

Abstract
Lung cancer is a leading cause of death in both men and women, with over 1,000,000 new cases diagnosed worldwide annually and a 5-year survival rate of only 14%, a figure that has improved little in the past thirty years. This poor prognosis suggests a need for novel approaches for the treatment and prevention of lung cancer. The renin-angiotensin system is an established, primary regulator of blood pressure, homeostasis, and natriuresis; however, compelling evidence indicates that the angiotensin peptides also play a role in cell proliferation and inflammation. Angiotensin II is a vasoconstrictor, a mitogen, and an angiogenic factor, while angiotensin-(1-7) has vasodilator, anti-proliferative, and anti-angiogenic properties. This review focuses on studies examining the renin-angiotensin system in pulmonary cancers and whether clinical intervention of this pathway may serve as an effective chemotherapeutic and/or chemopreventive modality for lung cancer.
Keywords: Renin-angiotensin system, angiotensin, lung cancer, pulmonary cancer, angiotensin-(1-7), angiotensin converting enzyme, angiogenesis, angiotensin receptor blocker
Lung cancer is one of the most frequent human cancers with over one million people diagnosed worldwide each year. The mortality from lung cancer among men and women in developed countries approaches almost 200,000 new cases and 160,000 deaths annually in the United States [1-4]. Cigarette smoking accounts for 80-90% of the lung cancer cases. Chronic exposure to tobacco consumption products is the leading cause of premature mortality in industrialized countries, resulting in about 50% of deaths in people between 35 - 65 years old. Exposure to asbestos, radon, and other environmental agents, as well as genetic factors contribute to the remainder of the cases. Despite improvements in treatment modalities, the 5-year lung cancer survival rate has improved to only 14% in the past thirty years. The high mortality is due to the frequent presence of advanced stage metastasis at the initial diagnosis, with more than two-thirds of patients showing lymph-node metastases at the time of presentation. This grim prognosis indicates the need for novel therapies targeting unique signaling pathways to reduce lung cancer deaths. This review summarizes studies investigating the role of the renin-angiotensin system (RAS) in lung carcinogenesis as well as explores potential RAS targets for chemotherapy and chemoprevention.
THE RAS
The RAS is a physiological regulator of blood pressure, homeostasis, and cell proliferation, as reviewed in [5-8]. The parent compound angiotensinogen, produced predominantly in the liver and secreted into the circulation, is degraded by circulating renin to the decapeptide angiotensin I (Ang I), as shown in Fig. (1). The cascade diverges with the proteolytic processing of Ang I to the peptide hormones, Ang II and Ang-(1-7), products with different carboxy termini and contrasting biological actions. Angiotensin converting enzyme (ACE), produced primarily in the epithelial cells of the lung, catalyzes the conversion of Ang I to Ang II and attenuation of the enzyme actions by selective inhibitors, such as enalopril, captopril, perindopril, is among the therapeutic modalities commonly used to treat patients with high blood pressure. Blockade of ACE activity prevents the formation of the vasoconstrictor and mitogen Ang II and the degradation of the vasodilators bradykinin and Ang-(1-7), leading to a reduction in blood pressure as well as the myriad of beneficial effects attributed to ACE inhibition. Ang-(1-7), a heptapeptide with anti-proliferative properties, is normally present in the circulation at concentrations similar to Ang II and is primarily derived from Ang I by tissue peptidases, including neprilysin, thimet oligopeptidase and prolyl endopeptidase (as reviewed in [9]). The two arms of the pathway are bridged by the moncarboxypeptidase angiotensin converting enzyme 2 (ACE2) which generates Ang-(1-7) from Ang II [10, 11]. While ACE inhibitors have no direct effect on ACE2 activity [12, 13], we found a marked up-regulation of ACE2 mRNA in Lewis rats treated with lisinopril [14] as well as an increase in cardiac ACE2 activity [14]. This study showed that ACE inhibitors indirectly affect ACE2 activity by a transcriptional regulatory mechanism and supports a role for Ang-(1-7) in the cardioprotective effects of ACE inhibitors. Ang-(1-7) is also a substrate for ACE [15], suggesting that ACE inhibition not only elevates Ang-(1-7) by increasing Ang I, a substrate for Ang-(1-7) production, but also by preventing Ang-(1-7) degradation (Fig. 2).
Ang II acts through two pharmacological classes of seven transmembrane, G protein-coupled receptors—the angiotensin type 1 (AT1) and angiotensin type II (AT2) receptors [16, 17] (Fig. 1). In rodents, there are two AT1 receptors (AT1a and AT1b) which have similar functions but have differential localization, while humans have a single AT1 receptor. The majority of physiological effects associated with Ang II, including vasoconstriction, natriuresis, diuresis, and mitogenesis, are mediated by the AT1 receptor and antagonists selective for blockade of AT1 receptor function (angiotensin receptor blockers or ARBs) are also commonly used in the treatment of hypertension [18]. The AT2 receptor has a more limited tissue distribution but is present in increased amounts in prenatal tissues and following tissue injury, such as myocardial infarction. Proposed functions for the AT2 receptor include stimulation of apoptosis and inhibition of cell growth [19-23].
Ang-(1-7) is a poor competitor at the prototypical AT1 or AT2 receptor [24, 25] and the majority of responses to Ang-(1-7) were not blocked by an AT1 or AT2 receptor antagonist [25, 26]. [D-alanine7]-angiotensin-(1-7) ([D-Ala7]-Ang-(1-7)), a modified form of Ang-(1-7) in which proline at position 7 is replaced by D-alanine, selectively blocked responses to Ang-(1-7), was a poor competitor at the AT1 or AT2 receptor, and did not block pressor or contractile responses to Ang II [27-30]. We showed that the inhibition of mitogen-stimulated VSMC growth by Ang-(1-7) was not prevented by AT1 or AT2 receptor antagonists but [D-Ala7]-Ang-(1-7) effectively blocked growth inhibition by Ang-(1-7) [31, 32]. Santos et al. [33] reported that the orphan G protein-coupled receptor mas is an Ang-(1-7) receptor. Ang-(1-7) competed for binding to a [D-Ala7]-Ang-(1-7)-sensitive receptor and stimulated arachidonic acid production in stably transfected cell lines containing the mas gene.
Besides serving as an endocrine system, the RAS has paracrine as well as autocrine functions. RAS complexity is compounded by localization in tissues, resulting in the synthesis, release, and action of the angiotensin peptides [9, 34, 35]. A functional tissue RAS was identified in every organ and tissue investigated, although some components of the pathway, particularly renin, are not always synthesized locally but are acquired from the endocrine RAS. In addition to normal tissues, RAS components are expressed in a variety of tumor cells, including carcinomas of the bladder, brain, cervix, colon, kidney, liver, lung, pancreas, prostate, skin, and stomach as previously reviewed [5, 36-38] as well as additional articles in this issue. The RAS as a chemotherapeutic target for cancer garnered attention in recent years since Ang II is implicated in cell proliferation and migration, angiogenesis, inflammation, and extracellular matrix formation. In addition, anti-hypertensive drugs currently on the market that manipulate the RAS have strong safety profiles and are relatively inexpensive.
ANGIOTENSIN PEPTIDE SYNTHESIZING ENZYMES AND LUNG CANCER
A role for RAS in lung cancer was suggested by studies initiated in the 1980s demonstrating that patients with pulmonary tumors had reduced concentrations of circulating ACE. Romer found that the serum ACE levels were significantly lower in 141 patients newly diagnosed with primary lung cancer as compared to healthy controls [39]. This finding was corroborated by others, demonstrating low serum ACE levels in patients with lung tumors as compared to patients with other pulmonary diseases or carcinomas [40, 41]. The lowest serum ACE levels correlated with poor prognosis and higher relapse rate [39, 42, 43] as well as metastatic disease, suggesting that quantification of serum ACE may be a prognostic indicator for lung cancer [44]. Further, ACE activity increased in patients with bronchial carcinoma following chemotherapy or radiotherapy [41, 43, 45] as well as in patients in clinical remission [44]. Prochazka et al. observed a reduction in ACE activity in primary human lung tumors as compared to normal lung tissue [46]; a decrease in serum ACE in patients with lung cancer is likely reflective of increased tumor burden, leading to reduced pulmonary epithelial cells which are the primary source for circulating ACE, as well as diminished ACE production by the lung cancer cells. Treatment of patients with chemotherapy or radiotherapy reduces the tumor load resulting in increased ACE production [41, 43]. Taken together, these results suggest that plasma ACE activity may serve as an effective biomarker for patients with poor prognosis as well as an indicator of responders to therapeutic intervention. With the advent of more sophisticated imaging techniques, ACE is no longer assessed as a biomarker for lung cancer; however, these studies indicate a perturbation of the RAS with lung carcinogenesis. Circulating concentrations of ACE are still quantified as a clinical measure of pulmonary sarcodosis [47].
Neprilysin [CD10, neutral endopeptidase 24.11, common acute lymphoblastic leukemia antigen (CALLA)] is a cell surface, zinc metalloprotease that cleaves peptide bonds on the amino side of hydrophobic amino acids. This enzyme catalyzes the conversion of a number of endogenous peptides, including bradykinin, substance P, neurotensin, oxytocin, atrial natriuretic factor, endothelin, Met-enkephalin, and Leu-enkephalin. As shown in Fig. (1), neprilysin also hydrolyzes the precursor RAS peptide Ang I to Ang-(1-7). Neprilysin is expressed at high levels in the lung, particularly in pulmonary epithelial cells, and regulates broncho-constriction and tissue responses to peptides. Cigarette smoke inactivates neprilysin in the lung, leading to the accumulation of mitogenic peptides in the bronchoalveolar lavage fluid [48-50]. The enzyme is reduced in small cell and non-small cell lung cancer cell lines and tumors [49] as well as in bronchioalveolar lavage fluid of lung cancer patients [50]. Transfection of human lung cancer cells with an expression vector containing the neprilysin gene resulted in reduced proliferation in vitro and in vivo in athymic mouse xenografts [51]. In several of these reports, the authors proposed that the ability of neprilysin to degrade mitogenic peptides was important in the growth regulation of lung cancer cells. This suggests that the lack of Ang II degradation as well as reduced production of Ang-(1-7) by neprilysin may account, at least in part, for the aberrant growth of lung cancer cells. A summary of the pre-clinical data discussed in this review suggesting that the RAS plays a role in lung cancer is found in Table 1.
ANG II AND LUNG CANCER CELL GROWTH
There are limited studies examining the Ang II-mediated signaling pathways activated in lung cancer cells. A dose-dependent, transient increase in cytosolic free calcium was observed in human A549 lung cancer cells following treatment with Ang II [54]. Losartan blocked the effect, indicating that AT1 receptor activation was involved in the process. Ang II had no effect on calcium levels in normal lung cells or small cell lung cancer cell lines, suggesting that the elevation in free calcium in the A549 cells by the octapeptide may play a role in non-small cell lung carcinogenesis. Ang II increased COX-2 and prostaglandin E2 production in normal lung fibroblasts in a dose-dependent manner but not in the human lung adenocarcinoma A549 cell line; this difference may be due to an enhanced level of COX-2 in the cancer cells, thereby limiting the regulatory effect of the angiotensin peptide [66-69]. Further studies are needed to corroborate these findings in vitro and in vivo.
Bruce Uhal and colleagues demonstrated that the ACE inhibitors captopril or lisinopril blocked apoptosis induced by activation of the Fas receptor in human and rat alveolar epithelial cells as well as human A549 lung cancer cells [55, 56]. Conversely, direct Ang II treatment stimulated apoptosis in these cells and the effect was blocked by losartan, a specific AT1 receptor blocker but not by the selective AT2 receptor antagonists PD-123319 or PD-126055 [70]. The activation of Fas as well as the stimulation of apoptosis by either bleomycin or amiodarone resulted in increased Ang II concentration and angiotensinogen mRNA in both rat alveolar epithelial cells and human A549 lung cancer cells which was blocked by anti-sense oligonucleotides to angiotensinogen, ACE inhibitors, or ARBs [56, 71]. These results suggest that Ang II promotes apoptosis in lung cancer cells; treatment with ACE inhibitors that prevent Ang II synthesis or ARBs that block the actions of Ang II inhibits this response. Conversely, the ACE inhibitor captopril induced apoptosis in LNM35 lung cancer cells [72], the ARB losartan increased apoptosis in C6 glioma cells or tumors [73] as well as human pancreatic cells [74], telmisartan, an AT1 receptor antagonist, induced early apoptosis and DNA fragmentation in prostate cancer cells [75] and ACE inhibitors or ARBs enhance programmed cell death in leukemic cell lines [76]. As the studies from Uhal and colleagues also conflict with existing literature demonstrating that Ang II promotes proliferation in a variety of vascular and cancer cells, further study is warranted to determine the balance between the proliferative and apoptotic actions of Ang II in lung cancer cells. The discrepancy observed may be due to differences in the ratio of the AT1 to AT2 receptor on the cancer cell lines examined. Receptor binding assays using pharmacological antagonists for the two receptor subtypes are needed to begin to understand the regulation of apoptosis in various cancers by Ang II.
EFFECT OF ACE INHIBITORS AND ARBS ON LUNG CARCINOGENESIS
In a retrospective study of 5207 patients in Scotland, the relative risks of incident and fatal cancer among the 1559 patients treated with ACE inhibitors were significantly reduced, to 0.72 and 0.65, respectively [77]. The relative risk was lowest in patients with lung, colon, or sex-specific cancer, as compared with other sites. Inhibition of ACE activity decreases the vasoconstrictor and growth stimulator Ang II and increases Ang-(1-7), a heptapeptide with vasodilator and anti-proliferative properties [78, 79] (Fig. 2). This suggests that the reduced risk of cancer could result not only from decreased Ang II but also from the elevation in Ang-(1-7). Since this clinical study demonstrated a reduced incidence of lung cancer, the alteration in the angiotensin peptide levels by ACE inhibitors may not only inhibit the growth of lung cancer cells, but also prevent lung tumor formation. In a similar retrospective study, patients with advanced non-small-cell lung cancer that received a therapy regimen of an ACE inhibitor or ARB with a first-line platinum-based chemotherapeutics had a 3.1 month longer median survival than patients not receiving the anti-hypertensive medication [80]. These results suggest that ACE inhibitors or ARBs may provide enhanced efficacy for lung cancer in combination with chemotherapeutics.
Pre-clinical studies with ACE inhibitors or ARBs also demonstrate a role for the RAS in lung carcinogenesis as well as prevention of lung tumor formation. Prontera et al. showed that the combination of the matrix metalloproteinase inhibitor batimastat and the ACE inhibitor captopril markedly reduced the mean volume, mean metastasis and survival time of Lewis lung tumors in syngeneic C57BL/6 mice as compared to control animals [59]. In addition, cyclosporin-enhanced pulmonary metastases were reduced to control levels in mice following treatment with the AT1 receptor antagonist losartan [62]. Attoub et al. reported that the ACE inhibitor captopril reduced the growth of human LNM35 lung tumors as well as lymph node metastases in mice by more than 50% as compared to tumors in control animals with no appreciable side-effects [72]. Immunohistochemical analysis of tumor tissue sections showed that captopril administration markedly reduced the cell proliferation marker Ki67. Captopril induced apoptotic morphological changes in LNM35 lung cancer cells, suggesting that the anti-proliferative effect of this ACE inhibitor was due to induction of programmed cell death. Taken together, these studies suggest that administration of ACE inhibitors or ARBs may effectively reduce lung cancer proliferation as well as lung tumor metastases to prevent further tumor formation.
EFFECT OF ACE INHIBITORS AND ARBS ON LUNG TUMOR ANGIOGENESIS
Ang II increased the growth of vascular smooth muscle cells in vitro and stimulated blood vessel formation in several in vivo models of angiogenesis [81-84]; conversely, ACE inhibitors that block endogenous Ang II production or ARBs that attenuate Ang II activity reduce angiogenesis. The AT1 receptor antagonist candesartan (TCV-116, CV11974) significantly inhibited the growth of pulmonary metastases from renal cell carcinoma with an associated decrease in vascular endothelial growth factor (VEGF) and inhibition of angiogenesis as compared to control animals [63]. In similar studies, the lung metastasis of intravenously injected Lewis lung carcinoma cells was markedly inhibited following candesartan or ACE inhibitor lisinopril treatment with a significant decrease in tumor-associated blood vessel formation [61]. A significant reduction in VEGF-A mRNA and protein in association with attenuated tumor growth was also observed in Lewis lung tumors following candesartan administration [52], Incubation of Lewis lung carcinoma cells with Ang II caused a significant increase in VEGF-A mRNA and protein which was prevented by co-administration of an AT1 receptor antagonist. While these studies demonstrate that Ang II stimulates angiogenesis in lung tumors through activation of AT1 receptors, the molecular signaling pathways involved in the process are not known.
AT2 RECEPTOR AND LUNG CARCINOGENESIS
As illustrated in Fig. (1), Ang II activates both AT1 and AT2 receptors, two distinct pharmacological classes of seven transmembrane, G protein-coupled receptors [16, 17]. While the physiological role of the AT1 receptor in blood pressure regulation, diuresis, and mitogenesis is established, the precise function of the AT2 receptor is not clear for many tissues. Several studies demonstrate that Ang II binding to the AT2 receptor may be involved in lung carcinogenesis. Kanehira et al. showed that AT2 receptor-null mice had reduced tumor number and multiplicity following treatment with NNK [4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone] as compared to control [64]. Co-incubation of A549 human lung cancer cells with lung fibroblasts from the AT2 receptor-null mice decreased colony count with an associated increase in transforming growth factor-β (TGF-β) production, suggesting that the AT2 receptor on lung fibroblasts may be involved in chemical carcinogen-induced lung tumorigenesis. The immunostain for both the AT1 and AT2 receptors was enhanced in NNK-induced tumor sections, [65] indicating a potential role not only for the angiotensins receptors on lung cancer cells but also on cells of the tumor microenvironment in lung carcinogenesis. In support, the volume of Lewis lung tumor xenografts was significantly reduced in mice following administration of the AT2 receptor antagonist PD123,319 (20 mg/kg/day) with an associated reduction in VEGF [60]. Inhibition of the parent cell growth was also observed following blockade of the AT2 receptor with an associated reduction in VEGF. These studies suggest that activation of the AT2 receptor may promote lung tumorigenesis by increasing cell proliferation and tumor angiogenesis. Conversely, Pickel et al. [85] found that over-expression of the AT2 receptor using nanoparticle vectors reduced the growth, increased the number of apoptotic cells and activated caspase 3 in the human adenocarcinoma cell line A549 and bronchioalveolar carcinoma line H358. These results suggest that activation of the AT2 receptor may inhibit lung cancer proliferation and are in conflict with the studies described above in carcinogen-induced lung tumorigenesis and in the Lewis lung tumor xenografts. The differences may be reflective of the AT2 receptor number as an 80-fold increase in the AT2 receptor mRNA was detected in the A549 transfected cell line [85]. While the protein concentration of the receptor was not determined, the mRNA data suggest that the level of the AT2 receptor achieved by transfection using the nanoparticle vectors may be considerably higher than occurs by normal, physiological regulation. Further studies are certainly needed to rectify this conflicting data before the AT2 receptor could be considered as a target for lung cancer intervention.
ANG-(1-7) AND LUNG CANCER
As discussed above, Ang-(1-7) is an endogenous, seven amino acid peptide hormone of the RAS with vasodilator, anti-proliferative, and anti-thrombotic properties. Ang-(1-7) mediates biological properties through activation of a unique, G-protein-coupled AT(1-7) receptor, mas. We showed that this heptapeptide inhibits the growth of human lung cancer cells through a reduction in MAP kinase [53]. Ang-(l-7) caused a significant decrease in serum-stimulated growth of human SK-LU-1, A549, and SK-MES-1 lung cancer cells with a dose- and time-dependent reduction in DNA synthesis and IC50’s in the sub-nanomolar range. Other angiotensin peptides, Ang I, Ang II, Ang-(2-8), Ang-(3-8) and Ang-(3-7), did not attenuate mitogen-stimulated DNA synthesis of SK-LU-1 cells, demonstrating that Ang-(l-7) selectively inhibits the growth of these human cancer cells. The Ang-(l-7) receptor antagonist [D-Ala7]-Ang-(l-7) blocked the attenuation of serum-stimulated DNA synthesis in SK-LU-1 cells by Ang-(l-7), while neither AT1 nor AT2 angiotensin receptor subtype antagonists prevented the response to the heptapeptide. Mas mRNA and protein were detected in the three lung cancer cell lines, suggesting that mas mediated the anti-proliferative response. Pretreatment of SK-LU-1 cells with 10 nM Ang-(l-7) reduced serum-stimulated phosphorylation of ERK1 and ERK2 (by 61% and 68%, respectively), indicating that the anti-proliferative effects may occur, at least in part, through inhibition of the ERK signal transduction pathway. These results suggest that Ang-(l-7) inhibits lung cancer cell growth through activation of an angiotensin peptide receptor and may represent a novel therapeutic and/or preventive treatment for lung cancer.
Ang-(1-7) administration markedly attenuated the growth of human A549 lung cancer xenografts [57]. Tumor volume was reduced by 30% following medication with the heptapeptide as compared with the size before treatment; in contrast, tumor size in the saline-treated animals increased 2.5-fold. No adverse side effects, including changes in heart and body weight, heart rate or blood pressure, were observed following Ang-(1-7) administration. The tumor inhibition correlated with a reduction in the proliferation marker Ki67 as well as cyclooxygenase 2 (COX-2) mRNA and protein in tumors from the Ang-(1-7)-medicated animals as compared with the saline control tumor tissue. In contrast, the heptapeptide had no effect on COX-1 mRNA in xenograft tumors. Similar results for COX regulation were observed with the parent A549 human lung cancer cells in tissue culture. These results suggest that Ang-(1-7) may decrease COX-2 activity and pro-inflammatory prostaglandins to inhibit lung tumor growth. Because Ang-(1-7) reduces growth through activation of a selective AT(1-7) receptor, the heptapeptide may serve as a first-in-class, targeted therapy for lung cancer by reducing COX-2.
COXs, the key enzymes in the conversion of arachidonic acid to prostaglandins (PGs) and other bioactive lipids, are up-regulated by a wide variety of mitogens and tumor promoting agents involved in the regulation of normal growth responses and in aberrant cellular growth. Selective COX-2 inhibitors provided a promising treatment for lung cancer. COX-2 is over-expressed in lung tumors [86-89] COX-2 inhibitors prevent lung cancer in experimental animals, [86, 87, 90, 91] and epidemiological studies suggest that regular use of NSAIDs can reduce incidence of lung cancer [92]. Unfortunately, a number of these drugs were withdrawn from the market due to an increased risk of cardiovascular events with long-term drug treatment for colon cancer [93]. The APPROVe (Adenomatous Polyp Prevention on VIOXX) study was stopped after 18 months since patients taking rofecoxib had twice the risk of a myocardial infarction compared with those receiving placebo. Increased incidence of thrombotic events (myocardial infarction, angina, stroke, transient ischemic events, etc.) was reported in previous clinical trials with Vioxx (VIGOR, with 8076 patients) and Celecoxib (CLASS, with 8059 patients); additionally, in 23,407 patients in primary prevention trials, the increased incidence of cardiovascular events was 0.74% and 0.80% with rofecoxib and celecoxib, respectively, compared to the normal population (0.52%) [93]. Since Ang-(1-7), an endogenous peptide hormone, causes a significant but not complete reduction in COX-2, treatment with the heptapeptide or drugs that elevate endogenous Ang-(1-7) may represent a novel mechanism to reduce COX-2 activity and inhibit lung cancer cell growth and tumor formation. Ang-(1-7) has anti-thrombotic properties, which will oppose any increase in thrombosis by reduced COX-2 activity. Ang-(1-7) caused a decrease in thrombus weight following vena cava occlusion as well as reduced collagen adhesion to platelets, in 2-kidney, 1-clip hypertensive rats [94]. Yoshida et al. [95] reported an increase in plasminogen activated inhibitor-1 (PAI-1) and tissue plasminogen activator in cultured human umbilical vein endothelial cells treated with Ang-(1-7). These results suggest that Ang-(1-7) may reduce COX-2 to inhibit lung cancer cell growth as well as provide anti-thrombotic protection against cardiovascular events due to decreased COX-2 in blood vessels.
ANG-(1-7) AND ANGIOGENESIS
Ang-(1-7) inhibited angiogenesis in a murine sponge model, a technique representative of the formation of new blood vessels from pre-existing blood vessels during wound healing [82]. In this model, a cannulated sponge disc was implanted subcutaneously in the dorsa of mice to induce a wound repair response. Infusion of Ang-(1-7) reduced hemoglobin content, blood flow, and proliferative activity in the disc, as compared to a disc-containing vehicle. The anti-angiogenic effect of the heptapeptide in this model was regulated by a [D-Ala7]-Ang-(1-7)-specific receptor and was blocked by pre-incubation with either aminoguanidine or NG-nitro-Z-arginine methyl ester (L-NAME), indicating that the response was mediated by nitric oxide [96]. While this is a model of angiogenesis during wound healing, it suggests that Ang-(1-7) may inhibit the angiogenesis that occurs during tumor formation, to supply necessary nutrients for tumor growth.
We showed that subcutaneous injection of Ang-(1-7) not only caused a significant reduction in human A549 lung tumor xenograft growth in mice but also markedly decreased vessel density [58], suggesting that the heptapeptide inhibits angiogenesis to reduce tumor size. VEGF-A, a primary angiogenic factor, was reduced approximately 85% in lung tumor xenografts from mice medicated with Ang-(1-7) as compared to tumors from saline-treated animals. An associated decrease in VEGF-A mRNA suggested that a transcriptional regulatory mechanism was involved in the reduction of angiogenic factor by the heptapeptide. Similar results were obtained with the parent A549 human lung cancer cells in culture with a maximal decrease in VEGF-A after 12 h treatment with Ang-(1-7). Antagonists specific for the Ang-(1-7) receptor completely blocked the reduction in VEGF-A by the heptapeptide. We previously showed that intravenous infusion of 24 μg/kg/h of Ang-(1-7) for 28 days markedly attenuated human A549 lung cancer xenografts [57]. As shown in Fig. (3), VEGF-A was also reduced in the tumors from animals administered Ang-(1-7), suggesting that the decrease in the angiogenic factor by the heptapeptide was not affected by different drug scheduling modalities. Taken together, these results suggest that Ang-(1-7) attenuates tumor angiogenesis by reducing VEGF-A.
Additional in vitro and in vivo models of angiogenesis were used to assess the anti-angiogenic properties of Ang-(1-7).[58] A decrease in human endothelial cell tubule formation in Matrigel was observed following a 16 h incubation with Ang-(1-7), with a maximal reduction at a 10 nmol/L concentration. The Ang-(1-7)-mediated effects were blocked by the specific Ang-(1-7) receptor antagonist [D-proline(7)]-Ang-(1-7). Similar results were obtained using human EA.hy.926 cells, a transformed human endothelial cell line derived from umbilical vein endothelial cells.[97] EA.hy.926 cells retain their endothelial phenotype and were previously used to study angiogenesis.[98,99] The cells were seeded onto Matrigel with or without 10 nM Ang-(1-7) and after 16 h, the EA.hy.926 cells were photographed to visualize and quantify tubule formation. As shown in Panel A of Fig. (4), EA.hy.926 cells formed multiple tube-like structures on Matrigel. In contrast, EA.hy.926 cells seeded in the presence of Ang-(1-7) formed fewer tube-like structures and the length of the tubes decreased compared to those in cells cultured in the absence of the heptapeptide. Tube formation was reduced significantly in the presence of increasing concentrations of Ang-(1-7) (Fig. 4, Panel B), suggesting that the heptapeptide inhibits angiogenesis in a dose-dependent manner. The chorioallantoic membrane (CAM) assay also was used to assess the effect of Ang-(1-7) on vascularization in vivo.[58] Control embryos displayed extensive neovascularization, while a marked reduction in vessel formation and branching was observed following 2-day incubation with 100 nM Ang-(1-7). These effects were blocked completely by the specific Ang-(1-7) receptor antagonist [D-proline(7)]-Ang-(1-7), suggesting that the anti-angiogenic actions of the heptapeptide were mediated by an AT(1-7) receptor. Taken together, these preclinical studies suggest that Ang-(1-7) may be a first-in-class compound for the treatment of lung cancer, providing combination therapy as a selective COX-2 and angiogenic inhibitor, targeting a specific AT(1-7) receptor mas.
PERSPECTIVE
The studies discussed above suggest that the RAS not only may be an effective target for chemotherapeutic/chemopreventive intervention but also may be involved in lung carcinogenesis. Undoubtedly, controversies prevail and additional research is required. Since ACE inhibitors and ARBs are currently prescribed for hypertension control and the safety profile of these drugs is known, properly designed clinical trials with lung cancer patients assessing these two medications is certainly feasible. Of note, there are multiple formulations of ACE inhibitors and ARBs and their efficacy may differ depending on the drug structure chosen. It is unlikely that ACE inhibitors or ARBs will be a single drug treatment for lung cancer; however, these medications may provide synergistic effects to existing chemotherapies by reducing Ang II-mediated mitogenesis and angiogenesis as well as increasing the anti-proliferative and anti-angiogenic effects of Ang-(1-7). In addition, the tortuous structure of tumor blood vessels leads to vasoconstriction, thereby limiting drug delivery. Administration of either ACE inhibitors or ARBs could cause dilation of the tumor vessels, leading to improved overall drug delivery. The use of these two anti-hypertensive agents in combination with other chemotherapeutic agents is worthy of investigation.
Both arms of the RAS must be considered in the development of new therapeutic medications. Drugs that limit the synthesis or activity of Ang II will potentially decrease tumor growth by attenuating the mitogenic and angiogenic properties of the peptide. On the other hand, increased production of Ang-(1-7), administration of the heptapeptide, or activation of the mas receptor with synthetic agonists should inhibit tumor growth by initiating signaling pathways that reduce cell growth and new blood vessel formation. Since Ang-(1-7) counteracts the anti-proliferative effects of Ang II as well as other mitogens [31], drugs that activate the Ang-(1-7) arm of the RAS may be more effective. Based on our preclinical data discussed above, we initiated a Phase I clinical trial to examine the toxicity of Ang-(1-7) in patients with solid tumors. The Phase I trial was completed in 18 months and the results were published in Clinical Cancer Research [100]. The drug was well-tolerated with no hypertension, bleeding disorders, or drug-related deaths. Of the 15 evaluable patients treated with the heptapeptide, 4 patients displayed clinical benefit with an associated reduction in circulating placental growth factor, while the remainder of the patients did not. This is in agreement with our preclinical data demonstrating the anti-angiogenic effects of Ang-(1-7) [58]. These results suggest that Ang-(1-7), a drug that would increase endogenous synthesis of Ang-(1-7), or a synthetic agonist of the mas receptor may represent a novel cancer treatment. However, it is essential that the cardiovascular effects are considered when designing a chemotherapeutic drug targeting components of the RAS, as this pathway plays a critical role in blood pressure regulation, homeostasis and natriuresis.
Fig. (1).

The renin-angiotensin system.
Fig. (2).

Effect of ACE inhibition on angiotensin peptides.
Fig. (3).
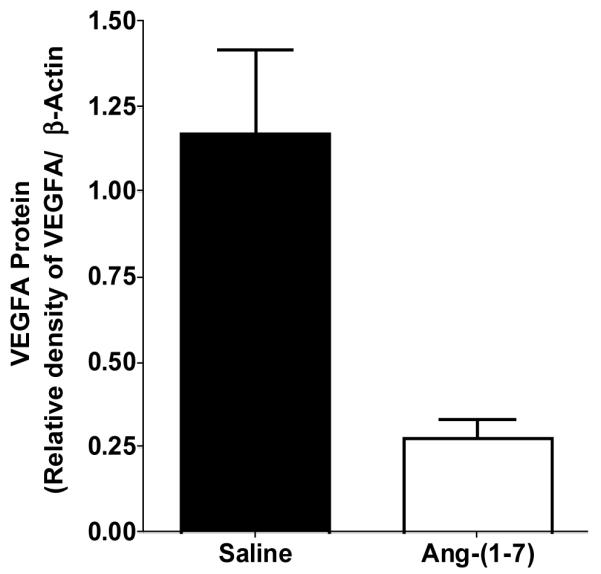
Reduction of VEGF-A in human A549 lung tumor xenografts infused with saline or Ang-(1-7). n = 3 for each group.
Fig. (4).

Ang-(1-7) inhibition of endothelial cell tubule formation. Panel A – A representative photograph of endothelial cells incubated with or without Ang-(1-7) on Matrigel. Panel B – Quantification of tubule number following 16 h treatment with various doses of Ang-(1-7). *p<0.05; n = 3-5.
Table 1.
Summary of Pre-Clinical Studies Supporting a Role for the RAS in Lung Cancer
| Source | Key Finding | Reference |
|---|---|---|
| Human NCI-H345, H209, H146, H82, H69, H187, H2122 and A549 |
Neprilysin is reduced in small cell and non-small cell lung cancer lines; transfection of the neprilysin gene resulted in reduced proliferation. |
[49] |
| Murine Lewis lung carcinoma cells | Ang II increased Lewis lung cell VEGF-A which was prevented by addition of an AT1 receptor antagonist CV11974. |
[52] |
| Human SK-LU-1, A549, SK-MES-1 lung cancer cells |
Ang-(l-7) caused a significant decrease in serum-stimulated growth of human lung cancer cells with an associated reduction in MAP kinase. |
[53] |
| Human A549 lung tumor cells | Ang II activation of the AT1 receptor increased cytosolic free calcium in human A549 lung cancer cells. | [54] |
| Ang II promoted apoptosis in lung cancer cells; treatment with ACE inhibitors or ARBs inhibited the response. |
[55,56] | |
| Human A549 lung tumor xenografts | Ang-(1-7) reduced COX-2 mRNA and protein in human A549 lung cancer cells. | [57] |
| Subcutaneous injection of Ang-(1-7) markedly decreased vessel density with a concomitant reduction in VEGF. |
[58] | |
| Murine Lewis lung carcinoma xenograft |
Batimastat and AT1 receptor blocker captopril reduced the mean volume and mean metastasis of Lewis lung tumors. |
[59] |
| Attenuated lung tumor growth and a decrease in VEGF-A was observed following ACE inhibitor candesartan administration. |
[52] | |
| Tumor xenografts were reduced following AT2 receptor antagonist treatment with an associated reduction in VEGF. |
[60 ] | |
| Lewis lung carcinoma metastasis was inhibited following candesartan or lisinopril treatment with a decrease in blood vessel formation. |
[61] | |
| Murine lung metastasis from renal cell carcinoma |
Cyclosporin-enhanced pulmonary metastases were reduced in mice following treatment with losartan. |
[62] |
| Candesartan inhibited the growth of pulmonary metastases with a decrease in VEGF and inhibition of angiogenesis. |
[63] | |
| Chemically-induced mouse lung tumor |
AT2 receptor-null mice had reduced tumor number and multiplicity following treatment with NNK. | [64] |
| AT2 receptor on lung fibroblasts may be involved in chemical carcinogen-induced lung tumorigenesis. |
[64] | |
| Immunostaining for the AT1 and AT2 receptors was enhanced in NNK-induced tumor sections. | [65] |
ACKNOWLEDGEMENTS
Funding was provided by NIH grant HL-51952 as well as support from the Farley-Hudson Foundation and the Wake Forest University Comprehensive Cancer Center. P. E. Gallagher and E. A. Tallant hold a patent for the treatment of cancer with Ang-(1-7).
ABBREVIATIONS
- RAS
renin-angiotensin system
- Ang
angiotensin
- ACE
angiotensin converting enzyme
- ACE2
angiotensin converting enzyme 2
- AT1
angiotensin type 1
- AT2
angiotensin type 2
- ARB
angiotensin receptor blocker
- [D-Ala7]-Ang-(1-7)
[D-alanine7]-angiotensin-(1-7)
- CACCA
common acute lymphoblastic leukemia antigen
- COX2
cyclooxygenase 2
- VEGF
vascular endothelial growth factor
- NNK
[4-methylnitosamino-1-(3-pyridyl)-1-butone]
- TGF-β
transforming growth factor-β
- APPROVe
adenomatous polyp prevention on VIOXX
- PAI-1
plasminogen activated inhibitor – 1
- L-NAME
NG-nitro-2-arginine methyl ester
REFERENCES
- [1].Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J. Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- [2].Dowell JE. Small cell lung cancer: Are we making progress? Am. J. Med. Sci. 2010;339:68–76. doi: 10.1097/MAJ.0b013e3181bccef5. [DOI] [PubMed] [Google Scholar]
- [3].Egleston BL, Meireles SI, Flieder DB, Clapper ML. Population-based trends in lung cancer incidence in women. Semin. Oncol. 2009;36:506–515. doi: 10.1053/j.seminoncol.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cokkinides V, Bandi P, McMahon C, Jemal A, Glynn T, Ward E. Tobacco control in the united states--recent progress and opportunities. CA Cancer J. Clin. 2009;59:352–365. doi: 10.3322/caac.20037. [DOI] [PubMed] [Google Scholar]
- [5].Tallant EA, Menon J, Soto-Pantoja DR, Gallagher PE. Angiotensin peptides and cancer. In: Kastin AJ, editor. The Handbook of Biologically Active Peptides. Elsevier; 2006. Chapter 66. [Google Scholar]
- [6].Varagic J, Trask AJ, Jessup JA, Chappell MC, Ferrario CM. New angiotensins. J. Mol. Med. 2008;86:663–671. doi: 10.1007/s00109-008-0340-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fyhrquist F, Saijonmaa O. Renin-angiotensin system revisited. J. Intern. Med. 2008;264:224–236. doi: 10.1111/j.1365-2796.2008.01981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Iwai M, Horiuchi M. Devil and angel in the renin-angiotensin system: ACE-angiotensin II-AT1 receptor axis vs. ACE2-angiotensin-(1-7)-mas receptor axis. Hypertens. Res. 2009;32:533–536. doi: 10.1038/hr.2009.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ferrario CM, Chappell MC, Tallant EA, Brosnihan KB, Diz DI. Counterregulatory actions of angiotensin-(1-7) Hypertension. 1997;30(part 2):535–541. doi: 10.1161/01.hyp.30.3.535. [DOI] [PubMed] [Google Scholar]
- [10].Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang K, Godbout K, Parsons T, Baronas E, Hsieh F, Acton S, Patane M, Nichols A, Tummino P. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002;277:14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- [11].Turner AJ, Tipnis SR, Guy JL, Rice GI, Hooper NM. ACEH/ACE2 is a novel mammalian metallocarboxypeptidase and a homologue of angiotensin-converting enzyme insensitive to ace inhibitors. Can. J. Physiol. Pharmacol. 2001;80:346–353. doi: 10.1139/y02-021. [DOI] [PubMed] [Google Scholar]
- [12].Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- [13].Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000;87:E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- [14].Ferrario CM, Jessup JA, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, Diz DI, Gallagher PE. Effect of angiotensin converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin converting enzyme 2. Circulation. 2005;111:2605–2610. doi: 10.1161/CIRCULATIONAHA.104.510461. [DOI] [PubMed] [Google Scholar]
- [15].Chappell MC, Pirro NT, Sykes A, Ferrario CM. Metabolism of angiotensin-(1-7) by angiotensin-converting enzyme. Hypertension. 1998;31(part 2):362–367. doi: 10.1161/01.hyp.31.1.362. [DOI] [PubMed] [Google Scholar]
- [16].Bumpus FM, Catt KJ, Chiu AT, DeGasparo M, Goodfriend T, Husain A, Peach MJ, Taylor DG, Jr., Timmermans PBMWM. Nomenclature for angiotensin receptors. a report of the nomenclature committee of the council for high blood pressure research. Hypertension. 1991;17:720–721. doi: 10.1161/01.hyp.17.5.720. [DOI] [PubMed] [Google Scholar]
- [17].De Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 2000;52:415–472. [PubMed] [Google Scholar]
- [18].Tallant EA, Ferrario CM. Biology of angiotensin II receptor inhibition with a focus on losartan: a new drug for the treatment of hypertension. Exp. Opin. Invest. Drugs. 1996;5:1201–1214. [Google Scholar]
- [19].Nakajima M, Hutchinson HG, Fujinaga M, Hayashida W, Morishita R, Zhang L, Horiuchi M, Pratt RE, Dzau VJ. The angiotensin II type 2 (AT2) receptor antagonizes the growth effects of the AT1 receptor: Gain-of-function study using gene transfer. Proc. Natl. Acad. Sci. USA. 1995;92:1-663–10667. doi: 10.1073/pnas.92.23.10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Timmermans PB, Smith RD. The diversified pharmacology of angiotensin II-receptor blockade. Blood Press Suppl. 1996;2:53–61. [PubMed] [Google Scholar]
- [21].Stoll M, Steckelings UM, Paul M, Bottari SP, Metzger R, Unger T. The angiotensin AT2-receptor mediates inhibition of cell proliferation in coronary endothelial cells. J. Clin. Invest. 1995;95:651–657. doi: 10.1172/JCI117710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wolf G, Harendza S, Schroeder R, Wenzel U, Zahner G, Butzmann U, Freeman RS, Stahl RA. Angiotensin II’s antiproliferative effects mediated through AT2-receptors depend on down-regulation of SM-20. Lab Invest. 2002;82:1305–1317. doi: 10.1097/01.lab.0000029207.92039.2f. [DOI] [PubMed] [Google Scholar]
- [23].Li H, Qi Y, Li C, Braseth LN, Gao Y, Shabashvili AE, Katovich MJ, Sumners C. Angiotensin type 2 receptor-mediated apoptosis of human prostate cancer cells. Mol. Cancer Ther. 2009;8:3255–3265. doi: 10.1158/1535-7163.MCT-09-0237. [DOI] [PubMed] [Google Scholar]
- [24].Jaiswal N, Jaiswal RK, Tallant EA, Diz DI, Ferrario CM. Alterations in prostaglandin production in spontaneously hypertensive rat smooth muscle cells. Hypertension. 1993;21:900–905. doi: 10.1161/01.hyp.21.6.900. [DOI] [PubMed] [Google Scholar]
- [25].Jaiswal N, Tallant EA, Jaiswal RK, Diz DI, Ferrario CM. Differential regulation of prostaglandin synthesis by angiotensin peptides in porcine aortic smooth muscle cells: subtypes of angiotensin receptors involved. J. Pharmacol. Exp. Ther. 1993;265:664–673. [PubMed] [Google Scholar]
- [26].Jaiswal N, Diz DI, Tallant EA, Khosla MC, Ferrario CM. Characterization of angiotensin receptors mediating prostaglandin synthesis in C6 glioma cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1991;260:R1000–R1006. doi: 10.1152/ajpregu.1991.260.5.R1000. [DOI] [PubMed] [Google Scholar]
- [27].Fontes MAP, Silva LCS, Campagnole-Santos MJ, Khosla MC, Guertzenstein PG, Santos RAS. Evidence that angiotensin-(1-7) plays a role in the central control of blood pressure at the ventro-lateral medulla acting through specific receptors. Brain Res. 1994;665:175–180. doi: 10.1016/0006-8993(94)91171-1. [DOI] [PubMed] [Google Scholar]
- [28].Santos RAS, Campagnole-Santos MJ, Baracho NCV, Fontes MAP, Silva LCS, Neves LAA, Oliveira DR, Caligiorne SM, Rodrigues ARV, Gropen C, Jr., Carvalho WS, Simoes E Silva AC, Khosla MC. Characterization of a new angiotensin antagonist selective for angiotensin-(1-7): Evidence that the actions of angiotensin-(1-7) are mediated by specific angiotensin receptors. Brain Res. Bull. 1994;35:293–298. doi: 10.1016/0361-9230(94)90104-x. [DOI] [PubMed] [Google Scholar]
- [29].Britto RR, Santos RAS, Fagundes-Moura CR, Khosla MC, Campangnole-Santos MJ. Role of angiotensin-(1-7) in modulation of the baroreflex in renovascular hypertensive rats. Hypertension. 1997;30:549–556. doi: 10.1161/01.hyp.30.3.549. [DOI] [PubMed] [Google Scholar]
- [30].Oliveira DR, Santos RAS, Santos GFP, Khosla MC, Campagnole-Santos MJ. Changes in the baroreflex control of heart rate rpoduced by central infusion of selective angiotensin antagonists in hypertensive rats. Hypertension. 1996:1284–1290. doi: 10.1161/01.hyp.27.6.1284. [DOI] [PubMed] [Google Scholar]
- [31].Freeman EJ, Chisolm GM, Ferrario CM, Tallant EA. Angiotensin-(1-7) inhibits vascular smooth muscle cell growth. Hypertension. 1996;28:104–108. doi: 10.1161/01.hyp.28.1.104. [DOI] [PubMed] [Google Scholar]
- [32].Tallant EA, Diz DI, Ferrario CM. Antiproliferative actions of angiotensin-(1-7) in vascular smooth muscle. Hypertension. 1999;34(part 2):950–957. doi: 10.1161/01.hyp.34.4.950. [DOI] [PubMed] [Google Scholar]
- [33].Santos RAS, Simoes E Silva AC, Maric C, Silva DMR, Machado RD, DuBuhr I, Heringer-Walther S, Pinheiro SV, Lopes MT, Bader M, Mendes EP, Lemos VS, Campagnole-Santos MJ, Schultheiss H-P, Speth R, Walther T. Angiotensin-(1-7) is an endogenous ligand for the g protein-coupled receptor mas. Proc. Natl. Acad. Sci. 2003;100:8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chappell MC, Diz DI, Gallagher PE. The renin-angiotensin system and the exocrine pancreas. J. Pancreas. 2001;2:33–39. [PubMed] [Google Scholar]
- [35].Allred AJ, Diz DI, Ferrario CM, Chappell MC. Pathways for angiotensin-(1-7) metabolism in pulmonary and renal tissues. Am. J. Physiol. Renal Physiol. 2000;279:F841–F850. doi: 10.1152/ajprenal.2000.279.5.F841. [DOI] [PubMed] [Google Scholar]
- [36].Ager EI, Neo J, Christophi C. The renin-angiotensin system and malignancy. Carcinogenesis. 2008;29:1675–1684. doi: 10.1093/carcin/bgn171. [DOI] [PubMed] [Google Scholar]
- [37].Rosenthal T, Gavras I. Angiotensin inhibition and malignancies: a review. J. Hum. Hypertens. 2009;23:623–635. doi: 10.1038/jhh.2009.21. [DOI] [PubMed] [Google Scholar]
- [38].Hanif K, Bid HK, Konwar R. Reinventing the ACE inhibitors: some old and new implications of ACE inhibition. Hypertens. Res. 2010;33:11–21. doi: 10.1038/hr.2009.184. [DOI] [PubMed] [Google Scholar]
- [39].Romer FK. Angiotensin-converting enzyme and its association with outcome in lung cancer. Br. J. Cancer. 1981;43:135–142. doi: 10.1038/bjc.1981.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mansfield CM, Kimler BF, Henderson SD, Vats TS, Svoboda DJ. Angiotensin-I-converting enzyme in cancer patients. J. Clin. Oncol. 1984;2:452–456. doi: 10.1200/JCO.1984.2.5.452. [DOI] [PubMed] [Google Scholar]
- [41].Schweisfurth H, Heinrich J, Brugger E, Steinl C, Maiwald L. The value of angiotensin-I-converting enzyme determinations in malignant and other diseases. Clin. Physiol Biochem. 1985;3:184–192. [PubMed] [Google Scholar]
- [42].Siefkin AD, Parsons GH, Patwell SW, Hollinger MA. The value of serial serum angiotensin converting enzyme determinations in hospitalized patients with lung disease. Am. J. Med. Sci. 1984;288:200–207. doi: 10.1097/00000441-198412000-00002. [DOI] [PubMed] [Google Scholar]
- [43].Roulston JE, Galloway PJ, Douglas G. Plasma angiotensin-converting enzyme activity in patients with bronchial carcinoma. Br. J. Dis. Chest. 1986;80:229–234. doi: 10.1016/0007-0971(86)90057-4. [DOI] [PubMed] [Google Scholar]
- [44].Varela AS, Bosco Lopez Saez JJ. Utility of serum activity of angiotensin-converting enzyme as a tumor marker. Oncology. 1993;50:430–435. doi: 10.1159/000227224. [DOI] [PubMed] [Google Scholar]
- [45].Nussinovitch N, Peleg E, Yaron A, Ratt P, Rosenthal T. Angiotensin converting enzyme in bleomycin-treated patients. Int. J. Clin. Pharmacol. Ther. Toxicol. 1988;26:310–313. [PubMed] [Google Scholar]
- [46].Prochazka J, Krepela E, Sedo A, Viklicky J, Fiala P. Aminopeptidases and angiotensin I-converting enzyme activities in primary human lung tumors and lung parenchyma. Neoplasma. 1991;38:501–508. [PubMed] [Google Scholar]
- [47].Dubrey SW, Falk RH. Diagnosis and management of cardiac sarcoidosis. Prog. Cardiovasc. Dis. 2010;52:336–346. doi: 10.1016/j.pcad.2009.11.010. [DOI] [PubMed] [Google Scholar]
- [48].Dusser DJ, Djokic TD, Borson DB, Nadel JA. Cigarette smoke induces bronchoconstrictor hyperresponsiveness to substance P and inactivates airway neutral endopeptidase in the guinea pig. possible role of free radicals. J. Clin. Invest. 1989;84:900–906. doi: 10.1172/JCI114251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Shipp MA, Tarr GE, Chen CY, Switzer SN, Hersh LB, Stein H, Sunday ME, Reinherz EL. CD10/Neutral endopeptidase 24.11 hydrolyzes bombesin-like peptides and regulates the growth of small cell carcinomas of the lung. Proc. Natl. Acad. Sci. U. S. A. 1991;88:10662–10666. doi: 10.1073/pnas.88.23.10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cohen AJ, Franklin WA, Magill C, Sorenson J, Miller YE. Low neutral endopeptiase levels in bronchoalveolar lavage fluid of lung cancer patients. Am. J. Respir. Crit. Care Med. 1999;159:907–910. doi: 10.1164/ajrccm.159.3.9806062. [DOI] [PubMed] [Google Scholar]
- [51].Bunn PA, Jr., Helfrich BA, Brenner DG, Chan DC, Dykes DJ, Cohen AJ, Miller YE. Effects of recombinant neutral endopeptidase (EC 3.4.24.11) on the growth of lung cancer cell lines in vitro and in vivo. Clin. Cancer Res. 1998;4:2849–2858. [PubMed] [Google Scholar]
- [52].Imai N, Hashimoto T, Kihara M, Yoshida S, Kawana I, Yazawa T, Kitamura H, Umemura S. Roles for host and tumor angiotensin II type 1 receptor in tumor growth and tumor-associated angiogenesis. Lab Invest. 2007;87:189–198. doi: 10.1038/labinvest.3700504. [DOI] [PubMed] [Google Scholar]
- [53].Gallagher PE, Tallant EA. Inhibition of lung cancer cell growth by angiotensin-(1-7) Carcinogenesis. 2004;25:2045–2052. doi: 10.1093/carcin/bgh236. [DOI] [PubMed] [Google Scholar]
- [54].Batra VK, Gopaladrishnan V, McNeill JR, Hickie RA. Angiotensin II elevates cytosolic free calcium in human lung adenocarcinoma cells via activation of AT1 receptors. Cancer Lett. 1994;76:19–24. doi: 10.1016/0304-3835(94)90129-5. [DOI] [PubMed] [Google Scholar]
- [55].Uhal BD, Gidea C, Bargout R, Bifero A, Ibarra-Sunga O, Papp M, Flynn K, Filippatos G. Captopril inhibits apoptosis in human lung epithelial cells: a potential antifibrotic mechanism. Am. J. Physiol. 1998;275:L1013–L1017. doi: 10.1152/ajplung.1998.275.5.L1013. [DOI] [PubMed] [Google Scholar]
- [56].Wang R, Zagariya A, Ang E, Ibarra-Sunga O, Uhal BD. Fas-induced apoptosis of alveolar epithelial cells requires ANG II generation and receptor internalization. Am. J. Physiol. Lung Cell Mol. Physiol. 1999;277:L1245–L1250. doi: 10.1152/ajplung.1999.277.6.L1245. [DOI] [PubMed] [Google Scholar]
- [57].Menon J, Soto-Pantoja DR, Callahan MF, Cline JM, Ferrario CM, Tallant EA, Gallagher PE. Angiotensin-(1-7) inhibits growth of human lung adenocarcinoma xenografts in nude mice through a reduction in cyclooxygenase-2. Cancer Res. 2007;67:2809–2815. doi: 10.1158/0008-5472.CAN-06-3614. [DOI] [PubMed] [Google Scholar]
- [58].Soto-Pantoja DR, Menon J, Gallagher PE, Tallant EA. Angiotensin-(1-7) inhibits tumor angiogenesis in human lung cancer xenografts with a reduction in vascular endothelial growth factor. Mol. Cancer Ther. 2009;8:1676–1683. doi: 10.1158/1535-7163.MCT-09-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Prontera C, Mariani B, Rossi C, Poggi A, Rotilio D. Inhibition of gelatinase A (MMP-2) b batimastat and captopril reduces tumor growth and lung metastases in mice bearing lewis lung carcinoma. Int. J. Cancer. 1999;81:761–766. doi: 10.1002/(sici)1097-0215(19990531)81:5<761::aid-ijc16>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- [60].Clere N, Corre I, Faure S, Guihot AL, Vessieres E, Chalopin M, Morel A, Coqueret O, Hein L, Delneste Y, Paris F, Henrion D. Deficiency or blockade of angiotensin II type 2 receptor delays tumorigenesis by inhibiting malignant cell proliferation and angiogenesis. Int. J. Cancer. 2010 doi: 10.1002/ijc.25234. [DOI] [PubMed] [Google Scholar]
- [61].Fujita M, Hayashi I, Yamashina S, Itoman M, Majima M. Blockade of angiotensin AT1a receptor signaling reduces tumor growth, angiogenesis, and metastasis. Biochem. Biophys. Res. Commun. 2002;294:441–447. doi: 10.1016/S0006-291X(02)00496-5. [DOI] [PubMed] [Google Scholar]
- [62].Maluccio M, Sharma V, Lagman M, Konijn G, Suthanthiran M. Angiotensin II receptor blockade: a novel strategy to prevent immunosuppressant-associated cancer progression. Transplant. Proc. 2001;33:1820–1821. doi: 10.1016/s0041-1345(00)02696-8. [DOI] [PubMed] [Google Scholar]
- [63].Miyajima A, Kosaka T, Asano T, Asano K, Seta K, Kawai T, Hayakawa M. Angiotensin II type I antagonist prevents pulmonary metastasis of murine renal cancer by inhibiting tumor angiogensis. Cancer Res. 2002;62:4176–4179. [PubMed] [Google Scholar]
- [64].Kanehira T, Tani T, Takagi T, Nakano Y, Howard EF, Tamura M. Angiotensin II type 2 receptor gene deficiency attenuates susceptibility to tobacco-specific nitrosamine-induced lung tumorigenesis: involvement of transforming growth factor-beta-dependent cell growth attenuation. Cancer Res. 2005;65:7660–7665. doi: 10.1158/0008-5472.CAN-05-0275. [DOI] [PubMed] [Google Scholar]
- [65].Tamura M, Yan H, Zegarra-Moro O, Edl J, Oursler S, Chard-Bergstrom C, Andrews G, Kanehira T, Takekoshi S, Mernaugh R. Specific single chain variable fragment (ScFv) antibodies to angiotensin II AT(2) receptor: Evaluation of the angiotensin II receptor expression in normal and tumor-bearing mouse lung. J. Mol. Histol. 2008;39:351–358. doi: 10.1007/s10735-008-9172-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Matsuzuka T, Miller K, Pickel L, Doi C, Ayuzawa R, Tamura M. The synergistic induction of cyclooxygenase-2 in lung fibroblasts by angiotensin II and pro-inflammatory cytokines. Mol. Cell Biochem. 2009;320:163–171. doi: 10.1007/s11010-008-9918-y. [DOI] [PubMed] [Google Scholar]
- [67].Ostman A, Augsten M. Cancer-associated fibroblasts and tumor growth--bystanders turning into key players. Curr. Opin. Genet. Dev. 2009;19:67–73. doi: 10.1016/j.gde.2009.01.003. [DOI] [PubMed] [Google Scholar]
- [68].Anton K, Glod J. Targeting the tumor stroma in cancer therapy. Curr. Pharm. Biotechnol. 2009;10:185–191. doi: 10.2174/138920109787315088. [DOI] [PubMed] [Google Scholar]
- [69].Haviv I, Polyak K, Qiu W, Hu M, Campbell I. Origin of carcinoma associated fibroblasts. Cell Cycle. 2009;8:589–595. doi: 10.4161/cc.8.4.7669. [DOI] [PubMed] [Google Scholar]
- [70].Papp M, Li X, Zhuang J, Wang R, Uhal BD. Angiotensin receptor subtype AT1 mediates alveolar epithelial cell apoptosis in response to ANG II. Am. J. Physiol. Lung Cell Mol. Physiol. 2002;282:L713–L718. doi: 10.1152/ajplung.00103.2001. [DOI] [PubMed] [Google Scholar]
- [71].Li X, Zhang H, Soledad-Conrad V, Zhuang J, Uhal BD. Bleomycin-induced apoptosis of alveolar epithelial cells requires angiotensin synthesis do novo. Am. J. Physiol. Cell Physiol. 2003;284:L501–L507. doi: 10.1152/ajplung.00273.2002. [DOI] [PubMed] [Google Scholar]
- [72].Attoub S, Gaben AM, Al-Salam S, Al Sultan MA, John A, Nicholls MG, Mester J, Petroianu G. Captopril as a potential inhibitor of lung tumor growth and metastasis. Ann. N. Y. Acad. Sci. 2008;1138:65–72. doi: 10.1196/annals.1414.011. [DOI] [PubMed] [Google Scholar]
- [73].Arrieta O, Guevara P, Escobar E, Garcia-Navarrete R, Pineda B, Sotelo J. Blockade of angiotensin II type 1 receptor decreases the synthesis of growth factors and induces apoptosis in C6 cultured cells and C6 rat glioma. Br. J. Cancer. 2005;92:1247–1255. doi: 10.1038/sj.bjc.6602483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Gong Q, Davis M, Chipitsyna G, Yeo CJ, Arafat HA. Blocking angiotensin II type 1 receptor triggers apoptotic cell death in human pancreatic cancer cells. Pancreas. 2010 doi: 10.1097/MPA.0b013e3181c314cd. [DOI] [PubMed] [Google Scholar]
- [75].Funao K, Matsuyama M, Kawahito Y, Sano H, Chargui J, Touraine JL, Nakatani T, Yoshimura R. Telmisartan is a potent target for prevention and treatment in human prostate cancer. Oncol. Rep. 2008;20:295–300. [PubMed] [Google Scholar]
- [76].De l. I., Lopez-Jorge CE, Gomez-Casares MT, Lemes CA, Martin CP, Lopez BJ, Suarez CA, Molero LT. Induction of apoptosis in leukemic cell lines treated with captopril, trandolapril and losartan: a new role in the treatment of leukaemia for these agents. Leuk. Res. 2009;33:810–816. doi: 10.1016/j.leukres.2008.09.029. [DOI] [PubMed] [Google Scholar]
- [77].Lever AF, Hole DJ, Gillis CR, McCallum IRMGT, MacKinnon PL, Meredith PA, Murray LS, Reid JL, Robertson MJ. Do inhibitors of angiotensin-I-converting enzyme protect against risk of cancer? The Lancet. 1998;352:179–184. doi: 10.1016/S0140-6736(98)03228-0. [DOI] [PubMed] [Google Scholar]
- [78].Ferrario CM, Jaiswal N, Yamamoto K, Diz DI, Schiavone MT. Hypertensive mechanisms and converting enzyme inhibitors. Clin. Cardiol. 1991;14(Suppl. IV):IV-56–IV-62. doi: 10.1002/clc.4960141809. [DOI] [PubMed] [Google Scholar]
- [79].Santos RAS, Brosnihan KB, Chappell MC, Resquero J, Chernicky CL, Greene LJ, Ferrario CM. Converting enzyme activity and angiotensin metabolism in the dog brainstem. Hypertension. 1988;11(Suppl I):I-153–I-157. doi: 10.1161/01.hyp.11.2_pt_2.i153. [DOI] [PubMed] [Google Scholar]
- [80].Wilop S, von HS, Crysandt M, Esser A, Osieka R, Jost E. Impact of angiotensin I converting enzyme inhibitors and angiotensin II type 1 receptor blockers on survival in patients with advanced non-small-cell lung cancer undergoing first-line platinum-based chemotherapy. J. Cancer Res. Clin. Oncol. 2009;135:1429–1435. doi: 10.1007/s00432-009-0587-3. [DOI] [PubMed] [Google Scholar]
- [81].Nadal JA, Scicli GM, Carbini LA, Scicli G. Angiotensin II stimulates migration of retinal microvascular pericytes: Involvement of TGF-b and PDGF-BB. Am. J. Physiol. Heart Circ. Physiol. 2002;282:H738–H748. doi: 10.1152/ajpheart.00656.2001. [DOI] [PubMed] [Google Scholar]
- [82].Machado RD, Santos RA, Andrade SP. Opposing actions of angiotensins on angiogenesis. Life Sci. 2000;66:67–76. doi: 10.1016/s0024-3205(99)00562-7. [DOI] [PubMed] [Google Scholar]
- [83].Tamarat R, Silvestre JS, Durie M, Levy BI. Angiotensin II angiogenic effect in vivo involves vascular endothelial growth factor- and inflammation-related pathways. Lab Invest. 2002;82:747–756. doi: 10.1097/01.lab.0000017372.76297.eb. [DOI] [PubMed] [Google Scholar]
- [84].Otani A, Takagi H, Oh H, Suzuma K, Matsumura M, Ikeda E, Honda Y. Angiotensin II-stimulated vascular endothelial growth factor expression in bovine retinal pericytes. Invest Ophthalmol. Vis. Sci. 2000;41:1192–1199. [PubMed] [Google Scholar]
- [85].Pickel L, Matsuzuka T, Doi C, Ayuzawa R, Maurya DK, Xie SX, Berkland C, Tamura M. Overexpression of angiotensin II type 2 receptor gene induces cell death in lung adenocarcinoma cells. Cancer Biol. Ther. 2010;9 doi: 10.4161/cbt.9.4.10643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Brown JR, Dubois RN. Cycloxygenase-2 in lung carcinogenesis and chemoprevention. Chest. 2004;125:134S–140S. doi: 10.1378/chest.125.5_suppl.134s-a. [DOI] [PubMed] [Google Scholar]
- [87].Castelao JE, Bart RD, III, DiPerna CA, Sievers EM, Bremmer RM. Lung cancer and cyclooxygenase-2. Ann. Thorac. Surg. 2003;76:1327–1335. doi: 10.1016/s0003-4975(03)00334-5. [DOI] [PubMed] [Google Scholar]
- [88].Wang D, Dubois RN. Prostaglandins and cancer. Gut. 2006;55:115–122. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].van Zandwijk N. Chemoprevention in lung carcinogenesis--an overview. Eur. J. Cancer. 2005;41:1990–2002. doi: 10.1016/j.ejca.2005.05.011. [DOI] [PubMed] [Google Scholar]
- [90].Rioux N, Castonguay A. Prevention of NNK-induced lung umorigenesis in A/J Mice by acetylsalicylic acid and NS-398. Cancer Res. 1998;58:5354–5360. [PubMed] [Google Scholar]
- [91].Moody TW, Leyton J, Zakowicz H, Hida T, Kang Y, Jakowlew S, You L, Ozbun L, Zia H, Yougberg J, Malkinson A. Indomethacin reduces lung adenoma number in A/J mice. Anticancer Res. 2001;21:1749–1755. [PubMed] [Google Scholar]
- [92].Schreinemachers DM, Everson RB. Aspirin Use and lung, colon, and breast cancer incidence in a prospective study. Epidemiology. 1994;5:138–146. doi: 10.1097/00001648-199403000-00003. [DOI] [PubMed] [Google Scholar]
- [93].Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated wih selective COX-2 inhibitors. JAMA. 2001;286:954–959. doi: 10.1001/jama.286.8.954. [DOI] [PubMed] [Google Scholar]
- [94].Kucharewicz I, Chabielska E, Pawlak D, Matys T, Rolkowski R, Buczko W. The Antithrombotic effect of angiotensin-(1-7) closely resembles that of losartan. J. Renin. Angiotensin. Aldosterone. Syst. 2000;1:268–272. doi: 10.3317/jraas.2000.041. [DOI] [PubMed] [Google Scholar]
- [95].Yoshida M, Naito Y, Urano T, Takada A, Takada Y. L-158,809 and (D-Ala(7))-Angiotensin I/II (1-7) decrease PAI-1 release from human umbilical vein endothelial cells. Thromb. Res. 2002;105:531–536. doi: 10.1016/s0049-3848(02)00056-7. [DOI] [PubMed] [Google Scholar]
- [96].Machado RDP, Santos RAS, Andrade SP. Mechanisms of angiotensin-(1-7)-induced inhibition of angiogenesis. Am. J. Physiol. Regulatory Integrative Comp. Physiol. 2001;280:994–1000. doi: 10.1152/ajpregu.2001.280.4.R994. [DOI] [PubMed] [Google Scholar]
- [97].Edgell CJ, McDonald CC, Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc. Natl. Acad. Sci. U. S. A. 1983;80:3734–3747. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Bauer J, Margolis M, Schriner C, Edgell CJ, Azizkhan J, Laxarowski E, Juliano RL. In vitro model of angiogenesis using a human endothelium-derived permanent cell line: Contributions of induced gene expression, g-proteins, and integrins. J. Cell Physiol. 1992;153:437–449. doi: 10.1002/jcp.1041530302. [DOI] [PubMed] [Google Scholar]
- [99].Schonherr E, Schafer L, O’Connell BC, Kresse H. Matrix Metalloproteinase expression by endothelial cells in collagen lattices changes during co-culture with fibroblasts and upon induction of decorin expression. J. Cell Physiol. 2001;187:37–47. doi: 10.1002/1097-4652(2001)9999:9999<::AID-JCP1048>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- [100].Petty WJ, Miller AA, McCoy TP, Gallagher PE, Tallant EA, Torti FM. Phase I and pharmacokinetic study of angiotensin-(1-7), an endogenous antiangiogenic hormone. Clin. Cancer Res. 2009;15:7398–7404. doi: 10.1158/1078-0432.CCR-09-1957. [DOI] [PMC free article] [PubMed] [Google Scholar]