on comparison of sequence data from two strains will miss some polymorphic loci. Indeed, the results provided by the approach rely upon the phylogenetic distance between the two strains being compared. If the strains are very closely related, only a few TRs will be found different between them, but these tandem repeats will probably be the most polymorphic ones. Conversely, if the strains are distant in the phylogenetic tree, a larger number of polymorphic TRs will be found, some of them will be only moderately polymorphic. Obviously, when a few well-selected strains have been sequenced, it is likely that very few polymorphic tandem repeats are undetected in the Strain Comparison pages.
It is of course still going to be very important to determine the TR allele frequency for isolates carefully selected to be representative of the global diversity of a given pathogen before suggesting the configuration of an MLVA assay to use in subsequent studies. In addition, those TR markers that are highly polymorphic in diverse test panels of isolates may be monomorphic when applied to isolates responsible for local outbreaks. The configuration of TR markers used to make up an assay needs to be determined empirically with representative local isolates and tailored to the study population and study questions.
Polymorphic tandem repeats selection for species with only one sequenced strain
The identification of simple criteria able to predict tandem repeat polymorphism when genome sequence data is available for only one strain would indeed greatly facilitate the development of MLVA assays. It would seem reasonable for instance to expect that the number of copies and the internal homogeneity of tandem arrays are strong predictors [23]. We take advantage here of the many strain comparisons which are made available via the strain comparison pages to evaluate such criteria.
We have analyzed bacteria with at least three sequenced genomes (Staphylococcus aureus: 6 strains, Escherichia coli: 4 strains, Streptococcus pyogenes: 4 strains and Salmonella typhi and typhimurium: 3 strains). We assume that in such cases, only a few polymorphic tandem repeats are missed in the comparisons. We compared the distribution of tandem repeats sequence characteristics among the group of "polymorphic" loci (differing in at least two of the strains compared, excluding length differences between strains that resulted from microdeletions in the flanking sequences) and the others. Comparisons were performed for the following sequence characteristics: unit length, copy number, total length, %GC, GC bias (=|%G-%C|/(%G+%C)), %matches, and HistoryR (a score derived from tandem repeat history reconstruction algorithm [44] as described in [23]). None of the variables were normally distributed, as tested with Kolmogorov-Smirnov test, so a non-parametric Wilcoxon test was used to compare the distributions, which were judged significantly different at the .05 level of the statistic (2 tailed). Distributions were significantly different for all 4 species studied for %matches, total length and copy number. As shown on Figure 6, polymorphic TRs have a higher internal conservation and total length than monomorphic ones. Copy number, which is correlated with total length, is also higher among polymorphic TRs.
Figure 6.
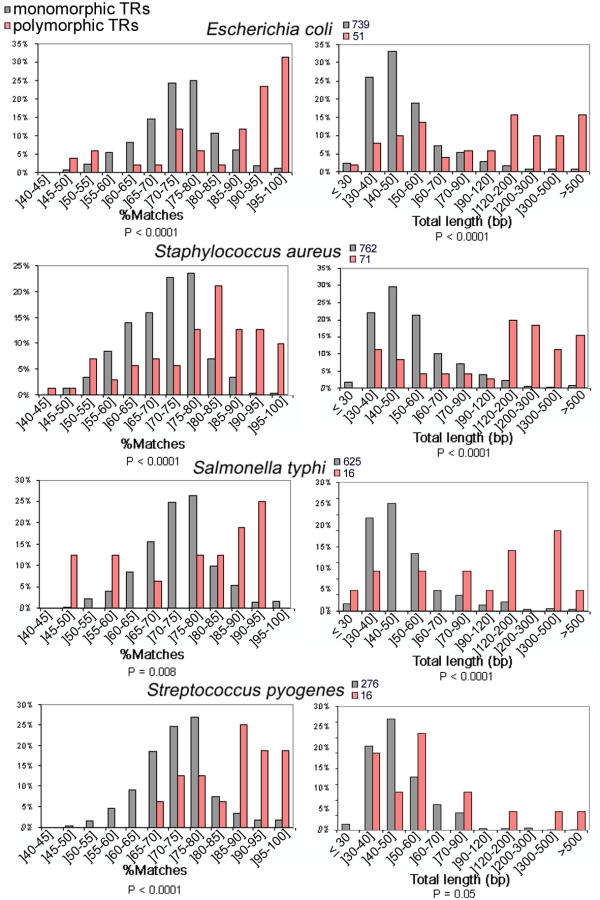
Proportion of predicted polymorphic (pink) and monomorphic (grey) tandem repeats according to different parameters (internal homogeneity of the repeat array (%matches) or total length). P-values obtained for the non-parametric Wilcoxon tests appear below each histogram.
Selecting the longest and most conserved tandem repeats should thus improve polymorphic TRs identification. Table 1 illustrates the query "total length ≥ 80 bp and %matches ≥ 80%" applied to the four species used to find predictive criteria. For all four species, the group fulfilling the criterion is, as expected, enriched in polymorphic (at least two alleles) tandem repeats: in Staphyloccocus aureus, polymorphic tandem repeats represent only 8.5% of the whole population of tandem repeat loci but are predominant (87%) in the criterion positive group. The enrichment is even greater for highly polymorphic TRs, i.e. with 3 alleles or more: for example from 4.5% in the whole set to 66% in the positive group for Staphylococcus aureus. However this simple criterion misses more than half of the polymorphic loci. In addition, the efficiency of the criterion is highly variable in the different species: it is relatively satisfying in Staphylococcus aureus (54% of polymorphic tandem repeats would be missed) but very inefficient in Streptococcus pyogenes (almost 90% are missed). The results for highly polymorphic loci (3 alleles or more) are more consistent (the proportion of TRs with 3 alleles or more detected by the criterion ranges from 58% for Escherichia coli to 100% for Salmonella).
Table 1.
Use of the criterion "total length ≥ 80 bp and %matches ≥ 80%" on 4 species for which 3 strains or more were compared. The number of monomorphic, polymorphic (2 alleles or more) and highly polymorphic (3 alleles or more) TRs in whole set, and positive and negative groups are listed. (a) "criterion" refers to the selection of TRs with L ≥ 80 bp and %M ≥ 80%
| Comparison (total number of TRs) | Whole set (proportion of total number) | Tandem repeats with L≥80 bp AND %M≥80% (proportion among the set) | Tandem repeats with L<80 bp OR %M<80% (proportion among the set) | % of the polymorphic TRs (2 alleles or more) that were detected by criteriona | % of the TRs with 3 alleles or more that were detected by criteriona | % of all TRs that fulfil the criteriona | ||||||
| 1 allele | 2 alleles or more | 3 alleles or more | 1 allele | 2 alleles | 3 alleles or more | 1 allele | 2 alleles or more | 3 alleles or more | ||||
| S aureus (833 TRs) | 762 (91.5%) | 71 (8.5%) | 38 (4.5%) | 5 (13%) | 8 (20%) | 25 (66%) | 757 (95%) | 25 (3.5%) | 13 (1.5%) | 46% | 66% | 7.23% |
| E coli (790 TRs) | 739 (93.5%) | 51 (6.5%) | 12 (1.5 %) | 12 (38%) | 13 (40%) | 7 (22%) | 727 (96%) | 26 (3.5%) | 5 (0.5%) | 39% | 58% | 4.86% |
| S typhi / typhimurium (641 TRs) | 625 (97.5%) | 16 (2.5%) | 2 (0.3%) | 13 (68%) | 4 (22%) | 2 (10%) | 612 (98%) | 10 (2%) | 0 (0%) | 37.5% | 100% | 3.27% |
| S pyogenes (292 TRs) | 276 (94.5%) | 16 (5.5%) | 3 (1%) | 4 (67%) | 0 (0%) | 2 (33%) | 272 (95%) | 14 (4.7%) | 1 (0.3%) | 12.5% | 67% | 2.71% |
It is tempting to speculate that these observations are applicable to other species. Subsequently, we applied the criterion to ten of the 2-strains comparisons available on the Strain Comparison Page (Table 2). In all ten instances, the criterion positive group is enriched in TRs with different lengths between the two strains, compared to the whole set. This proportion varies from less than 3% in Streptococcus agalactiae to more than 20% in Xylella fastidiosa in the whole set. It is increased to 33% and 93% respectively among the set of loci which satisfy the criterion (these percentages correspond to the predictor's specificity), but the vast majority of polymorphic loci will be missed (90% and 80% respectively). Sensitivity, that is % of the TRs with different lengths that were detected by criterion varies from 6.90% for Brucella to 44.26% for Mycobacterium tuberculosis.
Table 2.
Use of the criterion "total length ≥ 80 bp and %matches ≥ 80%" on 10 species for which 2 strains were compared. The numbers of tandem repeats with equal lengths and different lengths between the two strains in the whole set, and positive and negative groups are listed.
| Comparison (total number of TRs loci) | Whole set (proportion) | Criterion + (L≥80 bp, %M≥80%) | Criterion - | Sensitivity (% of the TRs with different lengths that were detected by criterion) | Specificity (% of the TRs predicted by the criterion that have different length) | % of all TRs that fulfil the criterion | |||
| equal length | different length | equal length | different length | equal length | different length | ||||
| H pylori 26695/J99 (624 TRs) | 506 (81%) | 118 (19%) | 0 | 11 | 506 | 107 | 9% | 100% | 2% |
| N meningitidis MC58/Z2491 (642 TRs) | 528 (82%) | 114 (18%) | 10 | 23 | 518 | 91 | 20% | 70% | 5% |
| M tuberculosis H37Rv/CDC1551(1502 TRs) | 1441 (96%) | 61 (4%) | 35 | 27 | 1406 | 34 | 44% | 44% | 4% |
| L monocytogenes EGD-e/L innocua Clip11262 (576 TRs) | 553 (96%) | 23 (4%) | 2 | 3 | 551 | 20 | 13% | 60% | 1% |
| S agalactiae NEM316/2603 (398 TRs) | 387 (97%) | 11 (3%) | 2 | 1 | 385 | 10 | 9% | 33% | 1% |
| S pneumoniae TIGR4/R6 (406 TRs) | 339 (83%) | 67 (17%) | 14 | 29 | 325 | 38 | 43% | 67% | 10% |
| Y pestis CO-92/KIM5 P12 (1499 TRs) | 1372 (92%) | 127 (8%) | 44 | 19 | 1328 | 108 | 15% | 30% | 4% |
| R prowazekii Madrid E/R conorii malish 7 (316 TRs) | 290 (92%) | 26 (8%) | 0 | 2 | 290 | 24 | 8% | 100% | 1% |
| Brucella suis 1330/ Brucella melitensis 16 M (739 TRs) | 681 (92%) | 58 (8%) | 2 | 4 | 679 | 54 | 7% | 67% | 1% |
| X fastidiosa 9a5c/grape Temecula1 (573 TRs) | 440 (77%) | 133 (23%) | 2 | 28 | 438 | 105 | 21% | 93% | 5% |
The finding that polymorphic tandem repeats have, on average, a higher internal conservation, total length, and copy number than monomorphic ones is in agreement with previous observations that TR polymorphism is correlated with conservation in Yersinia pestis and with total length in Bacillus anthracis [9]. It is also reminiscent of the behavior of microsatellites (also called short sequence repeats: SSR, see [45] for review), which are stabilized by internal variations [46] and by reduction of the number of repeats [47]. Unfortunately, we show here that such simple prediction criteria may miss a very large proportion of polymorphic tandem repeats, and provide highly variable results in different species. This indicates that, in the absence of sequence data from two strains or more, the systematic testing of tandem repeats polymorphism across a set of relevant strains remains the most appropriate way to develop an MLVA assay. Consequently, the Strain Comparison page is of great use when two strains or more have been sequenced.
Conclusions
Bacterial strain typing at the subspecies level is essential for epidemiological issues in the context of disease control. This can be used to determine if an S. aureus or P. aeruginosa infection for instance has been acquired in an hospital environment or not. On a larger scale, it can be used to trace the emergence of new, more virulent or drug resistant M. tuberculosis strains. It is also of interest in the field of bioterrorism and bioweapons control, as was shown by the investigations following the 2001 B. anthracis attacks. Tandem repeats typing has recently emerged as one way to address this issue. Indeed, in the case of a number of highly monomorphic bacterial species, including B. anthracis and Y. pestis, tandem repeats typing is the method of choice for subspecies typing. In addition to the fact that these loci represent an important fraction of the existing polymorphism, it offers a number of practical advantages, including the ease of typing, and of data exchanges among different countries. It is hoped that the tools which are described here will help evaluate the potential of tandem repeats typing assays for a larger range of pathogens.
Availability
All the tools presented are freely available from http://minisatellites.u-psud.fr.
List of abbreviations used
ASP: active server pages
MLVA: multiple locus VNTR analysis
PCR: polymerase chain reaction
TR: tandem repeat
TRF: tandem repeats finder
Authors contributions
FD is the developer of the database and web site, and the curator of the database. GV participated in the development of the initial procedure for the tandem repeat size comparisons between two genomes. The two authors contributed equally to the writing.
Acknowledgments
Acknowledgments
This work was funded by grants from Délégation Générale de l'Armement (DGA, France) aimed at facilitating the typing of dangerous pathogens.
Contributor Information
France Denœud, Email: France.Denoeud@igmors.u-psud.fr.
Gilles Vergnaud, Email: Gilles.Vergnaud@igmors.u-psud.fr.
References
- van Belkum A. High-throughput epidemiologic typing in clinical microbiology. Clin Microbiol Infect. 2003;9:86–100. doi: 10.1046/j.1469-0691.2003.00549.x. [DOI] [PubMed] [Google Scholar]
- Enright MC, Spratt BG. Multilocus sequence typing. Trends Microbiol. 1999;7:482–7. doi: 10.1016/S0966-842X(99)01609-1. [DOI] [PubMed] [Google Scholar]
- Gutacker MM, Smoot JC, Migliaccio CA, Ricklefs SM, Hua S, Cousins DV, Graviss EA, Shashkina E, Kreiswirth BN, Musser JM. Genome-wide analysis of synonymous single nucleotide polymorphisms in Mycobacterium tuberculosis complex organisms: resolution of genetic relationships among closely related microbial strains. Genetics. 2002;162:1533–43. doi: 10.1093/genetics/162.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achtman M, Zurth K, Morelli G, Torrea G, Guiyoule A, Carniel E. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc Natl Acad Sci U S A. 1999;96:14043–8. doi: 10.1073/pnas.96.24.14043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Belkum A, Scherer S, van Leeuwen W, Willemse D, van Alphen L, Verbrugh H. Variable number of tandem repeats in clinical strains of Haemophilus influenzae. Infect Immun. 1997;65:5017–27. doi: 10.1128/iai.65.12.5017-5027.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frothingham R, Meeker-O'Connell WA. Genetic diversity in the Mycobacterium tuberculosis complex based on variable numbers of tandem DNA repeats. Microbiology. 1998;144:1189–1196. doi: 10.1099/00221287-144-5-1189. [DOI] [PubMed] [Google Scholar]
- Supply P, Mazars E, Lesjean S, Vincent V, Gicquel B, Locht C. Variable human minisatellite-like regions in the Mycobacterium tuberculosis genome. Mol Microbiol. 2000;36:762–771. doi: 10.1046/j.1365-2958.2000.01905.x. [DOI] [PubMed] [Google Scholar]
- Adair DM, Worsham PL, Hill KK, Klevytska AM, Jackson PJ, Friedlander AM, Keim P. Diversity in a variable-number tandem repeat from Yersinia pestis. J Clin Microbiol. 2000;38:1516–9. doi: 10.1128/jcm.38.4.1516-1519.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Flèche P, Hauck Y, Onteniente L, Prieur A, Denoeud F, Ramisse V, Sylvestre P, Benson G, Ramisse F, Vergnaud G. A tandem repeats database for bacterial genomes: application to the genotyping of Yersinia pestis and Bacillus anthracis. BMC Microbiol. 2001;1:2. doi: 10.1186/1471-2180-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Flèche P, Fabre M, Denoeud F, Koeck JL, Vergnaud G. High resolution, on-line identification of strains from the Mycobacterium tuberculosis complex based on tandem repeat typing. BMC Microbiol. 2002;2:37. doi: 10.1186/1471-2180-2-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanier JG, Jones SJ, Cleary P. Small DNA deletions creating avirulence in Streptococcus pyogenes. Science. 1984;225:935–8. doi: 10.1126/science.6089334. [DOI] [PubMed] [Google Scholar]
- Hollingshead SK, Fischetti VA, Scott JR. Size variation in group A streptococcal M protein is generated by homologous recombination between intragenic repeats. Mol Gen Genet. 1987;207:196–203. doi: 10.1007/BF00331578. [DOI] [PubMed] [Google Scholar]
- Keim P, Kalif A, Schupp J, Hill K, Travis SE, Richmond K, Adair DM, Hugh-Jones M, Kuske CR, Jackson P. Molecular evolution and diversity in Bacillus anthracis as detected by amplified fragment length polymorphism markers. J Bacteriol. 1997;179:818–24. doi: 10.1128/jb.179.3.818-824.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keim P, Price LB, Klevytska AM, Smith KL, Schupp JM, Okinaka R, Jackson PJ, Hugh-Jones ME. Multiple-Locus Variable-Number Tandem Repeat Analysis Reveals Genetic Relationships within Bacillus anthracis. J Bacteriol. 2000;182:2928–2936. doi: 10.1128/JB.182.10.2928-2936.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvestre P, Couture-Tosi E, Mock M. Polymorphism in the collagen-like region of the Bacillus anthracis BclA protein leads to variation in exosporium filament length. J Bacteriol. 2003;185:1555–63. doi: 10.1128/JB.185.5.1555-1563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supply P, Lesjean S, Savine E, Kremer K, van Soolingen D, Locht C. Automated high-throughput genotyping for study of global epidemiology of Mycobacterium tuberculosis based on mycobacterial interspersed repetitive units. J Clin Microbiol. 2001;39:3563–3571. doi: 10.1128/JCM.39.10.3563-3571.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergnaud G, Denoeud F. Minisatellites: Mutability and Genome Architecture. Genome Res. 2000;10:899–907. doi: 10.1101/gr.10.7.899. [DOI] [PubMed] [Google Scholar]
- Pourcel C, Vidgop Y, Ramisse F, Vergnaud G, Tram C. Characterization of a Tandem Repeat Polymorphism in Legionella pneumophila and Its Use for Genotyping. J Clin Microbiol. 2003;41:1819–1826. doi: 10.1128/JCM.41.5.1819-1826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onteniente L, Brisse S, Tassios PT, Vergnaud G. Evaluation of the polymorphisms associated with tandem repeats for Pseudomonas aeruginosa strain typing. J Clin Microbiol. 2003;41:4991–7. doi: 10.1128/JCM.41.11.4991-4997.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOLD Genomes OnLine Database http://ergo.integratedgenomics.com/GOLD/
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Tandem Repeats Finder http://tandem.bu.edu/trf/trf.html
- Denoeud F, Vergnaud G, Benson G. Predicting Human Minisatellite Polymorphism. Genome Res. 2003;13:856–867. doi: 10.1101/gr.574403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ActiveState Programmer Network (ASPN) ActivePerl download page http://www.activestate.com/products/ActivePerl
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The NCBI BLAST ftp site ftp://ftp.ncbi.nih.gov/blast/
- The tandem repeats database http://minisatellites.u-psud.fr
- The Strain Comparison Page http://minisatellites.u-psud.fr/comparison
- Parkhill J, Wren BW, Thomson NR, Titball RW, Holden MT, Prentice MB, Sebaihia M, James KD, Churcher C, Mungall KL, Baker S, Basham D, Bentley SD, Brooks K, Cerdeno-Tarraga AM, Chillingworth T, Cronin A, Davies RM, Davis P, Dougan G, Feltwell T, Hamlin N, Holroyd S, Jagels K, Karlyshev AV, Leather S, Moule S, Oyston PC, Quail M, Rutherford K, Simmonds M, Skelton J, Stevens K, Whitehead S, Barrell BG. Genome sequence of Yersinia pestis, the causative agent of plague. Nature. 2001;413:523–7. doi: 10.1038/35097083. [DOI] [PubMed] [Google Scholar]
- Deng W, Burland V, Plunkett G, 3rd, Boutin A, Mayhew GF, Liss P, Perna NT, Rose DJ, Mau B, Zhou S, Schwartz DC, Fetherston JD, Lindler LE, Brubaker RR, Plano GV, Straley SC, McDonough KA, Nilles ML, Matson JS, Blattner FR, Perry RD. Genome sequence of Yersinia pestis KIM. J Bacteriol. 2002;184:4601–11. doi: 10.1128/JB.184.16.4601-4611.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radnedge L, Agron PG, Worsham PL, Andersen GL. Genome plasticity in Yersinia pestis. Microbiology. 2002;148:1687–98. doi: 10.1099/00221287-148-6-1687. [DOI] [PubMed] [Google Scholar]
- Fleischmann RD, Alland D, Eisen JA, Carpenter L, White O, Peterson J, DeBoy R, Dodson R, Gwinn M, Haft D, Hickey E, Kolonay JF, Nelson WC, Umayam LA, Ermolaeva M, Salzberg SL, Delcher A, Utterback T, Weidman J, Khouri H, Gill J, Mikula A, Bishai W, Jacobs WR, Jr, Venter JC, Fraser CM. Whole-Genome Comparison of Mycobacterium tuberculosis Clinical and Laboratory Strains. J Bacteriol. 2002;184:5479–5490. doi: 10.1128/JB.184.19.5479-5490.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Makino K, Ohnishi M, Kurokawa K, Ishii K, Yokoyama K, Han CG, Ohtsubo E, Nakayama K, Murata T, Tanaka M, Tobe T, Iida T, Takami H, Honda T, Sasakawa C, Ogasawara N, Yasunaga T, Kuhara S, Shiba T, Hattori M, Shinagawa H. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 2001;8:11–22. doi: 10.1093/dnares/8.1.11. [DOI] [PubMed] [Google Scholar]
- Perna NT, Plunkett G, 3rd, Burland V, Mau B, Glasner JD, Rose DJ, Mayhew GF, Evans PS, Gregor J, Kirkpatrick HA, Posfai G, Hackett J, Klink S, Boutin A, Shao Y, Miller L, Grotbeck EJ, Davis NW, Lim A, Dimalanta ET, Potamousis KD, Apodaca J, Anantharaman TS, Lin J, Yen G, Schwartz DC, Welch RA, Blattner FR. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature. 2001;409:529–33. doi: 10.1038/35054089. [DOI] [PubMed] [Google Scholar]
- Welch RA, Burland V, Plunkett G, 3rd, Redford P, Roesch P, Rasko D, Buckles EL, Liou SR, Boutin A, Hackett J, Stroud D, Mayhew GF, Rose DJ, Zhou S, Schwartz DC, Perna NT, Mobley HL, Donnenberg MS, Blattner FR. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci U S A. 2002;99:17020–4. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Blast in the tandem repeats database page http://minisatellites.u-psud.fr/Blast
- The Bacterial Genotyping Page http://bacterial-genotyping.igmors.u-psud.fr
- Tettelin H, Masignani V, Cieslewicz MJ, Eisen JA, Peterson S, Wessels MR, Paulsen IT, Nelson KE, Margarit I, Read TD, Madoff LC, Wolf AM, Beanan MJ, Brinkac LM, Daugherty SC, DeBoy RT, Durkin AS, Kolonay JF, Madupu R, Lewis MR, Radune D, Fedorova NB, Scanlan D, Khouri H, Mulligan S, Carty HA, Cline RT, Van Aken SE, Gill J, Scarselli M, Mora M, Iacobini ET, Brettoni C, Galli G, Mariani M, Vegni F, Maione D, Rinaudo D, Rappuoli R, Telford JL, Kasper DL, Grandi G, Fraser CM. Complete genome sequence and comparative genomic analysis of an emerging human pathogen, serotype V Streptococcus agalactiae. Proc Natl Acad Sci U S A. 2002;99:12391–6. doi: 10.1073/pnas.182380799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F, Hou S, Layman D, Leonard S, Nguyen C, Scott K, Holmes A, Grewal N, Mulvaney E, Ryan E, Sun H, Florea L, Miller W, Stoneking T, Nhan M, Waterston R, Wilson RK. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature. 2001;413:852–6. doi: 10.1038/35101614. [DOI] [PubMed] [Google Scholar]
- Jin Q, Yuan Z, Xu J, Wang Y, Shen Y, Lu W, Wang J, Liu H, Yang J, Yang F, Zhang X, Zhang J, Yang G, Wu H, Qu D, Dong J, Sun L, Xue Y, Zhao A, Gao Y, Zhu J, Kan B, Ding K, Chen S, Cheng H, Yao Z, He B, Chen R, Ma D, Qiang B, Wen Y, Hou Y, Yu J. Genome sequence of Shigella flexneri 2a: insights into pathogenicity through comparison with genomes of Escherichia coli K12 and O157. Nucleic Acids Res. 2002;30:4432–41. doi: 10.1093/nar/gkf566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricker BJ, Ewalt DR, Halling SM. Brucella 'Hoof-Prints': strain typing by multi-locus analysis of variable number tandem repeats (VNTRs) BMC Microbiol. 2003;3:15. doi: 10.1186/1471-2180-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodner B, Hinkle G, Gattung S, Miller N, Blanchard M, Qurollo B, Goldman BS, Cao Y, Askenazi M, Halling C, Mullin L, Houmiel K, Gordon J, Vaudin M, Iartchouk O, Epp A, Liu F, Wollam C, Allinger M, Doughty D, Scott C, Lappas C, Markelz B, Flanagan C, Crowell C, Gurson J, Lomo C, Sear C, Strub G, Cielo C, Slater S. Genome sequence of the plant pathogen and biotechnology agent Agrobacterium tumefaciens C58. Science. 2001;294:2323–8. doi: 10.1126/science.1066803. [DOI] [PubMed] [Google Scholar]
- Wood DW, Setubal JC, Kaul R, Monks DE, Kitajima JP, Okura VK, Zhou Y, Chen L, Wood GE, Almeida NF, Jr, Woo L, Chen Y, Paulsen IT, Eisen JA, Karp PD, Bovee D, Sr, Chapman P, Clendenning J, Deatherage G, Gillet W, Grant C, Kutyavin T, Levy R, Li MJ, McClelland E, Palmieri A, Raymond C, Rouse G, Saenphimmachak C, Wu Z, Romero P, Gordon D, Zhang S, Yoo H, Tao Y, Biddle P, Jung M, Krespan W, Perry M, Gordon-Kamm B, Liao L, Kim S, Hendrick C, Zhao ZY, Dolan M, Chumley F, Tingey SV, Tomb JF, Gordon MP, Olson MV, Nester EW. The genome of the natural genetic engineer Agrobacterium tumefaciens C58. Science. 2001;294:2317–23. doi: 10.1126/science.1066804. [DOI] [PubMed] [Google Scholar]
- Benson G, Dong L. Reconstructing the duplication history of a tandem repeat. Proc Int Conf Intell Syst Mol Biol. 1999:44–53. [PubMed] [Google Scholar]
- van Belkum A, Scherer S, van Alphen L, Verbrugh H. Short-sequence DNA repeats in prokaryotic genomes. Microbiol Mol Biol Rev. 1998;62:275–93. doi: 10.1128/mmbr.62.2.275-293.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher S, Fuchs RP, Bichara M. Two distinct models account for short and long deletions within sequence repeats in Escherichia coli. J Bacteriol. 1997;179:6512–7. doi: 10.1128/jb.179.20.6512-6517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bolle X, Bayliss CD, Field D, van de Ven T, Saunders NJ, Hood DW, Moxon ER. The length of a tetranucleotide repeat tract in Haemophilus influenzae determines the phase variation rate of a gene with homology to type III DNA methyltransferases. Mol Microbiol. 2000;35:211–22. doi: 10.1046/j.1365-2958.2000.01701.x. [DOI] [PubMed] [Google Scholar]