

Streptomyces cattleya is unusual in that it elaborates organofluorine metabolites and in particular secretes fluoroacetate and 4-fluorothreonine as secondary metabolites when incubated in a medium supplemented with fluoride ions.[1,2] The first committed step on the biosynthetic pathway to these fluorometabolites involves the enzymatic mediated synthesis of 5′-fluoro-5′-deoxyadenosine (5′-FDA) from S-adenosyl-L-methionine (SAM) and fluoride ions.[3-5] After depurination, 5′-FDA is metabolized first to 5-fluoro-5-deoxyribose-1-phosphate (5′-FRP)[6] and then to fluorocetaldehyde.[7] Fluoroacetaldehyde is converted separately into fluoroacetate[8] and 4-fluorothreonine[9] (Scheme 1).
Scheme 1.

A summary of the biosynthesis of fluoroacetate and 4-fluorothreonine highlighting the fluorinase and the other characterized enzymes on the pathway.
The fluorination enzyme, which has been the subject of a recent structural study,[10] has been isolated and fully characterized, the gene has been cloned, and the enzyme has been overexpressed. A stereochemical study that involved (5′S)-[5′-2H]-SAM has indicated that the C–F bond of 5′-FDA replaces the C–S bond of SAM with an inversion of configuration.[11,12] This observation, as well as a theoretical study[13] and structural analysis[10] all suggest an SN2 reaction mechanism for the fluorinase. Despite the high bond-dissociation energy of the C–F bond (the strongest bond in organic chemistry[14]), we herein report that the fluorinase operates in the reverse direction and that it utilizes a chloride ion. Incubation of 5′-FDA with L-(13C-methyl)methionine resulted in the formation of 13C-methyl-SAM, which was identified by HPLC–ES-MS. A similar experiment with L-selenomethionine (L-Se-met) and 5′-FDA resulted in a sixfold more-efficient reaction to generate L-Se-SAM, which was readily identified through the mass-spectrometry fragmentation product of methylselenoadenosine (MeSeAdo) by its signature isotope fingerprint in HPLC–ES-MS. Comparison with the Vmax values that were calculated at saturating kinetics (assays run in each direction) indicated that the equilibrium lies in favor of 5′-FDA by a factor of three (see Scheme 2).
Scheme 2.

The fluorinase operates in reverse with both 5′-FDA and 5′-ClDA.
Efforts were made to replace the fluoride ion with a chloride ion as a substrate for the fluorinase by using standard assay protocols. Incubation with chloride ions in the presence of SAM has never resulted in the production of 5′-chloro-5′-deoxyadenosine (5′-ClDA) (by HPLC). It now emerges, however, that the failure to detect 5′-ClDA is due to the equilibrium of the chloride reaction lying significantly in favor of substrates over products rather than an inherent inability of the fluorinase to activate the chloride ions towards nucleophilic attack. This was revealed in two separate coupled-enzyme assays that were designed to shift the equilibrium of the reaction towards the organochlorine product. The first of these involved an assay in which the fluorinase was coupled to an L-amino acid oxidase (Bothrops atrox venom; purchased from Sigma Chem. Co. Ltd.). Removal of L-methionine, the coproduct of halide substitution by enzymatic oxidation, inhibits the reverse reaction. This assay therefore generated 5′-ClDA and allowed, for the first time, the detection of this product from SAM and chloride ions (see Scheme 3.)
Scheme 3.

Coupled enzyme assays with 5′-ClDA driving organochlorine synthesis.
A related experiment involved a coupled fluorinase/adenyl acid deaminase (Aspergillus sp.)[15] assay. In this reaction the resultant 5′-ClDA was converted into 5′-chloro-5′-deoxyinosine (5′-ClDI). The production of 5′-ClDI was readily identified by HPLC–ES-MS against a reference compound.[16] These experiments indicated that the fluorinase can process chloride ions in a manner similar to that of fluoride ions. A set of fluorinase/L-methionine oxidase coupled assay experiments that were conducted at high concentrations of SAM (1 mM) and halide ion (25 mM) indicated a rate preference for F− over Cl− by a factor of 120. This preference was also demonstrated in a fluorinase-mediated trans-halogenation reaction as illustrated in Figure 1. Incubation of 5′-ClDA with fluoride ion, L-selenomethionine (catalyst), and the fluorinase resulted in an efficient and progressive conversion of 5′-ClDA to 5′-FDA, a reaction that proceeds through L-Se-SAM as a transient but detectable (by using HLPC) intermediate (see Figure 1).
Figure 1.

HPLC profile monitoring the fluorinase mediated transhalogenation reaction of 5′-ClDA to 5′-FDA with time and using L-Se-methionine as a co-catalyst. L-Se-SAM accumulates as a minor product.
The increased size of the chloride ion over the fluoride ion raises the legitimate question of the ability of chloride ions or 5′-ClDA to coordinate to the fluoride- and 5′-FDA-binding sites of the protein. To explore this aspect further, 5′-ClDA was cocrystallised with overexpressed fluorinase (without added L-methionine). This resulted in a structure with 5′-ClDA bound to the active site of the enzyme as illustrated in Figure 2. There are three monomers in the asymmetric unit and all three have essentially the same structure although there are some small differences owing to crystal packing. Only one subunit of the trimer is discussed.
Figure 2.
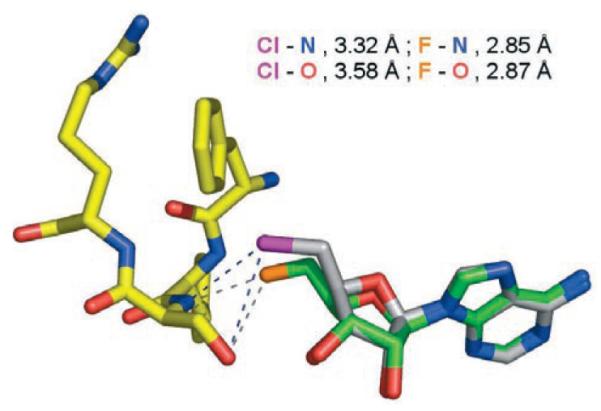
Structure of the 5′-ClDA-fluorinase co-complex with contacts and distances between the halogens and Ser-156 N and O atoms shown. For 5′-ClDA, C=gray, N=blue, O=red, and Cl=purple. The protein is similarly colored except C=yellow. Shown superimposed over 5′-ClDA is 5′-FDA with C=green, F=orange, and N=blue. As can be seen, the Cl atom lies further away from the backbone amide of Ser-156.
The difference electron density shows that the C5′–Cl bond adopts two orientations that locate the organochlorine atom in one of two positions. One conformer (shown in Figure 2) is similar to that observed for 5′-FDA when bound to the fluorinase, however, the chlorine atom is displaced by 1.3 Å relative to the location of the fluorine in the 5′-FDA complex. The chlorine atom is anchored to the main chain amide of Ser-156 by a hydrogen bond and a polar contact to the side chain hydroxy group of the same residue. Similar noncovalent contacts are also observed between the fluorine atom and Ser-156 although the distances are shorter for the 5′-FDA structure. We attribute this change in position to be a consequence of the larger van der Waals radius of the Cl atom relative to the F atom. The protein underwent no significant change in response to the larger atom, rather the substrate adjusts its position which perhaps suggests a rigid ligand binding site. The altered position of the halogen does not, however, prevent the enzyme from catalyzing the reaction.
In the second conformation (not shown) the chlorine rotated out of the halogen binding site into the “empty” L-methionine sulfur binding site. Of course in the catalytic situation, L-methionine will occupy the space above the ribose ring and there will be no possibility of chlorine adopting this second orientation.
This study reveals that the fluorinase will catalyze the combination of chloride ions and SAM to generate 5′-ClDA. The reaction is only observed in coupled-enzyme assays that prevent the reverse reaction from occurring. No organohalogen products were detected when Br− and I− were used in these reactions. This reaction extends the repertoire of the fluorinase and reveals a novel enzymatic chlorination reaction which operates by nucleophilic substitution rather than through the electrophilic hypochlorite[17] or flavin dependent enzymes[18] or by a recently reported enzymatic chlorine radical process.[19] A combination of nucleophilic chloride ion and SAM is responsible for the production of chloromethane in fungi.[20,21] This is the only other enzymatic reaction we are aware of in which Cl− acts as a nucleophile, interestingly with a different regiochemistry on the same substrate.
Experimental Section
HPLC and NMR spectroscopy time course: The reaction mixtures for HPLC were incubated at 37°C with 5′-ClDA (0.3 mM), L-Se-methionine (0.08 mM), KF (75 mM), and fluorinase (6 mg mL−1, 500 μL) in a final volume of 650 μL. Samples for HPLC analysis (60 μL) were boiled at 95°C (3 min) and the precipitated protein was then removed by centrifugation. An aliquot (20 μL) of the clear supernatant was used for HPLC analysis (Vivian 9012 UV/Vis detector at 260 nm and 9050 solvent delivery module).
Samples for 19F NMR (500 MHz) analysis were incubated at 37°C with 5′-FDA (0.5 mM), L-methionine (40 mM), D2O (10%) and fluorinase (6 mg mL−1, 500 μL) in a final volume of 700 μL.
Preparations of ESI-MS sample: A sample (250 mL) for ESI-MS analysis was incubated at 37°C overnight with 5′-FDA (0.4 mM), methionine (L-methionine-13C-methyl or selenomethionine; 15 mM) and fluorinase (6 mg mL−1) and then were boiled at 95°C (3 min) and the precipitated protein was then removed by centrifugation. The supernatant was injected directly into the ESI-MS (MicroMass, Manchester, UK) with MeCN as the solvent. The SAM analogue molecules were detected in positive ion mode. Se-SAM underwent fragmentation to 5′-methylselenoadenosine during ESI-MS analyses (see the Supporting Information).
Crystallization of the 5-ClDA co-complex: FDAS (4 mgmL−1) was incubated overnight with 5′-ClDA (20 mM) at room temperature. The solution was then crystallized by vapor diffusion against a reservoir containing PEG 1000 (32%), phosphate-citrate buffer solution (0.1M, pH 4.2), and Li2SO4 (0.2M). A single crystal was selected and flash cooled to 100 K in a nitrogen stream. A data set was recorded at the European synchrotron radiation facility by using the ID14 EH2 beamline. The structure was solved by molecular replacement with the original FDAS structure and refined by using Refmac. The final R-factor was 19.0% with an R-free of 24.7%.
Supplementary Material
Footnotes
We thank the BBSRC for funding (H.D. and D.A.R.) and GSK and the Pro-Bio-Faraday for a Studentship (R.P.M.).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- [1].Sanada M, Miyano T, Iwadare S, Williamson JM, Arison BH, Smith JL, Douglas AW, Liesch JM, Inamine E. J. Antibiot. 1984;39:259–265. doi: 10.7164/antibiotics.39.259. [DOI] [PubMed] [Google Scholar]
- [2].O’Hagan D, Harper DB. Nat. Prod. Rep. 1994;11:123–133. doi: 10.1039/np9941100123. [DOI] [PubMed] [Google Scholar]
- [3].O’Hagan D, Schaffrath C, Cobb S, Hamilton JTG, Murphy CD. Nature. 2002;416:279. doi: 10.1038/416279a. [DOI] [PubMed] [Google Scholar]
- [4].Schaffrath C, Deng H, O’Hagan D. FEBS Lett. 2003;547:111–114. doi: 10.1016/s0014-5793(03)00688-4. [DOI] [PubMed] [Google Scholar]
- [5].Schaffrath C, Cobb SL, O’Hagan D. Angew. Chem. 2002;114:4069–4071. doi: 10.1002/1521-3773(20021018)41:20<3913::AID-ANIE3913>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2002;41:3913–3915. doi: 10.1002/1521-3773(20021018)41:20<3913::AID-ANIE3913>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- [6].Cobb SL, Deng H, Hamilton JTG, McGlinchey RP, O’Hagan D. Chem. Commun. 2004:592–593. doi: 10.1039/b400754a. [DOI] [PubMed] [Google Scholar]
- [7].Deng H, O’Hagan D, Schaffrath C. Nat. Prod. Rep. 2004;21:773–784. doi: 10.1039/b415087m. [DOI] [PubMed] [Google Scholar]
- [8].Murphy CD, Moss SJ, O’Hagan D. Appl. Environ. Microbiol. 2001;67:4919–4921. doi: 10.1128/AEM.67.10.4919-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Murphy CD, O’Hagan D, Schaffrath C. Angew. Chem. 2001;113:4611–4613. doi: 10.1002/1521-3773(20011203)40:23<4479::aid-anie4479>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2001;40:4479–4481. doi: 10.1002/1521-3773(20011203)40:23<4479::aid-anie4479>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- [10].Dong CJ, Huang FL, Deng H, Schaffrath C, Spencer JB, O’Hagan D, Naismith JH. Nature. 2004;427:561–565. doi: 10.1038/nature02280. [DOI] [PubMed] [Google Scholar]
- [11].Cadicamo CD, Courtieu J, Deng H, Meddour A, O’Hagan D. ChemBioChem. 2004;5:685–690. doi: 10.1002/cbic.200300839. [DOI] [PubMed] [Google Scholar]
- [12].O’Hagan D, Goss RJM, Meddour A, Courtieu J. J. Am. Chem. Soc. 2003;125:379–387. doi: 10.1021/ja026654k. [DOI] [PubMed] [Google Scholar]
- [13].Senn H, O’Hagan D, Thiel W. J. Am. Chem. Soc. 2005;127:13643–13655. doi: 10.1021/ja053875s. [DOI] [PubMed] [Google Scholar]
- [14].Smart BE. Properties of Fluorinated Organic Compounds. Vol. 1. ACS; 1995. pp. 979–1010. [Google Scholar]
- [15].Margolin AL, Borcherding DR, Wolfkugel D, Margolin N. J. Org. Chem. 1994;59:7214–7218. [Google Scholar]
- [16].Cobb SL. Ph.D thesis. University of St Andrews; 2005. [Google Scholar]
- [17].Butler A, Carter-Franklin JN. Nat. Prod. Rep. 2004;21:180–188. doi: 10.1039/b302337k. [DOI] [PubMed] [Google Scholar]
- [18].Dong C, Flecks S, Unversucht S, Haupt C, van Pée K-H, Naismith JH. Science. 2005;309:2216–2219. doi: 10.1126/science.1116510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vaillancourt FH, Yeh E, Vosburg DA, O’Connor SE, Walsh CT. Nature. 2005;436:1191–1194. doi: 10.1038/nature03797. [DOI] [PubMed] [Google Scholar]
- [20].Saxena D, Aouad S, Attieh J, Saini H. Appl. Environ. Microbiol. 1998;64:2831–2835. doi: 10.1128/aem.64.8.2831-2835.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ni X, Hager LP. Proc. Natl. Acad. Sci. USA. 1999;96:3611–3615. doi: 10.1073/pnas.96.7.3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.