

Abstract
Slits are a group of secreted glycoproteins that act as molecular guidance cues in cellular migration. Recently, several studies demonstrated that Slit-2 can operate as candidate tumour suppressor protein in various tissues. In this study, we show Slit-2 expression in basal cell layers of normal oral mucosa colocalized with P-cadherin expression. In contrast, there is a loss of Slit-2 and P-cadherin expression in mucosa of oral squamous cell carcinoma (OSCC). Our in vitro investigations reveal a correlation of P-cadherin and Slit-2 expression: OSCC cells with induced P-cadherin expression (PCI52_PC) display an increased Slit-2 expression. However, abrogating P-cadherin function with a function-blocking antibody decreases Slit-2 secretion confirming a direct link between P-cadherin and Slit-2. Moreover, experiments with OSCC cells show that Slit-2 interferes with a Wnt related signalling pathway, which in turn affects Slit-2 expression in a feedback loop. Functionally, transwell migration assays demonstrate a Slit-2 dose-dependent decrease of PCI52_PC cell migration. However, there is no influence on migration in mock control cells. Responsible for this migration block might be an interaction of P-cadherin with Roundabout (Robo)-3, a high affinity receptor of Slit-2. Indeed, proximity ligation assays exhibit P-cadherin/Robo-3 interactions on PCI52_PC cells. Additionally, we detect a modulation of this interaction by addition of recombinant Slit-2. Down-regulation of Robo-3 expression via small interfering RNA neutralizes Slit-2 induced migration block in PCI52_PC cells. In summary, our experiments show antitumorigenic effects of Slit-2 on P-cadherin expressing OSCC cells supposedly via modulation of Robo-3 interaction.
Introduction
Oral squamous cell carcinoma (OSCC) is the most frequent malignant tumour of the oral cavity. Despite improved therapeutic intervention the 5 years survival rate is still only 50% (1). This underlines the importance to explore novel molecular markers, which could help to improve diagnosis and treatment of OSCC.
Cadherins belong to an important family of glycosylated Ca2+-dependent adhesion molecules (2). They are comprised of a large extracellular domain responsible for homophilic cell–cell interactions, a transmembrane domain and a highly conserved cytoplasmatic tail that is bridged to the actin cytoskeleton by binding directly and indirectly to various cytoplasmatic proteins (3). Cadherins are localized to adherens junctions, participate in the maintenance of cell–cell contacts and control diverse morphogenetic events. In pathological processes, they play a dominant role in tumour metastasis and cell migration (4). The best characterized and most widely distributed members of the cadherin superfamily are the classical cadherins. P-cadherin, an important epithelial molecule, is one of the classical cadherins. It is exclusively expressed in the regenerative basal and suprabasal cell layers of the epithelium (5), but the functional role of P-cadherin in tissue regeneration and homeostasis is largely unknown. Our main goal is to determine the role of P-cadherin and its association with other molecules in malignant oral epithelial cells.
In a previous study, we demonstrated that P-cadherin is an indispensible component in reconfiguring mesenchymal OSCC cells with epithelial features by affecting the glycogen synthase kinase (GSK) 3-beta-signalling pathway. Induced expression of P-cadherin leads to an increased GSK3-beta activation, a nuclear decrease of the E-cadherin repressor Snail via phosphorylation by GSK3-beta and subsequent cytoplasmatic translocation of Snail in OSCC cells (6). GSK3-beta is a serine/threonine kinase that phosphorylates many proteins including proteins of the Wnt-signalling pathway (7). A further key component of the canonical and non-canonical Wnt signalling is beta-catenin which binds and activates members of the Lef-1/Tcf family of DNA-binding proteins and is also affected by GSK3-beta. Furthermore, beta-catenin functions as a part of the cadherin complex, which regulates in this way cell–cell adhesion and cell migration (8). In normal cells, most beta-catenin is retained at the cell membrane or it undergoes rapid turnover by the multiprotein destruction complex containing GSK3-beta, hence there is less free beta-catenin available for nuclear shuttling (9).
Initially, the Slit/Roundabout (Robo)-signalling pathway was described in the nervous system where it regulates axon guidance, branching and neural migration (10,11). In non-neuronal cells, Slit has been found to inhibit chemotaxis of leukocytes, Langerhans cells and vascular smooth muscle cells (12–14), whereas endothelial cells and breast cancer cells are attracted by Slit (15,16). Slits are large multidomain leucine-rich repeat-containing proteins with three known members in mammals: Slit-1, Slit-2 and Slit-3, which are secreted (17). Slit ligands bind to Robo receptors, a membrane-protein family that all share a single transmembrane domain. The Robo family constitutes four Robos (Robo-1, Robo-2, Robo-3 and Robo-4) (18).
Recently, several studies revealed that Slit-2 affecting cell migration especially exhibits tumour-suppressive effects in various human cancers (19–22). A recent article reported that Slit-2 inhibits growth and metastasis of squamous cell carcinoma (23). However, little is known about the antitumorigenic effects of Slit-2 in OSCC.
In this study, we show for the first time a connection of Slit-2 and P-cadherin expression. We demonstrate that Slit-2 expression and secretion depends on P-cadherin expression and function. Furthermore, our results suggest a feedback loop of signalling events where Slit-2 affects GSK3-beta signalling resulting in Slit-2 expression. Functionally, addition of Slit-2 to OSCC cells expressing P-cadherin can slow down cell migration. In vitro, we show an interaction of P-cadherin with Robo-3, a high affinity receptor of Slit-2 (24), on aberrant oral epithelial keratinocytes, which can be modulated by addition of Slit-2. Altogether, our results indicate that Robo-3 receptor may act as a modulator of P-cadherin function meaning Slit-2-induced P-cadherin/Robo-3 complex formation on the one hand decreases cell migration, on the other hand helps to maintain appropriate amounts of Slit-2 expression levels in oral epithelium.
Material and methods
Cell lines and culture conditions
The OSCC cell line PCI 52, derived from a larynx carcinoma (pT2N0M0G2), was stably transfected with a full length complementary DNA (cDNA) clone of P-cadherin as described by Bauer et al. (6). Clones were designated as PCI52_PC1-3. PCI52_PC and PCI52_mock cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Pan-Biotech, Aidenbach, Germany) with 10% fetal calf serum (Gibco, Karlsruhe, Germany), 1% penicillin/streptomycin (Gibco) and 1% L-glutamine (Gibco). All cells were grown under selection with G418 (Gibco).
Tissue samples
For immunohistochemical studies, archival formalin-fixed and paraffin-embedded OSCC tissues and healthy control samples from the same OSCC patients were obtained from the Department of Pathology of the University Hospital Regensburg.
Immunohistochemistry
Paraffin-embedded tissue sections were deparaffined and rehydrated. The slides were incubated with 0.05% protease XXIV (Roth, Karlsruhe, Germany), with 0.1% hyaluronidase (Roth), with 3% peroxidase (Roth) and blocked with 3% bovine serum albumin (Sigma–Aldrich, Steinheim, Germany) in phosphate buffered saline (Roth) containing 0.1% Tween 20 (Sigma, Steinheim, Germany) (phosphate buffered saline with Tween-20) for 1 h at 37°C. Goat polyclonal antibody anti-Slit-2 (1:5, Santa Cruz Biotechnology, Heidelberg, Germany) was used as primary antibody for 16 h at 4°C. Afterwards, the paraffin sections were incubated with the secondary antibody donkey anti-goat IgG-horseradish peroxidase (Santa Cruz Biotechnology) (1:400) for 1 h at 37°C and diaminobenzidine solution (DakoCytomation, Hamburg, Germany). The cell nuclei were counterstained with hematoxylin (Roth). All images were taken using the microscope Olympus BX 61. Modifications for P-cadherin immunohistochemistry: the slides were incubated with mouse monoclonal anti-P-cadherin antibody (BD Transduction Laboratories, Philadelphia, NJ) (1:100) for 1 h.
Immunocytochemistry
PCI52_PC and PCI52_mock cells were plated on glass coverslips and fixed. After blocking with 3% peroxidase and 3% bovine serum albumin in phosphate buffered saline with Tween-20 goat polyclonal anti-Slit-2 antibody (1:5) was used for 16 h at 4°C. Afterwards, the cells were incubated with the secondary antibody donkey anti-goat IgG-horseradish peroxidase (1:400) for 1 h at 37°C and diaminobenzidine.
Isolation of RNA from snap frozen tissues
Snap frozen tissues of normal and OSCC mucosa (250 mg) were cut with a scalpel, placed in 2.5 ml, −80°C pre-chilled RNAlater-ICE (Ambion, Austin, TX) and incubated for 16 h at −20°C. Afterwards, RNA was isolated using RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions and was used for real-time polymerase chain reaction analyses.
Real-time polymerase chain reaction analyses
For real-time polymerase chain reaction, total cellular RNA was isolated from PCI52_PC and PCI52_mock cells using RNeasy Mini Kit according to the manufacturer’s instructions. Reverse transcription of RNA to cDNA was performed using transcriptor high fidelity cDNA synthesis kit (Roche, Penzberg, Germany) according to the manufacturer’s protocol. Quantitative gene expression was done using Brilliant II Fast quantitative polymerase chain reaction Master Mix from Stratagene (Agilent Technologies, Santa Clara, CA) with Taqman UPL probes (Roche) (Beta-actin: 5′-ATTGGCAATGAGCGGTTC-3′ and 5′-TGAAGGTAGTTTCGTGGATGC-3′; Robo-3: 5′-ACATCCCTCAGGAGATCTGG-3′ and 5′-TCACTTTGCCTCCCTTGG-3′; Slit-1: 5′-GGCCCAGTGTATGGATGAAG-3′ and 5′-GGATCTCACAGAGCTGTCCAC-3′; Slit-2: 5′-CATGGAGGAACTTGCCACTTA-3′ and 5′-TCCATCAGCACAAATACACCA-3′; Slit-3: 5′-ACCGCTTCCAGTGCAAAG-3′ and 5′-GCATGTCCCGTTATTCTTGC-3′ and P-cadherin: 5′-GCTGGGGAAAGTATTCATGG-3′ and 5′-CCTTCAGTGACCTTCTTTCCTG-3′). Beta-actin messenger RNA (mRNA) was used for normalization.
Beta-catenin/Tcf inhibitor FH535 treatment
PCI52_mock cells were incubated with 20, 40 and 100 μM or without FH535 beta-catenin/Tcf inhibitor (Calbiochem, Nottingham, UK) for 24 h (25). For control, PCI52_mock cells were incubated with dimethyl sulfoxide. After FH535 beta-catenin/Tcf inhibitor treatment total cellular RNA was isolated.
Function blocking P-cadherin assay
PCI52_PC cells were seeded in 96-well plates and incubated with function blocking mouse monoclonal antibody Cadherin P (1:10; Thermo Scientific, Bonn, Germany), which binds to the N-terminal domain of P-cadherin, or with IgG1 control antibody (Abcam, Cambridge, UK) for 16 h. Supernatant human Slit-2 levels were detected with an enzyme-linked immunosorbent assay for human Slit-2 according to the manufacturer’s instructions (USCN Life Science, Wuhan, China). This experiment was repeated eight times. To visualise the impact of P-cadherin function blocking antibody, a proximity ligation assay (PLA) was performed with PCI52_PC cells after function blocking P-cadherin antibody treatment.
Proximity ligation assay
For detection of protein interactions, the PLA (Olink Bioscience, Uppsala, Sweden) was performed. PCI52_PC cells were seeded with or without 100 ng recombinant mouse (rm) Slit-2 (R&D Systems, Wiesbaden, Germany) on glass coverslips. After reaching 80% confluence, the assay was performed according to the manufacturer’s instructions. Briefly, after fixation and blocking, the cells were incubated with the primary antibodies rabbit polyclonal anti-P-cadherin C-terminal (Abnova, Heidelberg, Germany) and goat polyclonal anti-Robo-3 C-terminal (antibodies-online GmbH, Aachen, Germany) (1:50). Subsequently, the cells were incubated with secondary antibodies conjugated with oligonucleotides. After hybridization and ligation of the oligonucleotides, the DNA was amplified by addition of an amplification mixture. A detection mixture detected the amplicons resulting in red fluorescence signals. In the final step, the nuclei were counterstained with 4′,6-diamidino-2-phenylindole and the cells were mounted with mounting medium (Vectashield; Vectorlabs, Peterborough, UK). As control, the cells were incubated without any antibody and with an antibody directed against the N-terminus of P-cadherin, respectively. Additionally, PLAs were performed with human oral keratinocytes (ScienCell, San Diego, CA). Paraffin-embedded tissue sections of normal oral mucosa were also used for PLAs. Paraffin sections were deparaffined, rehydrated and demasked with 10 mM sodium citrate buffer (Roth). Therefore, PLA proceeds as described above. For evaluation, the microscope Olympus BX 61 was used. To quantify the number of P-cadherin/Robo-3 interactions 42 and 160 cells, respectively, were counted. Quantification: counts/nucleus.
Immunoprecipitation experiment
PCI52_PC cells were incubated with 100 ng rm Slit-2 for 16 h. For control, PCI52_mock and PCI52_PC cells without Slit-2 treatment were used. After Slit-2 incubation, cell lysates were prepared using radioimmunoprecipitation assay buffer (Sigma). For immunoprecipitation experiments, cell lysates were mixed with 4 μg goat polyclonal anti-Robo-3 antibody and 100 μl magnetic protein A/G microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) and purified using μMACS columns (Miltenyi Biotec) according to the manufacturer’s instructions. The samples eluted from μMACS columns with sodium dodecyl sulfate sample buffer (Roth) were analysed by western blotting.
Western blot analyses
For western blot analysis, immunoprecipitates were separated with sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Blot membranes were blocked, subsequently incubated with primary mouse monoclonal anti-P-cadherin antibody (1:1000) for 16 h at 4°C. As loading control, goat polyclonal anti-Robo-3 antibody (1:1000) was used to confirm the P-cadherin/Robo-3 interaction. All appropriate secondary antibodies conjugated with horseradish peroxidase were purchased from Pierce (Rockford, IL). Modifications for P-cadherin and Slit-2, respectively, western blot analysis: blots were incubated with primary mouse monoclonal anti-P-cadherin (1:1000) and goat polyclonal anti-Slit-2 antibody (1:500), respectively, for 16 h at 4°C. Equal loading was verified with rabbit monoclonal antibody to beta-actin (1:10 000; Abcam). Modifications for beta-catenin western blot analysis: PCI52_mock cells were incubated with 100 ng rm Slit-2 for 15, 30, 60 and 120 min or without rm Slit-2 and subsequently, cell lysates were prepared. Blots were incubated with primary rabbit monoclonal anti-phospho-beta-catenin antibody (This antibody detects endogenous levels of beta-catenin only when phosphorylated at serines 33, 37 or threonine 41.) (1:1000; Cell Signaling Technology, Danvers, MA), mouse anti-beta-catenin (1:1000; BD Transduction Laboratories) or rabbit monoclonal anti-beta-actin for 16 h at 4°C.
Transient transfection of small interfering RNA-Robo-3
PCI52_PC and PCI52_mock cells were transfected with small interfering RNA (siRNA) specific to human Robo-3 (Thermo Scientific). Cells were seeded the day before transfection in six-well plates in DMEM without antibiotics. Cells were transfected with 20 μM siRNA or 20 μM of control siRNA (non-targeting, Thermo Scientific) using Dharmafect 1 (Thermo Scientific). After 48 h, the extent of mRNA knockdown was determined by quantitative reverse transcription–polymerase chain reaction (qRT–PCR). After 3 days, siRNA transfected cells were used for migration assays.
Migration assay
Migration assays were performed using 24-well dishes and ThinCert cell culture inserts (Greiner Bio-One, Frickenhausen, Germany). Inserts were placed in wells of a 24-well plate forming a migration chamber. The migration chamber consisted of an upper and lower compartment separated by a membrane with 8 μm pore diameter. The lower 24-well compartment was filled with DMEM containing 10% fetal calf serum. In the upper compartment, PCI52_PC or PCI52_mock cells were seeded with 10 and 100 ng or without rm Slit-2 and siRNA-transfected cells, respectively, in DMEM containing 0.2% bovine serum albumin. After incubation for 4.5 h at 37°C cells which migrated to the reverse side of the membrane were stained with calcein-AM (Calbiochem), released into the lower compartment by trypsinization for 20 min. Quantification of cells was performed measuring the relative fluorescent units with a microplate reader (Tecan, Crailsheim, Germany).
Statistical analyses and evaluation
All statistical analyses were done using the Mann–Whitney-U-test. For evaluation of the quantitative polymerase chain reaction and migrations experiments, raw data were set to 100% and standard deviation was calculated in subsequent experiments.
Results
Our area of interest is to investigate the role of P-cadherin and its association with other molecules in OSCC. Novel studies report on a connection of cadherins with the Slit/Robo system (26–30). For this reason, we examined if P-cadherin acts in concert with this receptor system.
Expression of Slit-2 was decreased in OSCC
To address this issue, immunohistochemical stainings of P-cadherin and Slit-2 were performed on paraffin sections of healthy oral mucosa and patients suffering from OSCC. Immunohistochemical stainings of tissue sections from different patients showed that in normal oral mucosa, Slit-2 was localized in basal and suprabasal cell layers (Figure 1a–c). It is known that P-cadherin is also expressed in these epithelial cell layers (Figure 1d). However, during neoplastic transformation of OSCC, Slit-2 revealed a rather low and dyslocalized expression pattern in the oral mucosa (Figure 1e–g). It is suggested that there is a strong P-cadherin expression in dysplasia, whereas its membrane bound expression is lost during OSCC invasion (Figure 1h, arrows) (6). To confirm the immunohistochemical results, we investigated Slit-2 and P-cadherin mRNA expression of OSCC patients and healthy controls. qRT–PCR of cDNA obtained from snap frozen tissues demonstrated a significant decrease of Slit-2 expression (P = 0.0032) in patients suffering from invasive OSCC compared with control patients (Figure 1i). Interestingly, qRT–PCR experiments of cDNA from snap frozen tissues of patients with progressed OSCC revealed a significant decrease of P-cadherin expression (P = 0.0012) in contrast to healthy controls as well (Figure 1j).
Fig. 1.
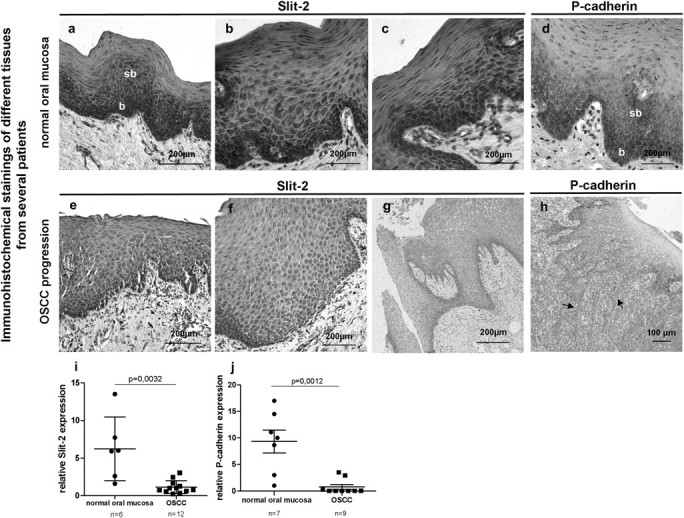
Representative immunohistochemical stainings of different tissues from several patients show that in normal oral mucosa, Slit-2 expression (a–c) is localized in basal and suprabasal epithelial cell layers like P-cadherin (d). During neoplastic transformation of OSCC, Slit-2 reveals a rather low and dislocalized expression pattern in the oral mucosa (e–g). Tissue sections of OSCC patients display a loss of membrane-bound P-cadherin at the invasion front of OSCC (h). qRT–PCR of cDNA obtained from snap frozen tissues confirms a significant decrease of Slit-2 (i) and P-cadherin (j) expression in OSCC patients compared with healthy controls (b, basal; sb, suprabasal cell layers of normal oral mucosa; arrows, invasion front).
P-cadherin overexpression induced Slit-2 expression in the OSCC cell line PCI 52
To analyse the role of Slit-2 and P-cadherin in detail, we used the highly undifferentiated OSCC cell line PCI 52 deficient of the classical cadherins as an in vitro model system. This cell line was stably transfected with full length P-cadherin. The chosen cell clones were designated as PCI52_PC1-3. Figure 2a shows the P-cadherin expression of the selected PCI52_PC cell clones and PCI52_mock control cells in a western blot analysis. Immunohistochemical stainings of PCI52_PC cells revealed that P-cadherin was localized at the cell surface membrane, whereas in PCI52_mock control cells, there was no visible P-cadherin expression (6).
Fig. 2.

Western blot analysis displays a strong P-cadherin expression in the stably transfected OSCC cell line PCI 52 (PCI52_PC1-3), whereas no P-cadherin expression can be seen in mock transfected OSCC cells (PCI52_mock1-2) (a). After P-cadherin overexpression Slit-2 expression increases significantly in PCI52_PC cells compared with PCI52_mock cells (b), whereas both Slit-1 and Slit-3 expression reveal no correlation with P-cadherin expression (c and d). Western blot analysis shows a higher Slit-2 protein level in PCI52_PC cells than in PCI52_mock cells (e). Immunocytochemical stainings affirm a stronger Slit-2 expression in PCI52_PC cells contact each other (f, see arrows) in contrast to sparse Slit-2 expression in a few PCI52_mock single cells (g, see arrows). Blocking P-cadherin function with function blocking (fb) P-cadherin antibody results in a significant decrease of Slit-2 secretion in PCI52_PC cells compared with cells with IgG control antibody treatment (h). Within an hour after Slit-2 incubation phosphorylation of beta-catenin appears in PCI52_mock cells, which have only trace amounts of phospho-beta-catenin (i). Inhibition of the beta-catenin/Lef/Tcf complex with FH535 inhibitor in PCI52_mock cells results in a dose-dependent increase of Slit-2 expression (j).
When we analysed the Slit expression in PCI52_PC and PCI52_mock cells by qRT–PCR, we discovered a correlation between P-cadherin and Slit-2 expression. Figure 2b depicts a significantly increased Slit-2 expression (P < 0.01) in stable P-cadherin transfected OSCC cell clones (PCI52_PC1-3) compared with mock control cells (PCI52_mock1-2). In contrast, no correlation could be seen between P-cadherin and Slit-1 and Slit-3 expression, respectively (Figure 2c and d). Indeed, western blot analysis demonstrated a higher Slit-2 protein level in PCI52_PC cells than in PCI52_mock cells (Figure 2e). Additionally, immunocytochemical stainings confirm a stronger Slit-2 expression in PCI52_PC cells than in PCI52_mock cells (Figure 2f and g). In a confluent monolayer, Slit-2 is located pericellular in cell patches of PCI52_PC cells (arrows in Figure 2f), whereas in PCI52_mock cells, only a few single cells show Slit-2 expression (arrows in Figure 2g). Hence, the qRT–PCR data as well as the western blot results and immunocytochemical studies suggest a supposable link between P-cadherin and Slit-2 expression.
Slit-2 secretion was enhanced in the OSCC cell line PCI 52 by P-cadherin overexpression
To show the direct connection between Slit-2 and P-cadherin, we blocked P-cadherin with a function blocking P-cadherin antibody in confluently grown PCI52_PC cells. Subsequently, the Slit-2 concentration in the supernatant was measured by enzyme-linked immunosorbent assay. The function-blocking antibody binds to the N-terminal domain of P-cadherin and prevents P-cadherin function but does not detach the cells from the culture plate. In our experiment, we observed that blocking P-cadherin function resulted in a significant decrease of Slit-2 secretion (P < 0.01) compared with cells with IgG control antibody treatment (Figure 2h).
Slit-2 induced phosphorylation of beta-catenin in the OSCC cell line PCI 52
Screening for signalling pathways that could be involved in the Slit-2 expression, we identified the Wnt related GSK3-beta-signalling pathway. In a previous study (6), we showed that GSK3-beta is activated in PCI52_PC cells and, consequently, the transcription factor Snail is translocated from the nucleus into the cytoplasm in contrast to PCI52_mock cells. Additionally, beta-catenin, which is another important part in this pathway is abundant in the cytoplasm in our PCI52_mock cells (6). Interestingly, western blot analysis demonstrated that in PCI52_mock cells, beta-catenin is temporarily phosphorylated within an hour after Slit-2 incubation indicating that Slit-2 influenced in some way beta-catenin phosphorylation (Figure 2i). However, after prolonged incubation of Slit-2 for >1 h led to a decrease of beta-catenin phosphorylation (Figure 2i).
Slit-2 expression was influenced in the OSCC cell line PCI 52 by blocking of beta-catenin
On the other hand, we found that beta-catenin also modifies the Slit-2 expression. Inhibition of beta-catenin/Lef/Tcf complex with the FH535 inhibitor resulted in a dose-dependent increase of Slit-2 mRNA expression (P = 0.011) in PCI52_mock cells pointing to an involvement of beta-catenin together with further transcription factors in Slit-2 expression (Figure 2j).
Slit-2 decreased migration of the P-cadherin overexpressing OSCC cell line PCI 52
In our next experiments, we investigated the functional role of Slit-2. In a previous study (6), we showed that the migration of P-cadherin overexpressing cell clones is decreased compared with mock control cells. Here, we demonstrated a further dose-dependent decrease (∼50%, P < 0.05 and P < 0.01) in cell migration after addition of extra Slit-2 to P-cadherin overexpressing OSCC cells (Figure 3a). However, addition of Slit-2 to PCI52_mock cells did not show any effect on cell migration (Figure 3a). Furthermore, no Slit-2 influence on chemoinvasion could be observed (data not shown).
Fig. 3.

After addition of Slit-2 PCI52_PC cells display a dose-dependent decrease in cell migration, whereas PCI52_mock cells do not show any reaction on cell migration after Slit-2 addition (a). PLAs demonstrate an interaction of P-cadherin with Robo-3 receptor in vitro on PCI52_PC cells (b). After addition of Slit-2 to PCI52_PC cells, an increased number of P-cadherin/Robo-3 interactions can be observed (c and d). P-cadherin/Robo-3 interactions can also be seen in human oral keratinocytes (HOK) (e) and in basal cell layers of normal oral mucosa (f). Coimmunoprecipitation experiments confirm the occurrence of increased P-cadherin/Robo-3 interactions following Slit-2 treatment (g) (b = basal, sb = suprabasal cell layers of normal oral mucosa).
Slit-2 amplified P-cadherin/Robo-3 interactions in the P-cadherin overexpressing OSCC cell line PCI 52
To explain the effect of Slit-2 exerted on P-cadherin overexpressing OSCC cells, we were curious as to whether there was a link between P-cadherin and Robo receptors. It is known that Slit proteins are ligands for these receptors. During our experiments, we observed that Robo-3, a high affinity receptor of Slit-2, is expressed in the same basal and suprabasal cell layers of oral epithelium (data not shown). Therefore, we concentrated on Robo-3 and its interaction with Slit-2 and P-cadherin.
To detect protein interactions between P-cadherin and Robo-3, we performed PLAs. This assay is a new method to depict and to quantify protein interactions in an immunohistological way. At the end of this assay, red-fluorescent signals display protein interactions. With this experiment, we detected interactions of P-cadherin with Robo-3 on PCI52_PC cells distributed over the whole cell (Figure 3b). Interestingly, after addition of Slit-2 to PCI52_PC cells, we observed an increase of these P-cadherin/Robo-3 interactions (Figure 3c). We counted the fluorescent signals of 42 cells and noticed a significant rise of P-cadherin/Robo-3 interactions (P < 0.001) in cells incubated with Slit-2 (Figure 3d). As additional cells for PLAs, we used human oral keratinocytes. In these normal cells, we also detected some P-cadherin/Robo-3 interactions spread across the whole cell (Figure 3e). Indeed, in basal cell layers of normal oral mucosa, P-cadherin interacted with Robo-3 (Figure 3f). Coimmunoprecipitation experiments confirmed this enhanced P-cadherin/Robo-3 association in PCI52_PC cells following Slit-2 treatment. Subsequent western blot analyses of P-cadherin after Robo-3 precipitation revealed an increase of P-caderin/Robo-3 signals indicating increased interactions after Slit-2 incubation (Figure 3g).
Inhibition of P-cadherin function resulted in decreased P-cadherin/Robo-3 interactions
Quantifying P-cadherin/Robo-3 interactions in a PLA experiment after treatment of PCI52_PC cells with function blocking P-cadherin antibody, we observed 40% decrease of P-cadherin/Robo-3 interactions (Figure 4a–c, P < 0.01). In the function blocking experiment, the N-terminal P-cadherin antibody prevents cell adhesion, which results in a reduced Slit-2 secretion as shown above in Figure 2h. As a result of this, the decreased Slit-2 concentration caused diminished P-cadherin/Robo-3 interactions as demonstrated with PLA assays (Figure 4a–c).
Fig. 4.

After blocking P-cadherin function with function blocking (fb) P-cadherin antibody, PCI52_PC cells reveal a significant decrease of P-cadherin/Robo-3 interactions by PLAs (a–c). Knockdown of Robo-3 with siRNA (d) in PCI52_PC and PCI52_mock cells results in a significant increase in cell migration in Slit-2 producing PCI52_PC cells. However, in PCI52_mock cells, there is no significant migration effect after Robo-3 knockdown (e) (n.t. = non-targeting).
Knockdown of Robo-3 induced migration in the P-cadherin overexpressing OSCC cell line PCI 52
To analyse the role of the Robo-3 receptor during migration of the OSCC cells, we performed a knockdown of the Robo-3 receptor via transfection of siRNA-Robo-3. Transfection with siRNA against Robo-3 resulted in a ∼60% knockdown of Robo-3-mRNA (Figure 4d, P < 0.01). Subsequently, cell migration assays were performed. Interestingly, after Robo-3 knockdown in Slit-2 producing PCI52_PC cells, we observed a significant increase in cell migration (Figure 4e, P < 0.05). However, in PCI52_mock cells, there was no significant migration effect after Robo-3 knockdown (Figure 4e). These data indicate that knockdown of Robo-3 receptor neutralized Slit-2 induced migration block in PCI52_PC cells (as shown above in Figure 3a).
Discussion
In the present study, we show antitumorigenic effects of the secreted glycoprotein Slit-2 on P-cadherin expressing OSCC cells mediated by the Robo-3 receptor. Our experiments demonstrate how P-cadherin might act in concert with the cell membrane receptor Robo-3 to induce a stationary phenotype by means of Slit-2 induced P-cadherin/Robo-3 complex formation in OSCC cells. In several reports, interactions of N-cadherin with Robo receptors have already been discussed (26,27,29).
In our study, Slit-2 was immunolocalized in basal and suprabasal epithelial cell layers of normal oral mucosa. The same localization could be observed for the adhesion molecule P-cadherin. Slit-2 showed a low expression and diffuse localization in dysplastic stratified cell layers or at the onset of invasion. Kim et al. (23) detected Slit-2 mRNA in normal human esophageal mucosa by in situ hybridization, whereas Slit-2 expression was also reduced in esophageal squamous cell carcinoma.
Moreover, our results displayed a correlation of Slit-2 and P-cadherin expression in P-cadherin overexpressing OSCC cells and a dependency of Slit-2 secretion on P-cadherin function. Those findings suggest that the cell adhesion molecule P-cadherin stabilizes Slit-2 expression and facilitates Slit-2 secretion by a still unknown mechanism. A hypothetical model of the P-cadherin-Slit-2-mechanism derived from our data demonstrates Figure 5. It shows how Slit-2 might induce P-cadherin/Robo-3 complex formation and subsequent effects on Slit-2 transcription and secretion.
Fig. 5.

Hypothetical model of the formation of Slit-2 induced P-cadherin/Robo-3 complex and subsequent effects on Slit-2 transcription and secretion: Activation of Robo-3 by Slit-2 results in the formation of a complex between Robo-3 and P-cadherin mediated by still unknown molecules (a, small picture). Extracellular Slit-2 binds to Robo-3 receptor with subsequent abundant cytosolic beta-catenin phosphorylation and degradation. Consequently, further inhibiting transcription factors cannot be expressed thus supporting Slit-2 transcription in a feedback loop. Additionally, P-cadherin–P-cadherin interactions affect Slit-2 expression and facilitate Slit-2 secretion (a). In contrast, disruption of P-cadherin–P-cadherin interactions causes a diminished Slit-2 secretion and a decrease of P-cadherin/Robo-3 interactions. Because of the disrupted cell adhesion and decreased Slit-2 secretion, beta-catenin is not phosphorylated possibly due to GSK3-beta inactivation. Thus, beta-catenin is translocated to the nucleus where genes are activated which in turn might contribute to the inhibition of Slit-2 transcription (b).
Additionally, our experiments displayed a re-expression of Slit-2 in OSCC cells after inhibition of binding beta-catenin to Lef/Tcf. One would expect a decrease of gene expression after blocking the beta-catenin/Lef/Tcf complex. However, in recent publications Blauwkamp et al. (31) and Hoverter et al. (32) have demonstrated that Lef/Tcf gene transcription heavily depends on the equipment of transcription factors which in combination can have transcription repressor function. Therefore, our observed effect of Slit-2 increase points to an involvement of beta-catenin in combination with further transcription factors effecting Slit-2 expression.
Besides, Slit-2 treatment affects phosphorylation of beta-catenin in OSCC cells which have more phosphorylated GSK3-beta meaning a more inactive form resulting in stabilized beta-catenin (6). Recently, Prasad et al. (33) and Tseng et al. (30) have confirmed that Slit-2 mediated its tumour-suppressive effect by regulating beta-catenin and phosphoinositide-3-kinase/AKT pathway in breast cancer and lung cancer cells. In line with these data, our studies suggest a feedback loop of signalling events whereby Slit-2 may affect its own transcription. This may lead to enhanced phosphorylation and degradation of cytoplasmic beta-catenin, limiting the amount of beta-catenin for nuclear shuttling and regulation of Slit-2 transcription in OSCC cells (Figure 5).
Furthermore, we could demonstrate an interaction of P-cadherin with Robo-3, a high affinity receptor of Slit-2, in vitro on aberrant oral epithelial keratinocytes by PLAs. The interaction was modulated by addition of Slit-2. In contrast, in mouse fibroblast L-cells, N-cadherin interacted with Robo receptor resulting in the inhibition of cadherin function (26). However, Shiau et al. (29) reported that N-cadherin acted in concert with Robo-2 in placode-derived cranial sensory neurons and strengthened placodal cell adhesion required for proper gangliogenesis.
Slit, which mediates its function by binding to Robo regulates migration of neurons, leukocytes and cancer cells (12,34,35). In our study, we also demonstrated a dose-dependent antimigratory effect of Slit-2 on OSCC cells overexpressing P-cadherin but not on P-cadherin-deficient OSCC cells. On the other hand, our experiments showed that knockdown of Robo-3 expression induced enhanced cell migration predominantly in P-cadherin overexpressing OSCC cells. Likewise, in experiments of Mertsch et al. (36) knockdown of Robo-1 neutralized the repulsive effect of Slit-2 in glioma cell lines. That implies that P-cadherin is one component in a complex with Slit-2 and Robo-3 for slowing down cell migration. Our results suggest that P-cadherin, Slit-2 and Robo-3 form a complex (Figure 5a, small picture) strengthening epithelial architecture in vivo by transmitting antimigratory signalling. In line with our data, other publications verified that Slit-2 inhibits migration of tumour cells in vitro. In medulloblastoma, Slit-2 inhibited the cell invasion in a variety of in vitro models (37). Furthermore, in CXCL12/CXCR4-induced breast cancer, Slit treatment inhibited cell chemotaxis, chemoinvasion and adhesion (38). Moreover, Mertsch et al. (36) showed that Slit-2 was chemorepulsive for glioma cell lines in a modified Boyden chamber assay, the same experimental setup we used. In a report of Yiin et al. (39), Slit-2 down-regulated Cdc42 activation through its receptor Robo-1 and attenuated migration of glioma cells in vitro. Likewise, Slit-2 has antimigratory effects on dendritic cells. By allergen, sensitization in the skin activated Slit-2 down-regulated the migration of Langerhans cells (13).
In summary, our study indicates that the Robo-3 receptor may act as a modulator of P-cadherin function. This implies that Slit-2 induced P-cadherin/Robo-3 complex formation on the one hand decreases cell migration, on the other hand helps to maintain appropriate amounts of Slit-2 expression levels in oral epithelium.
Funding
Deutsche Forschungsgemeinschaft (BA3696/1-2) to R.B.
Acknowledgments
We thank Anja Reck for the excellent technical assistance.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- cDNA
complementary DNA
- DMEM
Dulbecco’s modified Eagle’s medium
- GSK
glycogen synthase kinase
- mRNA
messenger RNA
- OSCC
oral squamous cell carcinoma
- PLA
proximity ligation assay
- rm
recombinant mouse
- qRT–PCR
quantitative reverse transcription–polymerase chain reaction
- Robo
Roundabout
References
- 1.Bray F, et al. Estimates of cancer incidence and mortality in Europe in 1995. Eur. J. Cancer. 2002;38:99–166. doi: 10.1016/s0959-8049(01)00350-1. [DOI] [PubMed] [Google Scholar]
- 2.Takeichi M. Cadherins: a molecular family important in selective cell-cell adhesion. Annu. Rev. Biochem. 1990;59:237–252. doi: 10.1146/annurev.bi.59.070190.001321. [DOI] [PubMed] [Google Scholar]
- 3.Drees F, et al. Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell. 2005;123:903–915. doi: 10.1016/j.cell.2005.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wheelock MJ, et al. Cadherin-mediated cellular signaling. Curr. Opin. Cell Biol. 2003;15:509–514. doi: 10.1016/s0955-0674(03)00101-7. [DOI] [PubMed] [Google Scholar]
- 5.Shimoyama Y, et al. Cadherin cell-adhesion molecules in human epithelial tissues and carcinomas. Cancer Res. 1989;49:2128–2133. [PubMed] [Google Scholar]
- 6.Bauer K, et al. P-cadherin induces an epithelial-like phenotype in oral squamous cell carcinoma by GSK-3beta-mediated Snail phosphorylation. Carcinogenesis. 2009;30:1781–1788. doi: 10.1093/carcin/bgp175. [DOI] [PubMed] [Google Scholar]
- 7.Woodgett JR. Judging a protein by more than its name: GSK-3. Sci. STKE. 2001;2001:re12. doi: 10.1126/stke.2001.100.re12. [DOI] [PubMed] [Google Scholar]
- 8.Nelson WJ, et al. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483–1487. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen RH, et al. Wnt signaling to beta-catenin involves two interactive components. Glycogen synthase kinase-3beta inhibition and activation of protein kinase C. J. Biol. Chem. 2000;275:17894–17899. doi: 10.1074/jbc.M905336199. [DOI] [PubMed] [Google Scholar]
- 10.Battye R, et al. Repellent signaling by Slit requires the leucine-rich repeats. J. Neurosci. 2001;21:4290–4298. doi: 10.1523/JNEUROSCI.21-12-04290.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simpson JH, et al. Short-range and long-range guidance by slit and its Robo receptors. Robo and Robo2 play distinct roles in midline guidance. Neuron. 2000;28:753–766. doi: 10.1016/s0896-6273(00)00151-3. [DOI] [PubMed] [Google Scholar]
- 12.Wu JY, et al. The neuronal repellent Slit inhibits leukocyte chemotaxis induced by chemotactic factors. Nature. 2001;410:948–952. doi: 10.1038/35073616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guan H, et al. Neuronal repellent Slit2 inhibits dendritic cell migration and the development of immune responses. J. Immunol. 2003;171:6519–6526. doi: 10.4049/jimmunol.171.12.6519. [DOI] [PubMed] [Google Scholar]
- 14.Liu D, et al. Neuronal chemorepellent Slit2 inhibits vascular smooth muscle cell migration by suppressing small GTPase Rac1 activation. Circ. Res. 2006;98:480–489. doi: 10.1161/01.RES.0000205764.85931.4b. [DOI] [PubMed] [Google Scholar]
- 15.Wang B, et al. Induction of tumor angiogenesis by Slit-Robo signaling and inhibition of cancer growth by blocking Robo activity. Cancer Cell. 2003;4:19–29. doi: 10.1016/s1535-6108(03)00164-8. [DOI] [PubMed] [Google Scholar]
- 16.Schmid BC, et al. The neuronal guidance cue Slit2 induces targeted migration and may play a role in brain metastasis of breast cancer cells. Breast Cancer Res. Treat. 2007;106:333–342. doi: 10.1007/s10549-007-9504-0. [DOI] [PubMed] [Google Scholar]
- 17.Itoh A, et al. Cloning and expressions of three mammalian homologues of Drosophila slit suggest possible roles for Slit in the formation and maintenance of the nervous system. Brain Res. Mol. Brain Res. 1998;62:175–186. doi: 10.1016/s0169-328x(98)00224-1. [DOI] [PubMed] [Google Scholar]
- 18.Stein E, et al. Hierarchical organization of guidance receptors: silencing of netrin attraction by slit through a Robo/DCC receptor complex. Science. 2001;291:1928–1938. doi: 10.1126/science.1058445. [DOI] [PubMed] [Google Scholar]
- 19.Dallol A, et al. SLIT2, a human homologue of the Drosophila Slit2 gene, has tumor suppressor activity and is frequently inactivated in lung and breast cancers. Cancer Res. 2002;62:5874–5880. [PubMed] [Google Scholar]
- 20.Dallol A, et al. Frequent epigenetic inactivation of the SLIT2 gene in gliomas. Oncogene. 2003;22:4611–4616. doi: 10.1038/sj.onc.1206687. [DOI] [PubMed] [Google Scholar]
- 21.Dallol A, et al. SLIT2 axon guidance molecule is frequently inactivated in colorectal cancer and suppresses growth of colorectal carcinoma cells. Cancer Res. 2003;63:1054–1058. [PubMed] [Google Scholar]
- 22.Sundaresan V, et al. Homozygous deletions at 3p12 in breast and lung cancer. Oncogene. 1998;17:1723–1729. doi: 10.1038/sj.onc.1202103. [DOI] [PubMed] [Google Scholar]
- 23.Kim HK, et al. Slit2 inhibits growth and metastasis of fibrosarcoma and squamous cell carcinoma. Neoplasia. 2008;10:1411–1420. doi: 10.1593/neo.08804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabatier C, et al. The divergent Robo family protein rig-1/Robo3 is a negative regulator of slit responsiveness required for midline crossing by commissural axons. Cell. 2004;117:157–169. doi: 10.1016/s0092-8674(04)00303-4. [DOI] [PubMed] [Google Scholar]
- 25.Handeli S, et al. A small-molecule inhibitor of Tcf/beta-catenin signaling down-regulates PPARgamma and PPARdelta activities. Mol. Cancer Ther. 2008;7:521–529. doi: 10.1158/1535-7163.MCT-07-2063. [DOI] [PubMed] [Google Scholar]
- 26.Rhee J, et al. Activation of the repulsive receptor Roundabout inhibits N-cadherin-mediated cell adhesion. Nat. Cell Biol. 2002;4:798–805. doi: 10.1038/ncb858. [DOI] [PubMed] [Google Scholar]
- 27.Rhee J, et al. Cables links Robo-bound Abl kinase to N-cadherin-bound beta-catenin to mediate Slit-induced modulation of adhesion and transcription. Nat. Cell Biol. 2007;9:883–892. doi: 10.1038/ncb1614. [DOI] [PubMed] [Google Scholar]
- 28.Santiago-Martinez E, et al. Repulsion by Slit and Roundabout prevents Shotgun/E-cadherin-mediated cell adhesion during Drosophila heart tube lumen formation. J. Cell Biol. 2008;182:241–248. doi: 10.1083/jcb.200804120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shiau CE, et al. N-cadherin acts in concert with Slit1-Robo2 signaling in regulating aggregation of placode-derived cranial sensory neurons. Development. 2009;136:4155–4164. doi: 10.1242/dev.034355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tseng RC, et al. SLIT2 attenuation during lung cancer progression deregulates beta-catenin and E-cadherin and associates with poor prognosis. Cancer Res. 2010;70:543–551. doi: 10.1158/0008-5472.CAN-09-2084. [DOI] [PubMed] [Google Scholar]
- 31.Blauwkamp TA, et al. Novel TCF-binding sites specify transcriptional repression by Wnt signalling. EMBO J. 2008;27:1436–1446. doi: 10.1038/emboj.2008.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoverter NP, et al. A Wnt-fall for gene regulation: repression. Sci. Signal. 2008;1:pe43. doi: 10.1126/scisignal.139pe43. [DOI] [PubMed] [Google Scholar]
- 33.Prasad A, et al. Slit-2 induces a tumor-suppressive effect by regulating beta-catenin in breast cancer cells. J. Biol. Chem. 2008;283:26624–26633. doi: 10.1074/jbc.M800679200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen-Ba-Charvet KT, et al. Role of Slit proteins in the vertebrate brain. J. Physiol. Paris. 2002;96:91–98. doi: 10.1016/s0928-4257(01)00084-5. [DOI] [PubMed] [Google Scholar]
- 35.Ypsilanti AR, et al. Moving away from the midline: new developments for Slit and Robo. Development. 2010;137:1939–1952. doi: 10.1242/dev.044511. [DOI] [PubMed] [Google Scholar]
- 36.Mertsch S, et al. Slit2 involvement in glioma cell migration is mediated by Robo1 receptor. J. Neurooncol. 2008;87:1–7. doi: 10.1007/s11060-007-9484-2. [DOI] [PubMed] [Google Scholar]
- 37.Werbowetski-Ogilvie TE, et al. Inhibition of medulloblastoma cell invasion by Slit. Oncogene. 2006;25:5103–5112. doi: 10.1038/sj.onc.1209524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prasad A, et al. Slit protein-mediated inhibition of CXCR4-induced chemotactic and chemoinvasive signaling pathways in breast cancer cells. J. Biol. Chem. 2004;279:9115–9124. doi: 10.1074/jbc.M308083200. [DOI] [PubMed] [Google Scholar]
- 39.Yiin JJ, et al. Slit2 inhibits glioma cell invasion in the brain by suppression of Cdc42 activity. Neuro. Oncol. 2009;11:779–789. doi: 10.1215/15228517-2008-017. [DOI] [PMC free article] [PubMed] [Google Scholar]