

Abstract
Telomerase reactivation and expression of human telomerase gene [human telomerase reverse transcriptase (hTERT)] are hallmarks of unlimited proliferation potential of cancer cells. A polymorphic tandem repeats minisatellite of hTERT gene, termed MNS16A was reported to influence hTERT expression. To assess the role of MNS16A as potential biomarker for colorectal cancer (CRC), we investigated for the first time the association of MNS16A genotypes with risk of colorectal polyps and CRC. In the ongoing colorectal cancer study of Austria (CORSA), 3842 Caucasian participants were recruited within a large screening project in the province Burgenland including 90 CRC cases, 308 high-risk polyps, 1022 low-risk polyps and 1822 polyp free controls verified by colonoscopy. MNS16A genotypes were determined by polymerase chain reaction from genomic DNA. Associations of MNS16A genotypes with CRC risk were estimated by logistic regression analysis computing odds ratios (ORs) and 95% confidence intervals (CIs). We identified five different variable number of tandem repeats (VNTRs) of MNS16A including VNTR-364, a newly discovered rare variant. VNTR-274 allele was associated with a 2.7-fold significantly increased risk of CRC compared with the VNTR-302 wild-type (OR = 2.69; 95% CI = 1.11–6.50; P = 0.028). In our CORSA study, the medium length VNTR-274 was identified as risk factor for CRC. Although, this population-based study herewith reports the largest cohort size concerning MNS16A thus far, further large-scale studies in diverse populations are warranted to confirm hTERT MNS16A genotype as potential biomarker for assessment of CRC risk.
Introduction
Telomeres are highly conserved functional structures at the distal parts of eukaryotic chromosomes protecting them from degradation and end-to-end fusion (1). Telomerase gene [human telomerase reverse transcriptase (hTERT)], located on chromosome band 5p15.33 (2) encodes a ribonucleoprotein enzyme that extends chromosome ends, which have been shortened during successive cycles of cell division (3). Telomerase holoenzyme consists of a catalytic subunit (hTERT), a functional RNA subunit serving as template for telomeric DNA synthesis (human telomerase RNA), other telomerase components and several telomerase-associated proteins (e.g. reviewed in ref. 4). Telomerase activity is usually absent in somatic human cells but present in >90% of cancer cells and in vitro immortalized cells (5,6) underlining the importance of telomerase in tumorigenesis and cellular ageing.
Several studies correlated length of telomeres with hTERT expression in colorectal cancer (CRC) (7–14) and genome-wide association studies recently associated TERT-CLPTM1L locus with several cancer types including lung, urinary bladder, cervix and prostate cancer (15).
In 2003, Wang et al. (16) first reported in a lung cancer study the identification of a polymorphic tandem repeats minisatellite of hTERT termed MNS16A. This minisatellite is located down-stream of exon 16 of hTERT gene and up-stream in the putative promoter region of an antisense hTERT transcript. Detection of antisense messenger RNA of hTERT suggested a possible role in regulation of human telomerase expression. MNS16A was found to have two repeat elements forming a 23 bp core sequence or a 26 bp core sequence with a CAT insertion representing a transcription factor binding site for GATA-1. Two different variable number of tandem repeats (VNTRs) alleles, VNTR-302 and VNTR-243 were named on basis of their polymerase chain reaction (PCR) fragment size. In addition, two other rare alleles (VNTR-272 and VNTR-333) were discovered in cancer cell lines. For statistical analysis, a classification in short alleles (S alleles: VNTR-243 and VNTR-272) and long alleles (L alleles: VNTR-302 and VNTR-333) was introduced by Wang et al. (16). Promoter activity was demonstrated to depend on the number of tandem repeats implicating functionality of MNS16A genotype. It was proposed that this minisatellite might act as a repressor for the promoter of the hTERT antisense transcript and an influence of MNS16A on lung cancer susceptibility was suggested. Following studies investigated MNS16A as potential biomarker for glioblastoma (17–19), breast (20) and lung cancer (21,22) but with contradictory findings on clinical relevance regarding both risk and survival time.
In Austria, the incidence of CRC is in the top third within the European Union and CRC is the second most common cause of cancer-related death in women and men nationwide (GLOBOCAN 2008, http://globocan.iarc.fr/, accessed 30 September 2010). There is a remarkable decline in CRC incidence rates from east to west in Austria with the highest rates in the province Burgenland (23).
Our ongoing colorectal cancer study of Austria (CORSA) is nested within a large screening project covering the whole province Burgenland. This study comprised 3842 participants and is herewith the largest population concerning MNS16A thus far. Furthermore, in our CORSA study, we investigated MNS16A in the context of CRC for the first time. The purpose of this study was to examine the association of this polymorphic tandem repeats minisatellite with risk of colorectal polyps and CRC as potential biomarker for the identification of high-risk individuals to improve CRC screening.
Materials and methods
Study population
In the ongoing molecular epidemiology, CORSA 3842 Caucasian participants were recruited since May 2002 within a large province-wide screening project in the province Burgenland, Austria. This screening project ‘Burgenland against CRC’ uses faecal occult blood tests (FOBT) and all habitants of Burgenland aged between 40 and 80 years are invited to take part in the screening programme. Participants with a positive faecal occult blood testing receive further diagnostic workup such as colonoscopies. These persons are asked to participate in our ‘Molecular epidemiology study of CRC’. Results of colonoscopies are collected in a central database and standardized documentation guidelines are followed. Demographic and anthropometric factors as well as dietary and smoking habits are assessed by a short questionnaire. All subjects gave written informed consent. The study was approved by the institutional review board ‘Ethikkommission Burgenland’.
Between 2003 and 2008, the target group of the screening programme consisted of 698 951 female and male habitants aged between 40 and 80 years and a total of 33.6% (234 808) participated in the programme. 20 968 individuals (8.9%) were tested FOBT positive and 13 630 colonoscopies were performed. Within this time interval, 347 cases of CRC 1648 high-risk polyps, 2014 low-risk polyps and 816 hyperplastic polyps were diagnosed.
All participants (100%) in this study had positive FOBT and underwent complete colonoscopy. CRC cases were patients with histologically confirmed, previously untreated CRC, firstly diagnosed within this screening programme (n = 90). For statistical analysis, the polyp group consisting of 1330 patients was classified by their histopathology as high-risk (n = 308) and low-risk (n = 1022) polyps according to the amount of villous elements. Adenomatous villous, adenomatous tubulovillous and co-occurrence of adenomatous tubular with tubulovillous polyps were classified as high-risk polyps, whereas hyperplastic polyps which have no malignant potential and adenomatous tubular polyps were assigned to the low-risk group. The control group comprised 1822 individuals with positive FOBT who underwent colonoscopy but exhibited no pathological findings. Individuals with serious medical conditions at baseline including all malignant diseases were excluded from the study.
DNA purification and genotyping of MNS16A
Genomic DNA was purified from peripheral blood according to the QIAamp® DNA Blood Midi Spin Protocol (QIAGEN, Valencia, CA). Twenty microlitre PCR reactions contained 40 ng genomic DNA, GeneAmp 10× PCR Buffer II, MgCl2 Solution (1.5 mM MgCl2), deoxyribonucleotide Mix (150 nM dNTPs), AmpliTaq DNA Polymerase (0.3 U) (Applied Biosystems, Foster City, CA) and 350 nM forward and reverse primer (VBC-BIOTECH Service, Vienna, Austria). MNS16A primers were used as described by Wang et al. (16) PCR products were amplified on a 2720 Thermal Cycler (Applied Biosystems) by the following thermal profile: 95°C (5 min); 35 cycles of 95°C (30 s), 65°C (30 s), 72°C (30 s) and 72°C (10 min). MNS16A genotypes were determined by electrophoretical separation of PCR products in ethidium bromide stained 2.5% agarose gels. Genotyping was performed blinded to case–control status and 15% of randomly chosen samples were re-genotyped for quality control.
Several PCR products were also loaded on 5% non-denaturing polyacrylamide gels and electrophoretical separation was performed at constant 80 V (50 min). For detection, polyacrylamide gels were stained in a 1:10 000 dilution of Vistra Green™ Nucleic Acid Stain (Amersham Biosciences, Piscataway, NJ).
Cloning of MNS16A PCR products and allele confirmation by sequencing
For cloning of size-selected MNS16A PCR products, the TOPO TA Cloning® Kit containing pCR®II-TOPO® vector was applied as described by the manufacturer (Invitrogen, Carlsbad, CA). Plasmid DNA purification of positive clones was performed with Wizard® Plus SV Minipreps DNA Purification System (Promega, Madison, WI). For verification of insert sequence of different VNTRs a sequencing service was engaged (QIAGEN GmbH Genomic Services, Hilden, Germany). Sequence analysis was carried out with Clone Manager Professional version 9.0 (Scientific and Educational Software, Cary, NC, 1994–2007).
Statistical analysis
Genotypic counts for each patient group (CRC, high-risk polyp, low-risk polyp and control) were tested for Hardy–Weinberg equilibrium (HWE) using χ2-tests. Due to low cell counts caused by low allele frequencies, P values for the χ2 tests were simulated using a simulation/permutation method implemented in the statistical programming language R (function HWE.test, package genetics). Exact significance tests are not available for situations where more than two alleles occur.
Associations between MNS16A genotypes and CRC risk were estimated by computing odds ratios (ORs) and 95% confidence intervals (CIs) by logistic regression analysis with adjustment for age, sex, smoking (current, former and never) and education. MNS16A genotypes were included as factors with the most common genotype as reference category. Calculations were performed for single alleles, genotypes and SL converted system. For the SL classification system, VNTR-243 and VNTR-274 were regarded as short (S) and VNTR-302, VNTR-333 and VNTR-364 as long (L) variant alleles. All tests were two-sided and P values <0.05 were considered statistically significant. All statistical analyses were performed with R Version 2.11.1 (R Development Core Team, 2010).
Results
Study population
Characteristics of the study population are given in Table I. A total of 3803 participants of the CORSA study was analysed, consisting of 90 patients with CRC, 308 patients with high-risk polyps, 1022 patients with low-risk polyps and 1822 colonoscopy-negative controls. Reduction of sample number eligible to statistical analysis was due to incomplete histology or confounding data. The mean age of the CRC case group (66.8 years) was significantly higher than the mean age of the control group (61.3 years; F-test, P < 0.0001). The study population consisted of 45.0% females and 55.0% males. Male subjects were overrepresented in all three case groups (64.4% of CRC, 67.0% of high-risk and 65.2% of low-risk polyps; χ2-test, P < 0.0001). In total, 3.3% of men had developed CRC (2.2% of women), 11.6% high-risk and 37.5% low-risk polyps compared with 7.0 and 24.5% of females, respectively.
Table I.
Selected characteristics of the study population
| CRCa | High-risk polypb | Low-risk polypc | Controld | |
| N (%) | N (%) | N (%) | N (%) | |
| Age (years) | ||||
| ≤50 | 8 (1.5) | 45 (8.3) | 135 (24.9) | 354 (65.3) |
| 51–60 | 18 (2.4) | 63 (8.4) | 238 (31.7) | 433 (57.6) |
| 61–70 | 24 (2.2) | 115 (10.6) | 372 (34.2) | 576 (53.0) |
| 71–80 | 34 (4.3) | 80 (10.2) | 258 (32.8) | 414 (52.7) |
| ≥80 | 6 (14.3) | 2 (4.8) | 14 (33.3) | 20 (47.6) |
| Total | 90 (100) | 305 (100) | 1017 (100) | 1797 (100) |
| Mean (95% CI) | 66.8 (45.8–87.7) | 62.9 (43.2–82.7) | 63.2 (43.2–83.2) | 61.3 (39.4–83.3) |
| Sex | ||||
| Male | 58 (3.3) | 205 (11.6) | 663 (37.5) | 841 (47.6) |
| Female | 32 (2.2) | 101 (7.0) | 354 (24.5) | 957 (66.3) |
| Total | 90 (100) | 306 (100) | 1017 (100) | 1798 (100) |
| Education | ||||
| Elementary | 52 (3.2) | 155 (9.5) | 552 (33.7) | 879 (53.7) |
| Secondary modern | 28 (2.3) | 120 (9.9) | 359 (29.6) | 707 (58.2) |
| Matura | 5 (2.5) | 13 (6.6) | 56 (28.4) | 123 (62.4) |
| University | 3 (6.5) | 5 (10.9) | 15 (32.6) | 23 (50.0) |
| Total | 88 (100) | 293 (100) | 982 (100) | 1732 (100) |
| Smoking | ||||
| Current | 13 (2.6) | 56 (11.0) | 198 (39.0) | 241 (47.4) |
| Former | 37 (4.0) | 93 (10.1) | 310 (33.6) | 483 (52.3) |
| Never | 40 (2.3) | 148 (8.7) | 489 (28.7) | 1029 (60.3) |
| Total | 90 (100) | 297 (100) | 997 (100) | 1753 (100) |
CRC cases.
Adenomatous villous, tubulovillous and villous + tubulovillous polyps.
Adenomatous tubular and hyperplastic polyps.
Polyp-free controls.
Sequencing of MNS16A VNTRs
Sequencing of cloned MNS16A PCR products yielded in the identification of five individual MNS16A variant sequences displayed in Figure 1. With increasing fragment length, alleles gain one additional tandem repeat. The MNS16A VNTRs-243, -274, -302, -333 and -364 contain 3, 4, 5, 6 and 7 tandem repeats, respectively, with 2, 3, 3, 4 and 5 CAT trinucleotide insertions, respectively. The newly identified MNS16A VNTR-364 represents a ‘333 allele’ with an additional 31 bp insert of one tandem repeat carrying an extra (fifth) CAT trinucleotide. VNTR-364 thereby carries the highest number of GATA-1 transcription factor binding sites. Regarding VNTR-243 and VNTR-302, we found minute differences from the core sequence published by Wang et al. (16) apparent in two bases (indicated by grey highlighting, Figure 1). Through checking the sequencing chromatograms, we can exclude sequencing errors. This difference was found in all five fragments we sequenced coming from five different individuals. Thus, the insertion site of additional tandem repeats can be tracked easily.
Fig. 1.
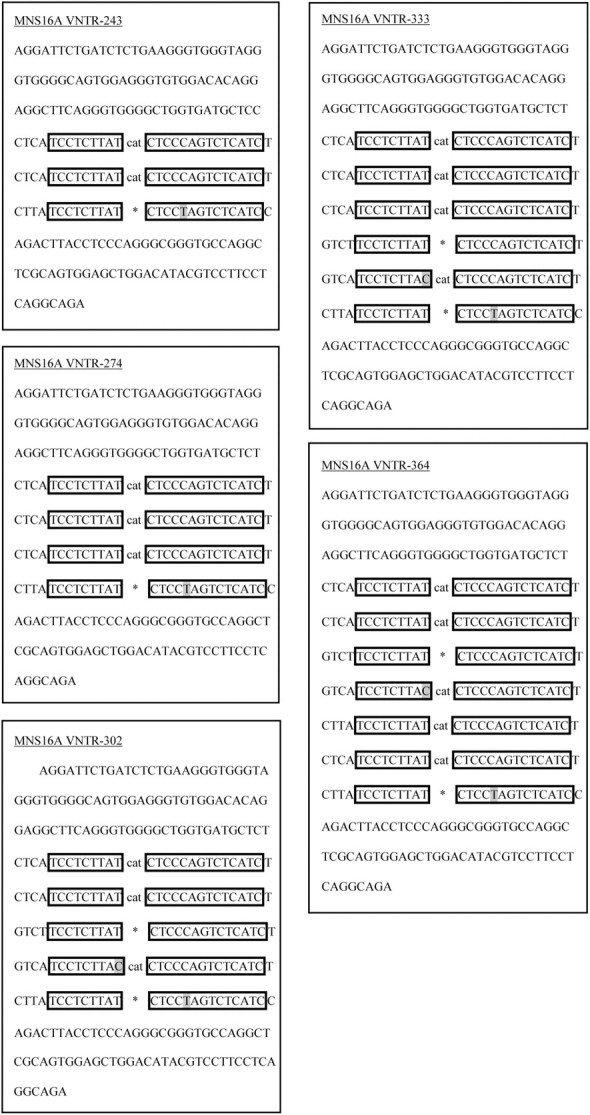
Sequences of hTERT MNS16A VNTRs. Alleles were named on basis of the amplicon length. CAT indicates insert of CAT trinucleotide between repeat elements, whereas * represents no CAT insert.
A BLAST search for the MNS16A VNTR-302 using Clone Manager led to a 100% match with hTERT sequence (NT_006576.16; accessed 02 March 2010). hTERT gene sequence including adjacent 3′ sequence was obtained from ‘nucleotide’ search and alignment in Clone Manager resulted in 100% match of VNTR-302 molecule with hTERT sequence (NG_009265.1; www.ncbi.nlm.nih.gov). The congruent region of NG_009265.1 with VNTR-302 was found between bases 47512-47813.
MNS16A genotyping
Three thousand eight hundred and forty-two individuals were genotyped for MNS16A allelotype by electrophoretical separation of PCR products. Genotyping was successful in 99.1% of all attempts. Fifteen per cent of randomly chosen samples were re-genotyped for quality control and results were in 100% accordance with previous findings. In total, five different MNS16A variants were detected emerging in ten different genotype patterns (Figure 2).
Fig. 2.

Genotype patterns of hTERT MNS16A. Five identified VNTRs were detected in 10 allele combinations named MNS16A genotype patterns 1–10. For SL classification, VNTR-243 and VNTR-274 were considered as short (S) and VNTR-302, VNTR-333 and VNTR-364 as long (L) variant alleles.
Heterozygotes of MNS16A frequently exhibited a weak third band above the two main band signals in agarose gels. When loaded on polyacrylamide gels, only heterozygous samples showed frequently a third band at ∼700 bp (data not shown). Also, a mixture of two homozygous samples previously found to exhibit only one distinct band was used as template for PCR, leading to the same additional band in agarose gels supporting the findings of conformational changes of PCR products only being responsible for the additional band signal (20). Furthermore, we demonstrated the existence of VNTR-333 by successful cloning and sequencing.
All genotyped samples with valid histology data were tested for HWE. P values of χ2-tests for each patient group were 0.980 (control), 0.943 (CRC), 0.884 (high-risk polyp) and 0.981 (low-risk polyp). The frequency distribution of genotypes of this study population was 42.36% for MNS16A genotype pattern 1, 10.07% for 2, 40.55% for 3, 2.50% for 4, 0.95% for 5, 2.47% for 6, 1.03% for 7, 0.03% for 8, 0.03% for 9 and 0.03% for 10 referring to the MNS16A genotype patterns stated in Figure 2. Genotypes 333/333, 302/364 and 274/333 are very rare variants and were found only once each. The variants 302/302 (42.36%) and 243/302 (40.55%) were similarly represented within the study population followed by the homozygous variant 243/243 (10.07%). The three rare variants, represented by only one individual each were excluded from further models. Genotype distribution by histology is given in Table II identifying the highest percentages of CRC for the MNS16A genotypes 243/274 (6.45%), 274/302 (5.56%) and 243/333 (5.71%). Remarkably, all 274 alleles eligible for statistical models were contained within this group of MNS16A genotypes with the highest amount of CRC.
Table II.
Genotype distributions of hTERT MNS16A in CRC, high-risk polyp, low-risk polyp and control group
| Genotypea | SL classification | CRC | High-risk polyp | Low-risk polyp | Control |
| N (%) | N (%) | N (%) | N (%) | ||
| 302/302 | LL | 35 (2.68) | 127 (9.72) | 411 (31.45) | 734 (56.16) |
| 243/243 | SS | 6 (1.97) | 22 (7.21) | 97 (31.80) | 180 (59.02) |
| 243/302 | SL | 38 (3.06) | 116 (9.35) | 395 (31.85) | 691 (55.73) |
| 302/333 | LL | 1 (1.47) | 11 (16.18) | 20 (29.41) | 36 (52.94) |
| 243/274 | SS | 2 (6.45) | 1 (3.23) | 13 (41.94) | 15 (48.39) |
| 274/302 | SL | 4 (5.56) | 8 (11.11) | 20 (27.78) | 40 (55.56) |
| 243/333 | SL | 2 (5.71) | 3 (8.57) | 14 (40.00) | 16 (45.71) |
Three very rare genotypes (333/333, 302/364 and 274/333) were excluded from statistical models.
Logistic regression models for the seven considered MNS16A genotypes (allele combinations) could not identify a significantly increased risk for distinct genotypes (Table III). Moreover, calculation of pooled genotype groups (SS, SL and LL groups) as applied in literature could not detect significant differences in genotype distribution of case and control groups.
Table III.
Association of hTERT MNS16A genotypes and CRC risk
| MNS16A | CRC |
CRC + high-risk polyp |
CRC + high-risk + low-risk polyp |
|||||||
| Controls | Cases |
Cases |
Cases |
|||||||
| Alleles | N = 3424, N (%) | N = 176, N (%) | OR (95% CI)a | P value | N = 752, N (%) | OR (95% CI)a | P value | N = 2692, N (%) | OR (95% CI)a | P value |
| 302 | 2235 (65.27) | 113 (64.20) | 1.00 | 502 (66.76) | 1.00 | 1759 (65.34) | ||||
| 243 | 1082 (31.60) | 54 (30.68) | 0.96 (0.69–1.35) | 0.825 | 218 (28.99) | 0.88 (0.74–1.05) | 0.160 | 834 (30.98) | 0.96 (0.86–1.08) | 0.510 |
| 274 | 55 (1.61) | 6 (3.41) | 2.69 (1.11–6.50) | 0.028 | 15 (1.99) | 1.29 (0.71–2.33) | 0.400 | 48 (1.78) | 1.13 (0.76–1.69) | 0.546 |
| 333 | 52 (1.52) | 3 (1.70) | 1.33 (0.40–4.45) | 0.640 | 17 (2.26) | 1.62 (0.92–2.87) | 0.098 | 51 (1.89) | 1.31 (0.88–1.96) | 0.185 |
| Genotypes | N = 1712, N (%) | N = 88, N (%) | N = 376, N (%) | N = 1346, N (%) | ||||||
| 302/302 | 734 (42.87) | 35 (39.77) | 1.00 | 162 (43.09) | 1.00 | 573 (42.57) | 1.00 | |||
| 243/243 | 180 (10.51) | 6 (6.82) | 0.68 (0.28–1.66) | 0.394 | 28 (7.45) | 0.68 (0.44–1.06) | 0.090 | 125 (9.29) | 0.85 (0.66–1.11) | 0.236 |
| 243/302 | 691 (40.36) | 38 (43.18) | 1.09 (0.68–1.77) | 0.713 | 154 (40.96) | 0.98 (0.76–1.25) | 0.846 | 549 (40.79) | 1.00 (0.85–1.17) | 0.968 |
| 302/333 | 36 (2.10) | 1 (1.14) | 0.66 (0.09–5.09) | 0.690 | 12 (3.19) | 1.62 (0.81–3.24) | 0.175 | 32 (2.38) | 1.19 (0.72–1.96) | 0.509 |
| 243/274 | 15 (0.88) | 2 (2.27) | 3.97 (0.84–18.77) | 0.082 | 3 (0.80) | 1.00 (0.28–3.61) | 0.995 | 16 (1.19) | 1.52 (0.73–3.16) | 0.265 |
| 274/302 | 40 (2.34) | 4 (4.55) | 2.48 (0.82–7.52) | 0.108 | 12 (3.19) | 1.41 (0.71–2.78) | 0.327 | 32 (2.38) | 1.00 (0.61–1.63) | 0.997 |
| 243/333 | 16 (0.93) | 2 (2.27) | 3.12 (0.65–14.94) | 0.154 | 5 (1.33) | 1.68 (0.60–4.76) | 0.326 | 19 (1.41) | 1.61 (0.81–3.20) | 0.177 |
| SL system | N = 1712, N (%) | N = 88, N (%) | N = 376, N (%) | N = 1346, N (%) | ||||||
| LL | 770 (44.98) | 36 (40.91) | 1.00 | 174 (46.28) | 1.00 | 605 (44.95) | 1.00 | |||
| SL | 747 (43.63) | 44 (50.00) | 1.21 (0.76–1.92) | 0.417 | 171 (45.48) | 0.98 (0.78–1.25) | 0.901 | 600 (44.58) | 1.00 (0.86–1.17) | 0.985 |
| SS | 195 (11.39) | 8 (9.09) | 0.87 (0.39–1.93) | 0.735 | 31 (8.24) | 0.69 (0.45–1.05) | 0.080 | 141 (10.48) | 0.89 (0.70–1.15) | 0.378 |
P values <0.05 were considered statistically significant (bold).
OR were adjusted for age, sex, smoking status and education.
But, logistic regression analysis for single alleles (effects of single alleles not biased by the partner allele) identified MNS16A VNTR-274 as risk factor for CRC. The four alleles (243, 274, 302 and 333) occurring in the seven considered allele combinations were distinctly added up, consequently, yielding in a doubled sample number (3058 allele combinations, 6116 alleles). The 274 allele was associated with a 2.7-fold increased risk of CRC compared with 302 wild-type allele (OR = 2.69; 95% CI = 1.11–6.50; P = 0.028).
Discussion
In our CORSA study, the hTERT MNS16A VNTR-274 was found to be associated with a significantly increased risk of CRC, suggesting an influence on CRC susceptibility. Also, a novel variant with an additional tandem repeat was discovered carrying a further binding site for the zinc finger transcription factor GATA-1 (16). MNS16A genotypes were determined in this study for the first time in an Austrian population and also for the first time ever in the context of CRC. Furthermore, the strength of this study is that it comprises the largest study population until now consisting of 3842 participants, investigating MNS16A genotypes. A further strength of CORSA is that controls were known to be free of polyps and CRC because all participants underwent complete colonoscopy. Such a high-quality control group, also referred to as ‘supercontrols’ (24) can enhance the statistical power of a study. However, a limitation of this study is, as expected in a population-based screening programme, that the number of CRC patients is rather small.
MNS16A was first identified in the course of a pilot scale hospital-based lung cancer study (16) comprising 53 cases and 72 cancer-free controls. The MNS16A LL genotype was associated with a >2-fold higher risk of lung cancer (OR = 2.18, 95% CI = 0.92–5.20), suggesting an influence of MNS16A genotype on lung cancer susceptibility.
Based on these findings, MNS16A was studied by the same author as potential biomarker for survival in 299 glioblastoma multiforme (GBM) patients (17). The SS genotype was associated with the longest median survival time. SL and LL genotypes were associated with increased risk of death (adjusted hazard ratio, HR = 1.78, 95% CI = 1.11–2.85).
A subsequent study in GBM by Carpentier et al. (18) examined MNS16A in 352 patients and 305 controls. The SS genotype was found to be more frequent in patients than in controls, however, in contrast to previous findings, no significant differences in survival times were found between SS, SL and LL genotypes.
In a large multinational GBM study (19), being part of the international Interphone study participants had been recruited in Denmark, Finland, Sweden and the United Kingdom. MNS16A genotypes of 648 glioma cases, 473 meningioma cases and 1359 controls were determined, but no significant differences in genotype distributions were observed. The HR for the SL genotype was significantly increased (HR = 2.44, 95% CI = 1.56–3.82) and for SS + SL the HR was 2.10 (95% CI = 1.41–3.1), not supporting a (linear) trend towards increasing HR with increasing number of S alleles. The LL genotype had a significantly better survival in GBM patients than the SS + SL group and also the HR for death was significantly increased for SL or SS + SL groups.
In a breast cancer study (20) of ethnic Han Chinese including 1029 cases and 1107 cancer-free controls, all three detected variant alleles were associated with a significantly increased risk of breast cancer. Furthermore, the 271/302 genotype was associated with a significantly elevated risk of axillary lymph node metastasis.
Recently, 808 non-Hispanic White patients with non-small cell lung cancer were genotyped and statistical analysis focused on 221 early-stage I or II non-small cell lung cancer patients displayed significantly shorter median survival times for carriers of SS and SL compared with LL genotypes (21).
The most recent MNS16A related lung cancer study (22) comprised 937 patients and 943 healthy controls. In this Korean study population, VNTR-243 was associated with an increased risk of lung cancer (OR = 1.55, 95% CI = 1.07–2.25), but a dual conflicting role of this MNS16A VNTR was observed because VNTR-243 also seemed to improve survival in patients.
In the Chinese breast cancer cohort (20), no VNTR-333 was detected, although frequently a third band in agarose gels (>302 bp) emerged that was shown to result from conformational changes of the heterozygote. We also found conformational changes of the PCR amplicons being responsible for the additional band signal and verified VNTR-333 by successful cloning and sequencing. Furthermore, we identified the very rare new MNS16A variant VNTR-364 containing an additional tandem repeat.
The highest percentages of CRC were observed for the three genotypes 243/274, 274/302 and 243/333. Interestingly, all 274 alleles eligible for statistical models were contained in this group, hinting a particular significance of this VNTR. Logistic regression models for the seven considered MNS16A genotypes did not identify genotypes with significantly increased risk of CRC. Moreover, no differences in genotype distribution in case and control groups were observed by following the conversion of alleles as applied in literature leading to combinations of short (S) and long (L) alleles (SS, SL and LL). But, logistic regression analysis for the single VNTRs identified MNS16A VNTR-274 as risk factor for CRC. The 274 allele was associated with a 2.7-fold increase in CRC risk compared with the 302 wild-type allele.
This finding of an S allele having malignant potential was strongly supported by results of the large Chinese breast cancer study (20) that found the 271/302 variant associated with the highest risk (OR = 1.77, 95% CI = 1.06–2.96) and by single allele consideration also associated the 271 (274 bp) variant with highest risk for breast cancer (OR = 1.67, 95% CI = 1.01–2.76).
Likewise, the results of the study of Andersson et al. (19) with the second largest population size supported our finding by associating the SS and SL groups with shortened survival times and increased risk of death, although, it remained unclear whether these effects were due to 274 alleles because the number of 274 alleles in SS and SL groups was not designated (19).
The findings of our study did not conflict with the overall suspicion against the S alleles but, in addition, specified a certain allele by identifying MNS16A VNTR-274 as risk allele for CRC. Regarding distinct alleles separately in calculation rather than combinations of alleles (genotypes) seemed to avoid a ‘diluting effect’ of the partner allele, particularly, in cases where a rare or risk-associated allele occurred with a common and unsuspicious variant such as 243 or 302. By avoidance of the established conversion of a four allele system into a two allele system, we identified a distinct risk allele and thereby offered a possible explanation for conflicting results of previous studies. This seemed to be particularly important since this study did not propose an extremely short (243) or long (333) allele as risk allele but one lying interjacently in terms of length and number of tandem repeats. These findings no longer support a linear correlation between length or number of tandem repeats and cancer risk (as ‘the shorter, the worse’ would describe), but a more complex correlation has to be considered.
In conclusion, the relatively strong significance of the finding of a medium length VNTR being a risk factor and not an extreme length of tandem repeats queries the established mode of statistical analysis using a classification system that combines alleles associated with diverse risks within the same genotype group. Reanalyses of existing genotyping data may be of interest. As the entities of hTERT antisense transcripts, MNS16A mode of regulating hTERT expression needs to be elucidated in detail. If the role of MNS16A polymorphism can be confirmed by large replication studies and also in other malignant diseases, this tandem repeats minisatellite can be expected to become a useful biomarker of risk, outcome or prediction in several malignancies.
Funding
‘Österreichische Nationalbank Jubiläumsfondsprojekt’ (12511).
Acknowledgments
We kindly thank all those who contributed to the screening project Burgenland against CRC. Furthermore, we are grateful to Doris Mejri and Monika Hunjadi for laboratory assistance.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- CI
confidence interval
- CORSA
colorectal cancer study of Austria
- CRC
colorectal cancer
- FOBT
faecal occult blood test
- GBM
glioblastoma multiforme
- HR
hazard ratio
- hTERT
human telomerase reverse transcriptase
- HWE
Hardy–Weinberg equilibrium
- NSCL
non-small cell lung cancer
- OR
odds ratio
- PCR
polymerase chain reaction
- VNTR
variable number of tandem repeat
References
- 1.Blackburn EH. The molecular structure of centromeres and telomeres. Annu. Rev. Biochem. 1984;53:163–194. doi: 10.1146/annurev.bi.53.070184.001115. [DOI] [PubMed] [Google Scholar]
- 2.Meyerson M, et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997;90:785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- 3.Greider CW, et al. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- 4.Blackburn EH. Switching and signaling at the telomere. Cell. 2001;106:661–673. doi: 10.1016/s0092-8674(01)00492-5. [DOI] [PubMed] [Google Scholar]
- 5.Shay JW, et al. A survey of telomerase activity in human cancer. Eur. J. Cancer. 1997;33:787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 6.Kim NW, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 7.Engelhardt M, et al. Relative contribution of normal and neoplastic cells determines telomerase activity and telomere length in primary cancers of the prostate, colon, and sarcoma. Clin. Cancer Res. 1997;3:1849–1857. [PubMed] [Google Scholar]
- 8.Engelhardt M, et al. Telomerase and telomere length in the development and progression of premalignant lesions to colorectal cancer. Clin. Cancer Res. 1997;3:1931–1941. [PubMed] [Google Scholar]
- 9.Garcia-Aranda C, et al. Correlations of telomere length, telomerase activity, and telomeric-repeat binding factor 1 expression in colorectal carcinoma. Cancer. 2006;106:541–551. doi: 10.1002/cncr.21625. [DOI] [PubMed] [Google Scholar]
- 10.Gertler R, et al. Telomere length and human telomerase reverse transcriptase expression as markers for progression and prognosis of colorectal carcinoma. J. Clin. Oncol. 2004;22:1807–1814. doi: 10.1200/JCO.2004.09.160. [DOI] [PubMed] [Google Scholar]
- 11.Hastie ND, et al. Telomere reduction in human colorectal carcinoma and with ageing. Nature. 1990;346:866–868. doi: 10.1038/346866a0. [DOI] [PubMed] [Google Scholar]
- 12.O'Sullivan J, et al. Telomere length in the colon declines with age: a relation to colorectal cancer? Cancer Epidemiol. Biomarkers Prev. 2006;15:573–577. doi: 10.1158/1055-9965.EPI-05-0542. [DOI] [PubMed] [Google Scholar]
- 13.Raynaud CM, et al. Telomere shortening is correlated with the DNA damage response and telomeric protein down-regulation in colorectal preneoplastic lesions. Ann. Oncol. 2008;19:1875–1881. doi: 10.1093/annonc/mdn405. [DOI] [PubMed] [Google Scholar]
- 14.Tatsumoto N, et al. High telomerase activity is an independent prognostic indicator of poor outcome in colorectal cancer. Clin. Cancer Res. 2000;6:2696–2701. [PubMed] [Google Scholar]
- 15.Rafnar T, et al. Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat. Genet. 2009;41:221–227. doi: 10.1038/ng.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, et al. Association of a functional tandem repeats in the downstream of human telomerase gene and lung cancer. Oncogene. 2003;22:7123–7129. doi: 10.1038/sj.onc.1206852. [DOI] [PubMed] [Google Scholar]
- 17.Wang L, et al. Survival prediction in patients with glioblastoma multiforme by human telomerase genetic variation. J. Clin. Oncol. 2006;24:1627–1632. doi: 10.1200/JCO.2005.04.0402. [DOI] [PubMed] [Google Scholar]
- 18.Carpentier C, et al. Association of telomerase gene hTERT polymorphism and malignant gliomas. J. Neurooncol. 2007;84:249–253. doi: 10.1007/s11060-007-9378-3. [DOI] [PubMed] [Google Scholar]
- 19.Andersson U, et al. MNS16A minisatellite genotypes in relation to risk of glioma and meningioma and to glioblastoma outcome. Int. J. Cancer. 2009;125:968–972. doi: 10.1002/ijc.24363. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, et al. A tandem repeat of human telomerase reverse transcriptase (hTERT) and risk of breast cancer development and metastasis in Chinese women. Carcinogenesis. 2008;29:1197–1201. doi: 10.1093/carcin/bgn099. [DOI] [PubMed] [Google Scholar]
- 21.Wang L, et al. A functional variant of tandem repeats in human telomerase gene was associated with survival of patients with early stages of non-small cell lung cancer. Clin. Cancer Res. 2010;16:3779–3785. doi: 10.1158/1078-0432.CCR-10-0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin G, et al. Dual roles of a variable number of tandem repeat polymorphism in the TERT gene in lung cancer. Cancer Sci. 2011;102:144–149. doi: 10.1111/j.1349-7006.2010.01782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vutuc C, et al. The burden of cancer in Austria. Eur. J. Cancer Prev. 1999;8:49–55. doi: 10.1097/00008469-199902000-00007. [DOI] [PubMed] [Google Scholar]
- 24.Tenesa A, et al. New insights into the aetiology of colorectal cancer from genome-wide association studies. Nat. Rev. Genet. 2009;10:353–358. doi: 10.1038/nrg2574. [DOI] [PubMed] [Google Scholar]