

Abstract
Two hereditary syndromes, lymphedema-distichiasis syndrome (LD) and blepharo-chelio-dontic (BCD) syndrome include the aberrant growth of eyelashes from the meibomian glands, known as distichiasis. LD is an autosomal dominant syndrome primarily characterized by distichiasis and the onset of lymphedema usually during puberty. Mutations in the forkhead transcription factor FOXC2 are the only known cause of LD. BCD syndrome consists of autosomal dominant abnormalities of the eyelid, lip, and teeth, and the etiology remains unknown. In this report, we describe a proband that presented with distichiasis, microcephaly, bilateral grade IV vesicoureteral reflux requiring ureteral re-implantation, mild intellectual impairment and apparent glomuvenous malformations. Distichiasis was present in three generations of the proband’s maternal side of the family. The glomuvenous malformations were severe in the proband, and maternal family members exhibited lower extremity varicosities of variable degree. A GLMN (glomulin) gene mutation was identified in the proband that accounts for the observed glomuvenous malformations; no other family member could be tested. TIE2 sequencing revealed no mutations. In the proband, an additional submicroscopic 265 kb contiguous gene deletion was identified in 16q24.3, located 609 kb distal to the FOXC2 locus, which was inherited from the proband’s mother. The deletion includes the C16ORF95, FBXO31, MAP1LC3B, and ZCCHC14 loci and 115 kb of a gene desert distal to FOXC2 and FOXL1. Thus, it is likely that the microcephaly, distichiasis, vesicoureteral and intellectual impairment in this family may be caused by the deletion of one or more of these genes and/or deletion of distant cis-regulatory elements of FOXC2 expression.
Keywords: FOXC2, FBXO31, MAP1LC3B, ZCCHC14, GLMN, distichiasis, vascular malformation, venous malformation, glomuvenous malformation
INTRODUCTION
Distichiasis is defined as a second row of eyelashes arising from the meibomian glands of the eyelid [Brooks et al., 2003]. Distichiasis can be inherited in an autosomal dominant manner as part of lymphedema-distichiasis syndrome (LD; OMIM 153400) or blepharo-chelio-dontic syndrome (BCD; OMIM 119580). LD is characterized by onset of bilateral lower limb lymphedema at puberty and congenital distichiasis, as well as other less penetrant clinical associations including varicose veins, congenital heart disease, cleft palate and ptosis [Bell et al., 2001; Brice et al., 2002; Erickson et al., 2001; Fang et al., 2000; Finegold et al., 2001]. FOXC2 mutations cause the majority of LD cases, although two LD families without FOXC2 mutations but with linkage to the FOXC2 region have been reported [Sholto-Douglas-Vernon et al., 2005]. Diagnostic criteria for BCD syndrome includes abnormalities of the eyelid (euryblepharon, ectropion of lower lid, distichiasis of the upper lid, lagophthalmia), lip (bilateral cleft lip and palate) and teeth (oligodontia and microdontia)[Gorlin et al., 1996]. To date, the molecular cause of BCD is unknown.
Vascular malformations are congenital lesions that exhibit a normal rate of endothelial cell proliferation, but result from inborn errors of vascular morphogenesis [Mulliken, 1988]. Vascular malformations are classified based on the affected vessel-type (artery, vein, capillary) and flow-characteristics (fast or slow) into venous malformations (VM), capillary malformations and arterial malformations. Some genetic factors responsible for vascular malformations have been elucidated (for review [Brouillard and Vikkula, 2007]). Most VM are sporadic, but some hereditary VM exhibit autosomal dominant inheritance [Boon et al., 2004]. Cutaneomucosal venous malformations (VMCM) are composed of dilated vessels with thin walls and poor smooth muscle cell coverage [Boon et al., 2004; Vikkula et al., 1996]. Linkage analysis led to the identification of mutations in the receptor tyrosine kinase TIE-2 (TEK) gene as the cause of CMVM [Vikkula et al., 1996]. Glomuvenous malformations (GVM) are vascular anomalies characterized by venous-like channels surrounded by abnormal smooth muscle-like cells known as glomus cells [Boon et al., 2004]. GLMN gene mutations were identified as the cause of glomuvenous malformations insert [Brouillard et al., 2002; Brouillard et al., 2005].
Here we report on a case that presented with distichiasis, microcephaly, bilateral grade IV vesicoureteral reflux, mild intellectual impairment and glomuvenous malformations. No lymphedema had developed in the proband and no FOXC2 coding exon mutations were detected. Clinical chromosomal microarray analysis identified a 16q24.3 submicroscopic contiguous gene deletion including the C16ORF95, FBXO31, MAP1LC3B, and ZCCHC14 loci. The deletion was further characterized by FISH, and the breakpoints were identified and sequenced. There were 2 bp of homology at the breakpoints, suggesting the deletion was formed by a non-homologous end joining (NHEJ) or template switching mechanism. Further genetic testing of known venous malformation gene candidates revealed that the proband was heterozygous for a known pathogenic 157delAAGAA GLMN mutation [Brouillard et al., 2002; Brouillard et al., 2005]. The 16q24.3 deletion may be a novel cause of a rare if not unique syndrome including distichiasis, microcephaly, bilateral grade IV vesicoureteral reflux, and intellectual impairment due to position effect on FOXC2 and/or deletion of one or more distal genes.
MATERIALS AND METHODS
ASCERTAINMENT
The proband was referred to the University of Michigan Pediatric Genetics Clinic for evaluation of a potential vascular malformation syndrome and was evaluated by one of the authors (J.W.I.). The presence of distichiasis suggested the features of the proband may be caused by a FOXC2 mutation. Therefore, informed consent for DNA testing was obtained from the proband and family members in accordance with the guidelines of the University of Michigan Medical School Institutional Review Board.
MR IMAGING
Imaging was performed on a 1.5 Tesla MR (Signa, GE Medical Systems). The MR images in Figure 3 were obtained under the following conditions: 3A-axial T1-weighted (TR 467 ms, TE 14 ms), 3B-Axial T2-weighted (TR 3400 ms, TE 79 ms), 3C-Coronal short tau inversion recovery (TR 4000 ms, TE 26.2 ms, TI 165 ms), 3D-Axial T1-weighted with fat saturation post gadolinium (TR 210 ms, TE 3.5 ms), 3E-Coronal T1-weighted with fat saturation post gadolinium (TR 255 ms, TE 3.3 ms).
Figure 3. MRI Imaging of the Vascular Anomalies of the Proband.
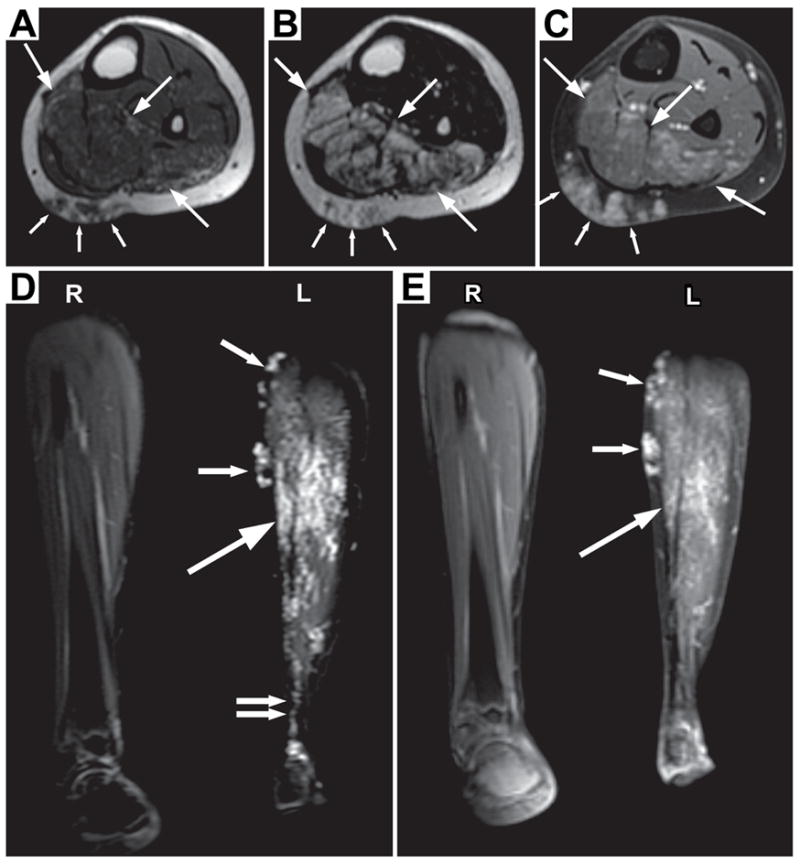
An infiltrative lesion (large arrows) is seen mainly within the soleus and flexor hallucis longus muscles, sparing the overlying distal medial gastrocnemius muscle. On T1 weighting (A), the intramuscular lesion is subtle and similar in signal intensity to the adjacent normal musculature. Scattered tiny areas of high signal may represent minimal fat within the lesion. On fluid sensitive sequences (B & D), the lesion is of high signal. After gadolinium is administered (C), there is mild enhancement within the intramuscular lesion which increases on later images (E). Best seen on the coronal images (D and E), the lesion appears composed of small vascular-like channels. The lesion extends into the adjacent subcutaneous fat (A–D, small arrows). The subcutaneous component also has the appearance of vascular-like channels. The overall appearance is most consistent with a vascular lesion, likely a venous malformation. The abnormality extends to the ankle (D, double arrows).
DNA ISOLATION AND ESTABLISHMENT OF THE LD-064 CELL LINE
Genomic DNA was extracted from 5ml blood samples using a guanidine HCl method for DNA extraction from blood [Ciulla et al., 1988]. The LD-064 cell line was established from a 5ml blood sample from the proband using the LSM Lymphocyte Separation Medium (ICN, Aurora OH) and immortalization with Epstein-Barr virus (EBV) [Gilbert, 2001].
FOXC2, GLOMULIN and TIE2 MUTATION SCREENING
FOXC2 was screened for mutations as previously described [Fang et al., 2000]. Deletion of the FOXC2 locus was tested for by fluorescent in situ hybridization (FISH) using BAC clone RP11-463O9 as probe. Sequencing of the GLMN and TIE2 genes was carried out as described [Brouillard et al., 2002; Vikkula et al., 1996].
CHROMOSOMAL ANALYSIS
The proband’s karotype was evaluated by the University of Michigan Clinical Cytogenetics Laboratory. Genomic microarray analysis using a BAC array (v4.1) was performed on DNA samples from the proband, the brother and mother of the proband at Baylor College of Medicine.
FISH
BAC clones RP11-463O9, RP11-235F18, RP11-178L8, RP11-482M8, RP11-264L1 were purchased from CHORI BACPAC Resource Center (Oakland, CA). FISH probes were generated using the Roche Biotin Nick Translation Kit (Indianapolis, IN) with 1 ug BAC DNA. FISH was performed as previously described [Wilke et al., 1996].
SNP GENOTYPING
SNPs surrounding the 16q24.3 deletion were chosen for genotyping using the UCSC genome browser and dbSNP [Karolchik et al., 2003; Sherry et al., 2001]. Primers for PCR were designed using Primer3 to flank the SNPs of interest [Rozen and Skaletsky, 2000]. All primers are listed in Table I. All PCRs were performed with Invitrogen Taq polymerase (Carlsbad, CA) using standard conditions (20mM Tris-HCl pH 8.4, 50mM KCl, 1.5mM MgCl2, 0.2mM dNTPs, 0.2 uM each sense and antisense primers) with a 60° C annealing temperature and an extension of 1 minute for 30 cycles. PCR products were separated by gel electrophoresis and subsequently purified with the Zymoclean gel DNA recovery kit (Zymo Research; Orange, CA). PCR products were sequenced by the University of Michigan DNA Sequencing Core (http://seqcore.brcf.med.umich.edu/).
Table I.
SNP Genotypes of the Proband
| SNP | Alleles | Forward primer (5′- 3′) | Reverse primer (5′- 3′) | Proband |
|---|---|---|---|---|
| rs11117242 | C/T | CCAATGAGGTAAGCGCTGTT | CAAGGGGACTGACTTTGGAA | T/T |
| rs12598171 | G/T | TTTTTATGGGTTAGTATCTTGCTCA | TGCAAGTCACTCACTATTCATAACTC | T/T |
| rs17697644 | A/G | AAAATGACTGTGGGTCCTTCA | ATGAGTAGGCTGCCCGTTTA | A/G |
| rs4843530 | A/G | TTGAGCCATCCCCAAGTTAC | GCATGTGCTCGTGGTAGAGA | G/G |
| rs7196293 | A/G | GTGTTTCAGATGCCGGTGAC | CACCCGTATGCTGTTGACC | A/G |
| rs8056012 | A/G | GGTGTGAGCACCACAGGAG | GATGGTGATGGTGACAATGG | A/G |
| rs4843542 | A/G | ACATTTCCCCTCCGTAAAAA | TTGGCTTAGAGAGCCTCCAG | A/G |
| rs1465452 | A/G | TCCATCCTGTAAAGGCAACC | GGCCAAGCCCATTTCTATTT | A/G |
| rs6540015 | C/G | GTTCAAATCCTGCCTTGCTC | TGGAATACGGGTGACAACAA | G/– |
| rs4843223 | C/T | TACTCATGGCCAAGGAATGG | TGCACACACAAGCACGTTTA | C/– |
| rs12447701 | C/G | TTCATCAAATGCTCCCTGTG | CATTTTGGTCTATTTGCTCTTGTT | G/– |
| rs12711472 | A/C | GCCATCTGTAGGTGACAGCA | AAAACACCCTCTAGGCAGCA | C/– |
| rs4843556 | C/T | GGCAGGATGTAGCAGAAAGC | TCAATTTCAGCATTGTTGGTG | C/– |
| rs6540016 | C/T | CAGCCCTAGGATGATGGAAA | GTGTTCCCCATCCACTTGTC | T/– |
| rs8047567 | C/G | TGCACTTACCACAGCCAGAG | TTTGCGTGAACCTCAGTTTG | G/– |
| rs4843549 | C/T | GTCAGGCGTCTGGTACAGGT | CCAAGAAGCAAGAGCAAACA | T/– |
| rs4843547 | A/C | GCAGCTGTTAAAGTCTCTTGTCC | TCTAGCCTGGGTGACAGAGC | C/– |
| rs7499277 | C/G | TGAAGGGAAACTCAGGTGCT | GGCCCAAGCCTCTCTCTTA | G/– |
| rs11640181 | A/G | TCAGAAAAATGGCCACAACA | CCCCTTGCTCAACTGTCATT | A/– |
| rs4843576 | C/T | CAGGGAGGGAGAGCCAAG | ATGCTACCAGCCTCCAGAAA | T/– |
| rs1966575 | A/G | AAGGAAAAGGAGGGAAGCAG | CTTCAAAGGAGGTGCAGAGC | A/– |
| rs7195872 | A/G | TGCAATGGATAAGTCCAGATT | CCTCTTGCCCAAGTTCACTG | A/– |
| rs3748391 | G/T | TTATGTGCTGAGTGGGTGGA | CTACCTGCCCGAGAGAGAAG | T/– |
| rs9308346 | A/T | ATCTGCAGCTCCCAGCTTTA | CCTTTCCTCCCCGGATAGT | T/– |
| rs6540031 | C/T | ACCCCCAGAGCTTGTTCTCT | CTCCCAAAGTGCTGGGACTA | C/– |
| rs889602 | C/G | AGGGACAGGGACACACAGAG | CAGCCCTACCAGCTGAACTC | G/– |
| rs933717 | C/T | TGTCCAGGATCTCACAGCAG | GCAGTTCCTCAAGTGGGATT | C/– |
| rs9938881 | A/C | AGCCACACTCATCACCACAG | CCAAGCAACACTGACTGCTC | C/– |
| rs9927125 | C/T | GGAGAAACTCCAAAACAGCAA | GACTTGGCTGCAGAGTTTCA | T/– |
| rs4843612 | A/G | TTTATTACCAGATAGTCCCTCAAGC | AAAATTAGTTTTCCCTCAACAATCA | G/A |
| rs4843613 | C/T | GCCCCCACTGTAAGTTGAAA | ACAGTCTGTGGCCTTTCCTG | T/C |
| rs4843242 | C/G | AGCCTCTCCCTCAGAGCTTC | TGTTGAGATTGGGTTGTTGG | G/C |
| rs7205226 | C/T | GCCTTTTCTGCTAGCTGCTC | CCCACCTGACCCATAAACAG | C/T |
| rs9928618 | C/G | CCCCCAGCATGTAGGAAGT | AAGCCCGAACCCTAACCTAA | G/G |
| rs8056477 | C/T | ATCGCAGGGGAGTACTTGG | CTTCCCTTTGATTCCCACAG | C/T |
INVERSE PCR
Inverse PCR was performed as previously described [Morrish et al., 2002; Wei et al., 2001]. Primers located distal to rs4843612 were chosen for inverse PCR using Primer3 [Rozen and Skaletsky, 2000]. 5 μg of genomic DNA from the proband were digested with SspI, SphI, PstI or HindIII and circularized with 3200 units of T4 DNA Ligase from NEB (Ipswich, MA) in a final volume of 1 ml. The resulting ligation products were PCR-amplified using primers Inverse-F (5′-TTACGCATGCTGTTTGGAAA-3′) and Inverse-R (5′-TGTGGTTTTGCCCTGGTCTT-3′). PCR conditions were as follows: initial cycle of 95°C for 5 min, followed by 35 cycles of 30s at 94°C, 30s at 60°C, 15min at 68°C, with a final step of 68°C for 10 min. PCR products were cloned into the Topo-XL cloning vector according to manufacturer’s instructions (Invitrogen; Carlsbad, CA). Topo-XL clones were end sequenced with M13Forward and Reverse primers by the University of Michigan DNA Sequencing Core. The deletion breakpoints were confirmed by PCR amplification across the breakpoint junction in a multiplex-PCR reaction that also included amplification of the wild type allele. Genomic DNA was amplified by PCR using standard conditions with oligos Wt-F (5′-AGCTCTAAAGTCCCCGAAGC-3′), Brkpt-R (5′-AGCCATCAGTGACCCCAGT-3′), Wt-R (5′-ACCGGTGCTTACTCTGAACG-3′), an annealing temperature of 65° C and an extension time of 1 minute for 35 cycles.
COPY NUMBER VARIATION
The Database of Genomic Variants (http://projects.tcag.ca/variation/) was used to identify copy number variants in the 16q24.3 region.
RESULTS
CLINICAL FINDINGS
The proband (Fig 1, III-2) was first seen at the University of Michigan at the age of 10 years and 2.5 months and was referred for evaluation of a possible vascular malformation syndrome and developmental delay. She was born at term after an uncomplicated pregnancy and weighed 2325 grams. Vascular malformations were noted at birth involving the right parietal area, upper left chest, left shoulder, left hand and left leg. Bayley Scales of Infant Development II testing at 32 months resulted in a mental development index below 50 and an overall developmental age of 19 months. Receptive and expressive communication skills were at the 24 month level. The proband had a history of tympanostomy tube placement at 2 years of age followed by normal tympanograms and normal speech recognition threshold hearing testing. In the first three years, she required casting for positioning of the left foot followed by heel cord release surgery. Subsequently, at 12 years, she had right equinus contracture requiring heel cord lengthening surgery. Past medical history also includes diagnoses of attention deficit hyperactivity disorder, learning disabilities, mild intellectual impairment, and recurrent urinary tract infections with bilateral grade IV vesicoureteral reflux requiring bilateral ureteral reimplantation surgery. The proband’s mother (Fig 1, II-2) also had a history of mental impairment and learning problems, which required special education services in school (formal testing was not performed). The proband, her mother and her maternal grandmother reported recurrent eye irritation due to the inward growth of eyelashes, which they treated themselves by eyelash removal (Fig 1, III-2, II-2, I-2 respectively). Furthermore, an extensive family history of eye irritation and eyelash removal was reported for the maternal side of the family (Fig 1, individuals III-3, III-4, II-2, II-4 and siblings of I-2 not shown in pedigree). A full brother (Fig 1, III-1) was unaffected by any of these aforementioned clinical conditions as verified by physical examination.
Figure 1. Transmission of the Phenotypes in the Family of the Proband.

Three generations of the proband’s family are shown by pedigree. Each generation has affected individuals indicating possible autosomal dominant inheritance of distichiasis and vascular malformations, but with variable expressivity for vascular malformations. Symbols used in the pedigree are shown in the legend.
Physical examination of the proband showed macrocephaly, with a head circumference measuring 48.9 cm (< 5%). An additional row of small eyelashes and pits from eyelash removal were apparent upon visual examination of the patient, her mother and maternal grandmother (data not shown). In addition to distichiasis, vascular anomalies were apparent in the proband and are pictured in Figure 2(A–F). The features of each lesion are described in Table II. The vascular anomalies were congenital and became more prominent with age. Some were macular (left chest, upper scalp) with blue and red colored vessels; others were deep blue, raised and soft in character. A skin biopsy was not obtained. An area of cutis aplasia was detected on the scalp to the left of the sagittal midline (10 × 2 cm) and was discolored by a macular vascular anomaly. Significantly, no vascular lesions were detected in the oral mucosa. Palpation of the lesions did not cause overt pain or blanching. No cleft lip or palate nor dental anomalies were detected. No lymphedema was observed in the proband or other family members. Renal ultrasound and echocardiogram revealed normal anatomy. Magnetic resonance imaging of the brain and skull revealed a vascular malformation of the right parietal scalp without intracranial involvement. Chest MRI revealed cutaneous and subcutaneous vascular anomaly of the left mid and anterior chest without intrathoracic extension. MRI of the left leg revealed an infiltrative lesion (large arrows, Fig 3A) mainly within the soleus and flexor hallucis longus muscles, sparing the overlying distal medial gastrocnemius muscle and extending to the ankle. The intramuscular lesion is subtle and similar in signal intensity to the adjacent normal musculature. Mild enhancement was observed within the intramuscular lesion which increased on later images and on coronal images (Fig. 3C and E) the lesion appears composed of small vascular-like channels. The lesion extends into the adjacent subcutaneous fat (Fig 3A–D, small arrows), and this subcutaneous component also has the appearance of vascular-like channels. The overall appearance is most consistent with a vascular lesion, likely a venous malformation. No histopathology was performed preventing exact identification of the ectatic vessels. Inheritance of the vascular malformations is uncertain, but the maternal grandmother of the proband had lower extremity varicosities and dilated superficial veins (Fig 2,G), and the father (Fig 1, II-1), who was unavailable for examination was reported by the maternal grandmother to have a prominent, discolored venous mass on his leg, larger than that observed in the proband. In addition, a similar leg mass was present in the paternal grandmother, who was also unavailable for examination.
Figure 2. Vascular Malformations.

VMs of right lateral brow (A), v-shaped reticular lesions on the left side of the chest extending from the scapula to below the sternum (B), lesions along the left arm (C), vascular malformations of the posterior lower left leg at the level of knee (D), vascular malformations of the left ankle and plantar surface of the foot (E), and lesions of the left second finger (F). Prominent superficial vasculature is exhibited in the left leg of the maternal grandmother of the proband (G).
Table II.
Qualitative Description of the Vascular Lesions of the Proband
| lesion | color | texture | appearance |
|---|---|---|---|
| scalp | purple; red | flat | spider-like |
| r. forehead | blue-purple | worm-like | prominent |
| l. forehead | blue | fleshy | punctate |
| chest | purple-red | peau d’orange | prominent |
| chest | blue | flat | macular |
| l. arm | blue-green | soft, fleshy | prominent |
| l. 2nd finger | blue-green | soft, fleshy | prominent |
| l. leg | blue-purple | soft | varicose, prominent |
| dorsum l. foot | blue-purple | soft | varicose, prominent |
CLINICAL TESTING
Standard G-banded chromosomes of the proband showed a normal karyotype with no gross chromosomal abnormalities. Fragile X DNA testing was normal. Hereditary distichiasis can be inherited as a prominent feature of lymphedema-distichiasis syndrome (LD; OMIM 153400) and varicose veins are a less penetrant clinical manifestation associated with LD [Mellor et al., 2007]. Therefore, based on the presence of both distichiasis and vascular malformations, FOXC2 was tested for mutations. Sequencing of the single FOXC2 coding exon did not detect a mutation (data not shown). Likewise, FISH using BAC probe RP11-463O9 that spans the FOXC2 gene demonstrated that the genomic region surrounding and including the FOXC2 region was not deleted (Fig 4). The genome of the proband was also analyzed for copy number alterations by chromosomal BAC microarray analysis (CMA). CMA detected a deletion of BAC clone RP11-106D4 (Fig 5), which is located 745 kb distal to the FOXC2 locus. The RP11-106D4 deletion was also detected in the mother of the proband (Fig 1, II-2), but was absent in her unaffected brother (Fig 1, III-1).
Figure 4. FOXC2 Deletion Screening.

A. Diagram of the genomic insert of BAC clone RP11-463O9 including the genes (gray boxes) and intergenic distances (kilobases). Exon/intron structure of MTHFSD is not shown for simplicity. B. FISH using RP11-463O9 as probe generated hybridization signals on both chromosome 16 homologs (white arrows) of the proband.
Figure 5. Mapping of the 16q24 Contiguous Gene Deletion.

A. Physical map of the 16q24 region showing the proximity of the deletion with respect to FOXC2. Genes are depicted by gray boxes ignoring exon/intron structure. The dashed lines show the relative position of a 612 kb segment extending from BAC clone RP11-235F18 to RP11-482M8. Genes in this magnified region are represented by arrows showing the direction of transcription for each gene. BAC clones used for FISH are shown as gray (1 signal-deleted) and black (2 signals-present) lines. SNPs rs1465452 and rs4843612 SNPs are marked by vertical black lines.
B–D. FISH on metaphase chromosomes from the proband. FISH was performed with BAC clone RP11-235F18 (B), RP11-178L8 (C), and RP11-482M8 (D). In panel C, RP11-264L1 (red) was co-hybridized with RP11-178L8 (green) as a control probe to mark each chromosome 16. White arrows mark hybridization signals. The red arrow marks the chromosome 16 carrying the deletion demonstrated by the absence of a green (RP11-178L8) hybridization signal (panel C).
IDENTIFICATION OF THE 16q24.3 DELETION BREAKPOINT JUNCTION
FISH was performed to confirm the 16q24.3 deletion in the proband. A FISH probe generated from BAC clone RP11-178L8, a clone that overlaps RP11-106D4, hybridized to only one copy of chromosome 16 on metaphase chromosomes from the proband confirming the deletion (Fig 5A,C). The size of the deletion was unknown due to the incomplete chromosome coverage of the BAC array. Therefore, BAC clones flanking RP11-106D4 and RP11-178L8 were also used as FISH probes. Proximal BAC clone RP11-235F18 and distal BAC clone RP11-482M8 both produced hybridization signals on both homologs of chromosome 16 roughly defining the size of the deletion (Fig 5B and D). To further refine the extent of the deletion, SNP genotyping was utilized to map a region of homozygosity surrounding the deletion. SNP genotypes from the 16q24.3 deletion region are summarized in Table I. rs1465452 and rs4843612 were the closest heterozygous SNPs flanking the deletion proximally and distally, respectively (Table I; Fig 5A). Thus, rs1465452 and rs4843612 narrowed the 16q24.3 deletion to approximately 280 kb.
To exactly determine the deletion boundaries, inverse PCR products were generated with primers distal to rs4843612, which were cloned and sequenced to identify the deletion breakpoint junction. Sequencing of one of the clones, MTB3-1-2, revealed the sequence of the 16q24.3 deletion breakpoint junction. There were 2 bp of homology at the breakpoints or any regions of extended homology in the 10 kb region flanking the breakpoints. The proximal and distal breaks occurred in an imperfect CAGAGA repeat sequence located at nucleotide positions 85,769,299 (NCBI Build 36) and in an old L1 retrotransposon (L1M2) at 86,034,334 respectively (Fig 6A). To confirm the positions of the breakpoints, genomic DNA isolated from a blood sample from the proband was used to PCR amplify the breakpoint region in both the wild type and 16q24.3 deletion allele (Fig 6B). The multiplex PCR assay generated both expected sizes for the wild type and 16q24.3 deletion alleles in the proband, whereas an unrelated control DNA generated PCR product corresponding to the wild type allele only. The deletion appears to result either from the simple ligation of proximal and distal DNA double strand breaks, consistent with formation by NHEJ or a template switching mechanism such as MMBIR and not from non-allelic homologous recombination (Fig 6A) [Hastings et al., 2009; Lieber et al., 2003]. Identification of the breakpoints showed that the submicroscopic 16q24.3 deletion includes 265,046 bp of sequence (Fig 6C). The genes for C16ORF95, FBXO31 (F-box protein 31) and MAP1LC3B (microtubule-associated protein 1 light chain 3 beta) and all but the first exon of the ZCCHC14 (zinc finger, CCHC domain containing 14) gene are deleted. In addition to these genes, the 16q24.3 deletion removes 115 kb of a gene desert located between FOXF1/FOXC2/FOXL1 gene cluster and C16ORF95.
Figure 6. Cloning of the 16q24 Deletion by Inverse PCR.

A. 16q24.3 deletion breakpoints. The genomic sequence surrounding the proximal and distal breakpoints of the 16q24.3 deletion is shown above a sequencing chromatogram from the pMTB3-1-2 inverse PCR clone (deleted sequence is shaded). UCSC genome browser coordinates are shown above the sequence. Breakpoints are indicated by red lines. Deleted sequence is shown in lowercase whereas present sequence is shown in uppercase.
B. Confirmation of 16q24 deletion breakpoints. A common forward primer (*) proximal to the 16q24 deletion in combination with reverse primers specific for the wild type allele (black) and the deletion allele (red) were used for PCR. The proband (P) was heterozygous for the 16q24 deletion whereas an unrelated control (C) was homozygous for the wild type allele. A control PCR reaction with no template was also performed (H).
C. The 16q24 deletion removes C16ORF95, FBXO31, MAP1LC3B, and ZCCHC14 loci. The 16q24 region is shown as a black horizontal line with vertical lines representing exons and arrows showing the direction of the corresponding transcripts. The 16q24 deletion is depicted below by a red line.
Three copy number variations (CNVs) in the 16q24.3 deletion region have been identified in the Database of Genomic Variants (Table III). Two of the 3 CNVs (variation 4970 and 8283) include gains with no observed deletion. The distal 8kb of the 16q24.3 deletion overlaps with a reported 168 kb deletion CNV, including the putative promoter region and first exon of ZCCHC14 [Wang et al., 2007]. Deletion of the centromeric 257 kb of the 16q24.3 deletion has not been detected in any reported CNV, or in any of >1000 high-resolution chromosomal microarray cases in the Michigan Medical Genetics Laboratories at the University of Michigan to date (unpublished results).
Table III.
Copy Number Variation in 16q24.3 Deletion Region
| variation | location | size (kb) | gain | loss |
|---|---|---|---|---|
| 4970 | 16: 85,906,951–86,092,333 | 185 | 3 | 0 |
| 4971 | 16: 86,025,064–86,192,948 | 167 | 1 | 8 |
| 114272 | 16: 85,945,085–86,000,336 | 2 | 0 |
GLMN and TIE2 MUTATION ANALYSIS
Physical evaluation of the vascular lesions in the proband (Fig 2; Table II) suggested that these lesions may be glomuvenous malformations. Most venous malformations are sporadic, but hereditary cutaneomucosal venous malformations and glomuvenous malformations are caused by mutations in TIE2 and GLMN, respectively [Brouillard et al., 2002; Brouillard et al., 2005; Vikkula et al., 1996; Wouters et al., 2010]. Allele-specific PCR demonstrated that the proband was heterozygous for the previously reported 157delAAGAA mutation (data not shown), whereas mutations were not detected in TIE2 [Brouillard et al., 2002; Vikkula et al., 1996]. Consequently, the vascular malformations of the proband are indeed glomuvenous malformations.
DISCUSSION
We have presented the clinical and genomic characterization of an individual with distichiasis, microcephaly, mild intellectual impairment, but without lymphedema, associated with an inherited 16q24.3 deletion that includes the C16ORF95, FBXO31, MAP1LC3B, and ZCCHC14 genes as well as 115 kb of a gene desert distal to the FOXF1/FOXC2/FOXL1 loci. The uniqueness of this case is illustrated by the ability to differentiate it from other syndromes. Distichiasis, the anomalous growth of eyelashes from the meibomian glands, can be inherited in lymphedema-distichiasis syndrome (LD). The absence of both lymphedema and a FOXC2 coding mutation, as well as absence of many of the other LD-associated features argues against diagnoses of LD. Similarly, the presence of microcephaly, bilateral grade IV vesicoureteral reflux, and intellectual impairment has not been reported to be associated with LD [Brice et al., 2002; Erickson et al., 2001; Finegold et al., 2001].
The proband was originally referred not only for developmental delay and microcephaly, but also for evaluation of the vascular anomalies noted in Figure 2 and Table II. Boon and colleagues described criteria for the differentiation between venous malformations, cutaneomucosal venous malformations (VMCM), and glomuvenous malformations [Boon et al., 2004]. Glomuvenous malformations have a cobblestoned appearance ranging in color from pink to purplish dark blue, consistent with characteristics of the proband’s lesions. Mutation testing determined that the proband is heterozygous for a deleterious GLMN mutation resulting in numerous venous malformations that grossly are consistent with congenital glomuvenous malformations. These venous malformations are distributed in many locations, although none were present in the oral mucosa and they did not blanch upon palpation, consistent with the characteristics of glomuvenous malformation. Although FOXC2 mutations have been associated with varicose veins, the presence of the GLMN mutation and resulting glomuvenous malformations makes it impossible to discern the effect of the 16q24.3 deletion upon the vasculature of the proband [Mellor et al., 2007; Ng et al., 2005]. The patients of Stankiewicz (D2, D5, and D8) carried larger deletions including the region deleted here, and there was no apparent venous malformations described although these patients had a myriad of congenital anomalies and short lifespans making a direct comparison tenuous. The GLMN mutation status of the mother and maternal grandmother is unknown. The proband’s father (unavailable for examination) was reported to have vascular anomalies similar to the proband, albeit a larger area of involvement on the leg. Consequently, the GLMN mutation most likely is responsible for vascular lesions of the proband and may have been inherited from her father, but it is uncertain whether the venous varicosities of other individuals on the maternal side of the family can be attributed to a GLMN mutation.
The absence of reports in the literature of 16q rearrangements producing 16q24.2–16q24.3 monosomy led to speculation that loss of this region is not tolerated [Callen et al., 1993]. More recently, Stankiewicz and colleagues have described individuals carrying large deletions that include the 16q24.3 deletion that we have delineated in this study [Stankiewicz et al., 2009]. Two of these patients (designated D2 and D5) had dilated renal pelvices and bilateral renal pelviectasis, respectively. Similarly, our proband had bilateral grade IV vesicoureteral reflux requiring bilateral ureteral reimplantation surgery. Taken together these observations suggest that the 16q24.3 deletion may be responsible for the renal anomaly in the proband.
Haploinsufficiency of one or a combination of the genes removed by the 16q24.3 deletion could cause the non-vascular phenotypic features of the proband. ZCCHC14 encodes a completely uncharacterized zinc finger protein. The first two exons and putative promoter region of ZCCHC14 are deleted in a CNV (variation 4971, Database of Genomic Variants) in 8 of 95 normal control samples arguing that haploinsufficiency of ZCCHC14 does not have a phenotypic consequence [Wang et al., 2007]. Like ZCCHC14, C16ORF95, a recently annotated gene located centromeric to FBXO31, has no known function limiting the speculation of its role in the phenotypic features of the proband.
FBXO31 is an F-box protein that has been reported to function as a tumor suppressor in breast cancer cell lines by associating with a SCF (Skp1-Cdc53-F-boxprotein) ubiquitin complex to target substrates for degradation to progress through the cell cycle; however, no role in ocular, neural or blood vascular development has been reported [Kumar et al., 2005]. Recently, FBXO31 was shown to target CYCLIND2 for degradation in response to DNA damage [Santra et al., 2009].
MAP1LC3B is one of three genes (MAP1LC3A, MAP1LC3B and MAP1LC3C) in the human genome homologous to the rat Map1lc3 and yeast Apg8 genes, both of which function in autophagy [He et al., 2003]. Conditional knockout of Atg5 and Atg7 has shown that impairment of autophagy leads to neurodegeneration in mice [Hara et al., 2006; Komatsu et al., 2006]. The accumulation of autophagic vesicles has also been found in a number of human neurodegenerative disorders including Parkinsonism and Huntington disease [Shintani and Klionsky, 2004]. Whether MAP1LC3B haploinsufficiency causes autophagic defects in humans is unknown and untested, but Map1lc3b transcripts are expressed in the central nervous system in mice during embryogenesis suggesting that MAP1LC3B is a candidate gene for the intellectual disability in our patient [Cann et al., 2008].
There is no support for a role of any of the three deleted genes in development of distichiasis. The relative proximity of the 16q24.3 deletion to the FOXC2 locus, and the fact that FOXC2 haploinsufficiency causes distichiasis, suggests the possibility that FOXC2 may be involved via deletion of long-range cis-regulatory element(s) controlling FOXC2 expression. There are many examples for such effects. A large non-coding region or “gene desert” spanning 747 kb is located distally to the forkhead gene cluster that includes FOXF1, FOXC2, and FOXL1. Conserved noncoding elements in gene deserts have been shown to have enhancer function in transgenic experiments [Nobrega et al., 2003]. Mapping the breakpoints of a balanced translocation (Y;16) to this region in a congenital lymphedema patient led to identification of FOXC2 mutations in LD families [Erickson et al., 1995; Fang et al., 2000]. The chromosome 16 translocation breakpoint occurred 120 kb telomeric to the FOXC2 locus suggesting that removal of chromosome 16 distal to the breakpoint and/or the addition of the Y chromosome disrupted FOXC2 expression. In our case, the 265 kb deletion is located 609 kb telomeric to FOXC2 and removes 151 kb of the gene desert. Therefore, the 16q24.3 deletion could remove cis regulatory element(s) responsible for FOXC2 expression in the developing eyelid, a site of normal Foxc2 expression during development [Dagenais et al., 2004; Iida et al., 1997; Kaestner et al., 1996; Pressman et al., 2000; Winnier et al., 1997].
In summary, we have precisely mapped a submicroscopic contiguous gene deletion in chromosome 16q24.3 in an individual with distichiasis, microcephaly, bilateral grade IV vesicoureteral reflux and mild intellectual impairment, who also has congenital glomuvenous malformations caused by a deleterious GLMN gene mutation. The 16q24.3 deletion causes haploinsufficiency of four genes, C16ORF95, FBXO31, MAP1LC3B, and ZCCHC14, and removes 115 kb of a gene desert distal to the forkhead gene cluster on chromosome 16. The nonvascular phenotypic features of the proband may result from haploinsufficiency of the deleted genes and/or by disruption of regulation of FOXC2 or other nearby gene expression.
Acknowledgments
The authors would like to thank the family that participated in this study. This study was supported by NIH grant HL71206 to T.W. Glover from the National Heart, Lung, and Blood Institute, National Institutes of Health and support to M.G. Butler from the American Heart Association Midwest Affiliate (Predoctoral Fellowship 0710064Z) and from the University of Michigan Program in Genetics Training Grant (GM007544-31). These studies were partially supported by the Interuniversity Attraction Poles initiated by the Belgian Federal Science Policy, network 6/05; Concerted Research Actions (A.R.C.) – Convention No 07/12-005 of the Belgian French Community Ministry; the National Institute of Health, Program Project P01 AR048564, and the F.R.S.-FNRS (Fonds de la Recherche Scientifique) (to M.V.). The authors thank Drs. Sau Wei Cheung and Carlos A. Bacino for conducting the original microarray experiments that identified the deletion in our patient and Dr. J.V. Moran for technical advice and assistance with inverse PCR.
References
- Bell R, Brice G, Child AH, Murday VA, Mansour S, Sandy CJ, Collin JR, Brady AF, Callen DF, Burnand K, Mortimer P, Jeffery S. Analysis of lymphoedema-distichiasis families for FOXC2 mutations reveals small insertions and deletions throughout the gene. Hum Genet. 2001;108:546–551. doi: 10.1007/s004390100528. [DOI] [PubMed] [Google Scholar]
- Boon LM, Mulliken JB, Enjolras O, Vikkula M. Glomuvenous malformation (glomangioma) and venous malformation: distinct clinicopathologic and genetic entities. Arch Dermatol. 2004;140:971–976. doi: 10.1001/archderm.140.8.971. [DOI] [PubMed] [Google Scholar]
- Brice G, Mansour S, Bell R, Collin JR, Child AH, Brady AF, Sarfarazi M, Burnand KG, Jeffery S, Mortimer P, Murday VA. Analysis of the phenotypic abnormalities in lymphoedema-distichiasis syndrome in 74 patients with FOXC2 mutations or linkage to 16q24. J Med Genet. 2002;39:478–483. doi: 10.1136/jmg.39.7.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BP, Dagenais SL, Nelson CC, Glynn MW, Caulder MS, Downs CA, Glover TW. Mutation of the FOXC2 gene in familial distichiasis. J Aapos. 2003;7:354–357. doi: 10.1016/s1091-8531(03)00144-7. [DOI] [PubMed] [Google Scholar]
- Brouillard P, Boon LM, Mulliken JB, Enjolras O, Ghassibe M, Warman ML, Tan OT, Olsen BR, Vikkula M. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”) Am J Hum Genet. 2002;70:866–874. doi: 10.1086/339492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillard P, Ghassibe M, Penington A, Boon LM, Dompmartin A, Temple IK, Cordisco M, Adams D, Piette F, Harper JI, Sved S, Boralevi F, Taieb A, Danda S, Baselga E, Enjolras O, Mulliken JB, Vikkula M. Four common glomulin mutations cause two thirds of glomuvenous malformations (“familial glomangiomas”): evidence for a founder effect. J Med Genet. 2005;42(2):e13. doi: 10.1136/jmg.2004.024174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillard P, Vikkula M. Genetic causes of vascular malformations. Hum Mol Genet. 2007;16(Spec No 2):R140–149. doi: 10.1093/hmg/ddm211. [DOI] [PubMed] [Google Scholar]
- Callen DF, Eyre H, Lane S, Shen Y, Hansmann I, Spinner N, Zackai E, McDonald-McGinn D, Schuffenhauer S, Wauters J, et al. High resolution mapping of interstitial long arm deletions of chromosome 16: relationship to phenotype. J Med Genet. 1993;30:828–832. doi: 10.1136/jmg.30.10.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann GM, Guignabert C, Ying L, Deshpande N, Bekker JM, Wang L, Zhou B, Rabinovitch M. Developmental expression of LC3alpha and beta: absence of fibronectin or autophagy phenotype in LC3beta knockout mice. Dev Dyn. 2008;237:187–195. doi: 10.1002/dvdy.21392. [DOI] [PubMed] [Google Scholar]
- Ciulla TA, Sklar RM, Hauser SL. A simple method for DNA purification from peripheral blood. Anal Biochem. 1988;174:485–488. doi: 10.1016/0003-2697(88)90047-4. [DOI] [PubMed] [Google Scholar]
- Dagenais SL, Hartsough RL, Erickson RP, Witte MH, Butler MG, Glover TW. Foxc2 is expressed in developing lymphatic vessels and other tissues associated with lymphedema-distichiasis syndrome. Gene Expr Patterns. 2004;4:611–619. doi: 10.1016/j.modgep.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Erickson RP, Dagenais SL, Caulder MS, Downs CA, Herman G, Jones MC, Kerstjens-Frederikse WS, Lidral AC, McDonald M, Nelson CC, Witte M, Blover TW. Clinical heterogeneity in lymphoedema-distichiasis with FOXC2 truncating mutations. J Med Genet. 2001;38:761–766. doi: 10.1136/jmg.38.11.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson RP, Hudgins L, Stone JF, Schmidt S, Wilke C, Glover TW. A “balanced” Y;16 translocation associated with Turner-like neonatal lymphedema suggests the location of a potential anti-Turner gene on the Y chromosome. Cytogenet Cell Genet. 1995;71:163–167. doi: 10.1159/000134099. [DOI] [PubMed] [Google Scholar]
- Fang J, Dagenais SL, Erickson RP, Arlt MF, Glynn MW, Gorski JL, Seaver LH, Glover TW. Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am J Hum Genet. 2000;67:1382–1388. doi: 10.1086/316915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finegold DN, Kimak MA, Lawrence EC, Levinson KL, Cherniske EM, Pober BR, Dunlap JW, Ferrell RE. Truncating mutations in FOXC2 cause multiple lymphedema syndromes. Hum Mol Genet. 2001;10:1185–1189. doi: 10.1093/hmg/10.11.1185. [DOI] [PubMed] [Google Scholar]
- Gilbert J. Establishment of permanent cell lines by Epstein-Barr virus transformation. Curr Protoc Hum Genet. 2001 doi: 10.1002/0471142905.hga03hs02. Appendix 3:Appendix 3H. [DOI] [PubMed] [Google Scholar]
- Gorlin RJ, Zellweger H, Curtis MW, Wiedemann HR, Warburg M, Majewski F, Gillessen-Kaesbach G, Prahl-Andersen B, Zackai E. Blepharo-cheilo-dontic (BCD) syndrome. Am J Med Genet. 1996;65:109–112. doi: 10.1002/(SICI)1096-8628(19961016)65:2<109::AID-AJMG5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Dang Y, Dai F, Guo Z, Wu J, She X, Pei Y, Chen Y, Ling W, Wu C, Zhao S, Liu JO, Yu L. Post-translational modifications of three members of the human MAP1LC3 family and detection of a novel type of modification for MAP1LC3B. J Biol Chem. 2003;278:29278–29287. doi: 10.1074/jbc.M303800200. [DOI] [PubMed] [Google Scholar]
- Iida K, Koseki H, Kakinuma H, Kato N, Mizutani-Koseki Y, Ohuchi H, Yoshioka H, Noji S, Kawamura K, Kataoka Y, Ueno F, Taniguchi M, Yoshida N, Sugiyama T, Miura N. Essential roles of the winged helix transcription factor MFH-1 in aortic arch patterning and skeletogenesis. Development. 1997;124:4627–4638. doi: 10.1242/dev.124.22.4627. [DOI] [PubMed] [Google Scholar]
- Kaestner KH, Bleckmann SC, Monaghan AP, Schlondorff J, Mincheva A, Lichter P, Schutz G. Clustered arrangement of winged helix genes fkh-6 and MFH-1: possible implications for mesoderm development. Development. 1996;122(6):1751–8. doi: 10.1242/dev.122.6.1751. [DOI] [PubMed] [Google Scholar]
- Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, Roskin KM, Schwartz M, Sugnet CW, Thomas DJ, Weber RJ, Haussler D, Kent WJ. The UCSC Genome Browser Database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Kumar R, Neilsen PM, Crawford J, McKirdy R, Lee J, Powell JA, Saif Z, Martin JM, Lombaerts M, Cornelisse CJ, Cleton-Jansen AM, Callen DF. FBXO31 is the chromosome 16q24.3 senescence gene, a candidate breast tumor suppressor, and a component of an SCF complex. Cancer Res. 2005;65:11304–11313. doi: 10.1158/0008-5472.CAN-05-0936. [DOI] [PubMed] [Google Scholar]
- Lieber MR, Ma Y, Pannicke U, Schwarz K. Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol. 2003;4:712–720. doi: 10.1038/nrm1202. [DOI] [PubMed] [Google Scholar]
- Mellor RH, Brice G, Stanton AW, French J, Smith A, Jeffery S, Levick JR, Burnand KG, Mortimer PS. Mutations in FOXC2 are strongly associated with primary valve failure in veins of the lower limb. Circulation. 2007;115:1912–1920. doi: 10.1161/CIRCULATIONAHA.106.675348. [DOI] [PubMed] [Google Scholar]
- Morrish TA, Gilbert N, Myers JS, Vincent BJ, Stamato TD, Taccioli GE, Batzer MA, Moran JV. DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat Genet. 2002;31:159–165. doi: 10.1038/ng898. [DOI] [PubMed] [Google Scholar]
- Mulliken JB. Vascular birthmarks: hemangiomas and malformations. Philadelphia, PA: Saunders; 1988. p. 483. [Google Scholar]
- Ng MY, Andrew T, Spector TD, Jeffery S. Linkage to the FOXC2 region of chromosome 16 for varicose veins in otherwise healthy, unselected sibling pairs. J Med Genet. 2005;42:235–239. doi: 10.1136/jmg.2004.024075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobrega MA, Ovcharenko I, Afzal V, Rubin EM. Scanning human gene deserts for long-range enhancers. Science. 2003;302(5644):413. doi: 10.1126/science.1088328. [DOI] [PubMed] [Google Scholar]
- Pressman CL, Chen H, Johnson RL. LMX1B, a LIM homeodomain class transcription factor, is necessary for normal development of multiple tissues in the anterior segment of the murine eye. Genesis. 2000;26:15–25. [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Santra MK, Wajapeyee N, Green MR. F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature. 2009;459:722–725. doi: 10.1038/nature08011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sholto-Douglas-Vernon C, Bell R, Brice G, Mansour S, Sarfarazi M, Child AH, Smith A, Mellor R, Burnand K, Mortimer P, Jeffery S. Lymphoedema-distichiasis and FOXC2: unreported mutations, de novo mutation estimate, families without coding mutations. Hum Genet. 2005;117:238–242. doi: 10.1007/s00439-005-1275-2. [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Sen P, Bhatt SS, Storer M, Xia Z, Bejjani BA, Ou Z, Wiszniewska J, Driscoll DJ, Maisenbacher MK, Bolivar J, Bauer M, Zackai EH, McDonald-McGinn D, Nowaczyk MM, Murray M, Hustead V, Mascotti K, Schultz R, Hallam L, McRae D, Nicholson AG, Newbury R, Durham-O’Donnell J, Knight G, Kini U, Shaikh TH, Martin V, Tyreman M, Simonic I, Willatt L, Paterson J, Mehta S, Rajan D, Fitzgerald T, Gribble S, Prigmore E, Patel A, Shaffer LG, Carter NP, Cheung SW, Langston C, Shaw-Smith C. Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet. 2009;84:780–791. doi: 10.1016/j.ajhg.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikkula M, Boon LM, Carraway KL, 3rd, Calvert JT, Diamonti AJ, Goumnerov B, Pasyk KA, Marchuk DA, Warman ML, Cantley LC, Mulliken JB, Olsen BR. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996;87:1181–1190. doi: 10.1016/s0092-8674(00)81814-0. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H, Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Gilbert N, Ooi SL, Lawler JF, Ostertag EM, Kazazian HH, Boeke JD, Moran JV. Human L1 retrotransposition: cis preference versus trans complementation. Mol Cell Biol. 2001;21:1429–1439. doi: 10.1128/MCB.21.4.1429-1439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke CM, Hall BK, Hoge A, Paradee W, Smith DI, Glover TW. FRA3B extends over a broad region and contains a spontaneous HPV16 integration site: direct evidence for the coincidence of viral integration sites and fragile sites. Hum Mol Genet. 1996;5:187–195. doi: 10.1093/hmg/5.2.187. [DOI] [PubMed] [Google Scholar]
- Winnier GE, Hargett L, Hogan BL. The winged helix transcription factor MFH1 is required for proliferation and patterning of paraxial mesoderm in the mouse embryo. Genes Dev. 1997;11:926–940. doi: 10.1101/gad.11.7.926. [DOI] [PubMed] [Google Scholar]
- Wouters V, Limaye N, Uebelhoer M, Irrthum A, Boon LM, Mulliken JB, Enjolras O, Baselga E, Berg J, Dompmartin A, Ivarsson SA, Kangesu L, Lacassie Y, Murphy J, Teebi AS, Penington A, Rieu P, Vikkula M. Hereditary cutaneomucosal venous malformations are caused by TIE2 mutations with widely variable hyper-phosphorylating effects. Eur J Hum Genet. 2010;18:414–420. doi: 10.1038/ejhg.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]