

Abstract
Protein synthesis in mammalian mitochondria produces 13 proteins that are essential subunits of the oxidative phosphorylation complexes. This review provides a detailed outline of each phase of mitochondrial translation including initiation, elongation, termination, and ribosome recycling. The roles of essential proteins involved in each phase are described. All of the products of mitochondrial protein synthesis in mammals are inserted into the inner membrane. Several proteins that may help bind ribosomes to the membrane during translation are described, although much remains to be learned about this process. Mutations in mitochondrial or nuclear genes encoding components of the translation system often lead to severe deficiencies in oxidative phosphorylation, and a summary of these mutations is provided.
Keywords: Mammal, Mitochondria, protein synthesis, initiation, elongation, termination
1. Protein Biosynthesis in Mammalian Mitochondria
1.1 Role of Mitochondria in Energy Metabolism
Mitochondria produce over 90 % of the energy used by mammalian cells through oxidative phosphorylation. They also carry out other critical functions including heme biosynthesis, a portion of the urea cycle, and play a role in apoptosis. Mitochondria are generally viewed as oblong-shaped organelles surrounded by two membranes. The outer membrane (OM) outlines the overall shape and forms an envelope, which presents a barrier that restricts passage to small molecules. The inner membrane (IM) is highly invaginated, forming cristae, and surrounds the interior soluble portion, the matrix. The IM is actually composed of two regions [1]. The inner membrane boundary (IMB) is closely associated with the OM with which it has a number of contact sites. The cristal membranes (CM) make up the majority of the surface of the IM. The IMB and the CM are connected by narrow, ring-like structures that form a barrier between the intracristal space and the intermembrane space. For simplicity, we will refer to the inner membrane (IM) throughout this manuscript as including both the IMB and the CM. The IM is the site of oxidative phosphorylation which generates most of the ATP used by aerobic cells.
Mammalian mitochondria have their own genome, consisting of about 16,000 base pairs of DNA encoding 2 rRNAs, 22 tRNAs and 13 polypeptides organized very compactly. The genes are generally directly adjacent to each other or are separated by only a few nucleotides. There is nearly a complete absence of non-coding regions. The genetic code has been slightly modified in animal mitochondria compared to the universal code [2]. For example UGA that normally serves as a stop codon is read as tryptophan in animal mitochondria. In addition to AUG, the AUA codon for isoleucine is read as methionine. While the AGA and AGG codons for arginine have been thought to act as stop codons, recent evidence suggests that they are used in −1 frameshifting (see Section 4.1) [3].
All of the proteins synthesized by the mitochondrial translational system are localized in the IM where they function as subunits in the electron transfer and ATP synthase complexes [4]. These include seven subunits of Complex I (NADH:ubiquinone oxidoreductase), one subunit from complex III (ubiquinone:cytochrome c oxidoreductase), three subunits from complex IV (cytochrome c:oxygen oxidoreductase) and 2 subunits of complex V (ATP synthase). The remaining 2,000 or so proteins present in mammalian mitochondria are the products of nuclear genes; they are synthesized in the cell cytosol and subsequently imported into the organelle. The synthesis of the oligomeric respiratory chain complexes requires the coordinate expression of genes in both the nuclear and mitochondrial genomes. How this process is regulated in not currently known.
1.2 General Features of Mammalian Mitochondrial Protein Synthesis
Despite many years of research, no in vitro translation system capable of correct initiation and synthesis of a mitochondrially encoded protein has been established from mammalian mitochondria, and much remains to be learned about this process. However, a number of the individual steps of mammalian mitochondrial protein synthesis have been successfully carried out in vitro providing information on specific features of this system. This review will focus on studies of the auxiliary factors required for mammalian mitochondrial protein synthesis. Brief mention will be made of major differences observed with the translational systems in lower eukaryotes.
Mammalian mitochondria contain a distinct set of ribosomes which, in mammals, sediment as 55S particles and consist of 28S small subunits and 39S large subunits [5]. These ribosomes have a molecular mass of about 2.7 × 106 daltons, roughly the size and shape of the Escherichia coli ribosome. Animal mitochondrial ribosomes have only two rRNA species, 12S in the small subunit and 16S in the large subunit, and are only 25 to 30% RNA [6]. A recent report, however, suggests that these ribosomes may also carry a 5S rRNA [7]. In contrast, bacterial ribosomes contain 60-70% RNA and eukaryotic cytoplasmic ribosomes are 50-60% RNA [7,9]. Comparisons of the primary sequences of the mitochondrial rRNAs with those of prokaryotes and eukaryotes show that there is only a small amount of identity between these rRNAs. The smaller mitochondrial rRNAs are not shortened at random positions throughout the primary sequence [10,11]. Rather, they lack either certain regions of secondary structure or entire domains [12].
Most of the mitochondrial ribosomal proteins have been identified using proteomics [13-18]. The small subunit of the bovine mitochondrial ribosome has about 29 proteins, of which 14 have homologs in prokaryotic ribosomes, while 15 are specific for mitochondrial ribosomes. Only six of these mitochondrial specific proteins have homologs in yeast mitochondrial ribosomes [19]. The large subunit has about 48 proteins. Of these, 28 are homologs of bacterial ribosomal proteins while the remaining 20 are unique to mitochondrial ribosomes. Again, only nine of these mitochondrial specific ribosomal proteins have homologs in yeast, indicating significant divergence between the protein composition of the mitochondrial ribosomes between the higher and lower eukaryotes. Many of the proteins with homologs in bacterial systems are significantly larger than their counterparts. Database analysis suggests that the human mitochondrial ribosome has a comparable spectrum of proteins as observed in the bovine system. For detailed information on mammalian mitochondrial ribosomes, the reader is referred to two recent reviews [12,20].
The 13 proteins coded for in the mitochondrial genome are translated from 9 monocistronic and 2 dicistronic mRNAs. Both dicistronic mRNAs contain overlapping reading frames [21,22]. With the exception of the two internal start sites found in the dicistronic mRNAs, the remaining 11 start sites are located at or near the 5′ end of each mRNA [23]. Thus, mitochondrial mRNAs lack the canonical Shine-Dalgarno sequence used in prokaryotes to help position the start codon at the P-site of the ribosome. Direct analysis of the 5′ ends of the eleven open reading frames located at the 5′ ends of the human mitochondrial mRNAs indicates that post-transcriptional processing completely eliminates the 5′ leader in all but three mRNAs [23] which have 1, 2 and 3 nucleotides 5′ to the start codon. None of the 5′ cistrons of Drosophila melanogaster mitochondrial mRNAs begin with noncoding nucleotides [24]. The start codon at the 5′ end can be either AUG or AUA, both of which encode methionine in mammalian mitochondria. These codons direct the insertion of formylmethionine during initiation and methionine during chain elongation. In bovine mitochondria, AUG serves as the start codon for 10 genes while AUA is used for the remaining three genes. AUA is the dominant codon for methionine during chain elongation.
The secondary structures at the 5′ ends of all the protein coding regions in the bovine mitochondrial transcriptome were analyzed at single nucleotide resolution using RNA SHAPE chemistry [25]. This analysis indicated that the 5′ ends of the mitochondrial mRNAs are quite unstructured [26] as indicated in the examples in Fig 1. The start codon tends to lie in a single-stranded region or in a very weak duplex stem.
Figure 1.
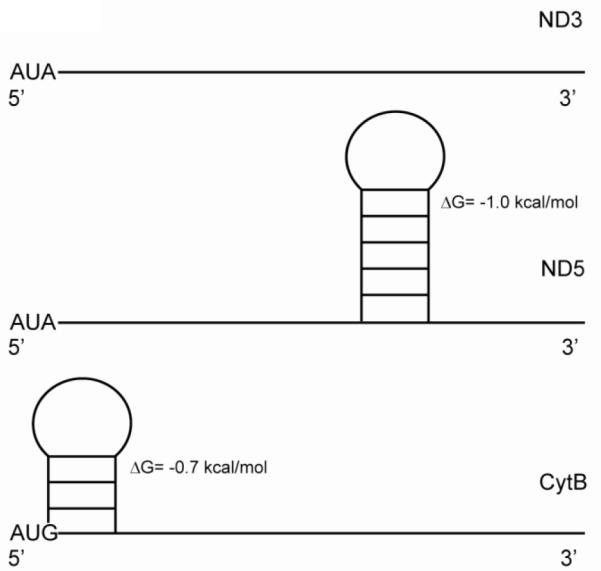
Secondary structures of three mitochondrial mRNAs. RNA SHAPE chemistry was used to analyze the structures of the 5′ ends of the mitochondrial mRNAs and indicated that most are largely unstructured [26]. The secondary structures of the NADH dehydrogenase subunits 3 (MTND3) and 5 (MTND5) mRNAs, as well as the cytochrome b (CytB) mRNA are shown. The free energy values for each stem-loop structure are indicated adjacent to the stem-loop.
Animal mitochondria have 22 tRNAs, one for each amino acid except for leucine and serine that have two each. The tRNAs found in mammalian mitochondria have a number of unusual features distinguishing them from canonical tRNAs. They are generally shorter that other tRNAs and often lack the conserved or semi-conserved nucleotides that play important roles in creating the L-shaped tertiary structure of prokaryotic and eukaryotic cytoplasmic tRNAs. Chemical and enzymatic probing has led to the idea that these tRNAs fold into the basic cloverleaf structure of canonical tRNAs, but lack a number of conserved tertiary interactions and have a weaker three-dimensional structure [27-31]. In particular, a number of the long-range interactions between the D-and T-arms of the tRNAs appear to be missing [32,28,27,33]. No direct structural information is available on a mitochondrial tRNA; however, cryo-EM of the mitochondrial ribosome shows a tRNA tightly bound at the P-site that is basically an L-shape with a caved-in elbow region [34]. The structure of this tRNA can be fitted into the crystallographic coordinates of canonical tRNAs except in the region of the elbow where the smaller sizes of the T- and D-loops of many mitochondrial tRNAs do not fit well into the structure of bacterial tRNAs.
The process of protein biosynthesis takes place in four phases, each requiring a set of auxiliary factors. During initiation, the start site on the mRNA is selected and the initiator tRNA (fMet-tRNA) is base-paired to the mRNA in the P-site of the ribosome. During elongation the codons in the mRNA are read sequentially while the amino acids are incorporated into the growing polypeptide chain. At termination and ribosome recycling, the completed polypeptide is released and ribosome complex is dissociated.
1.3 Experimental Approaches to studying mitochondrial translation
Despite a number of years of effort, no in vitro system has been established that will carry out the complete translation of a mammalian mitochondrial mRNA. Hence, most of what we currently understand about this process is based on partial reactions carried out using purified components. Initiation complex formation and the activities of the individual initiation factors are measured by the binding of [35S]fMet-tRNA to mitochondrial ribosomes or the small ribosomal subunit in the presence of poly(A,U,G) or an in vitro transcript of a mitochondrial mRNA [35,36]. Chain elongation is studied using purified mitochondrial ribosomes and elongation factors. The partial reactions of chain elongation are assayed by measuring the binding of [14C]Phe-tRNA to ribosomes in the presence of a poly(U) mRNA and the full elongation cycle is monitored by the polymerization of [14C]Phe into poly(Phe) as described in detail [37].
2. Initiation of Protein Synthesis in Mammalian Mitochondria
2.1 Model for Translational Initiation in Mammalian Mitochondria
Two mitochondrial initiation factors have been identified, mitochondrial initiation factor 2 (IF2mt) and mitochondrial initiation factor 3 (IF3mt). Using these two factors alone, it is possible in vitro to assemble an initiation complex on 55S ribosomes with fMet-tRNA correctly positioned at the start codon of a mitochondrial mRNA [36]. However, the process in vivo could be more complex and subject to significant regulation.
In the current working model for the initiation of translation in this minimal mitochondrial system (Fig. 2), the first step in initiation is the active dissociation of the 55S ribosome by IF3mt. In this step IF3mt interacts with the 55S particle loosening the interaction of the two subunits (Step 1), leading to the release of the 39S subunit and the formation of a 28S:IF3mt complex (Step 2). The timing of the binding of IF2mt:GTP is not clear at the present time and is show in Step 3 for convenience only. The mRNA feeds into the 28S subunit via a protein-rich mRNA entrance gate observed in the small subunit [34] (Step 4). The binding of the mRNA is thought to precede fMet-tRNA binding, because IF3mt has been shown to destabilize the fMet-tRNA bound to 28S subunits in the absence of mRNA (see Section 2.4.4). Toeprinting analysis [36] has indicated that when the first 17 nucleotides of the mRNA have entered the ribosome, the movement of the mRNA is paused while the 28S subunit inspects the codon at the 5′ end of the mRNA (inspection option in Step 4). During this pause, IF2mt:GTP can promote the binding of fMet-tRNA to the ribosome. If there is a start codon exposed in the P-site, codon:anticodon interactions between the fMet-tRNA and the 5′ AUG start codon lead to a stable initiation complex. If no codon:anticodon interactions can form due to a lack of fMet-tRNA and/or to the absence of a 5′ start codon, the mRNA resumes sliding through the small subunit and eventually dissociates. Following fMet-tRNA binding to the 5′ start codon, the large subunit joins the 28S initiation complex, IF2mt hydrolyzes GTP to GDP, and the initiation factors are released, resulting in a completed 55S initiation complex that is ready to enter elongation phase of protein synthesis (Step 5).
Figure 2.

Model for the initiation phase of mitochondrial translation. In the current model for the initiation of protein synthesis, mitochondrial initiation factor 3 (IF3mt) actively dissociates 55S ribosomes, forming a transient [IF3mt:55S] complex (Step 1) and leading to the formation of an IF3mt:28S complex (Step 2). Mitochondrial initiation factor 2 (IF2mt) bound to GTP binds to the small subunit (Step 3), followed by the fMet-tRNA and mRNA (Step 4), although the exact order of binding is not clear. The mRNA feeds into the mRNA entrance gate and the 5′ end pauses at the P-site of the ribosome for inspection of its 5′ start codon. In the presence of the correct start codon and fMet-tRNA, the mRNA is locked in place by codon:anticodon interactions to form the 28S initiation complex. If fMet-tRNA binds in the absence of mRNA, or if the mRNA does not contain a proper start codon, the inspection step fails. The failed inspection causes the mRNA to continue sliding through the ribosome to exit. Once the 28S initiation complex has formed, the large subunit joins, and along with the hydrolysis of GTP to GDP, the initiation factors exit (Step 5) leaving a 55S:fMet-tRNA:mRNA complex that is ready for the elongation phase of protein synthesis.
Consideration of the mechanism of polypeptide chain initiation in mammalian mitochondria must take into account the unusual properties of the mRNAs present, particularly the location of the start codon at or very near the 5′ end of the mRNA. To illustrate the importance of the leaderless nature of mitochondrial mRNAs, a series of 5′ extended mRNAs was prepared using the mRNA for subunit II of cytochrome oxidase containing 1, 2, 3, 6, 9 or 12 nucleotides prior to the 5′ start codon [36]. The formation of the initiation complex was slightly reduced by the addition of a single nucleotide prior to the 5′ AUG and the presence of only 3 nucleotides preceding the AUG codon led to more than a 40 % reduction in initiation complex formation. Additional nucleotides prior to the AUG start codon led to yet further decreases in initiation complex formation, with an 80% reduction in initiation complex formation observed when 12 nucleotides were present 5′ to the start codon. This result indicates that the ribosome is very inefficient in recognizing the start codon of mRNAs with more than 3 nucleotides 5′ to the AUG and suggests that post-transcriptional processing of the long primary transcript of the mitochondrial DNA is likely to occur prior to translation.
2.2. Mitochondrial Translational Initiation Factors 1 and 2
2.2.1 Background
As indicated in Section 2.1, the general mitochondrial translation machinery involves two initiation factors, IF2mt and IF3mt. Although IF1 has been viewed as a universal translational initiation factor, no corresponding factor has been identified in any mitochondrial system to date.
2.2.2. The IF2mt Gene
The gene for IF2mt (MIF2) is located on chromosome 2 and spans 33.5 kilobases [38]. It has 16 exons and 15 introns. Several single nucleotide polymorphisms are observed in this gene. The transcriptional start site is located 296 base pairs upstream of the AUG start codon. Three upstream AUG codons are present in the 5′ untranslated region of the IF2mt mRNA. Two of these are in-frame while the third is in a different reading frame. These extra AUG codons are likely to down-regulate the synthesis of IF2mt, which is estimated to be present in only 10-20 copies per mitochondrion. Transcription of the MIF2 gene generally correlates with the energy demands of the tissue [39]. No direct studies have been carried out on the regulation of expression of the IF2mt gene. However, the transcription start site contains binding sites for transcription factors Sp1, nuclear respiratory factor 2 (NRF-2) and the estrogen receptor [38] suggesting a complex pattern of regulation.
Native IF2mt was purified a number of years ago [40,41]. The purified factor is capable of promoting the binding of the initiator tRNA (fMet-tRNA) to mitochondrial 28S subunits or 55S ribosomes in the presence of a mRNA such as the AUG triplet or poly(A,U,G). This binding is stimulated significantly by GTP. Bovine IF2mt can stimulate the binding of fMet-tRNA to bacterial ribosomes in addition to mitochondrial ribosomes. However, the converse is not true, and E. coli IF2 is not active on mitochondrial ribosomes [41].
2.2.3. Characteristics of the IF2mt Coding Sequence
Several different nomenclatures have been used to describe the domain architecture of the IF2 from various organisms. For convenience we have used the nomenclature that divides E. coli IF2 into six domains (Fig. 3). Doman I in E. coli IF2 is present in the longest version of this factor (IF2α) but is absent in a shorter in vivo form (IF2β). The role of this domain is not known and it is absent in the IF2 species from many organisms. Domain II is thought to play a role in the interaction of IF2 with the small ribosomal subunit [42]. Domain III is poorly characterized but may also make contact with the small ribosomal subunit. The guanine nucleotide binding domain (G-domain, domain IV) is the most highly conserved region of the IF2 species from various organisms and carries the signature sequences for typical G-proteins. Domain V is also thought to play a role in ribosome binding. Finally domain VI is divided into 2 subdomains (C1 and C2). The C2 subdomain is directly involved in the interaction of IF2 with fMet-tRNA [43,44].
Figure 3.

Organization of initiation factor 2 (IF2). A. Domain alignment of E. coli, G. stearothermophilus, and M. thermoautotrophicum IF2, as well as mammalian IF2mt. B. Model for the 3-D structure of IF2mt, based on the cryo-EM map of IF2mt [63]. Domain 3 has been omitted from the structure, since cryo-EM images were not of sufficient resolution to obtain its coordinates, and no corresponding structures were available in the databases upon which to build a model. Domain IV is shown in blue, domain V is shown in purple, the insertion domain is shown in orange, domain VI-C1 is shown in green, and domain VI-C2 is shown in pink.
IF2mt begins with a mitochondrial import signal. Two natural variants of the IF2mt coding sequence have been noted and the first of these is found in the import sequence. The N-terminus of the mature protein is blocked and the actual start position of the mature form of this factor is unknown [45]. Initial predictions suggested that the import signal would encompass the first 28-29 amino acids and this is the current prediction using TargetP [46]. However, MitoProtII [47] predicts a 77 amino acid import signal and PSort2 predicts an 86 residue import signal [48]. Most of the work on the properties of IF2mt expressed in E. coli has been carried out on the version of the protein encompassing residues 78-727. This form is stably expressed in bacterial cells. Constructs based on a shorter import signal give rise to extensive proteolysis products and aberrant internal initiation products when expressed in E. coli [49]. The second variant of the IF2mt coding sequence is a conservative change of Val to Ile at amino acid 556.
Considering a mature form of IF2mt beginning at amino acid 78, the mitochondrial factor encompasses domains III-VI viewed on the basis of the six-domain model of E. coli IF2 (Fig. 3). Of these domains, the G-domain is the most highly conserved. For a detailed analysis of the conservation of these domains, the reader is referred to a previous review [50]. Of particular interest is the presence of a small insert of 37 amino acids present in the mitochondrial factor but not observed in the prokaryotic factors (discussed in 2.2.6) [51]. This insertion is located between domains V and VIC1 and its role is discussed below. It should be noted that there is no closer relationship between mammalian IF2mt and yeast IF2mt than between the mammalian factor and the IF2 of bacteria. This observation reflects the substantial divergence of the mitochondrial translational systems of the higher and lower eukaryotes.
2.3.4. Interaction of IF2mt with (f)Met-tRNA
Mammalian mitochondria are unique in having a single species of tRNAMet. Other translational systems, including the mitochondrial systems of plants and the lower eukaryotes, have separate tRNAMet species for initiation and elongation. Mitochondrial tRNAMet is aminoacylated with methionine and the Met-tRNA interacts with elongation factor Tu (EF-Tumt) for use in translational elongation. A portion of the Met-tRNA is formylated to serve in initiation. Mammalian mitochondria must, therefore, have a method to partition the single Met-tRNA into the initiation and elongation processes. Bovine mitochondrial Met-tRNA transformylase has been cloned and the protein has been studied biochemically [52,53]. The partitioning of Met-tRNA between initiation and elongation is postulated to arise through competition between the transformylase and EF-Tumt. If the Met-tRNA is formylated, it interacts with IF2mt and participates in chain initiation [54]. IF2mt shows a nearly 50-fold preference for the formylated initiator tRNA. If the Met-tRNA interacts with EF-Tumt•GTP, it is channeled to chain elongation. EF-Tumt does not interact with fMet-tRNA to a detectable extent. The region of IF2mt responsible for interacting with the fMet-tRNA has been mapped to the VIC2 subdomain [54].
In yeast mitochondria, formylation of Met-tRNA is not absolutely required for chain initiation, and yeast mutants that cannot formylate the initiator tRNA can carry out mitochondrial protein synthesis [55,56]. The ability of the yeast mitochondrial translational system to function without formylation of Met-tRNA is dependent on the presence of a factor designated Aep3p that is thought to interact with yeast IF2mt and facilitates its use of Met-tRNA [57]. There is no clear homolog of this protein in mammals although it has a weak homology to the small subunit ribosomal protein MRPS27 (e = 9.3×10−2).
2.2.5. Binding of IF2mt to the Small Subunit
The effects of various ligands on the binding of IF2mt to mitochondrial 28S subunits were examined using pull-down assays with biotinylated ribosomal subunits and streptavidin beads [51]. The mature form of IF2mt binds to the small subunit with a Kd of about 10-20 nM. This binding is strongly influenced by the presence of the non-hydrolyzable GTP analog GDPNP. Binding was reduced about 3-fold when GDP was used in place of GDPNP and over 10-fold when guanine nucleotides were omitted from the reaction mixture. For the full-length factor, the addition of fMet-tRNA and mRNA did not affect the binding of IF2mt to the 28S subunit.
The roles of the individual domains of IF2mt on the binding of this factor to mitochondrial 28S subunits were examined using a series of deletion derivatives [51]. Domain III plays an important role in the interaction of this factor with the ribosome and makes important contacts with the 28S subunit. Removal of domain IV, the G-domain, inactivates IF2mt as expected and also prevents effective binding to 28S subunits. Domain IV is not expected to interact with the small subunit since the G-domain should interact with the 39S subunit in the vicinity of the ribosomal L7/L12 stalk and the sarcin-ricin loop similar to the other translational G-proteins [58,59]. Presumably the presence of the G-domain is necessary for the correct arrangement of the domains to allow ribosome binding. Domain VI and in particular domain VIC2 is involved with the binding of the initiator tRNA. To achieve further insight into the function of the 37 amino acid insertion in IF2mt (Section 2.2.3), several residues in this amino acid stretch were mutated [51]. These mutations caused a significant reduction in the ability of IF2mt to bind to the 28S subunit indicating that this region plays an important role in the interaction of this factor with the small subunit.
2.2.6. Role of the Insertion Domain in IF2mt
IF1 is a small protein (71 amino acids in E. coli) that has been viewed as a “universal” initiation factor being present in both prokaryotic and eukaryotic cytoplasmic systems. No mitochondrial translational system has been shown to possess a factor equivalent to IF1. Studies with bovine IF2mt indicated that this factor is active on E. coli ribosomes but that its activity is not stimulated significantly by IF1 [60] suggesting that the mitochondrial factor might possess a region functionally equivalent to IF1. The best candidate for such a region is the 37 amino acid insertion present between domains V and VI of the mitochondrial factor (Fig. 3). To test this possibility, a strain of E. coli was constructed carrying a deletion of the infB gene encoding IF2 (ΔIF2). This deletion is lethal unless complemented by a source of functional IF2. Interestingly, an expression construct of IF2mt supported growth of the E. coli ΔIF2 strain, indicating that the mitochondrial factor is functional in this prokaryotic system. The infA gene encodes IF1 and its deletion is also lethal to E. coli. Interestingly, IF2mt was able to support the growth not only of the ΔIF2 strain but was also able to rescue cells carrying deletions in both the IF2 and the IF1 genes. This observation indicates that IF2mt plays the role of two initiation factors in mitochondria. When the 37 amino acid insertion in IF2mt was deleted, the truncated factor could still support the growth of the ΔIF2 strain but could not support growth of the ΔIF2ΔIF1 strain. Thus, it appears that the insertion in the mitochondrial protein serves the function of IF1 for mitochondria. Biochemical assays confirmed this conclusion [60].
E. coli IF1 binds to the bacterial 30S subunit near the aminoacyl-tRNA binding site, the A-site, blocking this site during polypeptide chain initiation [61,62]. To assess whether the 37 amino acid insertion in IF2mt interacts with the same binding site on the ribosome, cryo-electron microscopy was performed on an E. coli initiation complex formed with 70S ribosomes, IF2mt, fMet-tRNA, mRNA and GDPNP [63]. IF2mt makes extensive contacts with the interface sides of both ribosomal subunits extending from the lower body of the ribosome to the decoding center (Fig. 4A). Analysis of this complex indicates that domains III, V and VIC1 interact with the 30S subunit; the G-domain (IV), portions of domain VIC1 and parts of domain VIC2 contact the large subunit. Domain VIC2 also interacts with the -CCA end of the fMet-tRNA.
Figure 4.

Binding of IF2mt to the E. coli ribosome. This image was taken from the cryo-EM structure of the 70S:mRNA:fMet-tRNA:IF2mt:GDPNP complex [63] and shows the orientation of IF2mt on the E. coli ribosome. The bacterial 30S subunit is shown in light yellow and the bacterial 50S subunit is shown in blue. Defining features of each subunit are labeled, including the head, shoulder and spur of the small subunit and the central protuberance and L7/L12 stalk base on the large subunit. IF2mt is shown in red, and the initiator tRNA is shown in green. The authors thank Dr. Rajendra Agrawal, Wadsworth Center, New York State Dept. of Health, Albany, NY for the cryo-EM image.
The insertion domain of IF2mt occupies the same general position in the initiation complex as does E. coli IF1. It makes contact with portions of helices 18 and 44 as well as ribosomal protein S12. Although the insertion is only about half the size of IF1, its position in the 70S initiation complex clearly overlaps the position occupied by IF1. This data and the ability of IF2mt to replace both IF1 and IF2 in vivo argue strongly that a single initiation factor in mammalian mitochondria serves the role of two bacterial initiation factors. The interior of the mitochondrion is very densely packed and is thought to have a protein concentration of 800 mg/mL compared to about 300 mg/mL in bacterial cells [64]. Further, the invaginations in the inner membrane limit diffusion. The integration of the function of IF1 into IF2mt may have occurred during evolution to circumvent problems arising from the crowded and topologically complex structure of this organelle.
Examination of the electron density of the 70S initiation complex formed with IF2mt allowed the development of a three-dimensional model for the structure of this factor (Fig. 4B). This model does not include domain III. Although electron density for this region of IF2mt could be observed, no structural information on homologous proteins is known for this domain preventing the development of an accurate model for this region of the factor. The model for the remainder of IF2mt was based on the crystal structure of Methanobacterium thermoautotrophicum aIF2 and the NMR structures of the VIC1 and VIC2 domains of Bacillus (now Geobacillus) sterarothermophilus [63]. In this model, IF2mt resembles the structure of the archaeal and eubacterial factors with the exception of the 37 amino acid insertion. The insertion projects from the body of the protein between domains V and VIC1.
2.3. Mitochondrial Translational Initiation Factor 3
2.3.1 Background
Cybersearching of the human EST database with the sequences of a number of bacterial IF3 species did not reveal the presence of IF3mt. However, searches using the sequence of Mycoplasma IF3 or Euglena gracilis chloroplast IF3 as queries provided hits in the human and mouse ESTs [65].
2.3.2. Characteristics of the IF3mt Coding Sequence
The coding region of IF3mt encompasses 278 amino acids. Native mammalian mitochondrial initiation factor 3 (IF3mt) has never been purified and the actual N-terminus is not known. MitoProtII, PSort2 and TargetP all predict that this protein will be localized to mitochondria but all predict a different import signal cleavage site. For expression studies we have used the prediction by MitoProtII which suggests that human IF3mt will have a 31 amino acid import signal giving rise to a 247 amino acid mature form of the protein.
The organization of IF3mt based on a 31 amino acid import signal shows a central region with homology to the bacterial factors (Fig. 5) [65]. The prokaryotic factors are organized into an N-terminal domain and a C-terminal domain separated by a flexible linker of about 27 amino acids. Removal of the import signal of IF3mt leaves a 29 kDa protein that has regions with homology to the N- and C-terminal domains of bacterial IF3. The N-terminal homology domain is preceded by an extension of 31 amino acids and there is an extension of 33 amino acids following the C-terminal domain. Quite short N-terminal extensions compared to E. coli and related IF3 species are observed in a few prokaryotes. A long N-terminal extension is observed in Euglena gracilis chloroplast IF3 (about 150 amino acids).
Figure 5.

Organization of IF3mt. A. Domain organization of IF3mt. The regions of IF3mt with homology to bacterial IF3 are indicated. B. 3-D model of IF3mt based on the crystal structure of the N-terminal domain of G. stearothermophilus IF3 and the NMR structure of the C-terminal domain of mouse IF3mt [66]. The N-extension is shown using a dashed grey line and could not be modeled due to its predicted lack of structure. The N-domain is shown in yellow, the linker is shown in pink, and the C-domain is shown in blue. The C-extension is shown using a dashed green line and could not be modeled since no structure could be predicted for this region of IF3mt.
Clear homologs of human IF3mt are observed in the genomes of other mammals where the sequences are generally more than 60 % conserved. Lower conservation (about 35-40 %) is observed with the IF3mt of other vertebrates. It is more of a challenge to identify a putative IF3mt in the genomes of invertebrates. Reasonable candidates are observed in the Arthropoda including Drosophila and several mosquito genera. No convincing homolog is observed in Ceanorhabditis elegans although other nematodes such as Trichinella spiralis have reasonable candidates for IF3mt. Homologs of IF3mt can be observed in several fungi although no biochemical characterization of these proteins has been carried out to demonstrate their possible role in mitochondrial protein synthesis. Interestingly, no clear homolog of IF3mt is observed in the genome of Saccharomyces cerevisiae.
The homology domains of IF3mt are not particularly well conserved compared to bacterial IF3. In general, there is less than 25 % identity between the mammalian mitochondrial and bacterial factors. Overall, the C-terminal domain is more highly conserved than the N-terminal domain. The greatest homology to prokaryotic factors is observed with the IF3 from Elusimicrobium minutum and Dictyoglomus turgidum as well as several species of Mycoplasma. The evolutionary implications of the homology of IF3mt to various bacterial factors are not clear.
Although there is a low percent homology between G. stearothermophilus IF3 and the mammalian mitochondrial factor (23 %), the structure of the N-terminal homology domain of IF3mt can be modeled using Insight II based on the X-ray structure of G. stearothermophilus (formerly Bacillus) IF3 [66]. This region of IF3 is folded into a helical unit that is packed against a four stranded β-sheet (Fig. 5B). A second helical segment projects into the linker region. The N-terminal extension cannot be modeled and is predicted to be unstructured in solution. Presumably it assumes structure upon binding to the small ribosomal subunit.
The C-terminal domain of IF3mt has been modeled based on the NMR structure of the C-terminal domain of mouse IF3mt (PDB coordinates 2CRQ). This domain consists of two α-helices that lie on top of a 4-stranded β-sheet. The C-domain of the human IF3mt is 72 % identical to that of the mouse factor and this domain of human IF3mt is predicted to fold into a similar structure except that one of the β-strands breaks into two sections due to the presence of an internal proline residue. The NMR structure of the mouse factor does not include the C-terminal extension and no structure for this region could be predicted using Insight II.
The crystal structure of G. stearothermophilus IF3 indicates that the linker is α-helical and the model using this structure predicts a partially α-helical linker region. However, biochemical studies of IF3 indicate that the linker region has considerable flexibility and may assume a fixed structure only when IF3 binds the ribosome [67-69].
2.3.3. The IF3mt Gene
The gene for IF3mt covers nearly 15 kilobase pairs and is located on chromosome 13 (q12.2). A pseudogene is located on chromosome 5. A number of splice variants are observed. Four major mRNAs differing in the 5′ untranslated region (UTR) are listed in RefSeq at NCBI. These do not correspond exactly to the analysis of the gene in Emsembl but are close. The mRNAs range from 999 to 1098 nucleotides in length. They direct the synthesis of the same protein that is encoded by the last three exons of the gene. The differences in the 5′ UTR arise from the selection of different start sites and alternate splicing. Analysis of the 5′ UTRs indicate that all of the four major mRNAs have upstream open reading frames (uORFs) that could influence the level of expression of the IF3mt protein. In general, the uORFs are not present in a particularly good Kozak context [70] but could initiate to some extent. In all four variants, one would expect these uORFs to reduce the amount IF3mt synthesized. This observation is compatible with the idea that the amount of IF3mt is quite low, on the order of 10-20 molecules per mitochondrion.
Two natural polymorphisms are observed in human IF3mt. The first is a Thr to Leu variant located in the beginning of the N-terminal homology domain at position 68. A second variant is found at the beginning of the C-terminal extension (position 247) with a Phe to Leu change. It is unlikely that either of these changes would affect the properties of IF3mt. However, a silent mutation at nucleotide 798 of the coding region is correlated with Parkinson’s disease [71]. This mutation is believed to reduce the stability of the IF3mt mRNA leading to a reduction in the amount of this factor in mitochondria and, hence, to a reduction of mitochondrial protein synthesis. Defects in the assembly of the respiratory chain complexes could lead to oxidative stress to which dopaminergic neurons may be particularly sensitive.
2.4.4. Biological Roles of IF3mt and Analysis of Critical Regions of this Factor
IF3mt promotes the binding of fMet-tRNA to the mitochondrial 55S ribosome in the presence of IF2mt and mRNA. This stimulation is largely due to the ability of this factor to promote the dissociation of the ribosome into the large and small subunits allowing mRNA and initiator tRNA binding to the 28S subunit [65]. IF3mt has another interesting activity not observed in prokaryotic systems and that is the ability to dissociate fMet-tRNA bound to ribosomes in the absence of mRNA. This observation suggests that there may be a required order of assembly of the initiation complex with mRNA binding preceding the binding of the fMet-tRNA [72] (See model in Fig. 2). In eubacteria, IF3 is thought to proofread the initiation complex and to promote the dissociation of aminoacyl-tRNAs bound at non-AUG codons. However, the residues thought to be important for this activity in bacterial IF3 are not conserved in the mitochondrial factor [69,73,74] and no proofreading ability has been observed with this factor. It should be noted that mitochondrial ribosomes initiate translation with both AUG and AUA and that the lone tRNAMet species participates both in chain initiation and in chain elongation perhaps making a traditional proofreading activity for IF3mt unlikely.
The binding of IF3mt to mitochondrial 28S subunits has been studied by a number of methods including surface plasmon resonance, micron centrifugation and sucrose density gradient centrifugation [75]. The full-length mature form of this factor binds to the small subunit with a Kd of about 30 nM. The C-domain with the linker also binds tightly with a Kd of about 60 nM. When the linker region is removed, the binding affinity decreases to a Kd of about 95 nM. These observations indicate that there are strong contacts between the C-terminal domain and the 28S subunit and that the linker slightly enhances these interactions. The C-terminal domain is believed to bind to the platform region of the small subunit (section 2.3.5) where it would block several intersubunit bridges that form in the intact monosome. The strong affinity of the C-terminal domain for the small subunit is in contrast to observations made in the E. coli system in which the C-domain alone has a 100-fold lower affinity for the 30S subunit than the intact factor [73]. The C-domain of IF3mt alone is as active as the full-length mature form of the factor when tested for the ability to promote initiation complex formation with mitochondrial 55S ribosomes. This observation reflects the idea that this assay measures primarily the ability of IF3mt to promote ribosome dissociation. In independent tests, the C-domain has been shown to be about 9-fold less effective than the full-length factor in promoting the dissociation of 55S ribosomes. This decrease in ribosome dissociation is apparently not limiting for the initiation complex assay.
Mutagenesis has been used in an effort to define the regions of IF3mt that are important for its activity [66]. Mutations of residues 170-171 and of residue 175 to alanine in the C-terminal domain of IF3mt lead to an almost complete loss of activity in promoting initiation complex formation and in the dissociation of 55S ribosomes into subunits. Surprisingly, both of the inactive derivatives of IF3mt can bind to 28S subunits as well as the wild-type factor indicating that a defect is small subunit binding is not responsible for the loss of activity in promoting initiation complex formation. Rather, it appears that the mutations lead to a factor that is unable to promote the dissociation of 55S ribosomes into free 39S subunits and the 28S:IF3mt complex. This observation strongly suggests that IF3mt acts as an active ribosome dissociation factor and not as a passive anti-association factor (Fig. 2).
The N-terminal domain of IF3mt also binds rather well to the 28S subunit (Kd about 240 nM) in the absence of other factors [75]. This observation is in sharp contrast to those made in bacterial systems in which no binding of the N-terminal domain alone can be observed to the small subunit. The linker provides some of this interaction energy for the binding of the N-terminal domain of IF3mt to the small subunit and, when it is removed, the Kd is noticeably weaker (390 nM). The isolated N-terminal domain is not detectably active in promoting the dissociation of the 55S monosome and has little activity in promoting initiation complex formation. These results provide a picture of IF3mt as a dumbbell shaped molecule in which there are extensive contacts between both the N- and C-terminal domains and the small subunit. The linker optimizes the affinity of these domains for the 28S subunit making it more difficult for the large subunit to displace the factors from the 28S subunit.
The dissociation of the 55S ribosome by IF3mt requires the binding of this factor to the small subunit, thereby excluding its interaction with the 39S subunit. Full-length IF3mt has essentially no affinity for the large subunit (Kd = 1.5 μM, about 50-fold weaker than binding to the 28S subunit). However, deletion of the extensions results in about a 10-fold increase in the affinity of IF3mt for the large subunit. The N-terminal extension accounts for most of this effect with a small contribution from the C-terminal extension. This observation suggests that the N-terminal extension on IF3mt could have evolved to reduce the affinity of the factor for the large subunit, thereby promoting the proper interaction of the factor with the 28S subunit during initiation.
As indicated in the current working model for initiation in mammalian mitochondria (Fig. 2), mRNA binding is thought to precede the binding of fMet-tRNA. One of the roles of IF3mt is to promote the dissociation of any fMet-tRNA that binds to the 28S subunit prior to the mRNA. When the C-terminal extension is removed, this activity is almost completely lost, indicating that this extension may have evolved to promote premature binding of fMet-tRNA, thus, ensuring the proper sequence of events during initiation. Mutational analysis has identified two residues at positions 247 and 248 in the C-terminal extension critical for the ability of IF3mt to promote the dissociation of fMet-tRNA bound to 28S subunits in the absence of mRNA. This activity also requires the presence of the linker on the C-terminal domain [75]. The C-terminal extension is predicted to emerge from the C-terminal homology domain pointing toward the linker region (Fig. 5B) making it likely that these two regions of IF3mt work together to dissociate prematurely bound fMet-tRNA.
2.3.5. Binding Site of IF3mt on the Mitochondrial 28S Subunit
Mammalian mitochondrial ribosomes are protein-rich structures. About half of the proteins in these particles have homologs in bacterial systems while the other half are unique to mitochondrial ribosomes. A number of biochemical and biophysical studies have indicated that the C-terminal domain of E. coli IF3 binds to the platform region of the 30S subunit while other portions of this factor are located near the cleft and head of the subunit [76-80]. The platform region is one of the more highly conserved regions between bacterial and mitochondrial ribosomes [16,34]. However, the edges of the platform contain proteins specific to the mitochondrial ribosome. The head of the 28S subunit lacks a number of the bacterial ribosomal protein homologs and appears to contain several proteins specific to mitochondrial ribosomes. To obtain insights into the protein neighbors of IF3mt on the 28S subunit a chemical cross-linking procedure followed by mass spectrometry was undertaken [81]
Using this strategy, it was observed that IF3mt cross-links to mammalian mitochondrial homologs of the bacterial ribosomal proteins S5, S9, S10, and S18(2) (Fig. 6) and to unique mitochondrial ribosomal proteins MRPS29, MRPS32, MRPS36 and PTCD3 (Pet309). The cross-link to MRPS29 is somewhat surprising. This protein is also referred to as death associated protein 3 (DAP3) and is thought to play a role in apoptosis [82,83,84]. Immune electron microscopy has indicated that MRPS29 is located on the solvent side of the 28S subunit, on the opposite face of the subunit where IF3mt is thought to bind. However, MRPS29 is large for a ribosomal protein (nearly 44 kDa) and may have extensions that penetrate a considerable distance through or around the body of the subunit. PTCD3 is a pentatricopeptide domain protein known to associate with the small subunit of mitochondrial ribosomes [85]. Recent data has shown that PTCD3 is a small subunit ribosomal protein in mammals (E. Koc, manuscript in preparation). The location of this protein in the mitochondrial small subunit is not known. However, it cross-links extensively to IF3mt, suggesting that it may be at least partially located at the interface side of the 28S subunit.
Figure 6.
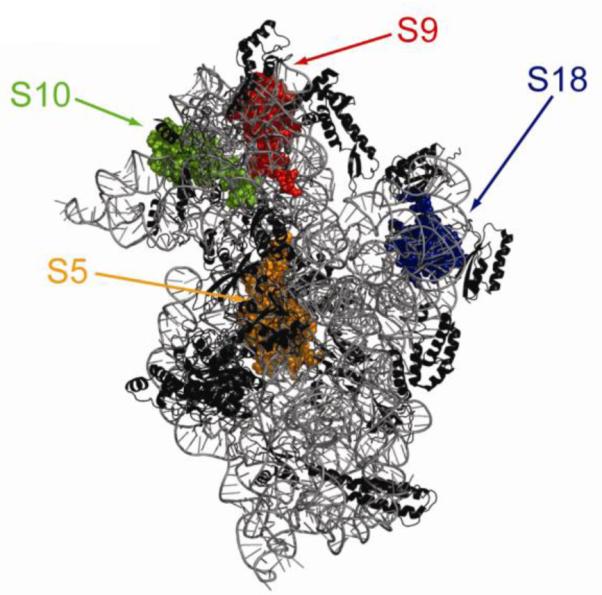
Interaction sites of IF3mt on the small ribosomal subunit. The structure of a “mock” mitochondrial 28S small subunit was prepared based on the crystal structure of the Thermus thermophilus 30S ribosomal subunit. The small subunit is shown with the interface side facing the reader and only homologous proteins and RNA segments present in the mitochondrial 28S subunit are shown. The ribosomal proteins that can cross-link to IF3mt are spaced-filled. Small subunit protein S5 is shown in orange, S9 in red, S10 in green, and S18 in blue.
Removal of the N- and C-terminal extensions of IF3mt resulted in the same cross-linking pattern as the full length IF3mt except that no cross-links were observed to MRPS36. Only one ribosomal protein (MRPS10 located in the head of the small subunit) was observed cross-linking to the N-terminal domain. The C-terminal domain (including the linker) cross-linked to MRPS5, MRPS9, MRPS10 and MRPS18(2) , all of which have bacterial homologs. Like the full-length factor, the C-terminal domain with the linker cross-linked to a number of proteins that do not have prokaryotic homologs including MRPS32 and MRPS36, MRPS29 and PTCD3.
The pattern of cross-links observed between IF3mt and 28S subunits is quite different from those observed with E. coli IF3 and 30 subunits indicating that contacts between IF3mt and mitochondrial small subunit are quite different from the contacts between bacterial IF3 and the small subunit of the prokaryotic ribosome.
3. Polypeptide Chain Elongation in Mammalian Mitochondria
3.1. Overview of the Cycle
The process of polypeptide chain elongation in mammalian mitochondria has many similarities to the process in prokaryotes [50,86]. This phase of translation has been more highly conserved during evolution than either the initiation or termination phases. In the basic sequence of events (Fig. 7), the GTP-bound, active, form of elongation factor Tu (EF-Tumt) binds aminoacyl-tRNA (aa-tRNA) forming a ternary complex (EF-Tumt• GTP•aa-tRNA). This ternary complex enters the A-site of the ribosome and is selected if cognate codon:anticodon interactions can take place (Step 1). Selection of the cognate ternary complex triggers the hydrolysis of GTP by EF-Tumt and the release of EF-Tu•GDP (Step 2). Elongation factor Ts (EF-Tsmt) promotes the exchange of GDP for GTP through the formation of an intermediate EF-Tumt•EF-Tsmt complex (Steps 3 and 4). The ribosome itself catalyzes peptide bond formation leaving a deacylated tRNA in the P-site and a peptidyl-tRNA one residue longer in the A-site of the ribosome (Step 5). Mitochondrial elongation factor G1 (EF-G1mt) then catalyzes the translocation step removing the deacylated tRNA from the P-site and moving the peptidyl-tRNA from the A-site to the P-site (Steps 6 and 7). No E-site is shown in this model since analysis of the cryo-EM of the mitochondrial ribosome suggests that mammalian mitochondrial ribosomes do not possess a site corresponding to the prokaryotic E-site [34].
Figure 7.

Model for the elongation phase of mitochondrial translation. The tRNA containing the growing polypeptide chain is located in the P-site of the ribosome. EF-Tumt brings the aa-tRNA to the A-site of the ribosome (Step 1). In concert with the hydrolysis of GTP to GDP, EF-Tumt leaves the ribosome (Step 2). EF-Tsmt binds to EF-Tumt, displacing the GDP molecule and forming an EF-Tumt·EF-Tsmt complex (Step 3). A GTP molecule displaces EF-Tsmt, and an EF-Tumt:GTP complex is formed (Step 4) which can then bind another aa-tRNA reforming the ternary complex. The large ribosomal subunit catalyzes peptide bond formation and the growing polypeptide chain is transferred to the tRNA in the A-site of the ribosome (Step 5). EF-G1mt:GTP binds to the ribosome at the A-site (Step 6) and catalyzes translocation of the ribosome, moving the deacylated tRNA out of the P-site and the peptidyl-tRNA from the A-site to the P-site (Step 7). A new cycle of elongation can then begin.
All of the elongation factors from mammals have been purified from bovine mitochondria and the properties of the native factors have been examined [37,87-91]. In addition, the bovine and human factors have been cloned, expressed in E. coli and further characterized [92-95]. The properties of the elongation factors have been extensively reviewed previously [50,96] and the reader should consult those reviews for more detailed information.
3.2: Elongation Factor EF-Tumt
3.2.1.General Properties
Native EF-Tumt has been isolated as a tight complex with EF-Tsmt from bovine liver. Unlike the prokaryotic system, no free EF-Tumt or free EF-Tsmt is observed in mitochondrial extracts [91]. The EF-Tumt• EF-Tsmt complex resists dissociation even at high concentrations of guanine nucleotides. This stability probably reflects the observation that guanine nucleotide binding to EF-Tumt is over 100-fold weaker than that observed with E. coli EF-Tu (Kd for GTP = 18 μM and Kd GDP = 1 μM) while binding to EF-Tsmt is quite tight (Kd = 5.5 nM) [90,97,98]. The strength of ternary complex formation depends on the aa-tRNA. For example the Kd for E. coli Phe-tRNA is about 18 nM while the Kd for bovine mitochondrial Phe-tRNA is about 75 nM [99].
3.2.2. The EF-Tumt Gene and Coding Region
The human gene for EF-Tumt (TUFM) was originally identified as producing a protein overexpressed in certain tumors [100] and portions of the native protein from Bos taurus were then sequenced from bovine liver [94]. The chromosomal location of the gene is at 16p11.2 and the gene contains 10 exons. A pseudogene is observed on chromosome 17. EF-Tumt has a mitochondrial import sequence of 43 amino acids and a mature form that is 409 residues in length with a native molecular mass of about 45,000 daltons. It is 55-60 % identical to the corresponding factors from bacteria (Fig. 8A).
Figure 8.

Regions of EF-Tumt responsible for interacting with aa-tRNA. A. Alignment of EF-Tumt and E. coli EF-Tu domain II [106]. The amino acid residues of domain II that interact with the 3′ acceptor stem region of the aminoacyl-tRNA are indicated with a •, and the residues that interact with the 5′ end of the tRNA are indicated with a B. Interaction of EF-Tu with Cys-tRNA based on the crystal structure of T. aquaticus EF-Tu:GDPNP:E. coli Cys-tRNACys (PDB# 1B23). C. Close-up image of the box in B. Image is rotated approximately 90 degrees. The Cys-tRNACys is shown in gray, EF-Tu is shown in orange, and GDPNP is shown in purple. The residues of EF-Tu that contact the tRNA are shown in pink, the 5′ G of the tRNA is shown in blue and the A of the 3′ -CCA end of the tRNA is green. The 5′ phosphate group of the tRNA is indicated by the black arrow.
The crystal structure of bovine EF-Tumt has been determined [101] and indicates that the mitochondrial factor is folded into three major domains as is bacterial EF-Tu (Fig. 8 and 9). Domain I is responsible for binding guanine nucleotides. Domain II, along with domain I, forms the binding site for the 3′ end of the aa-tRNA and contacts the small subunit of the ribosome. Domain III interacts with the extended acceptor-TψC helix of the aa-tRNA and all three domains form a pocket interacting with the 5′ end and a portion of the acceptor stem of the aa-tRNA (Fig. 8B and C). One of the major differences between the mitochondrial and bacterial factors is an extension of 11 amino acids at the C-terminus of EF-Tumt that may be involved in an interaction with the aa-tRNA in the ternary complex [50,101]. For a detailed discussion of the structure of EF-Tumt, and the binding of guanine nucleotides to this factor the reader is referred to a previous review [50].
Figure 9.

Crystal structure of the bovine EF-Tumt:EF-Tsmt complex. In the 3-D structure of the bovine EF-Tumt:EF-Tsmt complex (PDB coordinates 1XB2) [101], EF-Tumt is shown in orange and EF-Tsmt is in blue. The domains of each protein are labeled and the position of the insertion present in one isoform of EF-Tsmt is indicated by a red line.
3.2.3. Interaction of EF-Tumt with Mitochondrial aa-tRNA
The structures of several bacterial ternary complexes have been solved [102,103]. The residues of EF-Tu that form the binding pocket for the aa-tRNA have been largely conserved between the bacterial and mitochondrial factors (Fig. 8). However, several residues (Gln290, Arg335 and Pro269) are not conserved between bacterial EF-Tu and EF-Tumt. The potential importance of these residues in the ability of EF-Tumt to form a ternary complex with mitochondrial aa-tRNAs was tested by mutating the residues in the mitochondrial factor to those found in E. coli EF-Tu [104-106]. Of these residues, Pro269 did not appear to play a critical role; however, mutation of either Arg335 to Glu or Leu338 to Gln had a strong deleterious effect on the activity of EF-Tumt. Modeling of the structure of EF-Tumt suggests that Arg335 makes an important electrostatic contact with the 5′-phosphate of the aa-tRNA. Leu338 is present in a loop that helps form the binding pocket for the 5′ end of the aa-tRNA and clearly must play a role in the conformation of that loop to accommodate the formation of the ternary complex.
Some years ago [107] it was observed that, while EF-Tumt was active when supplied with E. coli aa-tRNAs, E. coli EF-Tu could not catalyze polypeptide chain elongation with mitochondrial aa-tRNAs. The bacterial factor was able to form a ternary complex with mitochondrial aa-tRNA but was unable to deliver them to the A-site of the ribosome. Domain exchanges between EF-Tumt and E. coli EF-Tu [108] indicated that the ability to deliver mitochondrial aa-tRNA to the ribosome resides primarily in domains I and II. When codon:anticodon interactions take place, a signal is sent through the body of the tRNA to domain I of EF-Tu, triggering GTP hydrolysis. The shorter mitochondrial aa-tRNAs may not be positioned quite correctly on the ribosome by the bacterial factor and fail to promote the GTPase activity of EF-Tu effectively. In the absence of GTP hydrolysis, EF-Tu cannot be used catalytically and, thus, is less active in promoting A-site binding [50].
3.3: Elongation Factor EF-Tsmt
3.3.1.Sequence Analysis
The gene for human EF-Tsmt (TSFM) maps to chromosomal location 12q14.1 and spans a bit over 20,000 base pairs. Four isoforms of EF-Tsmt are listed in RefSeq and NCBI while Expasy lists only two forms (isoforms 1 and 2). The NCBI isoforms 3 and 4 are extensively truncated at the C-terminus and are unlikely to represent functional forms. The nomenclature used by NBCI and Expasy for the two likely isoforms of EF-Tsmt is different leading to some potential confusion. In NCBI, human isoform 1 is listed as a 346 residue pre-protein containing a small additional exon compared to human isoform 2 which is 325 amino acids in length including the mitochondrial import sequence. Expasy defines the shorter 325 residue form (6 exons) as isoform 1 and refers to this as the ‘canonical’ sequence. The 325 residue human form aligns with the form of EF-Tsmt found in other mammals and is most likely the major active species.
3.3.2. Properties of EF-Tsmt
The most extensively studied form of EF-Tsmt is from Bos taurus. This protein has been purified from bovine liver as a tight complex with EF-Tumt. The amino terminus of the mature protein has been determined and this analysis indicates that there is a 55 residue mitochondrial import signal [93] giving a mature protein of 283 amino acids with a molecular mass of about 31,000 daltons. EF-Tsmt has been expressed in E. coli as a His-tagged protein [95]. Under these conditions, it forms a tight heterologous complex with E. coli EF-Tu that is very difficult to dissociate (Kd is about 10−11). This interaction is almost 100-fold tighter than the homologous E. coli EF-Tu•EF-Ts complex [95]. Purification of EF-Tsmt requires denaturation of this complex and renaturation of the remaining free EF-Tsmt.
3.3.3. Structure of the EF-Tu•EF-Tsmt Complex
Bacterial EF-Ts is organized into four domains: an N-terminal helical domain; a core domain divided into subdomain N and subdomain C of the core, a dimerization or coiled-coil domain and a C-terminal module [109]. The low degree of sequence conservation (25-30 %) between bacterial and mitochondrial EF-Ts made it difficult to align these proteins clearly until direct structural information became available on the bovine EF-Tumt•EF-Tsmt complex [101] (Figure 9). This structure indicates that EF-Tsmt has both similarities and differences compared to the bacterial factors. EF-Tsmt has an N-terminal domain with a similar fold to that of the prokaryotic factors. However, the core of EF-Tsmt, while forming basically a β-sandwich, differs in the number and organization of the β-strands. Most striking is the almost complete erosion of the coiled-coil domain and the C-terminal module found in bacterial EF-Ts.
As indicated in Section 3.1, the major role of EF-Ts is to promote guanine nucleotide exchange with EF-Tu. Three major areas of contact are observed in the EF-Tumt•EF-Tsmt complex [101]. The N-terminal domain and subdomain N of the core contact the G-domain (domain I) of EF-Tumt, while subdomain C of the core contacts domain III of EF-Tumt (Fig. 9). Detailed analysis of the effects of mutations in EF-Tsmt suggests that nucleotide exchange involves several critical processes [108,110-113]. These include disruption of the binding site for the Mg2+ ion, which stabilizes the bound guanine nucleotide, a destabilization of the interactions of the β-phosphate group with EF-Tumt and movements of portions of EF-Tumt that affect the interactions of the ribose and base with the factor. Thus, there are extensive changes in the structure of EF-Tumt when it interacts with EF-Tsmt, and substantial regions of both factors are involved in the nucleotide exchange process. For a more detailed analysis of the interactions between EF-Tumt and EF-Tsmt the reader is referred to the structural work and to a previous review [50,101].
3.4 Mitochondrial Elongation Factor G1
3.4.1. Gene
EF-G is generally viewed as the factor required for the translocation step of protein biosynthesis. Two forms of EF-Gmt are observed in mitochondrial systems [114,115]. EF-G1mt is involved in translocation while EF-G2mt appears to be used primarily during termination (see Section 4.3). The gene for EF-G1mt spans about 50,000 base pairs and encodes a mRNA of 3,468 nucleotides. It maps to chromosome 3q25. Expasy lists two isoforms in humans that are splice variants with the larger form (isoform 2) having an additional 19 amino acids. Isoform 1 (751 amino acids) is viewed as the canonical form and corresponds to the EF-G1mt generally found in mammals. For an analysis of the sequence of EF-G1mt, the reader is referred to a previous review [50].
3.4.2. Enzymatic Properties
Native EF-G1mt was purified about 14,000 fold to near homogeneity from bovine liver mitochondria a number of years ago [88]. The native protein has an apparent molecular mass of 80,000 daltons on SDS-polyacrylamide gel electrophoresis. Few detailed enzymatic studies have been carried out on this protein. The N-terminus is blocked. MitoProtII predicts a 35 amino acid mitochondrial import signal. Based on this prediction, residues 36-751 were cloned and expressed in E. coli as a His-tagged protein [92]. EF-G1mt is quite resistant to the antibiotic fusidic acid compared to other translocases. A number of residues in bacterial EF-G have been implicated in the sensitivity of this factor to fusidic acid. These residues are generally present in EF-G1mt and the resistance of this factor to inhibition remains to be understood. The native and the expressed protein are active with both mitochondrial 55S ribosomes and bacterial 70S ribosomes. In contrast, E. coli EF-G is not active on mitochondrial ribosomes [88,116]. This ribosome specificity appears to reside in the L7/L12 stalk of the large ribosomal subunit [117] that is involved in triggering the GTPase activity of the elongation factors.
4. Polypeptide Chain Termination and Ribosome Recycling in Mitochondria
4.1. Overview of the Process
Several comprehensive reviews covering the termination of translation in mammalian mitochondria have appeared in the past year and only a brief summary will be provided here [118-120]. Analysis of mitochondrial genomes indicates that both UAA and UAG serve as stop codons with the standard UGA stop codon reassigned to tryptophan. In humans two reading frames terminate with AGA and AGG leading to the initial assignment of these codons as stop codons. However, no release factor appears to recognize these codons and it is now believed that they promote a -1 frameshift moving a classical UAG codon into the A-site for termination [3]. In the current model for termination in mitochondria (Fig. 10), either the UAA or UGA stop codon appears in the A-site of the ribosome. This codon is recognized by a release factor mtRF1a (Step 1). Binding of this factor to the ribosome in the presence of GTP triggers the hydrolysis of the peptidyl-tRNA bond by the peptidyl transferase center on the 39S subunit and the release of the completed polypeptide (Step 2). It is not clear how mtRF1a exits the ribosome following polypeptide release. RRF1mt binds to the A-site of the ribosome along with RRF2mt (also known as EF-G2mt) (Step 3) and promotes ribosomal subunit dissociation and release of the deacylated tRNA and the mRNA (Step 4). After RRF1mt and RRF2mt are released from the ribosome (Step 5), another round of protein synthesis begins. The recycling of the ribosome requires the combined action of the mitochondrial ribosome recycling factor (RRF1mt ) and EF-G2mt, also referred to as RRF2mt [115,121,122]. There is no evidence for the presence of a Class II release factor such as bacterial RF3 in mammalian mitochondria. In bacteria, this factor is normally required for the removal of RF1 from the ribosome. Consequently, the mechanism by which mtRF1a is dissociated from mitochondrial ribosomes remains unknown.
Figure 10.

Model for the termination and ribosome recycling phases of mitochondrial protein synthesis. As the termination codon (UAG here) enters the A-site of the ribosome, mtRF1a and GTP bind to the A-site (Step 1) and promote GTP-dependent hydrolysis and release of the polypeptide chain (Step 2). How mtRF1a is released from the ribosome is not known. RRF1mt binds to the A-site of the ribosome (Step 3) and is joined by RRF2mt (also termed EF-G2mt). These factors promote the dissociation of the ribosomal subunits and release of the deacylated tRNA and the mRNA (Step 4). Following release of RRF1mt and RRF2mt (Step 5), the ribosome begins another round of protein synthesis.
4.2. Mitochondrial Release Factors
Database searching in the late 1990’s led to the identification of an open reading frame (mtRF1) with 30-40 % homology to bacterial release factors [121]. Subsequent database searches revealed the presence of three additional open reading frames with homology to RF1 (mtRF1a, C12orf65 and ICT1), all of which are localized to mitochondria. All of these factors have the classical GGQ motif that promotes termination by triggering hydrolysis of the peptidyl-tRNA bond [119]. However, mtRF1 has a variant on the PXT tripeptide motif expected to function in stop codon recognition while mtRF1a has the expected sequence [123,124].
Current studies have now demonstrated that mtRF1a functions in termination at UAA and UAG codons and is, therefore, the release factor used in the decoding process. The roles of the other factors with homology to class 1 release factors are less clear. Of these ICT1 [125,126] has been shown to function as a ribosome-dependent, codon-independent, peptidyl-tRNA hydrolase. In this respect, it may play a role in preventing the stalling of ribosomes on mRNAs that have been cleaved and lack a proper stop codon. When mitochondrial ribosomes stall at the 3′ end of such a mRNA, ICT1 cleaves the peptidyl-tRNA releasing the nascent chain and freeing the ribosome for proper recycling [123]. This system may, thus, play a role equivalent to the transfer-messenger RNA mechanism used in bacteria [126]. While the role of C12orf65 is not known, it has been suggested that this protein may play a role in recycling abortive peptidyl-tRNAs that are released from the ribosome during translational elongation [127].
4.3. Ribosome Recycling in Mammalian Mitochondria
As indicated in Fig. 10, the disassembly of the ribosome •mRNA•tRNA complex requires the action of the ribosome recycling factor (mtRRF) and EF-G2mt (Steps 3 and 4). Depletion of mtRRF in human cell lines is lethal, thus indicating that it is an essential protein [128]. Alignment of human mtRRF with the corresponding bacterial factor indicates that the mitochondrial factor has a long N-terminal extension. This factor is highly conserved in mammals and readily identified in the genomes of vertebrates. It is generally 30-40 % identical to the mtRRF of arthropods but less than 30 % identical to the factor in nematodes. Mammalian mtRRF species share only 25-30 % identity to the bacterial factors, yet mtRRF can bind to E. coli ribosomes indicating that certain contacts between this factor and the ribosome have been conserved during evolution [128]. Three variants are listed in NCBI while seven isoforms are listed in UniProt. These isoforms appear to arise primarily from alternative splicing; however, there is no experimental evidence that the shorter forms are expressed. The gene for mtRRF (MRRF) is located on chromosome 9 (9q33.2).
During termination, RRF works in conjunction with the translocase EF-G. As indicated in Section 4.1, analysis of mammalian genomes indicates the presence of two forms of mitochondrial EF-G (EF-G1mt and EF-G2mt). EF-G1mt functions as the translocase during polypeptide chain elongation while EF-G2mt (also referred to as RRF2mt) is thought to act primarily in chain termination [115]. However, it should be noted that over-expression of EF-G2mt slightly increases the levels of the respiratory chain complexes in cells carrying mutations in EF-G1mt suggesting that it may be able to function during elongation to a limited extent [129]. Many organisms have a single species of EF-G that functions in both elongation and termination. However, others have two forms as observed in mammalian mitochondria [130,131]. Some forms of EF-G2 can function in translocation while others are restricted to the termination phase of translation. EF-G2mt falls largely into this latter category. In bacterial termination, GTP hydrolysis by EF-G is required for ribosome recycling. In contrast, GTP hydrolysis by EF-G2mt is not necessary for the ribosomal splitting reaction directly. Rather, GTP hydrolysis appears to be required for the release of EF-G2mt and perhaps of mtRRF as well [115].
The sequence of EF-G2mt is a bit less highly conserved than that of EF-G1mt although greater than 80 % identity is generally observed with the mammalian species. The mammalian factor is usually 30-40 % identical to EF-G2mt of the lower eukaryotes. A number of shorter isoforms of the human factor are listed in UniProt based on various EST sequences, but there is no experimental evidence that they are functional.
5. Role of the Inner Membrane in the Synthesis of Mitochondrially Encoded Polypeptides
5.1 Background
There is clear evidence that mitochondrial ribosomes are preferentially associated with the IM and, in particular, are enriched in the intracristal regions [1]. The association of mitochondrial ribosomes with the IM allows the synthesis of mitochondrially encoded proteins near sites of their insertion into the membrane. Little is known about the proteins promoting the association of mammalian mitochondrial ribosomes with the IM. However, it is clear that both subunits of the mitochondrial ribosome can be found associated with the IM [132], and it is very likely that the insertion of these polypeptides into the respiratory chain complexes is a co-translational process [133-135].
5.2 Yeast Membrane Proteins Interacting with Mitochondrial Ribosomes
A number of proteins have been implicated in the interaction of yeast mitochondrial ribosomes with the IM (Table 1). The best studied of these is the protein Oxa1p which is involved in the insertion of a number of proteins including subunit II of cytochrome oxidase (Cox2) into the IM [134,136-142]. Oxa1p is an integral membrane protein with 5 transmembrane segments (TMS) [142]. It is the mitochondrial homolog of bacterial YidC which plays a role in both Sec-dependent and Sec-independent pathways in bacteria. The C-terminal 100 amino acids of Oxa1p are located in the mitochondrial matrix and interact with mitochondrial ribosomes [143-145]. Evidence from yeast suggests that Oxa1p can be cross-linked to the nascent chain while it is still on the ribosome and to the ribosomal protein homologous to L23 which is located at the exit site of the large subunit [140,144].
Table 1.
Proteins implicated in ribosome binding to mitochondrial membranes in yeast
| Yeast Protein |
Role | Human Homolog |
Representative Reference |
|---|---|---|---|
| Oxa1p | Co-translational insertion of mitochondrially synthesized proteins and post-translation insertion of imported proteins into the IM |
Oxa1L | [134] |
| Cox18p (Oxa2) |
Post-translational insertion of proteins into IM | Cox18 | [146] |
| Mss2 | Membrane insertion of specific proteins (Cox2) | None | [147] |
| Pnt1p | Membrane insertion of specific proteins (Cox2) | None | [196] |
| Mba1 | Membrane-bound ribosome receptor; | None | [149] |
| Mss51p | Binds newly synthesized Cox1 | None | [148] |
| Coa1p | Required for cytochrome oxidase assembly | None | [197] |
| Mdm38p | Binds ribosomes; Assembly of Complexes III and IV | LetM1 | [135] |
In addition to Oxa1p, a number of other yeast proteins have been implicated in the insertion of mitochondrial translation products into the IM and several of these are thought to interact with the mitochondrial ribosome. However, many of these do not have homologs in mammalian systems (Table 1), and there appear to be fundamental differences between yeast and mammals in the mechanism of synthesis and membrane insertion of the mitochondrial translation products [135,146-150].
In addition to Oxa1, mitochondria contain a homolog of the protein YidC2, (termed Cox18 or Oxa2), found in gram-positive bacteria. Cox18 (Oxa2) which has a homolog in humans, appears to act post-translationally [145]. The homolog of yeast Mdm38p in humans is LetM1 (also called LETM1), a 739 amino acid protein with a single TMS which is discussed in more detail below (Section 5.4).
5.3. Interaction of Mammalian Mitochondrial Ribosomes with Oxa1L
A homolog of Oxa1p (referred to as Oxa1L) is present in humans [151,152]. Human Oxa1L (like its yeast homolog), is believed to have 5 TMS and a C-terminal tail in the mitochondrial matrix which interacts with ribosomes. It has recently been reported that human Oxa1L exists as a 600-700 kDa heterooligomeric complex in mitochondria from human embryonic kidney cells [152]. In human cells, knock-down of Oxa1L leads to defects in the biogenesis of the F1F0-ATP synthase and of Complex I without affecting the levels of Complexes III or IV. These effects are different from those observed in yeast in which the assembly of Complex IV is strongly affected by mutations in Oxa1p [138]. The precise reason for the differences observed is not known. One major possibility is that the human protein may have evolved to possess specificity for the ATP synthase and Complex I. It is also possible that another protein may partially compensate for the low Oxa1L levels in the assembly of Complexes III and IV or that the knockdowns of Oxa1L achieved had not reached the threshold required to show a defect in the assembly of Complexes III and IV.
The C-terminal region encompassing approximately 100 amino acids of the tail of human Oxa1L (Oxa1L-CTT) has been cloned and expresses well in E. coli [153]. In contrast to suggestions made for yeast Oxa1p, the C-terminal tail of human Oxa1L does not form a coiled-coil helical structure in solution. Rather, this region of Oxa1L exists primarily as a monomer in solution, although both dimeric and tetrameric forms are observed at high salt concentrations. A number of techniques have demonstrated Oxa1L-CTT binds to bovine mitochondrial ribosomes and that this interaction is specific for the large subunit. This latter observation is expected since Oxa1L is thought to be involved in the insertion of mitochondrial translation products into the IM and the nascent chain emerges from the exit tunnel in the large subunit. The binding of Oxa1L-CTT to 39S subunits is an enthalpically driven process and is governed by a Kd of 0.3 to 0.8 μM. Stoichiometry calculations indicate that two copies of the C-terminal tail are bound to each 39S subunit [153]. Data derived from steady-state fluorescence quenching and fluorescence lifetime measurements indicate that there are extensive contacts between Oxa1L-CTT and the 39S subunit of the ribosome [154]. These studies also indicate that Oxa1L-CTT undergoes conformational changes and induced oligomer formation when it binds to the ribosome.
Analysis of proteins from the large subunit that cross-link to Oxa1L-CTT indicate that mammalian mitochondrial homologs of the bacterial ribosomal proteins L13, L20 and L28 are at or near the binding site of Oxa1L on the ribosome (Fig. 11). In addition, Oxa1L-CTT can be cross-linked to mammalian mitochondrial specific ribosomal proteins MRPL48, MRPL49 and MRPL51 suggesting that these proteins are located on the back of the large subunit in the vicinity of the exit tunnel. No cross-linking of Oxa1L-CTT to proteins decorating the conventional exit tunnel of the bacterial large ribosomal subunit (L22, L23, L24 and L29) is observed reflecting fundamental differences in the structures of the bacterial and mammalian mitochondrial ribosome as discussed in [153].
Figure 11.

Interaction sites of the C-terminal tail of Oxa1L (Oxa1L-CTT) on the large ribosomal subunit. The 3-D structure of the T. thermophilus 50S ribosomal subunit (PDB 2WRL) is shown viewed from the solvent side. Regions of the rRNA that are not present in the mitochondrial 39S subunit have been removed. Mammalian homologs of bacterial ribosomal proteins predicted to interact with Oxa1L-CTT based on cross-linking studies are shown as space-filled with L13 (red spheres), L20 (green spheres) and L28 (blue spheres). The proteins thought to make up the traditional polypeptide exit tunnel (L22, L23, L24, and L29) are shown in orange spheres and do not cross-link to Oxa1L-CTT. Other large subunit ribosomal subunits are shown in black.
5.4. LetM1in Humans
In yeast Mdm38p has been implicated in the interaction of ribosomes with the inner membrane [135]. This protein has a ribosome binding domain located in the mitochondrial matrix and a region anchored in the membrane itself [155]. Humans appear to have a homologous protein designated LetM1. This protein has been implicated in Wolf-Hirschhorn syndrome, a complex syndrome arising from deletions of the short arm of chromosome 4 [156]. Patients having this syndrome display developmental delays, are generally mentally retarded and often have epileptic seizures. The severity of the symptoms is strongly correlated with loss of the LetM1 gene. Depletion of LetM1 does not lead to a loss of respiratory chain function but does result in mitochondrial swelling and fragmentation [156]. Since yeast Mdm38p is thought to play a role in the interaction of mitochondrial ribosomes with the IM, it is possible that human LetM1 could also have this function. There is a report [157] that human LetM1 interacts with ribosomal protein MRPL36. This observation is perplexing since the bacterial homolog of this protein (L36) is a small protein that plays a role in organizing the tertiary structure of the 23S rRNA. It is located on the interface side of the large subunit and would not be expected to be in contact with the membrane. Mdm38p contacts the yeast mitochondrial ribosomal protein that is the homolog of bacterial L31. Mammalian mitochondrial ribosomes do not have this protein. Hence, the contacts of LetM1 with the mammalian mitochondrial ribosome, if they occur, will be different from those observed with the yeast ribosome. It should be pointed out that recent studies indicate that LetM1 is a Ca2+/H+ antiporter [158,159]. Thus, this protein is thought to have the dual function of controlling ion flux across the inner membrane and regulating mitochondrial translation by promoting the interaction of ribosomes with the IM [155].
6. Mitochondrial Protein Synthesis and Human Disease
6.1 General Comments
Several comprehensive reviews on defects in the human mitochondrial translational system have recently appeared and only a brief summary will be provided here [96,160-163]. Since mitochondria generate most of the ATP used by eukaryotic cells, defects in the synthesis of the respiratory chain components often lead to significant disease states in humans. These defects can arise either from mutations in the mitochondrial genome itself or from mutations in nuclear gene products required for the translation of mRNAs encoded in the mitochondrial genome. For an excellent summary, the reader is referred to Table 1 in Smit et al. [96].
6.2 Mutations in Mitochondrial tRNAs
Mammalian mitochondrial DNA encodes 22 tRNA species, one for each amino acid and two for Ser and Leu. This complement of tRNAs is sufficient to decode the genetic code used by mammalian mitochondria. More than 150 different mutations in the tRNA genes have been described. The diseases associated with mitochondrial tRNA mutations can lead to defects in Complexes I, III, IV and the ATP synthase. These defects may arise from a number of specific issues leading to translational defects, including the failure of the tRNA to be processed [164], reduced stability of the tRNA [165,166], reduction in aminoacylation [167-169], reduced ability of the mutated aa-tRNA to interact with EF-Tumt [167,170] or the failure of the tRNA to be correctly modified [171].
One of the most commonly mutated tRNAs is the tRNALeu responsible for reading the UUR codons. Mutations here lead to mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS). The best studied of these mutations is A3243G, which alters the highly conserved residue A14 in the D-stem of the tRNA leading, among other things, to structural alterations and a failure of the wobble base in the anticodon to be properly modified giving rise to translational errors [171]. A second major site for mutations is the tRNALys gene. For example, the A8344G mutation causes a change in position 55 of the TψC-loop. This mutation leads to myoclonus epilepsy associated with ragged-red fibers (MERRF) and arises from defects in aminoacylation and from the lack of taurine modification in the anticodon loop abolishing proper codon:anticodon interactions.
One of the most thoroughly studied mutations is T4409C in the gene for human mitochondrial tRNAMet. This mutation has been found to cause dystrophic muscles and exercise intolerance [172]. At the tRNA level, this mutation leads to the change of the “universal” U at position 8 to C at the junction of the acceptor arm and the dihydrouridine arm. This mutation results in a drastic disruption of the structure of the tRNAMet, significantly reducing its aminoacylation [173]. The small fraction of the tRNAMet that can be aminoacylated is not formylated by the mitochondrial Met-tRNA transformylase and thus does not function in initiation. It is also unable to form a stable ternary complex with elongation factor EF-Tu, which prevents any participation in chain elongation.
A carefully studied set of mutations in the tRNAPhe gene has also been analyzed [167]. These mutations are associated with a variety of disease states including myopathies, diabetes, encephalopathies, and deafness. Most of the mutations tested interfere with the three-dimensional folding of tRNAPhe and lead to defects in aminoacylation and in binding to EF-Tumt.
6.3 Mutations in Nuclear Genes Required for Mitochondrial Translation
All of the protein factors required for mitochondrial translation are the products of nuclear genes. Mutations in several of these have been characterized. These include mutations in the translational elongation and termination factors, ribosomal proteins and ribosome assembly factors, aa-tRNA synthetases and tRNA and rRNA modifying enzymes [96,162,163].
6.3.1. Mutations in Elongation Factors
Mutations in all three elongation factors have been identified. A lethal mutation of Arg336 to Gln in EF-Tumt was identified in an infant with lactic acidosis and fetal encephalopathy [174]. This residue is found in domain II of EF-Tumt and is in a region that interacts with the acceptor stem of the aa-tRNA in the ternary complex. This mutation leads to a defect in ternary complex formation with mitochondrial aa-tRNAs [170,175] resulting in a failure of mitochondrial protein synthesis. Interestingly, the R336Q mutation has only a mild effect on the interaction of EF-Tumt with E. coli aa-tRNAs. The difference observed between mitochondrial and bacterial aa-tRNAs most likely reflects the interaction of this residue with the 5′ phosphate of the aa-tRNA. This interaction is not important for bacterial aa-tRNAs but appears to be important for the more structurally fragile mitochondrial tRNAs [102,173].
Mutation of an Arg in EF-Tsmt to Trp (Arg312 in isoform 1, the canonical sequence, equivalent to Arg333 in isoform 2) is lethal leading to encephalopathy [176]. This mutation is predicted to disrupt the structure of subdomain C of the core of EF-Tsmt probably accounting for the reduced levels of EF-Tsmt in patients with this mutation [176]. Arg312 does not make direct contact with EF-Tumt but disruption of the structure of this region of EF-Tsmt is thought to reduce the interaction of subdomain C with domain III of EF-Tumt. The mutation reduces the ability of EF-Tsmt to bind EF-Tumt and reduces its nucleotide exchange activity [170].
Several different mutations have been observed in EF-G1mt [129,174]. For example, the mutation of Arg250 to Trp in the G’ subdomain of this factor leads to rapidly progressing mitochondrial encephalopathy. This mutation is expected to affect the ribosomal GTPase activity of EF-G1mt [177]. Interestingly, mitochondrial protein synthesis was affected in fibroblasts but not in muscle tissue from this patient reflecting the sometimes unexpected tissue-specific effects observed with mutations in mitochondrial translation. Unusual tissue-specific effects have also been observed in other EF-G1mt mutations and are believed to arise from imbalances in the levels of the elongation factors in the affected tissues [178].
6.3.2. Ribosomal Protein Mutations
Of the approximately 80 identified mitochondrial ribosomal proteins, mutations are known in only two, MRPS16 and MRPS22 [179,180]. The mutation in MRPS16 is a nonsense mutation (the Arg111 codon to a stop codon) and leads to agenesis of the corpus callosum, dysmorphism and fatal neonatal lactic acidosis. MRPS16 is a highly conserved ribosomal protein that has a clear homolog in bacteria. This mutation has an adverse effect on the assembly of the small ribosomal subunit leading to decreased levels of the 12S rRNA in the small subunit and a decrease in some but not all of the other ribosomal proteins of the 28S subunit [181].
A missense mutation in MRPS22 at position 170 changing a conserved Arg residue to His is lethal in humans. This mutation is associated with edema, fatal cardiomyopathy and tubulopathy [180]. MRPS22 has no counterpart in bacteria or yeast and its function is obscure. This mutation also leads to a defect in the assembly of the 28S subunit. A second mutation in MRPS22 (Leu 215 to Pro) has also been observed in a patient with Cornelia de Lange-like dysmorphic features, brain abnormalities and hypertrophic cardiomyopathy [182].
6.3.3. Mutations in Aminoacyl-tRNA Synthetases and RNA Modifying enzymes
All of the mitochondrial aminoacyl-tRNA synthetases are encoded by nuclear genes. Mutations in several of these (Asp, Arg, Ser, Lys and His) are known to lead to disease states. The reader is referred to Rötig [163] for a recent summary. Recently, two unrelated children with Leigh syndrome and combined deficiency in oxidative phosphorylation have been shown to have defects in the mitochondrial Met-tRNA transformylase [183]. This observation argues strongly that, unlike in yeast, formylation of Met-tRNA is required for the efficient initiation of translation in humans.
In general, mitochondrial tRNAs contain specific modified bases, and alterations in the modifying enzymes can produce defects in mitochondrial translation. The best studied example is mutation in the PUS1 gene that converts uridine in tRNAs to pseudouridine [184,185]. This minor base is thought to help stabilize the structure of the tRNA. PUS1 modifies both cytoplasmic and mitochondrial tRNAs and, as a result, the effects of mutations in this gene are somewhat difficult to sort out.
The TRMU gene encodes the mitochondria-specific enzyme that is responsible for the 2-thio modification of 5-taurinomethyl-2-thio-uridine in the wobble position of certain mitochondrial tRNAs. TRMU acts as a nuclear modulator of mutations in the 12S rRNA associated with deafness [186]. It should be pointed out that mutations in the 12S rRNA (A1555G and C1494T) are important determinants predisposing to deafness.
6.3.4. Other Mutations Affecting Mitochondrial Translation
6.3.4.1. Ribosome Assembly Factors
Little is currently known about the assembly of the mitochondrial ribosome. However, it is expected that numerous factors will be required for this process and that mutations in these factors will lead to disease states in humans. Several candidate genes important for ribosome assembly have been identified. These include two GTPases (NOA1 and ERAL1) which are likely to be required for the assembly of the small subunit [187,188]. ERAL1 has recently been shown to bind to the 3′-terminal stem-loop of the 12S rRNA in the small subunit and binding of ERAL1 to this site appears to promote the maturation of the 12S rRNA [189]. The proper assembly of the mitochondrial ribosome also requires methyltransferases possibly for the modification of the rRNA [190,191]. MERTF4, which has been implicated in mitochondrial transcription, also helps recruit the methylase NSUN4 to the large ribosomal subunit, an interaction that appears to be required for ribosome assembly [192].
6.3.4.2. Translational Activators and Other Factors
A number of factors that are required for the synthesis of at least some of the proteins encoded in the mitochondrial genome have been identified although currently their roles are poorly understood from a mechanistic point of view. Mutations in C12orf65 have been observed in patients with Leigh syndrome, optic atrophy and ophthalmoplegia [127]. The symptoms observed seem to arise from severe decreases in the levels of Complexes I, IV, and V and a smaller decrease in Complex III. This protein belongs to the release factor family and contains the GGQ motif characteristic of the class 1 release factors.
Little is known about the regulation of mitochondrial protein synthesis in mammalian systems. Yeast contains a number of gene-specific translational activators but, in general, these do not have homologs in mammals. A specific defect in the synthesis of Complex IV subunits has been found in a family with late onset Leigh syndrome [193]. This family has a mutation in the TACO1 gene. The role of TACO1 is not known, but it may play a role as a translational activator. A homolog of TACO1 is observed in the genomes of many bacteria, although the properties and function of this protein in bacteria also remain unknown.
Mutations in the LRPPRC gene have been shown to be the cause of the French Canadian variant of Leigh syndrome. The LRPPRC protein appears to be a distant relation of yeast pet309, a translational activator that also stabilizes the mRNAs for COXI and COXIII [194,195]. The relationship between these proteins appears to reside primarily in PPR motifs and the question of the function of LRPPRC in humans remains to be determined.
7. Emerging Issues
Much remains to be learned about mammalian mitochondrial protein synthesis. Basic mechanistic questions remain including how the mRNA is fed into the small ribosomal subunit at initiation and how the 5′ start codon is positioned in the P-site. While the factors required for the general processes in initiation, elongation, termination and ribosome recycling have been identified and a number of detailed studies on these factors have been carried out, additional factors have been implicated in mitochondrial translation and the roles of these factors in the translation of all or a subset of mitochondrial mRNAs remain to be investigated. Mitochondrial translation is certainly regulated and coordinated with other processes in this organelle including DNA replication and transcription; but the fundamental interactions involved are still being elucidated.
All of the products of mammalian mitochondrial protein synthesis are components of macromolecular complexes localized in the inner membrane. There is clear evidence that mitochondrial ribosomes are preferentially associated with the inner membrane and are enriched in the intracristal regions. The proteins required for this interaction in mammals are not well understood. One interesting possibility is that ribosomes synthesizing subunits of specific complexes are directed to sites where these complexes are being assembled. Such an interaction would permit the efficient assembly of the respiratory chain complexes and ATP synthase at specific sites since random insertion into the inner membrane of the hydrophobic polypeptides synthesized in mitochondria could lead to deleterious interactions. Understanding this process will require the identification of general and specific proteins necessary for the interaction of the mitochondrial ribosome with the inner membrane.
Highlights.
Mitochondria synthesis critical components of the oxidative phosphorylation system.
A biochemical perspective on mitochondrial protein synthesis is provided.
Mutations in this translational systems leads to human disease.
Acknowledgments
Funding for this project was provided by National Institutes of Health grant GM32734 to LLS. The funding sources had no role in this project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Vogel F, Bornhovd C, Neupert W, Reichert AS. Dynamic subcompartmentalization of the mitochondrial inner membrane. J. Cell Biol. 2006;175:237–247. doi: 10.1083/jcb.200605138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Osawa S, Jukes T, Watanabe K, Muto A. Recent evidence for evolution of the genetic code. Micro. Rev. 1992;56:229–264. doi: 10.1128/mr.56.1.229-264.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Temperley R, Richter R, Dennerlein S, Lightowlers RN, Chrzanowska- Lightowlers ZM. Hungry codons promote frameshifting in human mitochondrial ribosomes. Science. 2010;327:301. doi: 10.1126/science.1180674. [DOI] [PubMed] [Google Scholar]
- [4].Taanman J-W. The mitochondrial genome: structure, transcription, translation and replication. Biochim. Biophys. Acta. 1999;1410:103–123. doi: 10.1016/s0005-2728(98)00161-3. [DOI] [PubMed] [Google Scholar]
- [5].O’Brien TW. The general occurrence of 55 S ribosomes in mammalian liver mitochondria. J. Biol. Chem. 1971;246:3409–3417. [PubMed] [Google Scholar]
- [6].Pietromonaco S, Denslow N, O’Brien TW. Proteins of mammalian mitochondrial ribosomes. Biochimie. 1991;73:827–836. doi: 10.1016/0300-9084(91)90062-6. [DOI] [PubMed] [Google Scholar]
- [7].Smirnov A, Entelis N, Martin RP, Tarassov I. Biological significance of 5S rRNA import into human mitochondria: role of ribosomal protein MRP-L18. Genes Dev. 2011;25:1289–1305. doi: 10.1101/gad.624711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].van Holde K, W Hill, In: General physical properties of ribosomes. Nomura M, Tissieres A, Lengyel P, editors. Cold Spring Harbor NY; 1974. pp. 53–91. [Google Scholar]
- [9].Wittmann H. In: Structure of ribosomes. Hardesty B, Kramer G, editors. New York: 1986. pp. 1–27. [Google Scholar]
- [10].Zweib C, Glotz C, Brimacombe R. Secondary structure comparisons between small subunit ribosomal RNA molecules from six different species. Nuc. Acids Res. 1981;9:3621–3640. doi: 10.1093/nar/9.15.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Glotz C, Zweib C, Brimacombe R. Secondary structure of the large subunit ribosomal RNA from Escherichia coli, Zea mays chloroplasts and human and mouse mitochondrial ribosomes. Nuc. Acids Res. 1981;9:3287–3306. doi: 10.1093/nar/9.14.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Koc EC, Haque Md.E., Spremulli LL. Current views of the structure of the mammalian mitochondrial ribosome. Isr. J. Chem. 2010;50:45–59. [Google Scholar]
- [13].Goldschmidt-Reisin S, Kitakawa M, Herfurth E, Wittmann-Liebold B, Grohmann L, Graack H-R. Mammalian mitochondrial ribosomal proteins: N-terminal amino acid sequencing, characterization, and identification of corresponding gene sequences. J. Biol. Chem. 1998;273:34828–34836. doi: 10.1074/jbc.273.52.34828. [DOI] [PubMed] [Google Scholar]
- [14].Koc EC, Blackburn K, Burkhart W, Spremulli LL. Identification of a mammalian mitochondrial homolog of ribosomal protein S7. Biochem. Biophys. Res. Comm. 1999;266:141–146. doi: 10.1006/bbrc.1999.1785. [DOI] [PubMed] [Google Scholar]
- [15].Koc EC, Burkhart W, Blackburn K, Moseley A, Koc H, Spremulli LL. A proteomics approach to the identification of mammalian mitochondrial small subunit ribosomal proteins. J. Biol. Chem. 2000;275:32585–32591. doi: 10.1074/jbc.M003596200. [DOI] [PubMed] [Google Scholar]
- [16].Koc EC, Burkhart W, Blackburn K, Moseley A, Spremulli LL. The small subunit of the mammalian mitochondrial ribosome: Identification of the full complement of ribosomal proteins present. J. Biol. Chem. 2001;276:19363–19374. doi: 10.1074/jbc.M100727200. [DOI] [PubMed] [Google Scholar]
- [17].Koc EC, Burkhart W, Blackburn K, Koc H, Moseley A, Spremulli LL. Identification of four proteins from the small subunit of the mammalian mitochondrial ribosome using a proteomics approach. Prot. Sci. 2001;10:471–481. doi: 10.1110/ps.35301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Koc EC, Burkhart W, Blackburn K, Schlatzer DM, Moseley A, Spremulli LL. The large subunit of the mammalian mitochondrial ribosome: Analysis of the complement of ribosomal protein present. J. Biol. Chem. 2001;276:43958–43969. doi: 10.1074/jbc.M106510200. [DOI] [PubMed] [Google Scholar]
- [19].Graack H-R, Wittmann-Liebold B. Mitochondrial ribosomal proteins (MRPs) of yeast. Biochem. J. 1998;329:433–448. doi: 10.1042/bj3290433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Agrawal RK, Sharma MR, Yassin A, Lahiri I, Spremulli LL. In: Structure and Function of organellar ribosomes as revealed by cryo-EM in Ribosomes: Structure, function and dynamics. Rodnina M, Wintermeyer W, Green R, editors. Springer-Verlag; 2011. pp. 84–98. [Google Scholar]
- [21].Anderson S, de Brujin M, Coulson A, Eperon I, Sanger F, Young I. Complete sequence of bovine mitochondrial DNA: Conserved features of the mammalian mitochondrial genome. J. Mol. Biol. 1982;156:683–717. doi: 10.1016/0022-2836(82)90137-1. [DOI] [PubMed] [Google Scholar]
- [22].Anderson S, Bankier AT, Barrell BG, Debruijn MHL, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJH, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- [23].Montoya J, Ojala D, Attardi G. Distinctive features of the 5′-terminal sequences of the human mitochondrial mRNAs. Nature. 1981;290:465–470. doi: 10.1038/290465a0. [DOI] [PubMed] [Google Scholar]
- [24].Stewart JB, Beckenbach AT. Characterization of mature mitochondrial transcripts in Drosophila, and the implications for the tRNA punctuation model in arthropods. Gene. 2009;445:49–57. doi: 10.1016/j.gene.2009.06.006. [DOI] [PubMed] [Google Scholar]
- [25].Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE) J. Am. Chem. Soc. 2005;127:4223–4231. doi: 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- [26].Jones CN, Wilkinson KA, Hung KT, Weeks KM, Spremulli LL. Lack of secondary structure characterizes the 5′ ends of mammalian mitochondrial mRNAs. RNA. 2008;14:862–871. doi: 10.1261/rna.909208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Watanabe Y-I, Kawai G, Yokogawa T, Hayashi N, Kumazawa Y, Ueda T, Nishikawa K, Hirao I, Miura K-I, Watanabe K. Higher-order structure of bovine mitochondrial tRNASerUGA: chemical modification and computer modeling. Nuc. Acids Res. 1994;22:5378–5384. doi: 10.1093/nar/22.24.5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yokogawa T, Watanabe Y-I, Kumazawa Y, Ueda T, Hirao I, Miura K-I, Watanabe K. A novel cloverleaf structure found in mammalian mitochondrial tRNAser(UCN) Nuc. Acids Res. 1991;19:6101–6105. doi: 10.1093/nar/19.22.6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wakita K, Watanabe W, Yokogawa T, Kumazawa Y, Nakamura S, Ueda T, Watanabe K, Nishikawa K. Higher-order structure of bovine mitochondrial tRNAPhe lacking the ‘conserved’ GG and TYCG sequences as inferred by enzymatic and chemical probing. Nuc. Acids Res. 1994;22:347–353. doi: 10.1093/nar/22.3.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Helm M, Giege R, Florentz C. A Watson-Crick base pair disrupting methyl group (m1A) is sufficient for cloverleaf folding of human mitochondrial RNALys. Biochem. 1999;38:13338–13346. doi: 10.1021/bi991061g. [DOI] [PubMed] [Google Scholar]
- [31].Helm M, Brule H, Friede D, Giege R, Putz D, Florentz C. Search for characteristic structural features of mammalian mitochondrial tRNAs. RNA. 2000;6:1356–1379. doi: 10.1017/s1355838200001047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Steinberg S, Gautheret D, Cedergren R. Fitting the structurally diverse animal mitochondrial tRNAsSer to common three-dimensional constraints. J. Mol. Biol. 1994;236:982–989. doi: 10.1016/0022-2836(94)90004-3. [DOI] [PubMed] [Google Scholar]
- [33].Zagryadskaya EI, Kotlova N, Steinberg SV. Key Elements in maintenance of the tRNA L-shape. J. Mol. Biol. 2004;340:435–444. doi: 10.1016/j.jmb.2004.05.002. [DOI] [PubMed] [Google Scholar]
- [34].Sharma MR, Koc EC, Datta PP, Booth TM, Spremulli LL, Agrawal RK. Structure of the mammalian mitochondrial ribosome reveals an expanded functional role for its component proteins. Cell. 2003;115:97–108. doi: 10.1016/s0092-8674(03)00762-1. [DOI] [PubMed] [Google Scholar]
- [35].Grasso DG, Christian BE, Spencer AC, Spremulli LL. Over-expression and purification of mitochondrial translational initiation factor 2 and initiation factor 3. Methods Enzymology Translation Initiation: Reconstituted Systems and Biophysical Methods. 2007:59–78. doi: 10.1016/S0076-6879(07)30004-9. [DOI] [PubMed] [Google Scholar]
- [36].Christian BE, Spremulli LL. Preferential selection of the 5′-terminal start codon on leaderless mRNAs by mammalian mitochondrial ribosomes. J. Biol. Chem. 2010;285:28379–28386. doi: 10.1074/jbc.M110.149054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Schwartzbach C, Farwell M, Liao H-X, Spremulli LL. Bovine mitochondrial initiation and elongation factors. Meth. Enzymol. 1996;264:248–261. doi: 10.1016/s0076-6879(96)64025-7. [DOI] [PubMed] [Google Scholar]
- [38].Overman J, Enderle PJ, Farrow JM, Wiley JE, Farwell MA. The human mitochondrial translation initiation factor 2 gene (MTIF2): transcriptional analysis and identification of a pseudogene. Biochim. Biophys. Acta. 2003;1628:195–205. doi: 10.1016/s0167-4781(03)00144-1. [DOI] [PubMed] [Google Scholar]
- [39].Ma L, Spremulli LL. Cloning and sequence analysis of the human mitochondrial translational initiation factor 2 cDNA. J. Biol. Chem. 1995;270:1859–1865. doi: 10.1074/jbc.270.4.1859. [DOI] [PubMed] [Google Scholar]
- [40].Liao H-X, Spremulli LL. Initiation of protein synthesis in animal mitochondria: Purification and characterization of translational initiation factor 2. J. Biol. Chem. 1991;266:20714–20719. [PubMed] [Google Scholar]
- [41].Liao H-X, Spremulli LL. Identification and initial characterization of translational initiation factor 2 from bovine mitochondria. J. Biol. Chem. 1990;265:13618–13622. [PubMed] [Google Scholar]
- [42].Caserta E, Ferrara C, Milon P, Fabbretti A, Rocchetti A, Tomsic J, Pon CL, Gualerzi CO, La TA. Ribosomal interaction of Bacillus stearothermophilus translation initiation factor IF2: characterization of the active sites. J. Mol. Biol. 2010;396:118–129. doi: 10.1016/j.jmb.2009.11.026. [DOI] [PubMed] [Google Scholar]
- [43].Spurio R, Brandi L, Caserta E, Pon C, Gualerzi C, Misselwitz R, Krafft C, Welfle K, Welfle H. The C-terminal subdomain (IF2 C-2) contains the entire fMet-tRNA binding site of initiation factor IF2. J. Biol. Chem. 2000;275:2447–2454. doi: 10.1074/jbc.275.4.2447. [DOI] [PubMed] [Google Scholar]
- [44].Guenneugues M, Caserta E, Brandi L, Spurio R, Meunier B, Pon CL, Boelens R, Gualerzi CO. Mapping the fMet-tRNA binding site of initiation factor IF2. EMBO J. 2000;19:5233–5240. doi: 10.1093/emboj/19.19.5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ma J, Farwell M, Burkhart W, Spremulli LL. Cloning and sequence analysis of the cDNA for bovine mitochondrial translational initiation factor 2. Biochim. Biophys. Acta. 1995;1261:321–324. doi: 10.1016/0167-4781(95)00041-e. [DOI] [PubMed] [Google Scholar]
- [46].Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- [47].Claros MG, Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 1996;241:770–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- [48].Nakai K, Kanehisa M. A knowledge base for predicting protein localization sites in eukaryotic cells. Genomics. 1992;14:897–911. doi: 10.1016/S0888-7543(05)80111-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ma J, Spremulli LL. Expression, purification and mechanistic studies of bovine mitochondrial translational initiation factor 2. J. Biol. Chem. 1996;271:5805–5811. doi: 10.1074/jbc.271.10.5805. [DOI] [PubMed] [Google Scholar]
- [50].Spremulli LL, Coursey A, Navratil T, Hunter SE. Initiation and elongation factors in mammalian mitochondrial protein biosynthesis. Prog. Nucleic Acid Res. Mol. Biol. 2004;77:211–261. doi: 10.1016/S0079-6603(04)77006-3. [DOI] [PubMed] [Google Scholar]
- [51].Spencer AC, Spremulli LL. The interaction of mitochondrial translational initiation factor 2 with the small ribosomal subunit. Biochim. Biophys. Acta. 2005;1750:69–81. doi: 10.1016/j.bbapap.2005.03.009. [DOI] [PubMed] [Google Scholar]
- [52].Takeuchi N, Kawakami M, Omori A, Ueda T, Spremulli LL, Watanabe K. Mammalian mitochondrial Methionyl-tRNA transformylase from bovine liver: Purification, characterization and gene structure. J. Biol. Chem. 1998;273:15085–15090. doi: 10.1074/jbc.273.24.15085. [DOI] [PubMed] [Google Scholar]
- [53].Takeuchi N, Ueda T, Watanabe K. Expression and characterization of bovine mitochondrial Methionyl-tRNA transformylase. J. Biochem. 1998;124:1069–1071. doi: 10.1093/oxfordjournals.jbchem.a022222. [DOI] [PubMed] [Google Scholar]
- [54].Spencer AC, Spremulli LL. Interaction of mitochondrial initiation factor 2 with mitochondrial (f)Met-tRNA. Nuc. Acids Res. 2004;32:5464–5470. doi: 10.1093/nar/gkh886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Li Y, Holmes WB, Appling DR, RajBhandary UL. Initiation of protein synthesis in Saccharomyces cerevisiae mitochondria without formylation of the initiator tRNA. J. Bact. 2000;182:2886–2892. doi: 10.1128/jb.182.10.2886-2892.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tibbetts AS, Oesterlin L, Chan SY, Kramer G, Hardesty B, Appling DR. Mammalian mitochondrial initiation factor 2 supports yeast mitochondrial translation without formylated initiator tRNA. J. Biol. Chem. 2003;278:31774–31780. doi: 10.1074/jbc.M304962200. [DOI] [PubMed] [Google Scholar]
- [57].Lee C, Tibbetts AS, Kramer G, Appling DR. Yeast AEP3p is an accessory factor in initiation of mitochondrial translation. J. Biol. Chem. 2009;284:34116–34125. doi: 10.1074/jbc.M109.055350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bollen A, Heimark RL, Cozzone A, Traut RR, Hershey JW. Cross-linking of initiation factor IF-2 to Escherichia coli 30 S ribosomal proteins with dimethylsuberimidate. J. Biol. Chem. 1976;250:4310–4314. [PubMed] [Google Scholar]
- [59].La Teana A, Gualerzi CO, Dahlberg AE. Initiation factor IF-2 binds to the alpha- sarcin loop and helix 89 of Escherichia coli 23S ribosomal RNA. RNA. 2001;7:1173–1179. doi: 10.1017/s1355838201010366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gaur R, Grasso D, Datta PP, Krishna PDV, Das G, Spencer A, Agrawal RK, Spremulli L, Varshney U. A single mammalian mitochondrial translation initiation factor functionally replaces two bacterial factors. Mol. Cell. 2008;29:180–190. doi: 10.1016/j.molcel.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Allen GS, Zavialov A, Gursky R, Ehrenberg M, Frank J. The Cryo-EM structure of a translation initiation complex from Escherichia coli. Cell. 2005;121:703–712. doi: 10.1016/j.cell.2005.03.023. [DOI] [PubMed] [Google Scholar]
- [62].Carter AP, Clemons WM, Jr., Brodersen DE, Morgan-Warren RJ, Hartsch T, Wimberly BT, Ramakrishnan V. Crystal structure of an initiation factor bound to the 30S ribosomal subunit. Science. 2001;291:498–501. doi: 10.1126/science.1057766. [DOI] [PubMed] [Google Scholar]
- [63].Yassin AS, Haque ME, Datta PP, Elmore K, Banavali NK, Spremulli LL, Agrawal RK. Insertion domain within mammalian mitochondrial translation initiation factor 2 serves the role of eubacterial initiation factor 1. Proc. Natl. Acad. Sci U. S. A. 2011;108:3918–3923. doi: 10.1073/pnas.1017425108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Srere P. The infrastructure of the mitochondrial matrix. Trends Biochem. Sci. 1980;5:120–121. [Google Scholar]
- [65].Koc EC, Spremulli LL. Identification of mammalian mitochondrial translational initiation factor 3 and examination of its role in initiation complex formation with natural mRNAs. J. Biol. Chem. 2002;277:35541–35549. doi: 10.1074/jbc.M202498200. [DOI] [PubMed] [Google Scholar]
- [66].Christian B, Spremulli L. Evidence for an active role of IF3mt in initiation of translation in mammalian mitochondria. Biochem. 2009;48:3269–3278. doi: 10.1021/bi8023493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Moreau M, de Cock E, Fortier P-L, Garcia C, Albaret C, Blanquet S, Lallemand J-Y, Dardel F. Heteronuclear NMR studies of E. coli translation initiation factor IF3. Evidence that the inter-domain region is disordered in solution. J. Mol. Biol. 1997;266:15–22. doi: 10.1006/jmbi.1996.0756. [DOI] [PubMed] [Google Scholar]
- [68].Hua Y, Raleigh D. On the global architecture of initiation factor IF3: a comparative study of the linker regions from the Escherichia coli protein and the Bacillus stearothermophilus protein. J. Mol. Biol. 1998;278:871–878. doi: 10.1006/jmbi.1998.1736. [DOI] [PubMed] [Google Scholar]
- [69].de Cock E, Springer M, Dardel F. The interdomain linker of Escherichia coli initiation factor IF3: a possible trigger of translation initiation specificity. Mol. Microbiol. 1999;32:193–202. doi: 10.1046/j.1365-2958.1999.01350.x. [DOI] [PubMed] [Google Scholar]
- [70].Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999;234:187–208. doi: 10.1016/s0378-1119(99)00210-3. [DOI] [PubMed] [Google Scholar]
- [71].Anvret A, Ran C, Westerlund M, Thelander AC, Sydow O, Lind C, Hakansson A, Nissbrandt H, Galter D, Belin AC. Possible involvement of a mitochondrial translation initiation factor 3 variant causing decreased mRNA levels in Parkinson’s disease. Parkinsons Dis. 2010;2010:491751. doi: 10.4061/2010/491751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Bhargava K, Spremulli LL. Role of the N- and C-terminal extensions on the activity of mammalian mitochondrial translational initiation factor 3. Nuc. Acids Res. 2005;33:7011–7018. doi: 10.1093/nar/gki1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Petrelli D, LaTeana A, Garofalo C, Spurio R, Pon CL, Gualerzi CO. Translation initiation factor IF3: two domains, five functions, one mechanism? EMBO J. 2001;20:4560–4569. doi: 10.1093/emboj/20.16.4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sacerdot C, de Cock E, Engst K, Graffe M, Dardel F, Springer M. Mutations that alter initiation codon discrimination by Escherichia coli initiation factor IF3. J. Mol. Biol. 1999;288:803–810. doi: 10.1006/jmbi.1999.2737. [DOI] [PubMed] [Google Scholar]
- [75].Haque M, Spremulli LL. Roles of the N- and C-terminal domains of mammalian mitochondrial initiation factor 3 in protein biosynthesis. J. Mol. Biol. 2008;384:929–940. doi: 10.1016/j.jmb.2008.09.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].McCutcheon J, Agrawal R, Philips SM, Grassucci R, Gerchman S, Clemons WM, Ramakrishnan V, Frank J. Location of translational initiation factor IF3 on the small ribosomal subunit. Proc. Natl. Acad. Sci. U.S.A. 1999;96:4301–4306. doi: 10.1073/pnas.96.8.4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Dallas A, Noller HF. Interaction of translation initiation factor 3 with the 30S ribosomal subunit. Mol. Cell. 2001;8:855–864. doi: 10.1016/s1097-2765(01)00356-2. [DOI] [PubMed] [Google Scholar]
- [78].Pioletti M, Schlunzen F, Harms J, Zarivach R, Gluhmann M, Avila H, Bashan A, Bartels H, Auerbach T, Jacobi C, Hartsch T, Yonath A, Franceschi F. Crystal structures of complexes of the small ribosomal subunit with tetracycline, edeine and IF3. EMBO J. 2001;20:1829–1839. doi: 10.1093/emboj/20.8.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Moazed D, Samaha RR, Gualerzi C, Noller HF. Specific protection of 16 S rRNA by translational initiation factors. J. Mol. Biol. 1995;248:207–210. doi: 10.1016/s0022-2836(95)80042-5. [DOI] [PubMed] [Google Scholar]
- [80].Ehresmann C, Moine H, Mougel M, Dondon J, Grunberg-Manago M, Ebel JP, Ehresmann B. Cross-linking of initiation factor IF3 to Escherichia coli 30S ribosomal subunit by trans-diamminedichloroplatinum(II): characterization of two cross-linking sites in 16S rRNA; a possible way of functioning for IF3. Nuc. Acids Res. 1986;14:4803–4821. doi: 10.1093/nar/14.12.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Haque ME, Koc H, Cimen H, Koc EC, Spremulli LL. Contacts between mammalian mitochondrial translational initiation factor 3 and ribosomal proteins in the small subunit. Biochim. Biophys. Acta. 2011;1814:1779–1784. doi: 10.1016/j.bbapap.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Berger T, Brigl M, Herrmann JM, Vielhauer V, Luckow B, Schlondorff D, Kretzler M. The apoptosis mediator mDAP-3 is a novel member of a conserved family of mitochondrial proteins. J. Cell Sci. 2000;113:3603–3612. doi: 10.1242/jcs.113.20.3603. [DOI] [PubMed] [Google Scholar]
- [83].Koc EC, Ranasinghe A, Burkhart W, Blackburn K, Koc H, Moseley A, Spremulli LL. A new face on apoptosis: Death-associated protein 3 and PDCD9 are mitochondrial ribosomal proteins. FEBS Lett. 2001;492:166–170. doi: 10.1016/s0014-5793(01)02250-5. [DOI] [PubMed] [Google Scholar]
- [84].Han MJ, Chiu DT, Koc EC. Regulation of mitochondrial ribosomal protein S29 (MRPS29) expression by a 5′-upstream open reading frame. Mitochondrion. 2010;10:274–283. doi: 10.1016/j.mito.2009.12.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Davies SM, Rackham O, Shearwood AM, Hamilton KL, Narsai R, Whelan J, Filipovska A. Pentatricopeptide repeat domain protein 3 associates with the mitochondrial small ribosomal subunit and regulates translation. FEBS Lett. 2009;583:1853–1858. doi: 10.1016/j.febslet.2009.04.048. [DOI] [PubMed] [Google Scholar]
- [86].Agirrezabala X, Frank J. Elongation in translation as a dynamic interaction among the ribosome, tRNA, and elongation factors EF-G and EF-Tu. Q. Rev. Biophys. 2009;42:159–200. doi: 10.1017/S0033583509990060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Woriax V, Spremulli G, Spremulli LL. Nucleotide and aminoacyl-tRNA specificity of the mammalian mitochondrial elongation factor EF-Tu:Ts complex. Biochim. Biophys. Acta. 1996;1307:66–72. doi: 10.1016/0167-4781(95)00240-5. [DOI] [PubMed] [Google Scholar]
- [88].Chung HK, Spremulli LL. Purification and characterization of elongation factor G from bovine liver mitochondria. J. Biol. Chem. 1990;265:21000–21004. [PubMed] [Google Scholar]
- [89].Schwartzbach C, Spremulli LL. Bovine mitochondrial protein synthesis elongation factors: Identification and initial characterization of an elongation factor Tu-elongation factor Ts complex. J. Biol. Chem. 1989;264:19125–19131. [PubMed] [Google Scholar]
- [90].Schwartzbach C, Spremulli LL. Interaction of animal mitochondrial EF-Tu:EF-Ts with aminoacyl-tRNA, guanine nucleotides and ribosomes. J. Biol. Chem. 1991;266:16324–16330. [PubMed] [Google Scholar]
- [91].Woriax V, Bullard JM, Ma L, Yokogawa T, Spremulli LL. Mechanistic studies of the translational elongation cycle in mammalian mitochondria. Biochim. Biophys. Acta. 1997;1352:91–101. doi: 10.1016/s0167-4781(97)00002-x. [DOI] [PubMed] [Google Scholar]
- [92].Bhargava K, Templeton PD, Spremulli LL. Expression and characterization of isoform 1 of human mitochondrial elongation factor G. Protein Expre. Purif. 2004;37:368–376. doi: 10.1016/j.pep.2004.06.030. [DOI] [PubMed] [Google Scholar]
- [93].Xin H, Woriax VL, Burkhart W, Spremulli LL. Cloning and expression of mitochondrial translational elongation factor Ts from bovine and human liver. J. Biol. Chem. 1995;270:17243–17249. doi: 10.1074/jbc.270.29.17243. [DOI] [PubMed] [Google Scholar]
- [94].Woriax V, Burkhart W, Spremulli LL. Cloning, sequence analysis and expression of mammalian mitochondrial protein synthesis elongation factor Tu. Biochim. Biophys. Acta. 1995;1264:347–356. doi: 10.1016/0167-4781(95)00176-x. [DOI] [PubMed] [Google Scholar]
- [95].Xin H, Leanza K, Spremulli LL. Expression of bovine mitochondrial elongation factor Ts in Escherichia coli and characterization of the heterologous complex formed with prokaryotic elongation factor Tu. Biochim. Biophys. Acta. 1997;1352:101–112. doi: 10.1016/s0167-4781(97)00003-1. [DOI] [PubMed] [Google Scholar]
- [96].Smits P, Smeitink J, van den Heuvel L. Mitochondrial translation and beyond: processes implicated in combined oxidative phosphorylation deficiencies. J. Biomed. Biotechnol. 2010;2010:737385. doi: 10.1155/2010/737385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Cai Y-C, Bullard JM, Thompson NL, Spremulli LL. Interaction of mammalian mitochondrial elongation factor EF-Tu with guanine nucleotides. Prot. Sci. 2000;9:1791–1800. [PMC free article] [PubMed] [Google Scholar]
- [98].Cai Y-C, Bullard JM, Thompson NL, Spremulli LL. Interaction of mitochondrial elongation factor Tu with aminoacyl-tRNA and elongation factor Ts. J. Biol. Chem. 2000;275:20308–20314. doi: 10.1074/jbc.M001899200. [DOI] [PubMed] [Google Scholar]
- [99].Hunter SE, Spremulli LL. Interaction of mitochondrial elongation factor Tu with aminoacyl-tRNAs. Mitochondrion. 2004;4:21–29. doi: 10.1016/j.mito.2004.04.003. [DOI] [PubMed] [Google Scholar]
- [100].Wells J, Henkler F, Leversha M, Koshy R. A mitochondrial elongation factor-like protein is over-expressed in tumors and differentially expressed in normal tissues. FEBS Lett. 1995;358:119–125. doi: 10.1016/0014-5793(94)01403-n. [DOI] [PubMed] [Google Scholar]
- [101].Jeppesen MG, Navratil T, Spremulli LL, Nyborg J. Crystal structure of the bovine mitochondrial elongation factor Tu•Ts complex. J. Biol. Chem. 2005;280:5071–5081. doi: 10.1074/jbc.M411782200. [DOI] [PubMed] [Google Scholar]
- [102].Nissen P, Kjeldgaard M, Thirup S, Polekhina G, Reshetnikova L, Clark B, Nyborg J. Crystal structure of the ternary complex of Phe-tRNAphe, EF-Tu and a GTP analog. Science. 1995;270:1464–1472. doi: 10.1126/science.270.5241.1464. [DOI] [PubMed] [Google Scholar]
- [103].Nissen P, Thirup S, Kjeldgaard M, Nyborg J. The crystal structure of Cys-tRNACys- EF-Tu-GDPNP reveals general and specific features in the ternary complex and in tRNA. Struc. 1999;7:143–156. doi: 10.1016/s0969-2126(99)80021-5. [DOI] [PubMed] [Google Scholar]
- [104].Hunter SE, Spremulli LL. Effects of mutagenesis of residue 221 on the properties of bacterial and mitochondrial elongation factor EF-Tu. Biochim. Biophys. Acta. 2004;1699:173–182. doi: 10.1016/j.bbapap.2004.02.015. [DOI] [PubMed] [Google Scholar]
- [105].Hunter SE, Spremulli LL. Mutagenesis of Gln290 of Escherichia coli and mitochondrial elongation factor Tu affects interactions with mitochondrial aminoacyl- tRNA and GTPase activity. Biochem. 2004;43:6917–6927. doi: 10.1021/bi036068j. [DOI] [PubMed] [Google Scholar]
- [106].Hunter SE, Spremulli LL. Mutagenesis of Arg335 in bovine mitochondrial elongation factor Tu and the corresponding residue in the Escherichia coli factor affects interactions with mitochondrial aminoacyl-tRNAs. RNA Biology. 2004;2:95–102. doi: 10.4161/rna.1.2.1034. [DOI] [PubMed] [Google Scholar]
- [107].Kumazawa Y, Schwartzbach C, Liao H-X, Mizumoto K, Kaziro Y, Watanabe K, Spremulli LL. Interactions of bovine mitochondrial phenylalanyl-tRNA with ribosomes and elongation factors from mitochondria and bacteria. Biochim. Biophys. Acta. 1991;1090:167–172. doi: 10.1016/0167-4781(91)90097-6. [DOI] [PubMed] [Google Scholar]
- [108].Bullard JM, Cai Y-C, Zhang Y, Spremulli LL. Effects of domain exchanges between Escherichia coli and mammalian mitochondrial EF-Tu on interactions with guanine nucleotides, aminoacyl-tRNA and ribosomes. Biochim Biophys Acta. 1999;1446:102–114. doi: 10.1016/s0167-4781(99)00077-9. [DOI] [PubMed] [Google Scholar]
- [109].Kawashima T, Berthet-Colominas C, Wulff M, Cusack S, Leberman R. The structure of the Escherichia coli EF-Tu:EF-Ts complex at 2.5 Å resolution. Nature. 1996;379:511–518. doi: 10.1038/379511a0. [DOI] [PubMed] [Google Scholar]
- [110].Zhang Y, Li X, Spremulli LL. Role of the conserved aspartate and phenylalanine residues in prokaryotic and mitochondrial elongation factor Ts in guanine nucleotide exchange. FEBS Lett. 1996;391:330–332. doi: 10.1016/0014-5793(96)00789-2. [DOI] [PubMed] [Google Scholar]
- [111].Zhang Y, Sun V, Spremulli LL. Role of domains in Escherichia coli and mammalian mitochondrial elongation factor Ts in the interaction with elongation factor Tu. J. Biol. Chem. 1997;272:21956–21963. doi: 10.1074/jbc.272.35.21956. [DOI] [PubMed] [Google Scholar]
- [112].Zhang Y, Yu N-J, Spremulli LL. Mutational analysis of the roles of residues in Escherichia coli elongation factor Ts in the interaction with elongation factor Tu. J. Biol. Chem. 1998;273:4556–4562. doi: 10.1074/jbc.273.8.4556. [DOI] [PubMed] [Google Scholar]
- [113].Zhang Y, Spremulli LL. Roles of residues in mammalian mitochondrial elongation factor Ts in the interaction with bacterial and mitochondrial elongation factor Tu. J. Biol. Chem. 1998;273:28142–28148. doi: 10.1074/jbc.273.43.28142. [DOI] [PubMed] [Google Scholar]
- [114].Hammarsund M, Wilson W, Corcoran M, Merup M, Einhorn S, Grander D, Sangfelt O. Identification and characterization of two novel human mitochondrial elongation factor genes, hEFG2 and hEFG1, phylogenetically conserved through evolution. Hum. Genet. 2001;109:542–550. doi: 10.1007/s00439-001-0610-5. [DOI] [PubMed] [Google Scholar]
- [115].Tsuboi M, Morita H, Nozaki Y, Akama K, Ueda T, Ito K, Nierhaus K, Takeuchi N. EF-G2mt is an exclusive recycling factor in mammalian mitochondrial protein synthesis. Mol. Cell. 2009;35:502–510. doi: 10.1016/j.molcel.2009.06.028. [DOI] [PubMed] [Google Scholar]
- [116].Ulbrich B, Czempiel W, Bass R. Mammalian mitochondrial ribosomes. Studies on the exchangeability of polypeptide chain elongation factors from bacterial and mitochondrial systems. Eur. J. Biochem. 1980;108:337–343. doi: 10.1111/j.1432-1033.1980.tb04728.x. [DOI] [PubMed] [Google Scholar]
- [117].Terasaki M, Suzuki T, Hanada T, Watanabe K. Functional compatibility of elongation factors between mammalian mitochondrial and bacterial ribosomes: Characterization of GTPase activity and translation elongation by hybrid ribosomes bearing heterologous L7/12 proteins. J. Mol. Biol. 2004;336:331–342. doi: 10.1016/j.jmb.2003.12.034. [DOI] [PubMed] [Google Scholar]
- [118].Richter R, Pajak A, Dennerlein S, Rozanska A, Lightowlers RN, Chrzanowska-Lightowlers ZM. Translation termination in human mitochondrial ribosomes. Biochem. Soc. Trans. 2010;38:1523–1526. doi: 10.1042/BST0381523. [DOI] [PubMed] [Google Scholar]
- [119].Lightowlers RN, Chrzanowska-Lightowlers ZM. Terminating human mitochondrial protein synthesis: a shift in our thinking. RNA Biol. 2010;7:282–286. doi: 10.4161/rna.7.3.12023. [DOI] [PubMed] [Google Scholar]
- [120].Chrzanowska-Lightowlers ZM, Pajak A, Lightowlers RN. Termination of protein synthesis in mammalian mitochondria. J. Biol. Chem. 2011;286:34479–34485. doi: 10.1074/jbc.R111.290585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Zhang Y, Spremulli LL. Identification and cloning of human mitochondrial translational release factor 1 and the ribosome recycling factor. Biochim Biophys Acta. 1998;1443:245–250. doi: 10.1016/s0167-4781(98)00223-1. [DOI] [PubMed] [Google Scholar]
- [122].Christian B, Haque E, Spremulli L. Ribosome shifting or splitting: it is all up to the EF- G. Mol Cell. 2009;35:400–402. doi: 10.1016/j.molcel.2009.08.003. [DOI] [PubMed] [Google Scholar]
- [123].Soleimanpour-Lichaei HR, Kuhl L, Gaisne M, Passos JF, Wydro M, Rorbach J, Temperley R, Bonnefoy N, Tate W, Lightowlers R, Chrzanowska-Lightowlers Z. mtRF1a is a human mitochondrial translation release factor decoding the major termination codons UAA and UAG. Mol. Cell. 2007;27:745–757. doi: 10.1016/j.molcel.2007.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Nozaki Y, Matsunaga N, Ishizawa T, Ueda T, Takeuchi N. HMRF1L is a human mitochondrial translation release factor involved in the decoding of the termination codons UAA and UAG. Genes to Cells. 2008;13:429–438. doi: 10.1111/j.1365-2443.2008.01181.x. [DOI] [PubMed] [Google Scholar]
- [125].Richter R, Rorbach J, Pajak A, Smith PA, Wessels HJ, Huynen MA, Smeitink JA, Lightowlers RN, Chrzanowska-Lightowlers ZM. A functional peptidyl-tRNA hydrolase, ICT1, has been recruited into the human mitochondrial ribosome. EMBO J. 2010;29:1116–1125. doi: 10.1038/emboj.2010.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Haque ME, Spremulli LL. ICT1 comes to the rescue of mitochondrial ribosomes. EMBO J. 2010;29:1019–1020. doi: 10.1038/emboj.2010.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Antonicka H, Ostergaard E, Sasarman F, Weraarpachai W, Wibrand F, Pedersen AM, Rodenburg RJ, van der Knaap MS, Smeitink JA, Chrzanowska-Lightowlers ZM, Shoubridge EA. Mutations in C12orf65 in patients with encephalomyopathy and a mitochondrial translation defect. Am. J. Hum. Genet. 2010;87:115–122. doi: 10.1016/j.ajhg.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Rorbach J, Richter R, Wessels HJ, Wydro M, Pekalski M, Farhoud M, Kuhl I, Gaisne M, Bonnefoy N, Smeitink JA, Lightowlers RN, Chrzanowska-Lightowlers ZM. The human mitochondrial ribosome recycling factor is essential for cell viability. Nuc. Acids Res. 2008;36:5787–5799. doi: 10.1093/nar/gkn576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Coenen MJH, Antonicka H, Ugalde C, Sasarman F, Rossi R, Heister JGAM, Newbold RF, Trijbels FJMF, van den Heuvel LP, Shoubridge EA, Smeitink JAM. Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. New Engl. J. Med. 2004;351:2080–2086. doi: 10.1056/NEJMoa041878. [DOI] [PubMed] [Google Scholar]
- [130].Seshadri A, Samhita L, Gaur R, Malshetty V, Varshney U. Analysis of the fusA2 locus encoding EFG2 in Mycobacterium smegmatis. Tuberculosis. (Edinb.) 2009;89:453–464. doi: 10.1016/j.tube.2009.06.003. [DOI] [PubMed] [Google Scholar]
- [131].Connell SR, Takemoto C, Wilson DN, Wang H, Murayama K, Terada T, Shirouzu M, Rost M, Schuler M, Giesebrecht J, Dabrowski M, Mielke T, Fucini P, Yokoyama S, Spahn CM. Structural basis for interaction of the ribosome with the switch regions of GTP-bound elongation factors. Mol. Cell. 2007;25:751–764. doi: 10.1016/j.molcel.2007.01.027. [DOI] [PubMed] [Google Scholar]
- [132].Liu M, Spremulli LL. Interaction of mammalian mitochondrial ribosomes with the inner membrane. J. Biol. Chem. 2000;275:29400–29406. doi: 10.1074/jbc.M002173200. [DOI] [PubMed] [Google Scholar]
- [133].Ott M, Herrmann JM. Co-translational membrane insertion of mitochondrially encoded proteins. Biochim. Biophys. Acta. 2010;1803:767–775. doi: 10.1016/j.bbamcr.2009.11.010. [DOI] [PubMed] [Google Scholar]
- [134].Bonnefoy N, Fiumera HL, Dujardin G, Fox TD. Roles of Oxa1-related inner- membrane translocases in assembly of respiratory chain complexes. Biochim. Biophys. Acta. 2009;1793:60–70. doi: 10.1016/j.bbamcr.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Frazier AE, Taylor RD, Mick DU, Warscheid B, Stoepel N, Meyer HE, Ryan MT, Guiard B, Rehling P. Mdm38 interacts with ribosomes and is a component of the mitochondrial protein export machinery. J. Cell Biol. 2006;172:553–564. doi: 10.1083/jcb.200505060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Hell K, Hermann J, Pratije E, Neupert W, Stuart R. Oxalp mediates the export of the N- and C-termini of pCoxII from the mitochondrial matrix to the intermembrane space. FEBS Lett. 1997;418:367–370. doi: 10.1016/s0014-5793(97)01412-9. [DOI] [PubMed] [Google Scholar]
- [137].Hell K, Neupert W, Stuart RA. Oxa1p acts as a general membrane insertion machinery for proteins encoded by mitochondrial DNA. EMBO J. 2001;20:1281–1288. doi: 10.1093/emboj/20.6.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].He S, Fox TD. Membrane translocation of mitochondrially coded Cox2p: Distinct requirements for export of N and C termini and dependence on the conserved protein Oxa1p. Mol. Bio. Cell. 1997;8:1449–1460. doi: 10.1091/mbc.8.8.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Tokatlidis K, Schatz G. Biogenesis of mitochondrial inner membrane proteins. J. Biol. Chem. 1999;274:35285–35288. doi: 10.1074/jbc.274.50.35285. [DOI] [PubMed] [Google Scholar]
- [140].Hell K, Neupert W, Stuart RA. Oxa1p acts as a general membrane insertion machinery for proteins encoded by mitochondrial DNA. EMBO J. 2001;20:1281–1288. doi: 10.1093/emboj/20.6.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Hell K, Herrmann JM, Pratje E, Neupert W, Stuart RA. Oxa1p, an essential component of the N-tail protein export machinery in mitochondria. Proc. Natl. Acad, Sci. U.S.A. 1998;95:2250–2255. doi: 10.1073/pnas.95.5.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Herrmann JM, Neupert W, Stuart RA. Insertion into the mitochondrial inner membrane of a polytopic protein, the nuclear-encoded Oxa1p. EMBO J. 1997;16:2217–2226. doi: 10.1093/emboj/16.9.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Szyrach G, Ott M, Bonnefoy N, Neupert W, Herrmann JM. Ribosome binding to the Oxa1 complex facilitates co-translational protein insertion in mitochondria. EMBO J. 2003;22:6448–6457. doi: 10.1093/emboj/cdg623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Jia L, Dienhart M, Schramp M, McCauley M, Hell K, Stuart RA. Yeast Oxa1 interacts with mitochondrial ribosomes: the importance of the C-terminal region of Oxa1. EMBO J. 2003;22:6438–6447. doi: 10.1093/emboj/cdg624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Preuss M, Ott M, Funes S, Luirink J, Herrmann JM. Evolution of mitochondrial Oxa proteins from bacterial YidC: Inherited and acquired functions of a conserved protein insertion machinery. J. Biol. Chem. 2005;280:13004–13011. doi: 10.1074/jbc.M414093200. [DOI] [PubMed] [Google Scholar]
- [146].Gaisne M, Bonnefoy N. The COX18 gene, involved in mitochondrial biogenesis, is functionally conserved and tightly regulated in humans and fission yeast. FEMS Yeast Res. 2006;6:869–882. doi: 10.1111/j.1567-1364.2006.00083.x. [DOI] [PubMed] [Google Scholar]
- [147].Broadley SA, Demlow CM, Fox TD. Peripheral mitochondrial inner membrane protein, Mss2p, required for export of the mitochondrially coded Cox2p C tail in Saccharomyces cerevisiae. Mol. Cell. Biol. 2001;21:7663–7672. doi: 10.1128/MCB.21.22.7663-7672.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Zambrano A, Fontanesi F, Solans A, de Oliveira RL, Fox TD, Tzagoloff A, Barrientos A. Aberrant translation of cytochrome c oxidase subunit 1 mRNA species in the absence of Mss51p in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell. 2007;18:523–535. doi: 10.1091/mbc.E06-09-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Preuss M, Leonhard K, Hell K, Stuart RA, Neupert W, Herrmann JM. Mba1, a novel component of the mitochondrial protein export machinery of the yeast Saccharomyces cerevisiae. J. Cell Biol. 2001;153:1085–1096. doi: 10.1083/jcb.153.5.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Ott M, Prestele M, Bauerschmitt H, Funes S, Bonnefoy N, Herrmann JM. Mba1, a membrane-associated ribosome receptor in mitochondria. EMBO J. 2006;25:1603–1610. doi: 10.1038/sj.emboj.7601070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Bonnefoy N, Kermorgant M, Groudinsky O, Minet M, Slonimski PP, Dujardin G. Cloning of a human gene involved in cytochrome oxidase assembly by functional complementation of an Oxa1-mutation in Saccharomcyes cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 1994;91:11978–11982. doi: 10.1073/pnas.91.25.11978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Stiburek L, Fornuskova D, Wenchich L, Pejznochova M, Hansikova H, Zeman J. Knockdown of human Oxa1L impairs the biogenesis of F1Fo-ATP synthase and NADH:ubiquinone oxidoreductase. J. Mol. Biol. 2007;374:506–516. doi: 10.1016/j.jmb.2007.09.044. [DOI] [PubMed] [Google Scholar]
- [153].Haque ME, Elmore KB, Tripathy A, Koc H, Koc EC, Spremulli LL. Properties of the C-terminal tail of human mitochondrial inner membrane protein Oxa1L and its interactions with mammalian mitochondrial ribosomes. J. Biol. Chem. 2010;285:28353–28362. doi: 10.1074/jbc.M110.148262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Haque ME, Spremulli LL, Fecko CJ. Identification of protein-protein and protein- ribosome interacting regions of the C-terminal tail of human mitochondrial inner membrane protein Oxa1L. J. Biol. Chem. 2010;285:34991–34998. doi: 10.1074/jbc.M110.163808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Lupo D, Vollmer C, Deckers M, Mick DU, Tews I, Sinning I, Rehling P. Mdm38 is a 14-3-3-like receptor and associates with the protein synthesis machinery at the inner mitochondrial membrane. Traffic. 2011:1457–1466. doi: 10.1111/j.1600-0854.2011.01239.x. [DOI] [PubMed] [Google Scholar]
- [156].Dimmer KS, Navoni F, Casarin A, Trevisson E, Endele S, Winterpacht A, Salviati L, Scorrano L. LETM1, deleted in Wolf-Hirschhorn syndrome is required for normal mitochondrial morphology and cellular viability. Hum. Mol. Genet. 2008;17:201–214. doi: 10.1093/hmg/ddm297. [DOI] [PubMed] [Google Scholar]
- [157].Piao L, Li Y, Kim SJ, Byun HS, Huang SM, Hwang SK, Yang KJ, Park KA, Won M, Hong J, Hur GM, Seok JH, Shong M, Cho MH, Brazil DP, Hemmings BA, Park J. Association of LETM1 and MRPL36 contributes to the regulation of mitochondrial ATP production and necrotic cell death. Cancer Res. 2009;69:3397–3404. doi: 10.1158/0008-5472.CAN-08-3235. [DOI] [PubMed] [Google Scholar]
- [158].Waldeck-Weiermair M, Jean-Quartier C, Rost R, Khan MJ, Vishnu N, Bondarenko AI, Imamura H, Malli R, Graier WF. The leucine zipper EF hand-containing transmembrane protein 1 (LETM1) and uncoupling proteins- 2 and 3 (UCP2/3) contribute to two distinct mitochondrial Ca2+ uptake pathways. J. Biol. Chem. 2011;286:28444, 28555. doi: 10.1074/jbc.M111.244517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Jiang D, Zhao L, Clapham DE. Genome-wide RNAi screen identifies LetM as a mitochondrial Ca2+/H+ antiporter. Science. 2009;326:144–147. doi: 10.1126/science.1175145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Florentz C, Sohm B, Tryoen-Toth P, Putz J, Sissler M. Human mitochondrial tRNAs in health and disease. Cell Mol. Life Sci. 2003;60:1356–1375. doi: 10.1007/s00018-003-2343-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Perez-Toth X, Funes S, Camacho-Villasana Y, Marjavaara S, Tavares-Carreon F, Shingu-Vazquez M. Protein synthesis and assembly in mitochondrial disorders. Curr. Top. Med. Chem. 2008;8:1335–1350. doi: 10.2174/156802608786141124. [DOI] [PubMed] [Google Scholar]
- [162].Kemp JP, Smith PM, Pyle A, Neeve VCM, Tuppen HAL, Schara U, Talim B, Topaloglu H, Holinski-Feder E, Abicht A, Czermin B, Lochmiller H, McFarland R, Chinnery PF, Chrzanowska-Lightowlers ZMA, Lightowlers RN, Taylor RW, Horvath R. Nuclear factors involved in mitochondrial translation cause a subgroup of combined respiratory chain deficiency. Brain. 2011;134:183–195. doi: 10.1093/brain/awq320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Rotig A. Human diseases with impaired mitochondrial protein synthesis. Biochim. Biophys. Acta. 2011;1807:1198–1205. doi: 10.1016/j.bbabio.2011.06.010. [DOI] [PubMed] [Google Scholar]
- [164].Levinger L, Jacobs O, James M. In vitro 3′-end endonucleolytic processing defect in a human mitochondrial tRNA(Ser(UCN)) precursor with the U7445C substitution, which causes non-syndromic deafness. Nuc. Acids Res. 2001;29:4334–4340. doi: 10.1093/nar/29.21.4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Hao H, Moraes CT. A disease-associated G5703A mutation in human mitochondrial DNA causes a conformational change and a marked decrease in steady-state levels of mitochondrial tRNAAsn. Mol. Cell. Biol. 1997;17:6831–6837. doi: 10.1128/mcb.17.12.6831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Kelley SO, Steinberg SV, Schimmel P. Functional defects of pathogenic human mitochondrial tRNAs related to structural fragility. Nat. Struct. Biol. 2000;7:862–865. doi: 10.1038/79612. [DOI] [PubMed] [Google Scholar]
- [167].Ling J, Roy H, Qin D, Rubio MA, Alfonzo JD, Fredrick K, Ibba M. Pathogenic mechanism of a human mitochondrial tRNAPhe mutation associated with myoclonic epilepsy with ragged red fibers syndrome. Proc. Natl. Acad. Sci. U.S.A. 2007;104:15299–15304. doi: 10.1073/pnas.0704441104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Enriquez J, Chomyn A, Attardi G. MtDNA mutation in MERRF syndrome causes defective aminoacylation of tRNAlys and premature translation termination. Nat. Genet. 1995;10:47–55. doi: 10.1038/ng0595-47. [DOI] [PubMed] [Google Scholar]
- [169].Cenatiempo Y, Deville F, Dondon J, Grunberg-Manago M, Sacerdot C, Hershey JW, Hansen HF, Petersen HU, Clark BF, Kjeldgaard M, la Cour TFM, Mortensen KK, Nyborg J. The protein synthesis initiation factor 2 G-domain. Study of a functionally active C-terminal 65-kilodalton fragment of IF2 from Escherichia coli. Biochem. 1987;26:5070–5076. doi: 10.1021/bi00390a028. [DOI] [PubMed] [Google Scholar]
- [170].Akama K, Christian BE, Jones CN, Ueda T, Takeuchi N, Spremulli LL. Analysis of the functional consequences of lethal mutations in mitochondrial translational elongation factors. Biochim. Biophys. Acta. 2010;1802:692–698. doi: 10.1016/j.bbadis.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Kirino Y, Yasukawa T, Ohta S, Akira S, Ishihara K, Watanabe K, Suzuki T. Codon- specific translational defect caused by taurine-modification deficiency of mutant tRNA in MELAS, a human mitochondrial disease. Proc. Natl. Acad. Sci. U.S.A. 2004;101:15070–15075. doi: 10.1073/pnas.0405173101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Vissing J, Salamon MB, Arlien-Soborg P, Norby S, Manta P, DiMauro S, Schmalbruch H. A new mitochondrial tRNA(Met) gene mutation in a patient with dystrophic muscle and exercise intolerance. Neurology. 1998;50:1875–1878. doi: 10.1212/wnl.50.6.1875. [DOI] [PubMed] [Google Scholar]
- [173].Jones CN, Jones CI, Graham WD, Agris PF, Spremulli LL. A disease-causing point mutation in human mitochondrial tRNAMet results in tRNA misfolding leading to defects in translational initiation and elongation. J Biol. Chem. 2008;283:34445–34456. doi: 10.1074/jbc.M806992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Valente L, Tiranti V, Marsano RM, Malfatti E, Fernandez-Vizarra E, Mereghetti P, De Gioia L, Burlina A, Castellan C, Comi G, Salvasta S, Ferrero I, Zeviani M. Infantile encephalopathy and defective mtDNA translation in patients with mutations of mitochondrial elongation factors EFG1 and EF-Tu. Am. J. Hum. Genet. 2007;80:44–58. doi: 10.1086/510559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Valente L, Shigi N, Suzuki T, Zeviani M. The R336Q mutation in human mitochondrial EF-Tu prevents the formation of an active mt-EF-Tu.GTP.aa-tRNA ternary complex. Biochim. Biophys. Acta. 2009;1792:791–795. doi: 10.1016/j.bbadis.2009.06.002. [DOI] [PubMed] [Google Scholar]
- [176].Smeitink JAM, Elpeleg O, Antonicka H, Diepstra H, Saada A, Smits P, Sasarman F, Vriend G, Jacob-Hirsch J, Shaag A, Rechavi G, Welling B, Horst J, Rodenburg RJ, van den Heuvel B, Shoubridge EA. Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs. Am. J. Hum. Genet. 2006;79:869–877. doi: 10.1086/508434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Smits P, Antonicka H, van Hasselt PM, Weraarpachai W, Haller W, Schreurs M, Venselaar H, Rodenburg RJ, Smeitink JA, van den Heuvel LP. Mutation in subdomain G’ of mitochondrial elongation factor G1 is associated with combined OXPHOS deficiency in fibroblasts but not in muscle. Eur. J. Hum. Genet. 2011;19:275–279. doi: 10.1038/ejhg.2010.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Antonicka H, Sasarman F, Kennaway NG, Shoubridge EA. The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum. Mol. Genet. 2006;15:1835, 1846. doi: 10.1093/hmg/ddl106. [DOI] [PubMed] [Google Scholar]
- [179].Miller C, Saada A, Shaul N, Shabtai N, Ben den Heuvel E, Shaag A, Hershkowitz E, Elpeleg O. Defective mitochondrial translation caused by a ribosomal portein (MRPS16) mutation. Ann. Neurol. 2004;56:734–738. doi: 10.1002/ana.20282. [DOI] [PubMed] [Google Scholar]
- [180].Saada A, Shaag A, Arnon S, Dolfin T, Miller C, Fuchs-Telem D, Lombes A, Elpeleg O. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J. Med. Genet. 2007;44:784–786. doi: 10.1136/jmg.2007.053116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Haque Md.E., Grasso D, Miller C, Spremulli LL, Saada A. The effect of mutated mitochondrial ribosomal proteins S16 and S22 on the assembly of the small and large ribosomal subunits in human mitochondria. Mitochondrion. 2008;8:254–261. doi: 10.1016/j.mito.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Smits P, Saada A, Wortmann SB, Heister AJ, Brink M, Pfundt R, Miller C, Haas D, Hantschmann R, Rodenburg RJ, Smeitink JA, van den Heuvel LP. Mutation in mitochondrial ribosomal protein MRPS22 leads to Cornelia de Lange-like phenotype, brain abnormalities and hypertrophic cardiomyopathy. Eur. J. Hum. Genet. 2011;19:394–399. doi: 10.1038/ejhg.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Tucker EJ, Hershman SG, Kohrer C, Belcher-Timme CA, Patel J, Goldberger OA, Christodoulou J, Silberstein JM, McKenzie M, Ryan MT, Comptom AG, Jaffe JD, Carr S, Calvo SE, RajBhandary UL, Thorburn DR, Mootha VK. Mutations in MTFMT underlie a human disorder of formylation causing impaired mitochondrial translation. Cell Metab. 2011;14:428–434. doi: 10.1016/j.cmet.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Bykhovskaya Y, Mengesha E, Fischel-Ghodsian N. Pleiotropic effects and compensation mechanisms determine tissue specificity in mitochondrial myopathy and sideroblastic anemia (MLASA) Mol. Genet. Metab. 2007;91:148–156. doi: 10.1016/j.ymgme.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Fernandez-Vizarra E, Berardinelli A, Valente L, Tiranti V, Zeviani M. Nonsense mutation in pseudouridylate synthase 1 (PUS1) in two brothers affected by myopathy, lactic acidosis and sideroblastic anaemia (MLASA) J. Med. Genet. 2007;44:173–180. doi: 10.1136/jmg.2006.045252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Yan Q, Bykhovskaya Y, Li R, Mengesha E, Shohat M, Estivill X, Fischel-Vizarra N, Guan MX. Human TRMU encoding the mitochondrial 5- methylaminomethyl-2-thiouridylate-methyltransferase is a putative nuclear modifier gene for the phenotypic expression of the deafness-associated 12S rRNA mutations. Biochem. Biophys. Res. Commun. 2006;342:1130–1136. doi: 10.1016/j.bbrc.2006.02.078. [DOI] [PubMed] [Google Scholar]
- [187].Kolanczyk M, Pech M, Zemojtel T, Yamamoto H, Mikula I, Calvaruso MA, van den Brand M, Richter R, Fischer B, Ritz A, Kossler N, Thurisch B, Spoerle R, Smeitink J, Kornak U, Chan D, Vingron M, Martasek P, Lightowlers RN, Nijtmans L, Schuelke M, Nierhaus KH, Mundlos S. NOA1 is an essential GTPase required for mitochondrial protein synthesis. Mol. Biol. Cell. 2011;22:1–11. doi: 10.1091/mbc.E10-07-0643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Uchiumi T, Ohgaki K, Yagi M, Aoki Y, Sakai A, Matsumoto S, Kang D. ERAL1 is associated with mitochondrial ribosome and elimination of ERAL1 leads to mitochondrial dysfunction and growth retardation. Nuc. Acids Res. 2010;38:5554–5568. doi: 10.1093/nar/gkq305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Dennerlein S, Rozanska A, Wydro M, Chrzanowska den Brand ZM, Lightowlers RN. Human ERAL1 is a mitochondrial RNA chaperone involved in the assembly of the 28S small mitochondrial ribosomal subunit. Biochem. J. 2010;430:551–558. doi: 10.1042/BJ20100757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Metodiev MD, Lesko N, Park CB, Camara Y, Shi Y, Wibom R, Hultenby K, Gustafsson CM, Larsson NG. Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab. 2009;9:386–397. doi: 10.1016/j.cmet.2009.03.001. [DOI] [PubMed] [Google Scholar]
- [191].Shoubridge EA. Something old, something new, something borrowed. Cell Metab. 2009;9:307–308. doi: 10.1016/j.cmet.2009.03.010. [DOI] [PubMed] [Google Scholar]
- [192].Camara Y, Asin-Cayuela J, Park CB, Metodiev MD, Shi Y, Ruzzenente B, Kukat C, Habermann B, Wibom R, Hultenby K, Franz T, Erdjument-Cayuela H, Tempst P, Hallberg BM, Gustafsson CM, Larsson NG. MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab. 2011;13:527–539. doi: 10.1016/j.cmet.2011.04.002. [DOI] [PubMed] [Google Scholar]
- [193].Weraarpachai W, Antonicka H, Sasarman F, Seeger J, Schrank B, Kolesar JE, Lochmuller H, Chevrette M, Kaufman BA, Horvath R, Shoubridge EA. Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat Genet. 2009;41:833, 837. doi: 10.1038/ng.390. [DOI] [PubMed] [Google Scholar]
- [194].Mootha VK, Lepage P, Miller K, Bunkenborg J, Reich M, Hjerrild M, Delmonte T, Villeneuve A, Sladek R, Xu F, Mitchell GA, Morin C, Mann M, Hudson TJ, Robinson B, Rioux JD, Lander ES. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc. Natl. Acad. Sci U. S. A. 2003;100:605–610. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Xu F, Morin C, Mitchell G, Ackerley C, Robinson BH. The role of the LRPPRC (leucine-rich pentatricopeptide repeat cassette) gene in cytochrome oxidase assembly: mutation causes lowered levels of COX (cytochrome c oxidase) I and COX III mRNA. Biochem. J. 2004;382:331–336. doi: 10.1042/BJ20040469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Saracco SA, Fox TD. Cox18p is required for export of the mitochondrially encoded Saccharomyces cerevisiae Cox2p C-tail and interacts with Pnt1p and Mss2p in the inner membrane. Mol. Biol. Cell. 2002;13:1122–1131. doi: 10.1091/mbc.01-12-0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Pierrel F, Bestwick ML, Cobine PA, Khalimonchuk O, Cricco JA, Winge DR. Coa1 links the Mss51 post-translational function to Cox1 cofactor insertion in cytochrome c oxidase assembly. EMBO J. 2007;26:4335–4346. doi: 10.1038/sj.emboj.7601861. [DOI] [PMC free article] [PubMed] [Google Scholar]