

Abstract
Angiotensin-(1-7) [Ang-(1-7)] is an endogenous seven-amino acid peptide hormone with antiproliferative properties. Our previous studies showed that Ang-(1-7) inhibits the growth of human lung cancer cells in vitro and reduces the size of human lung tumor xenografts in vivo. In the current study, s.c. injection of Ang-(1-7) not only caused a significant reduction in human A549 lung tumor growth but also markedly decreased vessel density, suggesting that the heptapeptide inhibits angiogenesis to reduce tumor size. A decrease in human endothelial cell tubule formation in Matrigel was observed following a 16 h incubation with Ang-(1-7), with a maximal reduction at a 10 nmol/L concentration. Ang-(1-7) had similar antiangiogenic effects in the chick chorioallantoic membrane, causing a >50% decrease in neovascularization. The Ang-(1-7)-induced reduction in both endothelial cell tubule formation and vessel formation in the chick was completely blocked by the specific Ang-(1-7) receptor antagonist [d-proline7]-Ang-(1-7), suggesting that these biological actions are mediated by an AT(1-7) receptor. Ang-(1-7) significantly reduced vascular endothelial growth factor-A protein and mRNA in tumors from mice treated with the heptapeptide compared with saline controls as well as in the parent A549 human lung cancer cells in culture. These results suggest that Ang-(1-7) may attenuate tumor angiogenesis by reducing vascular endothelial growth factor-A, a primary proangiogenic protein. Taken together, this study shows that Ang-(1-7) exhibits significant antiangiogenic activity and may be a novel therapeutic agent for lung cancer treatment targeting a specific AT(1-7) receptor.
Introduction
Angiotensin-(1-7) [Ang-(1-7)] is a biologically active peptide hormone of the renin-angiotensin system with vasodilator, antiproliferative, and antithrombotic properties (1, 2). The catabolism of angiotensinogen by the enzyme renin forms angiotensin I, which is further processed to the octapeptide angiotensin II or the heptapeptide Ang-(1-7). Ang-(1-7) is formed from angiotensin I by endopeptidases such as neprilysin, prolyl endopeptidase, and thimet oligopeptidase; the heptapeptide also is generated from angiotensin II by the dipeptidyl carboxydipeptidase angiotensin-converting enzyme 2. Ang-(1-7) exerts its physiologic effects through activation of a unique G protein-coupled Ang-(1-7) [AT(1-7)] receptor encoded by the mas gene (3).
The antimitogenic effects of Ang-(1-7) were initially shown in vitro and in vivo in vascular smooth muscle cells (4) and cardiac myocytes (5, 6). Ang-(1-7) inhibited the proliferation of vascular smooth muscle cells (4) and reduced neointimal formation in the carotid artery following vascular injury (7) and in the abdominal aorta following stent implantation (8). Moreover, the heptapeptide decreased mitogen-stimulated protein synthesis in cultured neonatal cardiac myocytes (5) as well as reduced myocyte size and attenuated ventricular dysfunction following myocardial infarction (6). The Ang-(1-7)-mediated inhibition of vascular smooth muscle cell and cardiomyocyte growth suggests a potential role for the heptapeptide in the therapeutic treatment of cardiovascular diseases.
In previous studies, we showed that Ang-(1-7) also inhibits the proliferation of lung cancer cells in vitro (9). Ang-(1-7) caused a significant reduction in the serum-stimulated growth of three human lung adenocarcinoma cell lines. Treatment with the heptapeptide resulted in both dose- and time-dependent reduction in serum-stimulated DNA synthesis with IC50 values in the subnanomolar range. The Ang-(1-7) receptor antagonist [d-alanine7]-Ang-(1-7) blocked the attenuation of the serum-stimulated DNA synthesis in SK-LU-1 human lung cancer cells by Ang-(1-7), whereas neither AT1 nor AT2 angiotensin receptor antagonists prevented the response to the heptapeptide. mas mRNA and protein were detected in all three lung cancer cell lines, suggesting that the antiproliferative effect of Ang-(1-7) in lung cancer cells is mediated by the AT(1-7) receptor mas. Finally, Ang-(1-7) reduced serum-stimulated autophosphorylation and activation of the extracellular signal-regulated kinases 1 and 2, indicating that the anti-proliferative effects may occur, at least in part, through inhibition of mitogen-activated protein kinases.
More recently, we reported that Ang-(1-7) inhibited the growth of human lung tumors in preclinical studies in a mouse xenograft model (10). Athymic mice bearing A549 human lung tumors were infused with either saline or Ang-(1-7) for 28 days. Treatment with Ang-(1-7) significantly reduced tumor volume compared with the saline-treated controls. Immunohistochemical analysis of Ang-(1-7)-treated tumors showed a correlation between inhibition of tumor growth and a reduction in the proliferation marker Ki-67. Moreover, cyclooxygenase 2 (COX-2) activity was markedly reduced in tumors from mice infused with Ang-(1-7), suggesting that the heptapeptide may decrease the production of proinflammatory prostaglandins to inhibit lung tumor growth. Administration of Ang-(1-7) to athymic mice with human lung tumors not only inhibited tumor growth but also reduced tumor volume by >30% compared with the size of the tumors at the initiation of treatment. Because the reduction in tumor size by treatment with Ang-(1-7) was associated with a decrease in COX-2, the resultant decline in proliferative prostaglandins may have attenuated cell growth (10). However, the reduction in tumor size by Ang-(1-7) treatment could also result from an increase in apoptosis and/or a decrease in angiogenesis to reduce the blood supply to the tumor cells and promote cell death. The aim of this study was to determine whether Ang-(1-7) inhibits angiogenesis to prevent cancer cell proliferation and attenuate tumor growth.
Materials and Methods
Materials
Ang-(1-7) and [d-alanine7]-Ang-(1-7) were purchased from Bachem, whereas [d-proline7]-Ang-(1-7) or [d-Pro7]-Ang-(1-7) was synthesized by GenScript. DMEM, Ham’s F-12, penicillin, streptomycin, fetal bovine serum, and hypoxanthine-aminopterin-thymidine supplement were obtained from Life Technologies. Matrigel was purchased from BD Biosciences. Pooled human umbilical vein endothelial cells and endothelial cell growth medium-2 were obtained from Clonetics (Lonza) and A549 human lung adenocarcinoma cells (CCL-185) were purchased from the American Type Culture Collection.
Cell Culture
Human umbilical vein endothelial cells pooled from three individuals were grown in endothelial cell growth medium-2 in a humidified 37°C incubator with 5% CO2. A549 human lung adenocarcinoma cells, derived from a 58-year-old male Caucasian, were grown in Ham’s F-12 medium with 10% fetal bovine serum, 100 μg/mL penicillin, and 100 units/mL streptomycin; subconfluent monolayers of A549 cells were incubated for 24 h in medium without fetal bovine serum to remove growth factors.
Xenograft Model of Lung Cancer
Athymic mice were subjected to s.c. injections of human A549 lung cancer cells (1.0 × 106) in Matrigel (50:50) into the lower flank to induce tumor growth. Tumor size was measured twice a week during the treatment period using a caliper and tumor volume was calculated using the formula for a semi-ellipsoid: (4/3πr3)/2. After the tumors reached ~100 mm3, the mice were placed into two groups at random and the animals received s.c. injections of either saline or 1,000 μg/kg/d Ang-(1-7) in saline. The injections were administered daily for 5 days followed by a 2-day rest period for 6 cycles. The mice were anesthetized on day 42 and euthanized by decapitation. All procedures complied with the policies of the Wake Forest University Animal Care and Use Committee.
Immunohistochemistry
Tumors were fixed in 4% paraformaldehyde for 24 h and incubated in 70% ethanol for 48 h before embedding in paraffin. The embedded tumors were cut into 5-μm-thick sections and stained with H&E to determine morphology. Endothelial cells were highlighted by immunostaining with an antibody to CD34 (1:100; Abcam) using the streptavidin-biotin method as described previously (10, 11). Blood vessels were visualized by the presence of CD34-immunostained endothelial cells and identified by their morphology, as vessels cut in cross-section with visible lumens or vessels cut longitudinally with tube-like morphology (12). Tumor tissue sections were scanned at low power magnification (×50) with a Leica DM microscope (Leica Microsystems) to identify areas of the tumor with the highest density (“hotspots”) of CD34+ cells with vessel morphology. Selected sections were counted without the knowledge of treatment group at a ×200 magnification in a 0.3 mm2 field using the Simple PCI version 6.0 computer-assisted imaging software (Hamamatsu) and photographed with the QImaging Retiga 1300RCamera (QImaging). The number of vessels, as defined by Weidner (12), was expressed as the average of 6 fields (0.3 mm2) selected per tumor.
Endothelial Cell Tubule Formation Assay
The wells of a cooled 96-well plate were coated with 50 μL Matrigel and human umbilical vein endothelial cells were seeded onto the polymerized matrix at a density of 5 × 104 per well to visualize tubule formation. Cells were treated with Ang-(1-7) in the presence or absence of [d-Pro7]-Ang-(1-7) at the concentrations indicated to determine the effect of dose and to assess whether the response was receptor-mediated. Ang-(1-7) or [d-Pro7]-Ang-(1-7) was added to the culture medium, the cell suspension, and the Matrigel. After 16 h, tubule formation was assessed under a light microscope and quantified by counting the number of branch points.
Chick Chorioallantoic Membrane Assay
Chicken eggs obtained from a local farm (Tyson Farms) were incubated in a humidified atmosphere at 37°C. At day 4, the air cell of the egg shell was removed and covered with paraffin. After 2-day incubations, 7 mm methylcellulose disks containing saline, 100 nmol/L Ang-(1-7), 100 nmol/L Ang-(1-7), and 1.0 μmol/L AT(1-7) receptor antagonist [d-Pro7]-Ang-(1-7) or 1.0 μmol/L [d-Pro7]-Ang-(1-7) were placed onto the surface of the chick chorioallantoic membrane (CAM) in an avascular area. Neovascularization was examined 24 h later by counting branch points in pictures taken with a Nikon D200 with a 105 microlens.
Western Blot Hybridization
Cells lysates were obtained by solubilizing monolayers in Triton lysis buffer [100 mmol/L NaCl, 50 mmol/L NaF, 5 mmol/L EDTA, 1% Triton X-100, and 50 mmol/L Tris-HCl (pH 7.4) containing 0.01 mmol/L NaVO4, 0.1 mmol/L phenylmethylsulfonyl fluoride, and 0.6 μmol/L leupeptin]. Tumor tissue cut into 1 to 2 mm2 size pieces was homoge-nized in PBS [50 mmol/L NaPO4 (pH 7.2) and 100 mmol/L NaCl] and solubilized by boiling in 3% SDS-10% β-mercaptoethanol. Protein concentration in cell lysates or tissue homogenates was measured by a modification of the Lowry method (13). Proteins were separated by electrophoresis on 10% SDS-polyacrylamide gels and transferred to hydrophobic polyvinylidene difluoride membrane (Hybond-P; Amersham Biosciences). Nonspecific binding was blocked by incubation with Blotto [TBS containing 5% powdered milk and 0.1% Triton X-100]. Membranes were probed with primary antibodies to vascular endothelial growth factor-A (VEGFA; sc-507; 1:10,000; Santa Cruz Biotechnology) and treated with polyclonal horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences). Immunoreactive products were visualized using chemiluminescence reagents (SuperSignal Femto West or Pico West; Pierce Biotechnology) and quantified by densitometry. Protein loading was determined using an antibody to β-actin and quantified by densitometry. Cell or tissue content of VEGFA was expressed as the ratio of VEGFA/actin.
RNA Isolation and Reverse Transcription Real-time PCR
RNA was isolated from human lung tumor xenografts or cultured A549 cells using TRIzol (Life Technologies/Invitrogen) as directed by the manufacturer. The RNA was incubated with RQ1 DNase (Promega) to eliminate any residual DNA that would amplify during the PCR. The RNA concentration and integrity were assessed with an Agilent 2100 Bioanalyzer using a RNA 6000 Nano LabChip (Agilent Technologies). Total RNA (~1 μg) was reverse transcribed using avian myeloblastosis virus RT in a 20 μL reaction mixture containing deoxyribonucleotides, random hexamers, and RNase inhibitor in reverse transcription buffer. The reverse transcription reaction product was heated at 95°C to terminate the reaction. For real-time PCR, 2 μL of the resultant cDNA were added to TaqMan Universal PCR Master Mix (Applied Biosystems) with the VEGFA-specific primer/probe set (Applied Biosystems), and amplification was done on an ABI 7000 Sequence Detection System. The mixtures were heated at 50°C for 2 min and 95°C for 10 min followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. All reactions were done in triplicate and 18S rRNA, amplified with the use of the TaqMan rRNA control kit (Applied Biosystems), served as an internal control. The results were quantified as Ct values, where Ct is defined as the threshold cycle of PCR at which the amplified product is first detected and defined as relative gene expression (the ratio of target/control).
Statistics
All data are presented as the mean ± SE. Statistical differences were evaluated by Student’s t test or one-way ANOVA followed by Dunnett’s post hoc test. The criterion for statistical significance was set at P < 0.05.
Results
S.c. Injection of Ang-(1-7) Inhibits Lung Tumor Volume
Athymic mice bearing a human A549 lung tumor were administered s.c. injections of either saline or Ang-(1-7) at a concentration of 1,000 μg/kg/d. The mice were injected daily, for 5 days, followed by a 2-day rest period, and the injections were continued for 6 weeks. As shown in Fig. 1A, tumor volume in the two treatment groups was similar at the initiation of treatment [108.8 ± 3.9 mm3 in the saline-treated group compared with 110.5 ± 3.1 mm3 in the Ang-(1-7)-treated group; n = 5 in each group]. The tumors of saline-treated mice increased in size over time, whereas the growth of tumors from mice treated with Ang-(1-7) was reduced markedly by day 42 [752.5 ± 46.9 mm3 in the saline-treated group compared with 265.5 ± 27.4 mm3 in the Ang-(1-7)-treated group; n = 5 in each group; P < 0.0001]. The animals maintained their body weight as well as food and water consumption irrespective of treatment and showed no evidence of reduced motor function.
Figure 1.

Effect of Ang-(1-7) on human lung cancer xenograft growth. A, size of human A549 lung tumor xenografts from mice injected with saline or 1,000 μg/kg/d was measured using a caliper and volume was calculated using the formula for a semi-ellipsoid: (4/3πr3)/2. *, P < 0.05; n = 5. B, tumors from mice infused with either saline or Ang-(1-7) were weighed at the time of sacrifice. *, P < 0.0001; n = 5.
At the end of the study, the mice were euthanized and the tumors were removed and weighed. As shown in Fig. 1B, the tumors from mice treated with the heptapeptide weighed ~60% less than the tumors of mice infused with saline [0.72 ± 0.06 g in the Ang-(1-7)-treated group versus 1.66 ± 0.12 g in the saline-treated group; n = 5 in each group; P < 0.0001]. No gross pathologic abnormalities were observed in major organs following sacrifice, showing a lack of toxic side effects at the Ang-(1-7) dose given.
Ang-(1-7) Reduces Tumor Angiogenesis
S.c. lung tumors were immunostained for CD34, an endothelial cell marker, and intratumoral blood vessels highlighted by CD34+ immunoreactivity were identified by vessel morphology. Blood vessels were located throughout the tumor, but clusters of vessels or vessel “hotspots” were localized mainly in the periphery of the tumors. Tumors from animals administered saline or Ang-(1-7) showed a similar quantity of hotspots; however, the number of vessels in the clusters from mice injected with the heptapeptide was reduced significantly. Tumor tissue sections from saline-treated animals showed numerous blood vessels when compared with sections from Ang-(1-7)-treated animals (Fig. 2A). As shown in Fig. 2B, injection of Ang-(1-7) caused an ~50% reduction in intratumoral vessel density (average of 6 fields) when compared with sections from saline-treated animals [25.1 ± 4.9 vessels/field in the saline-treated group compared with 10.6 ± 2.1 vessels/field in the Ang-(1-7)-treated group; n = 5 in each group; P < 0.03], suggesting that the heptapeptide reduces angiogenesis to inhibit lung tumor growth.
Figure 2.

Inhibition of human lung tumor angiogenesis by Ang-(1-7). A, representative pictures of stained sections of tumors from mice treated with saline or Ang-(1-7) following incubation with an antibody to the endothelial cell marker CD34 at ×200 magnification. Blood vessels were identified based on their morphology in vessels highlighted by CD34-immunoreactive endothelial cells. B, average vessel density defined as the number of intratumoral vessels from six 0.3 mm2 fields in sections of tumors from mice treated with saline or Ang-(1-7). *, P < 0.03; n = 5.
Inhibition of Endothelial Cell Tubule Formation by Ang-(1-7)
The effect of Ang-(1-7) on the formation of tubules in Matrigel was determined to show a direct effect of the heptapeptide on endothelial cells. Human umbilical vein endothelial cells were plated at a density of 5 × 104/well into individual wells of a 96-well cluster dish coated with Matrigel in the presence or absence of Ang-(1-7). Untreated endothelial cells formed a network of multiple tube-like structures, as seen in Fig. 3A, whereas the number of tube-like structures was significantly decreased in the presence of 100 nmol/L Ang-(1-7) [52.8 ± 7.0 in the control group compared with 30.2 ± 3.6 in the Ang-(1-7)-treated group; n = 3; P < 0.05; Fig. 3B]. Endothelial cells also were incubated with 10 nmol/L Ang-(1-7) or with 10 nmol/L Ang-(1-7) in the presence of 100 nmol/L Ang-(1-7) receptor antagonist [d-Pro7]-Ang-(1-7) to determine whether the inhibition by the heptapeptide was a receptor-mediated process. As shown in Fig. 3, both 10 and 100 nmol/L Ang-(1-7) significantly reduced tubule formation and the number of tubules in the presence of 10 nmol/L Ang-(1-7) and the AT(1-7) receptor antagonist [d-Pro7]-Ang-(1-7) did not differ from the control, indicating that the inhibition of endothelial tubule formation required activation of an AT(1-7) receptor. The antagonist alone had no effect on tubule formation. Both mas mRNA and protein were detected in these cells by reverse transcription real-time PCR and Western blot hybridization, respectively, suggesting that mas may be involved in the process.
Figure 3.

Ang-(1-7) inhibition of endothelial cell tubule formation. Human umbilical vein endothelial cells were seeded onto Matrigel alone (control), with 10 or 100 nmol/L Ang-(1-7), with 10 nmol/L Ang-(1-7) and 100 nmol/L AT(1-7) receptor antagonist [d-Pro7]-Ang-(1-7) (D-Pro), or with 100 nmol/L [d-Pro7]-Ang-(1-7) alone. After 16 h, the cells were photographed to visualize and quantify tubule formation. A representative photograph of each treatment is shown in A and the quantification of tubule numbers is depicted in B. *, P < 0.05; n = 3.
Ang-(1-7) Reduction of Angiogenesis in the CAM
The antiangiogenic effect of Ang-(1-7) was also determined in vivo by measuring neovascularization during the development of the chick embryo. Methylcellulose discs containing saline or 100 nmol/L Ang-(1-7) were placed on the surface of the CAM in an avascular area. After 24 h of treatment, the discs were removed and the number of branch points was quantified. As shown in Fig. 4A, vessels continued to develop in the saline-treated control group, whereas vessel formation was significantly reduced in embryos treated with Ang-(1-7) [21.1 ± 2.0 branch points in the control group compared with 9.3 ± 1.1 branch points in the Ang-(1-7)-treated group; n = 9; P < 0.001]. In an additional group of chick embryos, Ang-(1-7) was added in the presence of the receptor blocker [d-Pro7]-Ang-(1-7) to prove that the inhibition of vessel formation was due to receptor activation. As shown in Fig. 4, the addition of the receptor antagonist completely blocked the response to Ang-(1-7), indicating that the reduction in neoangiogenesis was mediated by an AT(1-7) receptor. Incubation with 1 μmol/L [d-Pro7]-Ang-(1-7) alone had no effect on vessel formation in the CAM.
Figure 4.
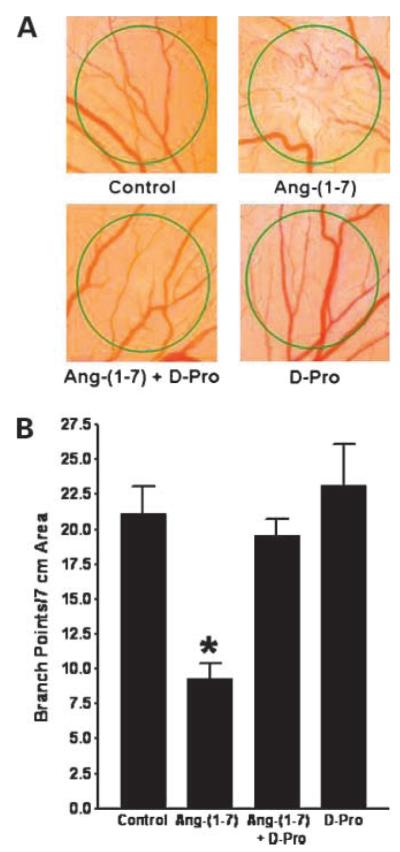
Inhibition of neovascularization in the CAM by Ang-(1-7). Methylcellulose disks containing saline, 100 nmol/L Ang-(1-7), 100 nmol/L Ang-(1-7) and 1.0 μmol/L AT(1-7) receptor antagonist [d-Pro7]-Ang-(1-7) (D-Pro), or 1.0 μmol/L [d-Pro7]-Ang-(1-7) alone were placed in an avascular area of the CAM. After 24 h, the disks were photographed and branch points were quantified to assess vessel formation. A representative photograph of each treatment is shown in A and the quantification of branch points is depicted in B. *, P < 0.001; n = 9 to 12.
Ang-(1-7) Reduces VEGFA in Human Lung Tumor Xenografts
VEGFA is the primary proangiogenic factor released from cancer cells to stimulate the growth of new blood vessels from preexisting vessels. VEGFA was measured by Western blot hybridization in total protein homogenates isolated from A549 human lung tumor xenografts to determine whether the heptapeptide reduces VEGFA in vivo. An immunoreactive band of 19 kDa was visible in tumor extracts from A549 lung tumor homogenates. Ang-(1-7) significantly reduced VEGFA protein in tumor xenografts from mice injected with Ang-(1-7) compared with tumors from saline-treated control animals [1.08 ± 0.18 relative protein expression in tumors from saline-treated mice compared with 0.15 ± 0.08 in tumors from Ang-(1-7) treated mice; n = 5 in each group; P < 0.002; Fig. 5A]. Total RNA was isolated from human lung tumor xenografts treated with saline or Ang-(1-7) and VEGFA mRNA was quantified by reverse transcription real-time PCR to identify the mechanism for the reduced VEGFA protein. A >50% decrease in VEGFA mRNA was observed in the human lung tumor xenografts from mice administered heptapeptide compared with controls [1.01 ± 0.10 relative gene expression in tumors from saline-treated mice compared with 0.43 ± 0.06 in tumors from Ang-(1-7)-treated mice; n = 5 in each group; P < 0.001; Fig. 5B], suggesting that the Ang-(1-7)-mediated reduction in VEGFA was due to either a transcriptional regulatory mechanism or a decrease in VEGFA mRNA stability.
Figure 5.

Reduction of VEGFA in human A549 lung tumor xenografts by Ang-(1-7). A, VEGFA protein was assessed by Western blot hybridization and quantified as the density of VEGFA immunoreactivity as a function of β-actin immunoreactivity in tumor tissue from human lung cancer xenografts treated with saline or Ang-(1-7). Inset, a representative gel. *, P < 0.002; n = 5 for each group. B, RNA was isolated from tumor tissue from mice injected with saline or Ang-(1-7) and VEGFA mRNA was quantified by reverse transcription real-time PCR. *, P < 0.001; n = 5 for each group.
Similarly, the parent human A549 lung cancer cells were incubated with Ang-(1-7) to assess the time-dependent inhibition of VEGFA by the heptapeptide. Actively growing A549 cells were treated with 100 nmol/L Ang-(1-7) for various periods between 4 and 24 h and VEGFA was quantified by Western blot hybridization. Treatment of the human lung cancer cells with the heptapeptide caused a marked decrease in VEGFA protein, with a maximal reduction of 55.5 ± 12.8% after a 12 h incubation with Ang-(1-7) (Fig. 6A). Administration of 100 nmol/L Ang-(1-7) also caused a time-dependent decrease in VEGFA mRNA in human A549 cells between 1 to 8 h of treatment (Fig. 6B). Treatment with the Ang-(1-7) receptor antagonists [d-Pro7]-Ang-(1-7) or [d-alanine7]-Ang-(1-7) completely blocked the Ang-(1-7)-mediated decrease in VEGFA mRNA, whereas the antagonists alone had no effect (Fig. 6C), indicating that the heptapeptide activated an AT(1-7) receptor to reduce VEGFA. These in vitro results provide further support that Ang-(1-7) may selectively decrease VEGFA to reduce angiogenesis.
Figure 6.

Reduction of VEGFA by Ang-(1-7) in human A549 cells. A549 cells were incubated with 100 nmol/L Ang-(1-7) from 4 to 24 h. VEGFA protein was assessed by Western blot hybridization and quantified as the density of VEGFA immunoreactivity as a function of β-actin immunoreactivity and expressed as a percentage of the control at time 0 in A. RNA was isolated from tumor tissue from mice injected with saline or Ang-(1-7) and VEGFA mRNA was quantified by reverse transcription real-time PCR in B. C, cells were incubated for 4 h with no addition (Con), 100 nmol/L Ang-(1-7) (A7), Ang-(1-7) in the presence of 1 μmol/L [d-Pro7]-Ang-(1-7) (DP), [d-Pro7]-Ang-(1-7) alone, Ang-(1-7) in the presence of 1 μmol/L [d-alanine7]-Ang-(1-7) (DA), or [d-alanine7]-Ang-(1-7) alone. *, P < 0.05; n = 4 for each group.
Discussion
The present study is the first to show that Ang-(1-7) reduces the growth of human lung tumor xenografts with a decrease in blood vessel density, suggesting that one mechanism whereby the heptapeptide decreases tumor cell proliferation is by reducing angiogenesis. In agreement, we showed inhibition of tubule formation by Ang-(1-7) as well as a reduction in the number of blood vessels in the CAM following treatment with the heptapeptide. More important, we found that Ang-(1-7) markedly reduced VEGFA in both human A549 lung tumor xenografts and the parent cells, suggesting that this potent growth factor is involved in the antiangiogenic response to the heptapeptide.
The results of the current study are supported by previous reports. Captopril, an antihypertensive drug that increases the level of Ang-(1-7) (14), inhibited neo-vascularization in the rat cornea induced by basic fibroblast growth factor and prevented capillary endothelial cell migration by blocking chemotaxis toward an inducer of angiogenesis (15). Ang-(1-7) also reduced angiogenesis in a murine sponge model of angiogenesis, a technique representative of the formation of new blood vessels from preexisting ones during wound healing (16). Infusion of the heptapeptide reduced hemoglobin content, blood flow, and proliferative activity compared with vehicle. The antiangiogenic effect of Ang-(1-7) in this model was regulated by an Ang-(1-7)-specific receptor (17), in agreement with our identification of an Ang-(1-7) receptor on endothelial cells from canine coronary artery and bovine aortic endothelial cells (18, 19). Further, the antiangiogenic response was blocked by preincubation with either aminoguanidine or NG-nitro-l-arginine methyl ester, indicating that the response is mediated by nitric oxide (17). Ang-(1-7) stimulates the release of nitric oxide from bovine aortic endothelial cells and causes the vasodilation of blood vessels through an endothelial release of nitric oxide (1). Whereas the studies by Machado et al. (16, 17) are in a model of angiogenesis during wound healing, we now show that Ang-(1-7) inhibits angiogenesis that occurs during tumor formation to supply necessary nutrients for tumor growth.
VEGFA, a robust stimulator of angiogenesis, has a variety of roles in this process, including stimulation of vascular permeability, induction of endothelial cell migration and division, promotion of endothelial cell survival, and stimulation of cell proliferation (20). A marked decrease in VEGFA was observed in the tumors from mice treated with Ang-(1-7) compared with tumors from control animals, suggesting that the heptapeptide may attenuate tumor angiogenesis by reducing VEGFA. The precise molecular mechanism(s) for the Ang-(1-7)-mediated decrease in VEGFA is not known. Because VEGFA mRNA was also reduced, Ang-(1-7) may activate signaling pathways to inhibit VEGFA transcription. The VEGFA promoter is regulated by a variety of effectors; however, the major regulator of VEGFA gene transcription in lung cancer cells is the hypoxia-inducible factor (HIF)-1 (21). HIF-1 is increased in response to hypoxia or conditions of low oxygen tension. Low oxygen tension in tumor cells located a distance away from blood vessels results in the production and release of VEGFA to initiate angiogenesis. As a consequence of the antiproliferative effects of Ang-(1-7), metabolic activity and oxygen consumption may be reduced to modify the tumor microenvironment and stabilize the HIF heterodimer. In addition, HIF-1α expression is regulated by other pathways. Mitogen-activated protein kinases are essential for the direct phosphorylation and activation of HIF-1α, which in turns induces VEGFA expression (22). Continuous activation of extracellular signal-regulated kinases 1 and 2 inhibits apoptosis, controls proliferation of endothelial cells, and promotes VEGFA expression by stimulating the activator protein-2/Sp-1 complex on the VEGFA promoter (22). We showed previously that Ang-(1-7) inhibits the activities of extracellular signal-regulated kinases 1 and 2 in lung cancer cells (9) and vascular smooth muscle cells (23), suggesting that the observed reduction in VEGFA mRNA and protein may be due to inhibition of mitogen-activated protein kinase signaling. Other growth factors also increase HIF-1α, the HIF-1 regulatory subunit. If HIF-1α is not regulated by Ang-(1-7) treatment, VEGFA may be controlled at transcription by other modulators, such as epidermal growth factor, transforming growth factor-β, or basic fibroblast growth factor (24). In addition, VEGFA expression is regulated at transcription by RNA stability, at translation by mRNA capping proteins, or at post-translation by glycosylation (24). The precise molecular mechanism(s) for the Ang-(1-7)-mediated decrease in VEGFA is currently under investigation.
In a previous study, we observed that administration of Ang-(1-7) decreased lung tumor growth and volume by >30%, whereas tumors of mice infused with saline continued to grow 2.5-fold at the end of the 28-day treatment (10). The decrease in tumor size caused by Ang-(1-7) was associated with a reduction in COX-2, suggesting that the heptapeptide may inhibit proinflammatory prostaglandins that promote lung tumor growth. Overexpression of COX-2 is associated with the initiation of angiogenesis through the production of both prostaglandins and endothelial cell growth factors, and COX-2 inhibitors block this process (25, 26). COX-2 inhibitors suppress the expression and secretion of the matrix metalloproteinases 2 and 9, suggesting that COX-2 also promotes tumor angiogenesis and tumor invasion. In addition, the downstream products of COX-2 stimulate VEGFA to promote angiogenesis (25). Taken together, these studies suggest that the decrease in angiogenesis by Ang-(1-7) may be due in part to a reduction in COX-2 mRNA and protein.
The proliferation of endothelial cells is a crucial step in the angiogenenic process. Endothelial cells are stimulated by tumor-released growth factors to migrate and divide at the tumor site, ultimately forming blood vessel tubes stabilized by smooth muscle cells. In this report, we showed that Ang-(1-7) inhibited endothelial cell tubule formation using a Matrigel assay. The inhibition of angiogenesis by Ang-(1-7) may be due in part to its antiproliferative properties to reduce critical endothelial cell division and prevent the formation of vessels. Ang-(1-7) also causes the receptor-mediated reduction in the proliferation of vascular smooth muscle cells in vitro and in vivo (4, 7, 27) through stimulation of prosta-cyclin production and activation of the cyclic AMP-dependent protein kinase. The reduction in vessel density observed in the human lung tumor xenografts or in the CAM may be due to a loss in structural support provided by vascular smooth muscle cells. Alternatively, the heptapeptide may block tube formation by inhibiting endothelial and/or vascular smooth muscle cell migration. Studies are currently under way to delineate the direct effect of Ang-(1-7) on the cellular components forming the vessel tube.
Lung cancer is a leading cause of cancer death among men and women in developed countries, approaching ~200,000 new cases each year and causing ~160,000 deaths annually in the United States (28). Despite improvements in treatment modalities, the 5-year survival rate has improved to only 15% in the past 30 years with >1,000,000 new cases of lung cancer diagnosed worldwide annually. This grim prognosis indicates the need for novel approaches to reduce the high lung cancer mortality. The results from our previous study and this report indicate that Ang-(1-7) has pleiotrophic effects that reduce the molecular mechanisms promoting unrestricted cell proliferation as well as tumor angiogenesis. Because the heptapeptide mediates biological effects by activation of a unique, G protein-coupled receptor, mas, Ang-(1-7) may serve as an effective, targeted chemotherapeutic agent for lung cancer.
Acknowledgments
We thank L. Tenille Howard, R. Lanning, Mark Landrum, and Hermina Borgerink for technical assistance.
Grant support: Susan G. Komen Breast Cancer Research Foundation; Department of Defense Breast Cancer Research predoctoral fellowship (D.R. Soto-Pantoja); NIH grants HL-051952 and HL-079498 (P.E. Gallagher and E.A. Tallant); and Unifi, Farley-Hudson Foundation, and Golfer’s Against Cancer of the Triad.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Ferrario CM, Averill DB, Brosnihan KB, et al. Angiotensin-(1-7). It’s contribution to arterial pressure control mechanisms. In: Unger T, Scholkens B, editors. Handbook of experimental pharmacology. Springer-Verlag Heidelberg; Germany: 2004. pp. 478–518. [Google Scholar]
- 2.Ferrario CM, Trask AJ, Jessup JA. Advances in biochemical and functional roles of angiotensin-converting enzyme 2 and angiotensin-(1-7) in regulation of cardiovascular function. Am J Physiol Heart Circ Physiol. 2005;289:H2281–90. doi: 10.1152/ajpheart.00618.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santos RA, Simoes e Silva AC, Maric C, et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A. 2003;100:8258–63. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freeman EJ, Chisolm GM, Ferrario CM, Tallant EA. Angiotensin-(1-7) inhibits vascular smooth muscle cell growth. Hypertension. 1996;28:104–8. doi: 10.1161/01.hyp.28.1.104. [DOI] [PubMed] [Google Scholar]
- 5.Tallant EA, Ferrario CM, Gallagher PE. Angiotensin-(1-7) inhibits growth of cardiac myocytes through activation of the mas receptor. Am J Physiol Heart Circ Physiol. 2005;289:H1560–6. doi: 10.1152/ajpheart.00941.2004. [DOI] [PubMed] [Google Scholar]
- 6.Loot AE, Roks AJ, Henning RH, et al. Angiotensin-(1-7) attenuates the development of heart failure after myocardial infarction in rats. Circulation. 2002;105:1548–50. doi: 10.1161/01.cir.0000013847.07035.b9. [DOI] [PubMed] [Google Scholar]
- 7.Strawn WB, Ferrario CM, Tallant EA. Angiotensin-(1-7) reduces smooth muscle growth after vascular injury. Hypertension. 1999;33:207–11. doi: 10.1161/01.hyp.33.1.207. [DOI] [PubMed] [Google Scholar]
- 8.Langeveld B, Van Gilst WH, Tio RA, Zijlstra F, Roks AJ. Angiotensin-(1-7) attenuates neointimal formation after stent implantation in the rat. Hypertension. 2005;45:138–41. doi: 10.1161/01.HYP.0000149382.83973.c2. [DOI] [PubMed] [Google Scholar]
- 9.Gallagher PE, Tallant EA. Inhibition of human lung cancer cell growth by angiotensin-(1-7) Carcinogenesis. 2004;25:2045–52. doi: 10.1093/carcin/bgh236. [DOI] [PubMed] [Google Scholar]
- 10.Menon J, Soto-Pantoja DR, Callahan MF, et al. Angiotensin-(1-7) inhibits growth of human lung adenocarcinoma xenografts in nude mice through a reduction in cyclooxygenase-2. Cancer Res. 2007;67:2809–15. doi: 10.1158/0008-5472.CAN-06-3614. [DOI] [PubMed] [Google Scholar]
- 11.Floyd HS, Farnsworth CL, Kock ND, et al. Conditional expression of the mutant Ki-rasG12C allele results in formation of benign lung adenomas: development of a novel mouse lung tumor model. Carcinogenesis. 2005;26:2196–206. doi: 10.1093/carcin/bgi190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weidner N. Current pathologic methods for measuring intratumoral microvessel density within breast carcinoma and other solid tumors. Breast Cancer Res Treat. 1995;36:169–80. doi: 10.1007/BF00666038. [DOI] [PubMed] [Google Scholar]
- 13.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 14.Luque M, Martin P, Martell N, Fernandez C, Brosnihan KB, Ferrario CM. Effects of captopril related to increased levels of prostacyclin and angiotensin-(1-7) in essential hypertension. J Hypertens. 1996;14:799–805. doi: 10.1097/00004872-199606000-00017. [DOI] [PubMed] [Google Scholar]
- 15.Volpert OV, Ward WF, Lingen MW, et al. Captopril inhibits angiogenesis and slows the growth of experimental tumors in rats. J Clin Invest. 1996;98:671–9. doi: 10.1172/JCI118838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Machado RD, Santos RA, Andrade SP. Opposing actions of angiotensins on angiogenesis. Life Sci. 2000;66:67–76. doi: 10.1016/s0024-3205(99)00562-7. [DOI] [PubMed] [Google Scholar]
- 17.Machado RD, Santos RA, Andrade SP. Mechanisms of angiotensin-(1-7)-induced inhibition of angiogenesis. Am J Physiol Regul Integr Comp Physiol. 2001;280:R994–1000. doi: 10.1152/ajpregu.2001.280.4.R994. [DOI] [PubMed] [Google Scholar]
- 18.Tallant EA, Lu X, Weiss RB, Chappell MC, Ferrario CM. Bovine aortic endothelial cells contain an angiotensin-(1-7) receptor. Hypertension. 1997;29:388–93. doi: 10.1161/01.hyp.29.1.388. [DOI] [PubMed] [Google Scholar]
- 19.Ferrario CM, Chappell MC, Tallant EA, Brosnihan KB, Diz DI. Counter-regulatory actions of angiotensin-(1-7) Hypertension. 1997;30:535–41. doi: 10.1161/01.hyp.30.3.535. [DOI] [PubMed] [Google Scholar]
- 20.Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002;20:4368–80. doi: 10.1200/JCO.2002.10.088. [DOI] [PubMed] [Google Scholar]
- 21.Nagy JA, Dvorak AM, Dvorak HF. VEGF-A and the induction of pathological angiogenesis. Annu Rev Pathol. 2007;2:251–75. doi: 10.1146/annurev.pathol.2.010506.134925. [DOI] [PubMed] [Google Scholar]
- 22.Berra E, Milanini J, Richard DE, et al. Signaling angiogenesis via p42/p44 MAP kinase and hypoxia. Biochem Pharmacol. 2000;60:1171–8. doi: 10.1016/s0006-2952(00)00423-8. [DOI] [PubMed] [Google Scholar]
- 23.Tallant EA, Clark MA. Molecular mechanisms of inhibition of vascular growth by angiotensin-(1-7) Hypertension. 2003;42:574–9. doi: 10.1161/01.HYP.0000090322.55782.30. [DOI] [PubMed] [Google Scholar]
- 24.Loureiro RMB, D’Amore PA. Transcriptional regulation of vascular endothelial growth factor in cancer. Cytokine Growth Factor Rev. 2005;16:77–89. doi: 10.1016/j.cytogfr.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 25.Gately S, Li WW. Multiple roles of COX-2 in tumor angiogenesis: a target for antiangiogenic therapy. Semin Oncol. 2004;31:2–11. doi: 10.1053/j.seminoncol.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 26.Iniguez MA, Rodriguez A, Volpert OV, Fresno M, Redondo JM. Cyclooxygenase-2: a therapeutic target in angiogenesis. Trends Mol Med. 2003;9:73–8. doi: 10.1016/s1471-4914(02)00011-4. [DOI] [PubMed] [Google Scholar]
- 27.Tallant EA, Diz DI, Ferrario CM. State-of-the-art lecture. Antiproliferative actions of angiotensin-(1-7) in vascular smooth muscle. Hypertension. 1999;34:950–7. doi: 10.1161/01.hyp.34.4.950. [DOI] [PubMed] [Google Scholar]
- 28.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]