

Abstract
Previous studies suggested that the cellular prion protein (PrPc) plays a critical role in the pathogenesis of Alzheimer's disease (AD). Specifically, amyloid-β (Aβ) oligomers were proposed to cause synaptic and cognitive dysfunction by binding to PrPc. To test this hypothesis, we crossed human amyloid precursor protein (hAPP) transgenic mice from line J20 onto a PrPc-deficient background. Ablation of PrPc did not prevent the premature mortality and abnormal neural network activity typically seen in hAPPJ20 mice. Furthermore, hAPPJ20 mice with or without PrPc expression showed comparably robust abnormalities in learning and memory and in other behavioral domains at 6–8 months of age. Notably, these abnormalities are not refractory to therapeutic manipulations in general: they can be effectively prevented by interventions that prevent Aβ-dependent neuronal dysfunction also in other lines of hAPP transgenic mice. Thus, at least in this model, PrPc is not an important mediator of Aβ-induced neurological impairments.
Introduction
Alzheimer's disease (AD) affects many millions of people and is on the rise worldwide (Thies and Bleiler, 2011). Amyloid-β (Aβ) peptides, released from the amyloid precursor protein (APP), are implicated in its pathogenesis. At high concentrations, Aβ peptides form diverse assemblies, among which Aβ oligomers may be the most pathogenic (Cheng et al., 2007; Shankar et al., 2008; Ashe and Zahs, 2010; Sakono and Zako, 2010). One of the most important unresolved questions is whether Aβ oligomers impair neuronal functions by interacting with specific cell surface receptors. Aβ oligomers bind a variety of receptors, including the receptor for advanced glycation end products (RAGE) (Origlia et al., 2009), α-7-nicotinic acetylcholine receptor (Dineley et al., 2002), cellular prion protein (PrPc) (Laurén et al., 2009; Balducci et al., 2010), and Ephrin-type B2 receptor (EphB2) (Renner et al., 2010; Cissé et al., 2011). Determining which interaction most affects cognitive functions is an important objective.
PrPc has been reported to mediate Aβ-induced deficits in hippocampal synaptic plasticity and cognitive impairments (Laurén et al., 2009; Gimbel et al., 2010). This membrane-anchored glycoprotein helps to maintain the brain's white matter and regulate its innate immune cells, responses to oxidative stress, and neurogenesis (Aguzzi et al., 2008). PrPc ablation prevented deficits in long-term potentiation (LTP) elicited by Aβ oligomers in acute hippocampal slices (Laurén et al., 2009) and behavioral impairments in APPswe/PSenΔE9 transgenic mice (Gimbel et al., 2010). However, others could not reproduce the electrophysiological rescue (Calella et al., 2010; Kessels et al., 2010), and one study reported that acute intracerebroventricular injection of synthetic Aβ oligomers caused similar deficits in learning and memory in mice with or without PrPc expression (Balducci et al., 2010).
Because the experimental design used in the latter study differs from the chronic exposure to Aβ oligomers that neurons experience in human APP (hAPP) transgenic mice and in AD, we set out to replicate the reported rescue of behavioral functions (Gimbel et al., 2010) in hAPPJ20 mice. hAPPJ20 mice have a robust phenotype with a range of AD-like alterations, including deficits in spatial and nonspatial learning and memory, behavioral abnormalities, synaptic impairments, changes in various synaptic activity-related proteins, amyloid plaques, dystrophic neurites, aberrant sprouting of axon terminals, astrocytosis, and microgliosis (Mucke et al., 2000; Cheng et al., 2007; Palop et al., 2007; Roberson et al., 2007, 2011; Meilandt et al., 2008; Harris et al., 2010; Cissé et al., 2011). We crossed hAPPJ20 mice onto a PrPc-deficient background (Büeler et al., 1992) and compared hAPPJ20 mice with or without PrPc expression at 6–8 months of age in a battery of behavioral tests and by video-EEG monitoring.
Materials and Methods
hAPPJ20 transgenic and Prnp knock-out mice
Hemizygous transgenic and NTG mice were from line J20, which expresses an alternatively spliced hAPPJ20 minigene encoding hAPP695, hAPP751, and hAPP770 with the Swedish and Indiana familial AD mutations directed by the PDGF β-chain promoter (Rockenstein et al., 1995; Mucke et al., 2000). PrPc-deficient Prnp knock-out mice (B6.129S7-Prnptm1Cwe/Orl) were acquired from the European Mutant Mouse Archive. hAPP mice and Prnp−/− mice were on a C57BL/6J background. Mice had access to food (Picolab Rodent Diet 20, Labdiet) and water ad libitum. For all experiments, groups were sex-balanced.
hAPP, C99, and Aβ measurements
For hAPP and C99 measurements, snap-frozen hippocampal samples were homogenized in PBS containing 0.5 mm EDTA, 1 mm DTT, 0.5% Triton, 0.1 m PMSF, Phosphatase Inhibitor Cocktails I and II (Sigma-Aldrich), and protease inhibitors (Roche). For Aβ measurements, the same lysates were diluted 1:2 with 7.5 m guanidine buffer. Total hAPP and C99 levels in hippocampal samples were assessed by Western blotting. The following antibodies were used: anti-hAPP (8E5, 1:1000; kindly provided by Elan Pharmaceuticals), anti-CTF (CT-15, 1:1000; kindly provided by Dr. Eddy Koo, University of California at San Diego, La Jolla, CA), and anti-tubulin (Sigma). Aβ1-x and Aβ1-42 levels were quantified by ELISA as described previously (Johnson-Wood et al., 1997; Mucke et al., 2000) and expressed relative to total protein concentration determined by Bradford assay.
Behavioral tests
Elevated plus maze.
The elevated plus maze has two open (without walls) and two enclosed (with walls) arms elevated 63 cm above the ground (Hamilton-Kinder). Mice were acclimated to the testing room under dim light for 1 h and tested as described previously (Harris et al., 2010; Cissé et al., 2011).
Open field.
Spontaneous locomotor activity in an open field was measured as described previously (Harris et al., 2010; Cissé et al., 2011).
Novel object recognition.
Mice were acclimated in the testing room for at least 1 h before testing. Testing was performed as described previously (Harris et al., 2010; Cissé et al., 2011) and the frequency of object interactions and time spent exploring each object were recorded with a video tracking system (Noldus) for analysis.
Morris water maze.
The apparatus and training protocol have been described previously (Harris et al., 2010; Cissé et al., 2011).
EEG recordings
Mice were implanted for video-EEG monitoring after anesthesia with Avertin (tribromoethanol, 250 mg/kg, i.p.). Teflon-coated silver wire electrodes (0.125 mm diameter) soldered to a multichannel electrical connector were implanted into the subdural space over the left frontal cortex (coordinates relative to the bregma were M/L, ±1 mm; A/P, ±1 mm) and the left and right parietal cortex (M/L, ±2 mm, A/P, ±2 mm). The left frontal cortex electrode was a reference. EEG recordings were performed at least 7 d after surgery on freely moving mice in a recording chamber. EEG activity was recorded with the Harmonie software (version 5.0b) for 48 h. Epileptic spikes were automatically detected by Gotman spike and seizure detectors (Harmonie) and confirmed by inspection of EEG recordings.
Statistical analyses
Statistical analyses were performed with GraphPad Prism. Differences between two means were assessed by paired or unpaired t test. Differences among multiple means were assessed, as indicated, by one-way, two-way, or repeated-measures ANOVA, followed by Bonferroni's or Tukey's post hoc test. Error bars represent SEM. Differences between observed and expected values were assessed by one- or two-sided χ2 test. Null hypotheses were rejected at the 0.05 level.
Results
PrPc ablation does not prevent behavioral abnormalities in hAPPJ20 mice
Like other hAPP transgenic models, hAPPJ20 mice display a disinhibition-like phenotype in the elevated plus maze and hyperactivity in arenas, such as the open field (Chin et al., 2005; Kobayashi and Chen, 2005; Cheng et al., 2007; Roberson et al., 2007; Meilandt et al., 2009; Harris et al., 2010; Cissé et al., 2011). To determine whether PrPc ablation prevents behavioral abnormalities, we generated hAPPJ20 mice with (Prnp+/+) or without (Prnp−/−) PrPc and compared them in these paradigms at 6–8 months, when behavioral abnormalities are readily detectable in hAPPJ20 mice on the Prnp+/+ background. hAPP/Prnp+/+ and hAPP/Prnp−/− mice spent more time in the open arms of the elevated plus maze than Prnp+/+ and Prnp−/− mice without hAPP (Fig. 1A), suggesting disinhibition or lower levels of anxiety. Ablation of PrPc had no effect. It also had no significant effect on the hyperactivity of hAPP mice in the open field (p = 0.132 by unpaired, one-tailed Student's t test) (Fig. 1B).
Figure 1.
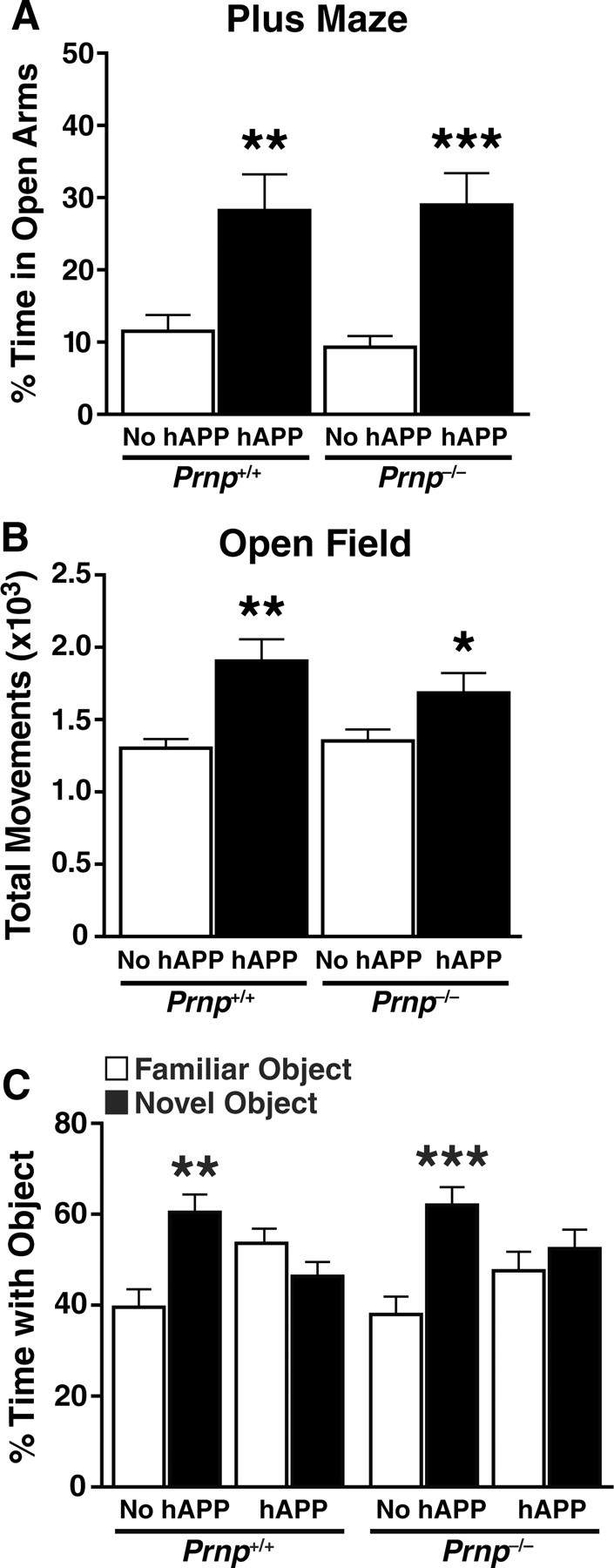
Ablation of PrPc in hAPPJ20 mice does not prevent their behavioral abnormalities in the elevated plus maze, open field, and object recognition test. A–C, Six- to eight-month-old Prnp+/+ and Prnp−/− mice with or without hAPP (n = 10–15 mice per genotype) were tested in the indicated behavioral paradigms. A, In the elevated plus maze, mice with hAPP spent more time in the open arms than mice without hAPP. hAPP/Prnp+/+ mice and hAPP/Prnp−/− mice showed a similar level of disinhibition. Two-way ANOVA revealed a significant effect of hAPP (p < 0.0001), but not of Prnp (p = 0.748), and no interaction between hAPP and Prnp (p = 0.607). B, In the open field, mice with hAPP were hyperactive compared to mice without hAPP. The subtle difference between hAPP/Prnp+/+ mice and hAPP/Prnp−/− mice was not significant even by one-tailed t test (p = 0.132). Two-way ANOVA revealed a significant effect of hAPP (p < 0.0001), but not of Prnp (p = 0.445), and no interaction between hAPP and Prnp (p = 0.223). C, Mice were analyzed in the novel object recognition test. Unlike littermates without hAPP, hAPPJ20/Prnp+/+ mice and hAPPJ20/Prnp−/− mice failed to spend more time with the novel than with the familiar object in test sessions. Two-way ANOVA of the average ratios of time spent with the novel versus the familiar object revealed a significant effect of hAPP (p < 0.005), but not of Prnp (p = 0.322), and no interaction between hAPP and Prnp (p = 0.561). *p < 0.05, **p < 0.005, ***p < 0.0005 versus mice without hAPP of the same Prnp genotype (Tukey test) (A, B) or versus familiar object (paired, two-tailed t test) (C). Data represent means ± SEM.
PrPc ablation does not prevent learning and memory deficits in hAPPJ20 mice
Like other hAPP transgenic mice, hAPPJ20 mice show deficits in spatial and nonspatial learning and memory (Cheng et al., 2007; Roberson et al., 2007, 2011; Meilandt et al., 2009; Harris et al., 2010; Cissé et al., 2011). Nonspatial learning and memory were assessed in the novel object recognition test. Unlike mice without hAPP, hAPP/Prnp+/+ and hAPP/Prnp−/− mice did not prefer the novel over the familiar object (Fig. 1C), suggesting deficits in recognizing or remembering the familiar object. Ablation of PrPc provided no benefit to hAPP mice.
Spatial learning and memory were tested in the Morris water maze. PrPc ablation failed to prevent deficits hAPPJ20 mice typically show in this test and even tended to worsen them. In the cued-platform task, all mice learned similarly (data not shown). In the hidden-platform (spatial) version, both hAPP/Prnp+/+ and hAPP/Prnp−/− mice showed significant learning deficits compared with Prnp+/+ and Prnp−/− mice lacking hAPP (Fig. 2A). hAPP mice without PrPc performed slightly worse than hAPP mice with PrPc (p < 0.05 by linear contrast and Bonferroni test). Swim speeds during the hidden platform training were comparable among all groups (data not shown).
Figure 2.

PrPc ablation in hAPPJ20 mice does not prevent deficits in spatial learning and memory. Six- to eight-month-old Prnp+/+ and Prnp−/− mice with or without hAPP (n = 10–15 mice per genotype) were trained in the Morris water maze for 4 d. Time (latency) and distance swum (data not shown) before reaching the platform were recorded. A probe trial (platform removed) was conducted 24 h after the last training. A, hAPP/Prnp+/+ and hAPP/Prnp−/− mice learned this task more poorly than mice without hAPP. Two-way repeated-measures ANOVA revealed a significant effect of hAPP (p < 0.0001), but not of Prnp (p = 0.0613), and no interaction between hAPP and Prnp (p = 0.632). B, C, During the probe trial, mice without hAPP, but not mice with hAPP, favored the target quadrant (B) and crossed the target location more often than corresponding places in nontarget quadrants (C). Two-way ANOVA of these data revealed a significant effect of hAPP (B, p < 0.05; C, p < 0.005), but not of Prnp (B, p = 0.8691; C, p = 0.647), and no interaction between hAPP and Prnp (B, p = 0.4620; C, p = 0.281). D, During the probe trial, mice with hAPP took longer to reach the target location than mice without hAPP. Two-way ANOVA revealed a significant effect of hAPP (p < 0.0001), but not of Prnp (p = 0.238), and no interaction between hAPP and Prnp (p = 0.483). One-way ANOVA and Bonferroni test revealed a significant difference between hAPP/Prnp+/+ and hAPP/Prnp−/− mice (p < 0.05). *p < 0.05, **p < 0.005, ***p < 0.0005 versus nontarget quadrants by paired, two-way t test (B, C) or versus mice without hAPP of the same Prnp genotype by Tukey test (D). Data represent means ± SEM.
At 24 h after the last training, spatial memory retention was assessed in a probe trial. The platform was removed, and mice were given 60 s to explore the pool. Unlike mice without hAPP, hAPP/Prnp+/+ and hAPP/Prnp−/− mice failed to spend significantly more time in the target quadrant (i.e., where the platform was) than in the other quadrants (Fig. 2B). hAPP/Prnp+/+ and hAPP/Prnp−/− mice also failed to cross the original platform location significantly more often than corresponding locations in nontarget quadrants (Fig. 2C). Finally, hAPP/Prnp+/+ and hAPP/Prnp−/− mice took longer to reach the original platform location than mice without hAPP (Fig. 2D). In all three outcome measures, hAPP/Prnp−/− mice tended to perform worse than hAPP/Prnp+/+ mice, although these differences did not reach statistical significance.
PrPc ablation does not change hippocampal levels of hAPP or Aβ in hAPPJ20 mice
We compared hippocampal Aβ levels by ELISA and hippocampal levels of hAPP and hAPP C-terminal fragment C99 by Western blots in 8- to 13-month-old hAPP/Prnp+/+ and hAPP/Prnp−/− mice (n = 11–13 per genotype, mean ± SEM, relative levels): Aβ1-x (148 ± 23 vs 100 ± 26), Aβ1-42 (108 ± 16 vs 78 ± 19), Aβ1-42/Aβ1-x ratio (0.67 ± 0.08 vs 0.81 ± 0.05), hAPP (4895 ± 498 vs 5007 ± 488), and C99 (8310 ± 1171 vs 6212 ± 989). None of these differences was statistically significant by unpaired t test.
PrPc ablation worsens premature mortality in hAPPJ20 mice
Like other hAPP transgenic lines, hAPPJ20 mice exhibit early mortality, which may be caused by epileptic seizures (Chin et al., 2004; Palop et al., 2007; Roberson et al., 2007, 2011; Meilandt et al., 2009). Prnp+/+, Prnp−/−, hAPP/Prnp+/+ and hAPP/Prnp−/− mice were born at comparable proportions (Fig. 3A). However, from birth to weaning (P27), more hAPP mice tended to die than mice without hAPP, independently of Prnp genotype (Fig. 3B). By 8 months of age, 8% of hAPP/Prnp+/+ mice died (Fig. 3C). Surprisingly, PrPc ablation worsened early mortality in hAPPJ20 mice, resulting in a 22% death rate in hAPP/Prnp−/− mice (Fig. 3C). During the observation period, only one death occurred in Prnp+/+ or Prnp−/− mice lacking hAPP.
Figure 3.
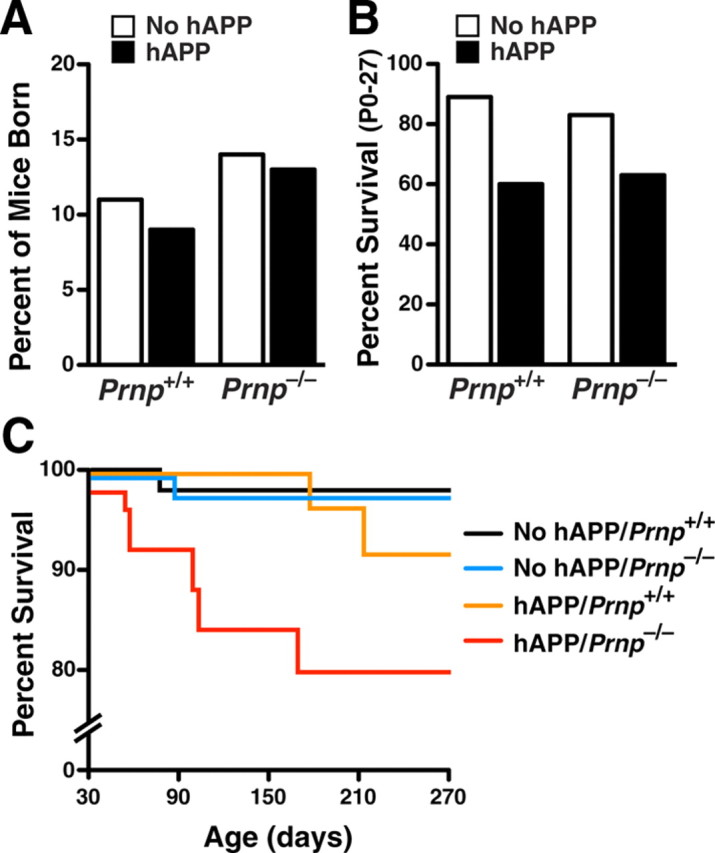
Ablation of PrPc worsens early mortality in hAPPJ20 mice. Death and survival were monitored in a total of 169 mice. A, Percentage of mice born by genotype. The difference between the number of mice obtained and expected for each genotype, based on Mendelian principles of inheritance (12.5%), was not significant (χ2 = 0.72; p = 0.86). B, Premature death reported from P0 to P27. Mortality tended to be higher in hAPP than NTG mice (one-sided χ2 = 1.9, p = 0.07), independently of their Prnp genotype (two-sided χ2 = 0.28, p = 0.59). C, Kaplan–Meier survival plot shows more mice with hAPP died prematurely than mice without hAPP after weaning (p < 0.01, Mantel–Cox test). Premature mortality was greater in hAPP/Prnp−/− mice than in hAPP/Prnp+/+ mice (p < 0.001, Mantel–Cox test).
PrPc ablation does not prevent spontaneous epileptiform activity in hAPPJ20 mice
EEG recordings from the neocortex and hippocampus of hAPPJ20 mice revealed spontaneous epileptiform activity (Palop et al., 2007), and similar findings have been obtained in other lines of hAPP mice (Minkeviciene et al., 2009; Roberson et al., 2011). Because Prnp−/− mice were reported as more resistant to proconvulsant drugs than Prnp+/+ mice (Ratté et al., 2011), we examined whether PrPc ablation makes hAPPJ20 mice more resistant to Aβ-induced epileptiform activity. Prnp+/+ and Prnp−/− mice with or without hAPP were compared by 48 h video and EEG recordings at 6–8 months of age. Bilateral recording electrodes over the parietal cortex revealed very few or no spikes in mice without hAPP (Fig. 4A) and an average of 7 spikes per hour in hAPPJ20/Prnp+/+ mice (Fig. 4B). Remarkably, PrPc ablation actually doubled the epileptiform activity recorded in hAPPJ20 mice (Fig. 4B). No seizures occurred during the recording period in any of the groups.
Figure 4.
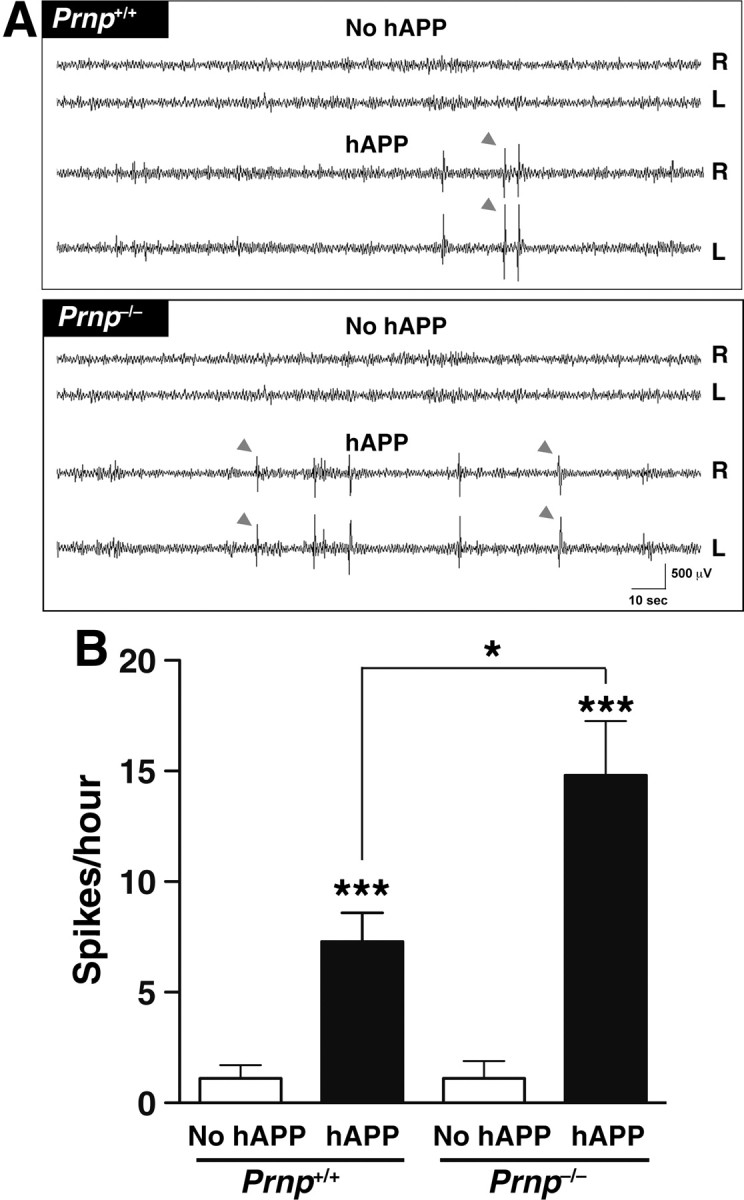
Ablation of PrPc worsens spontaneous epileptiform activity in hAPPJ20 mice. EEG was recorded over both parietal cortices for 48 h in 6- to 8-month-old mice (n = 3–6 mice per genotype). A, In contrast to mice lacking hAPP, hAPP/Prnp+/+ and hAPP/Prnp−/− mice showed frequent epileptiform spikes (see gray arrowheads). L, Left; R, right. B, Quantification of epileptiform spikes. Two-way ANOVA revealed effects of hAPP (p < 0.0001) and Prnp (p = 0.03), and an interaction between hAPP and Prnp (p = 0.036). *p < 0.01, ***p < 0.0001 versus mice without hAPP or as indicated by bracket (Bonferroni post hoc test). Values are means ± SEM.
Discussion
PrPc ablation did not prevent neuronal network dysfunction and behavioral abnormalities in hAPPJ20 transgenic mice. In fact, it doubled the early mortality rate and epileptiform activity in this line. The resulting selection process and survival of mice with relative resistance against the synergistic effects of hAPP/Aβ overexpression and PrPc ablation might have diminished our ability to detect an exacerbating effect of PrPc ablation on hAPP/Aβ-dependent behavioral abnormalities. Subtle signs of such an effect were evident in the probe trial of the Morris water maze test.
These results differ from studies showing that PrPc ablation prevents Aβ-induced LTP deficits in acute hippocampal slices (Laurén et al., 2009) and diminishes early mortality and impairments in spatial learning and memory in APPswe/PSenΔE9 transgenic mice (Gimbel et al., 2010). However, our findings agree with others showing that PrPc ablation does not affect Aβ-induced synaptic depression, reduction in spine density, and LTP deficits in acute hippocampal slices (Kessels et al., 2010), behavioral impairments from intracerebroventricular injection of Aβ (Balducci et al., 2010), and impairments of hippocampal synaptic plasticity (Calella et al., 2010).
hAPPJ20 mice and other hAPP lines show epileptiform activity (Palop and Mucke, 2010; Roberson et al., 2011). Genetic manipulations that prevent or counteract aberrant excitatory neuronal activity (e.g., ablation of Fyn, group IVA phospholipase A2, or tau) reduce premature mortality and/or cognitive deficits in hAPP-J20 mice (Chin et al., 2004, 2005; Roberson et al., 2007, 2011; Sanchez-Mejia et al., 2008). Interestingly, PrPc has also been implicated in epilepsy (Walz et al., 2002). However, PrPc ablation actually increased premature mortality and did not affect cognitive deficits in hAPPJ20 mice, making it unlikely that it protects against Aβ-induced seizure activity, which is closely linked to cognitive impairments in these mice (Palop et al., 2007; Palop and Mucke, 2010; Roberson et al., 2011). Indeed, PrPc ablation enhanced spontaneous epileptiform activity in hAPPJ20 mice, consistent with the increased susceptibility of Prnp−/− mice to kainate-induced seizures (Walz et al., 1999; Rangel et al., 2007).
We found that PrPc ablation doubled premature mortality in hAPPJ20 mice but had no effect on mice without hAPP, suggesting a specific interaction between hAPP/Aβ- and PrPc-dependent mechanisms. The lower seizure threshold of Prnp−/− mice might partially explain the increased early mortality of hAPPJ20/Prnp−/− mice. These results are in stark contrast to the increased survival of PrPc-deficient APPswe/PSenΔE9 mice, which were also on the C57BL/6J background (Gimbel et al., 2010).
The different effects of PrPc ablation on Aβ-induced deficits observed by different investigators might stem from differences in experimental protocols, hAPP lines and Prnp−/− strains. For example, if levels of pathogenic Aβ oligomers in relevant brain regions were higher in hAPPJ20 mice than in APPswe/PSenΔE9 mice, beneficial effects of PrPc ablation in hAPPJ20 mice might have been obscured. However, if PrPc were crucial in Aβ-induced cognitive deficits, its ablation should prevent those deficits in hAPPJ20 mice as did ablation of group IVA phospholipase A2 (Sanchez-Mejia et al., 2008) or tau (Roberson et al., 2007, 2011). Even partial reduction of these two molecules in hemizygous knock-out mice significantly ameliorated the deficits in hAPPJ20 mice (Roberson et al., 2007, 2011; Sanchez-Mejia et al., 2008). Thus, Aβ-induced cognitive deficits in hAPPJ20 mice are not so severe that they simply cannot be overcome by any intervention. In fact, neuronal and cognitive dysfunction in these mice could be reversed effectively by normalizing neuronal expression of EphB2, an alternative receptor for Aβ oligomers (Cissé et al., 2011).
Because Aβ binds to many molecules (Dineley et al., 2002; Laurén et al., 2009; Origlia et al., 2009; Balducci et al., 2010; Renner et al., 2010; Cissé et al., 2011), it is critical to determine which of these interactions have the greatest functional impact. Interactions of Aβ oligomers with the receptor for advanced glycation end products (Origlia et al., 2009) and with EphB2 (Cissé et al., 2011) have striking effects on neuronal and cognitive functions and, we suspect, may be more promising targets for therapeutic interventions in AD than PrPc.
Footnotes
The study was supported by NIH Grants NS065780, AG011385, and AG022074 to L.M. and National Center for Research Resources Grant RR18928 to the Gladstone Institutes. We thank Dr. E. Koo for the CT15 antibody, Dr. J. Palop for helpful comments, X. Wang for technical support, and G. Howard for editorial review; J. Carroll and Teresa Roberts for preparation of graphics; and M. Dela Cruz for administrative assistance. Behavioral data were obtained with the help of the Gladstone Institutes' Behavioral Core.
References
- Aguzzi A, Baumann F, Bremer J. The prion's elusive reason for being. Annu Rev Neurosci. 2008;31:439–477. doi: 10.1146/annurev.neuro.31.060407.125620. [DOI] [PubMed] [Google Scholar]
- Ashe KH, Zahs KR. Probing the biology of Alzheimer's disease in mice. Neuron. 2010;66:631–645. doi: 10.1016/j.neuron.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, Chiesa R, Gobbi M, Salmona M, Forloni G. Synthetic amyloid-{beta} oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A. 2010;107:2295–2300. doi: 10.1073/pnas.0911829107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp H, DeArmond S, Prusiner S, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, Mansuy IM, Aguzzi A. Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med. 2010;2:306–314. doi: 10.1002/emmm.201000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng IH, Scearce-Levie K, Legleiter J, Palop JJ, Gerstein H, Bien-Ly N, Puoliväli J, Lesné S, Ashe KH, Muchowski PJ, Mucke L. Accelerating amyloid-β fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem. 2007;282:23818–23828. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Yu G-Q, Kojima N, Masliah E, Mucke L. Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J Neurosci. 2004;24:4692–4697. doi: 10.1523/JNEUROSCI.0277-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Puoliväli J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, Mucke L. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2005;25:9694–9703. doi: 10.1523/JNEUROSCI.2980-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cissé M, Halabisky B, Harris J, Devidze N, Dubal DB, Sun B, Orr A, Lotz G, Kim DH, Hamto P, Ho K, Yu G-Q, Mucke L. Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature. 2011;469:47–52. doi: 10.1038/nature09635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Bell KA, Bui D, Sweatt JD. b-Amyloid peptide activates a7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Biol Chem. 2002;277:25056–25061. doi: 10.1074/jbc.M200066200. [DOI] [PubMed] [Google Scholar]
- Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Laurén J, Gimbel ZA, Strittmatter SM. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci. 2010;30:6367–6374. doi: 10.1523/JNEUROSCI.0395-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JA, Devidze N, Halabisky B, Lo I, Thwin MT, Yu GQ, Bredesen DE, Masliah E, Mucke L. Many neuronal and behavioral impairments in transgenic mouse models of Alzheimer's disease are independent of caspase cleavage of the amyloid precursor protein. J Neurosci. 2010;30:372–381. doi: 10.1523/JNEUROSCI.5341-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Nguyen LN, Nabavi S, Malinow R. The prion protein as a receptor for amyloid-beta. Nature. 2010;466:E3–E4. doi: 10.1038/nature09217. discussion E4–E5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi DT, Chen KS. Behavioral phenotypes of amyloid-based genetically modified mouse models of Alzheimer's disease. Genes Brain Behav. 2005;4:173–196. doi: 10.1111/j.1601-183X.2005.00124.x. [DOI] [PubMed] [Google Scholar]
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meilandt WJ, Yu G-Q, Chin J, Roberson ED, Palop JJ, Wu T, Scearce-Levie K, Mucke L. Enkephalin elevations contribute to neuronal and behavioral impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2008;28:5007–5017. doi: 10.1523/JNEUROSCI.0590-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meilandt WJ, Cisse M, Ho K, Wu T, Esposito LA, Scearce-Levie K, Cheng IH, Yu GQ, Mucke L. Neprilysin overexpression inhibits plaque formation but fails to reduce pathogenic Aβ oligomers and associated cognitive deficits in human amyloid precursor protein transgenic mice. J Neurosci. 2009;29:1977–1986. doi: 10.1523/JNEUROSCI.2984-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany T, Pitkänen A, Tanila H. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu G-Q, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Origlia N, Arancio O, Domenici L, Yan SS. MAPK, beta-amyloid and synaptic dysfunction: the role of RAGE. Expert Rev Neurother. 2009;9:1635–1645. doi: 10.1586/ern.09.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu G-Q, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel A, Burgaya F, Gavín R, Soriano E, Aguzzi A, Del Río JA. Enhanced susceptibility of Prnp-deficient mice to kainate-induced seizures, neuronal apoptosis, and death: role of AMPA/kainate receptors. J Neurosci Res. 2007;85:2741–2755. doi: 10.1002/jnr.21215. [DOI] [PubMed] [Google Scholar]
- Ratté S, Vreugdenhil M, Boult JK, Patel A, Asante EA, Collinge J, Jefferys JG. Threshold for epileptiform activity is elevated in prion knockout mice. Neuroscience. 2011;179:56–61. doi: 10.1016/j.neuroscience.2011.01.053. [DOI] [PubMed] [Google Scholar]
- Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, Triller A. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron. 2010;66:739–754. doi: 10.1016/j.neuron.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu G-Q, Mucke L. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu G-Q, Palop JJ, Noebels JL, Mucke L. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on Tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein EM, McConlogue L, Tan H, Power M, Masliah E, Mucke L. Levels and alternative splicing of amyloid b protein precursor (APP) transcripts in brains of transgenic mice and humans with Alzheimer's disease. J Biol Chem. 1995;270:28257–28267. doi: 10.1074/jbc.270.47.28257. [DOI] [PubMed] [Google Scholar]
- Sakono M, Zako T. Amyloid oligomers: formation and toxicity of Abeta oligomers. FEBS J. 2010;277:1348–1358. doi: 10.1111/j.1742-4658.2010.07568.x. [DOI] [PubMed] [Google Scholar]
- Sanchez-Mejia RO, Newman JW, Toh S, Yu G-Q, Zhou Y, Halabisky B, Cissé M, Scearce-Levie K, Cheng IH, Gan L, Palop JJ, Bonventre JV, Mucke L. Phospholipase A2 reduction ameliorates cognitive deficits in mouse model of Alzheimer's disease. Nat Neurosci. 2008;11:1311–1318. doi: 10.1038/nn.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thies W, Bleiler L. 2011 Alzheimer's disease facts and figures. Alzheimers Dement. 2011;7:208–244. doi: 10.1016/j.jalz.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Walz R, Amaral OB, Rockenbach IC, Roesler R, Izquierdo I, Cavalheiro EA, Martins VR, Brentani RR. Increased sensitivity to seizures in mice lacking cellular prion protein. Epilepsia. 1999;40:1679–1682. doi: 10.1111/j.1528-1157.1999.tb01583.x. [DOI] [PubMed] [Google Scholar]
- Walz R, Castro RM, Velasco TR, Carlotti CG, Jr, Sakamoto AC, Brentani RR, Martins VR. Cellular prion protein: implications in seizures and epilepsy. Cell Mol Neurobiol. 2002;22:249–257. doi: 10.1023/A:1020711700048. [DOI] [PMC free article] [PubMed] [Google Scholar]