

Abstract
Enantioselective copper-catalyzed cycliza-tion of γ-alkenylsulfonamides and a δ-alkenylsulfonamide in the presence of a range of vinyl arenes results in variously functionalized 2-substituted chiral nitrogen heterocycles via a formal alkene C–H functionalization process. Application of this reaction to the concise synthesis of a 5-HT7 receptor antagonist is demonstrated.
The C–H functionalization of alkenes is currently under intensive study as a direct carbon–carbon bond-forming method for the synthesis of higher substituted alkenes.1 The commonly employed Heck reaction2 enables direct C–H to C–C functionalization of alkenes with aryl and vinylhalides, but is rarely performed3 with alkyl halide coupling partners due to the propensity of the alkyl to undergo β-hydride elimination. The Heck-type coupling with alkyl halides has experienced some success with alternative methods that involve carbon radical intermediates, generally of low functionality.3b,4 New methods for the oxidative coupling of more functionalized alkyl groups with alkenes, however, are still required. Along these lines, the intermolecular oxidative Heck-type coupling of β-amino alkyl reagents with alkenes is unprecedented.5 Herein is reported the first intermolecular oxidative Heck-type coupling with a β-aminoalkyl intermediate.6 Furthermore, rather than starting from a preformed chiral β-amino alkyl halide, we envisioned the alkene could intercept a chiral β-aminoalkyl radical generated in situ from an enantioselective aminocupration of a γ-aminoalkene followed by C–Cu(II) homolysis. Under oxidizing reaction conditions, the resulting carbon radical coupling intermediate could be oxidized to an alkene, thus completing a net Heck-type reaction sequence (Scheme 1).
Scheme 1.

Alkene Amination–Heck-Type Coupling Cascade
We have previously shown that γ-pentenylsulfonamides undergo enantioselective Cu-catalyzed aminofunctionalizations, for example, carboamination,7 aminooxygenation,8 and diamination.9 These reactions involve carbon radical intermediates as evidenced by radical trapping experiments and regio-and stereoselectivity patterns of reactivity.10 The particular aminofunctionalization observed is a function of substrate structure and reaction components.11 The reaction disclosed herein significantly extends our previously reported doubly intramolecular carboamination methodology7 to an intermolecular C–C bond-forming process by using aryl-substituted alkenes as the otherwise unfunctionalized p-component.12 The resulting allyl-functionalized chiral indoline, pyrrolidine, and tetrahydroisoquinoline products (vide infra) should find application in drug discovery and organic synthesis.
The intermolecular carboamination reaction was first investigated with N-tosyl-2-allylaniline 1a using 1,1-diphenyl-ethylene (DPE) as the aryl alkene coupling partner.13 At the onset, we were uncertain if intermolecular radical addition to DPE would out-compete an intramolecular carboamination process, where the radical adds to the aryl ring of the tosyl group, generating sultam 3.7a While the reaction promoted by Cu(2-ethylhexanoate)2, a copper(II) source known to promote the alkene amination step,11 provided a low yield of 2a (Table 1, entry 1), the catalytic version, using Cu(OTf)2 (20 mol%), the 2,2′-bipyridine ligand, and MnO2 (3 equiv) as the stoichiometric oxidant provided a higher yield of 2a (Table 1, entry 2). Encouraged by these results, we further employed (R,R)-Ph-box as the ligand to enable an enantioselective variant. To our delight, this process was feasible, and chiral indoline 2a was formed in 62% yield and 73% ee (Table 1, entry 3). We found that use of activated 4 Å molecular sieves was beneficial to the yield and enantioselectivity (Table 1, entry 4). While an increase in diphenylethylene equivalents and reaction concentration did not further increase the yield of 2a, the DPE stoichiometry could be reduced to 3 equiv (Table 1, entries 5–7). The yield of 2a peaked around 75%, where the remaining mass was predominantly sultam 3 (15%). We further optimized the reaction by reducing reaction temperature and time (Table 1, entry 8). The catalyst loading could also be further reduced to 15 mol % Cu(OTf)2 for this substrate (Table 1, entry 9).
Table 1.
Alkene Amination–Heck-Type Coupling Optimizationa
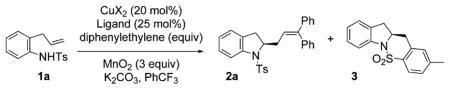 | |||||
|---|---|---|---|---|---|
| entry | CuX2·ligand | DPE equiv | time (h), temp (°C) | yieldb 2a (3) | eec |
| 1d,e | Cu(eh)2 | 5 | 24, 120 | 51 (40) | |
| 2e | Cu(OTf)2 ·Bipy | 5 | 24, 120 | 65 (30) | |
| 3e | Cu(OTf)2 ·(R,R)-Ph-box | 5 | 24, 120 | 62 (25) | 73 |
| 4e,f | Cu(OTf)2 ·(R,R)-Ph-box | 5 | 24, 120 | 75 (15) | 88 |
| 5f,g | Cu(OTf)2 ·(R,R)-Ph-box | 5 | 24, 120 | 76 (15) | 87 |
| 6f,g | Cu(OTf)2 ·(R,R)-Ph-box | 8 | 24, 120 | 75 (15) | nd |
| 7f,g | Cu(OTf)2 ·(R,R)-Ph-box | 3 | 24, 120 | 74 (15) | 89 |
| 8f,g | Cu(OTf)2 ·(R,R)-Ph-box | 3 | 8, 105 | 75 (15) | 92 |
| 9f,g,h | Cu(OTf)2 ·(R,R)-Ph-box | 3 | 8, 105 | 75 (15) | 91 |
Reactions run with 1a (0.174 mmol), Cu(II) (0.0348 mmol), ligand (0.0435 mmol), DPE (equiv), K2CO3 (0.174 mmol), and MnO2 (0.522 mmol) in CF3Ph (0.15 M) unless otherwise noted.
Isolated yield after flash chromatography on SiO2.
Determined by chiral HPLC analysis.
Cu(eh)2 (3 equiv) used in this reaction.
Reaction run at 0.10 M with respect to 1a.
Reaction carried out with activated 4 Å mol. sieves.
Reaction run at 0.15 M with respect to 1a.
Reaction run with 15 mol% Cu(OTf)2 and 19 mol% (R,R)-Ph-box. L = ligand, bipy = 2,2′-bipyridine, Cu(eh)2 = copper (2-ethylhexanoate)2, DPE = 1, 1-diphenylethylene.
nd = not determined.
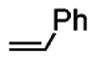
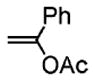
Following the optimized conditions (Table 1, entry 8), a series of substituted N-sulfonyl-2-allylanilines underwent the enantioselective alkene amination/Heck-type coupling reaction with vinylarenes. As illustrated in Table 2, the N-arylsulfonyl-anilines gave products with relatively higher enantiomeric excess than the N-mesyl- and N-trimethylsilylethylsulfonyl analogues (compare Table 2, entries 1–4 to entries 5 and 6). The yields and selectivities were relatively insensitive to the nature of the 4-substitution on the aniline (F, Cl, MeO, see Table 2, entries 7–9). Although diphenylethylene is generally the most reactive coupling partner, other vinylarenes such as styrene, α-methylstyrene, α-acetoxystyrene, α-pivaloxystyrene, benzofuran, and 3-methylbenzofuran also yielded coupling products. The coupling with styrene provided a mixture (4:1 E:Z) of alkene isomers (Table 2, entry 10). Ozonolysis of this mixture followed by reductive workup with NaBH4 provided one terminal alcohol product in 88% ee (see Supporting Information for details). Coupling of 1a with α-methylstyrene provided an inseparable 3:1 mixture of the internal and terminal alkenes, 2k and 2l, respectively (Table 2, entry 11). Ozonolysis of this mixture provided the respective aldehyde and ketone in 85% and 91% ee (see Supporting Information for details). The β-amino aldehyde derived from 2k has a lower %ee, possibly due to configurational instability (via reversible retro-Michael/Michael addition). Coupling of 1a with α-acetoxystyrene4e provided ketone 2m in 82% ee, albeit in only 20% yield (Table 2, entry 12). The remainder of the material was N-acyl-N-tosyl-2-allylaniline along with sultam 3. Changing to the α-pivaloxystyrene coupling partner provides 2m in 50% yield and 90% ee.
Table 2.
Indoline Synthesis Scopea
| entry | substrate | alkene | major product | yieldb | eec |
|---|---|---|---|---|---|
| 1 |
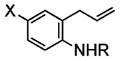 1a, X = H, R = Ts |
 (DPE) |
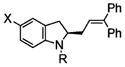 2a |
75 | 91 |
| 2 | 1b, X = H, R = Bs | DPE | 2b, X = H, R = Bs | 85 | 88 |
| 3 | 1c, X = H, R = Ns | DPE | 2c, X = H, R = Ns | 65 | 87 |
| 4 | 1d, X = H, R = 3,5-di-t-Bu-C6H3SO2 | DPE | 2d, X = H, R = 3,5-di-t-BuC6H3SO2 | 82 | 88 |
| 5 | 1e, X = H, R = Ms | DPE | 2e, X = H, R = Ms | 84 | 83 |
| 6 | 1f, X = H, R = SES | DPE | 2f, X = H, R = SES | 80 | 71 |
| 7 | 1g, X = F, R = Ts | DPE | 2g, X = F, R = Ts | 84 | 88 |
| 8 | 1h, X = OMe, R =Ts | DPE | 2h, X = OMe, R = Ts | 77 | 86 |
| 9 | 1i, X = Cl, R = Ts | DPE | 2i, X = Cl, R = Ts | 83 | 91 |
| 10 | 1a |
|
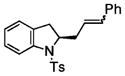 2j(E:Z = 4:1) |
71 | 88 |
| 11 | 1a |
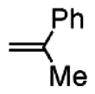
|
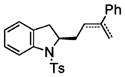 internal alkene 2k/terminal alkene 2l = 3:1) |
73 | 85/91 |
| 12d | 1a |
|
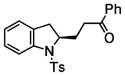 2m |
20 | 82 |
| 13d | 1a |
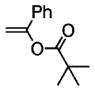
|
2m | 50 | 90 |
| 14ef | 1e |
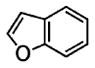
|
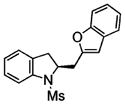 2n |
60 | 58 |
| 15e | 1e |
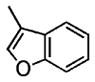
|
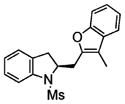 2o |
73 | 40 |
The same conditions as Table 1, entry 8, were used in these reactions. These conditions were more general than Table 1, entry 9. Yields and ee’s are the average of two runs.
Isolated yield.
Determined by chiral HPLC.
N-Acylation was a competing side reaction.
Reaction run at 120 °C for 24 h.
N-Mesyl-2-methylindoline, a hydroamination product, was a minor component of the crude mixture.
Coupling of N-mesyl-2-allylaniline 1e with benzofuran and 3-methylbenzofuran, respectively, provided the unique coupling products 2n and 2o, albeit is relatively lower %ee (Table 2, entries 14 and 15). In these reactions the N-tosyl substrate 1a was less effective, and intramolecular carboamination product 3 predominated (not shown). A comparison of Table 2, entries 5, 14, and 15, also illustrates that the reactivity of the coupling partner can affect the enantioselectivity. This seems to indicate that if the coupling partner is not highly reactive toward radical addition, the alkyl radical intermediate could ring open and close reversibly prior to coupling, resulting in erosion of enantioselectivity. It is also noteworthy that N-tosyl-2-allylani-line 1a gave the sultam 3 in low enantioselectivity in our doubly intramolecular carboamination reaction,7a while the majority of the intra-/intermolecular reactions in Table 2 provide indolines with very good %ee. This again seems to indicate that if the carbon radical addition is rapid, selectivity erosion via ring-opening becomes less problematic.
Electron-deficient alkene coupling partners such as acryloni-trile, methyl methacrylate, and 2(5H)-furanone either were not reactive, underwent aza-Michael addition (acrylonitrile), or produced what appeared to be polymers in reactions with 1a or 1e.
4-Pentenylsulfonamides 4 also underwent the coupling reaction in good yield and moderate to excellent enantio-selectivity (Table 3). These relatively less reactive substrates required longer reaction time (24 h) and higher temperature (120 °C). It is instructive that both the carbon backbone and the N-sulfonyl group have a significant effect on the enantioselectivity. For example, the N-tosyl substrate 4a provided the DPE coupling product 5a in 92% ee, while the N-mesyl substrate 4b gave 5b in only 55% ee (Table 3, entries 1 and 2). The N-tosyl-2,2-diphenyl-4-pentene 4c reacted with the highest enantioselectivity, 95% ee, while the N-mesyl-2,2-diphenyl-4-pentene 4d provided the coupled product in 90% ee (Table 3, entries 3 and 4). Clearly the 2,2-diphenyl substitution has a significant affect on the enantioselectivity. An example of a six-membered ring synthesis is shown in Table 3, entry 7, where a chiral isoquinoline 7 is formed in 51% yield and 79% ee. This substrate was less reactive and required a 48 h reaction time. It is noteworthy that most of the coupling products are crystalline, so their enantiopurity can in principle be increased by recrystallization.
Table 3.
Pyrrolidine and Tetrahydroisoquinoline Synthesisa
| entry | substrate | product | yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1 |
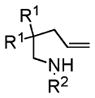 4a, R1 = Me, R2 = Ts |
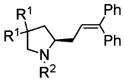 5a |
74 | 92 |
| 2 | 4b, R1 = Me, R2 = Ms | 5b, R1 = Me, R2 = Ms | 88 | 55 |
| 3 | 4c, R1 = Ph, R2 = Ts | 5c, R1 = Ph, R2 = Ts | 68 | 95 |
| 4 | 4d, R1 = Ph, R2 = Ms | 5d, R1 = Ph, R2 = Ms | 68 | 90 |
| 5 | 4e, R1 = H, R2 = Ts | 5e, R1 = H, R2 = Ts | 62 | 80 |
| 6 | 4f, R1 = H, R2 = 3,5-di-t-BuC6H3SO2 | 5f, R1 = H, R2 = 3,5-di-t-BuC6H3SO2 | 88 | ndd |
| 7e |
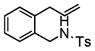 6 |
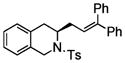 7 |
51 | 79 |
The same general reaction conditions as Table 1, entry 8, were used except the reactions were run at 120 °C for 24 h.
Isolated yields from flash chromatography on SiO2.
Enantiomeric excess determined by chiral HPLC analysis.
Not determined. The enantiomers could not be separated by chiral HPLC.
Reaction was run for 48 h.
The absolute stereochemistry of 2a was established by X-ray crystallography. The stereochemistry of 2b–2o, 4a–4f, and 6 was assigned by analogy to 2a and literature precedent.7,8
The proposed mechanism for this reaction is shown in Scheme 1. Ligand exchange provides the reactive R2N-[Cu] complex. Stereodetermining cis-aminocupration occurs via chairlike transition state A that places the N-substituent anti to the closest bis(oxazoline) phenyl substituent. The resulting unstable organocopper(II) species undergoes homolysis to provide Cu(I) and the corresponding primary carbon radical. Radical addition to the vinylarene and subsequent oxidation of the resulting benzylic radical provides the chiral nitrogen heterocycle.
An alternative mechanism for addition to the vinylarene could involve carbocupration of the organocopper intermediate followed by β-hydride elimination.2e–g To differentiate between carbon radical addition and carbocupration for this step, a competition experiment was performed where indoline 1a underwent the alkene amination/Heck-type cascade in the presence of 1.5 equiv of DPE and 1.5 equiv of styrene (eq 1).
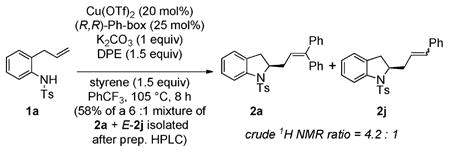 |
(1) |
We reasoned that a radical mechanism should favor addition to DPE over styrene since radicals generally add more rapidly to DPE.13 Conversely, in analogy to the migratory insertion step in the Pd-catalyzed Heck reaction, a carbocupration mechanism should favor addition to styrene, which is less hindered than DPE.2d (It is also noteworthy that styrene, but not diphenyl-ethylene, is a substrate for Cu-catalyzed Heck reactions.2e–g) This competition reaction led to formation of a 4.2:1 mixture of 2a and 2j (eq 1). Thus, it appears the radical mechanism is more likely.
Scheme 1 shows the stereochemistry-determining step to be the cis-aminocupration, yet in Table 2 we observe that the nature of the vinyl arene can affect the enantioselectivity of the reaction (vide supra). This could be indicating that the organocopper intermediate or the carbon radical can undergo some degree of ring-opening and unselective re-closing if the vinylarene is not a very reactive radical acceptor.
An illustration of the utility of this reaction is shown in the concise synthesis of a 5-HT7 receptor antagonist 9 (Scheme 2).14 Synthesis of sulfonamide 4g was performed by SN2 displacement on the commercially available 5-bromo-1-pentene with 3-methyl-benzenesulfonamide. The enantioselective amination/Heck-type cascade reaction provided pyrrolidine 5g in 66% yield and 85% ee. The (4S,5R)-di-Ph-box ligand was used in this example because it provided greater enantioselectivity than (S,S)-Ph-box. Oxidative cleavage of the alkene and subsequent reductive amination provided the 5-HT7 receptor antagonist 9. This synthesis was accomplished in 4 steps from commercially available reagents without the aid of protecting groups and is several steps shorter than its original synthesis.14
Scheme 2.

Concise Synthesis of a 5-HT7 Receptor Antagonist
In conclusion, we have developed an efficient and enantioselective alkyl Heck-type coupling cascade for the formation of functionalized chiral indolines, pyrrolidines and an isoquinoline from the respective acyclic γ- and δ-alkenylsulfonamides. The concise synthesis of a 5-HT7 receptor antagonist was accomplished to demonstrate the utility of the method. Sulfonamides and sultams are common moieties found in bioactive compounds.14,15 This method accesses such targets directly, enantioselectively and without the use of protecting groups. Removal of the N-sulfonyl groups can further provide valuable chiral amines. Further investigation into the heterocycle and alkene scope and application in the total synthesis of bioactive alkaloids is ongoing.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (NIGMS 078383) for support of this work. We thank William W. Brennessel and the X-ray Crystallographic Facility at the University of Rochester for the X-ray structure of 2a.
Footnotes
The authors declare no competing financial interest.
Experimental procedures, spectroscopic data, and crystallography data of 2a (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Recent examples: Ilies L, Asako S, Nakamura E. J Am Chem Soc. 2011;133:7672. doi: 10.1021/ja2017202.Schomaker JM, Boyd WC, Stewart IC, Toste FD, Bergman RG. J Am Chem Soc. 2008;130:3777. doi: 10.1021/ja800738d.Tsai AS, Brasse M, Bergman RG, Ellman JA. Org Lett. 2011;13:540. doi: 10.1021/ol102890k.Rakshit S, Grohmann C, Besset T, Glorius F. J Am Chem Soc. 2011;133:2350. doi: 10.1021/ja109676d.Shi BF, Zhang YH, Lam JK, Wang DH, Yu JQ. J Am Chem Soc. 2010;132:460. doi: 10.1021/ja909571z.Hatamoto Y, Sakaguchi S, Ishii Y. Org Lett. 2004;6:4623. doi: 10.1021/ol047971u.Xu YH, Lu J, Loh TP. J Am Chem Soc. 2009;131:1372. doi: 10.1021/ja8084548.Yu H, Jin W, Sun C, Chen J, Du W, He S, Yu Z. Angew Chem, Int Ed. 2010;49:5792. doi: 10.1002/anie.201002737.
- 2.Selected reviews: Oestrich M, editor. The Mizoroki-Heck Reaction. John Wiley & Sons; West Sussex, UK: 2009. Heck RF. Acc Chem Res. 1979;12:146.de Meijeres A, Meyer FE. Angew Chem, Int Ed. 1994;33:2379.Beletskaya IP, Cheprakov AV. Chem Rev. 2000;100:3009. doi: 10.1021/cr9903048.Cu-catalyzed Heck-type reactions: Iyer S, Ramesh C, Sarkar A, Wadgaonkar PP. Tetrahedron Lett. 1997;38:8113.Li JH, Wang DP, Xie YX. Tetrahedron Lett. 2005;46:4941.Peng Y, Chen J, Liu M, Gao W, Wu H. Synthesis. 2011:213.
- 3.(a) Firmansjah L, Fu GC. J Am Chem Soc. 2007;129:11340. doi: 10.1021/ja075245r. [DOI] [PubMed] [Google Scholar]; (b) Bloome KS, McMahen RL, Alexanian EJ. J Am Chem Soc. 2011;133:20146. doi: 10.1021/ja2091883. [DOI] [PubMed] [Google Scholar]; (c) Stowers KJ, Fortner KC, Sanford MS. J Am Chem Soc. 2011;133:6541. doi: 10.1021/ja2015586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heck-type couplings that proceed via alkyl radical intermediates: Branchaud BP, Detlefsen WD. Tetrahedron Lett. 1991;32:6273.Ikeda Y, Nakamura T, Yorimitsu H, Oshima K. J Am Chem Soc. 2002;124:6514. doi: 10.1021/ja026296l.Terao J, Kambe N. Bull Chem Soc Jpn. 2006;79:663.Lebedev SA, Petrov ES, Beletskaya IP. J Organomet Chem. 1988;344:253.Song CX, Cai GX, Farrell TR, Jiang ZP, Li H, Gan LB, Shi ZJ. Chem Commun. 2009:6002. doi: 10.1039/b911031c.Furst L, Matsuura BS, Narayanam JMR, Tucker JW, Stephenson CRJ. Org Lett. 2010;12:3104. doi: 10.1021/ol101146f.Weiss ME, Kreis LM, Lauber A, Carreira EM. Angew Chem, Int Ed. 2011;50:11125. doi: 10.1002/anie.201105235.
- 5.Intermolecular additionof β-aminoalkyl radicals to alkenes has been demonstrated: Maria EJ, Da Silva AD, Fourrey JL, Machado AS, Robert-Gero M. Tetrahedron Lett. 1994;35:3301.
- 6.Intramolecular Heck-type coupling of β-aminoalkyl intermediates with alkenes has been reported: Yip KT, Yang M, Law KL, Zhu N, Yang D. J Am Chem Soc. 2006;128:3130. doi: 10.1021/ja060291x.He W, Yip KT, Zhu NY, Yang D. Org Lett. 2009;11:5626. doi: 10.1021/ol902348t.Fuller PH, Chemler SR. Org Lett. 2007;9:5477. doi: 10.1021/ol702401w.
- 7.Cu-catalyzed doubly intramolecular carboamination: Zeng W, Chemler SR. J Am Chem Soc. 2007;129:12948. doi: 10.1021/ja0762240.Miao L, Haque I, Manzoni MR, Tham WS, Chemler SR. Org Lett. 2010;12:4739. doi: 10.1021/ol102233g.
- 8.Cu-catalyzed aminooxygenation: Fuller PH, Kim JW, Chemler SR. J Am Chem Soc. 2008;130:17638. doi: 10.1021/ja806585m.Paderes MC, Chemler SR. Eur J Org Chem. 2011;3679 doi: 10.1002/ejoc.201100444.
- 9.Cu-catalyzeddiamination: Sequeira FC, Turnpenny BW, Chemler SR. Angew Chem, Int Ed. 2010;49:6365. doi: 10.1002/anie.201003499.
- 10.(a) Sherman ES, Fuller SR, Kasi D, Chemler SR. J Org Chem. 2007;72:3896. doi: 10.1021/jo070321u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Paderes MC, Belding L, Fanovic B, Dudding T, Keister JB, Chemler SR. Chem—Eur J. 2012 doi: 10.1002/chem.201101703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chemler SR. J Organomet Chem. 2011;696:150. doi: 10.1016/j.jorganchem.2010.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Selected Pd-catalyzed intermolecular carboamination/cycliza-tion of alkenes with unfunctionalized arenes and functionalized alkenes and arenes: Rosewall CF, Sibbald PA, Liskin DV, Michael FE. J Am Chem Soc. 2009;131:9488. doi: 10.1021/ja9031659.Mai DN, Wolfe JP. J Am Chem Soc. 2010;132:12157. doi: 10.1021/ja106989h.Houlden CE, Bailey CD, Ford JG, Gagne MR, Lloyd-Jones GC, Booker-Milburn KI. J Am Chem Soc. 2008;130:10066. doi: 10.1021/ja803397y.Au-catalyzed intermolecular carboamination/cyclization of alkenes with functionalized arenes: Zhang G, Cui L, Wang Y, Zhang L. J Am Chem Soc. 2010;132:1474. doi: 10.1021/ja909555d.Brenzovich WE, Benitez D, Lackner AD, Shunatona HP, Tkatchouk E, Goddard WA, Toste FD. Angew Chem, Int Ed. 2010;49:5519. doi: 10.1002/anie.201002739.
- 13.(a) Choi J, Tang L, Norton JR. J Am Chem Soc. 2007;129:234. doi: 10.1021/ja066325i. [DOI] [PubMed] [Google Scholar]; (b) Fischer H, Radom L. Angew Chem, Int Ed. 2001;40:1340. doi: 10.1002/1521-3773(20010417)40:8<1340::aid-anie1340>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 14.Lovell PJ, Bromidge SM, Dabbs S, Duckworth DM, Forbes IT, Jennings AJ, King FD, Middlemiss DN, Rahman SK, Saunders DV, Collin LL, Hagan JJ, Riley GJ, Thomas DR. J Med Chem. 2000;43:342. doi: 10.1021/jm991151j. [DOI] [PubMed] [Google Scholar]
- 15.(a) Rotella DP, Sun Z, Zhu Y, Krupinski J, Pongrac R, Seliger L, Normandin D, Macor JE. J Med Chem. 2000;43:5037. doi: 10.1021/jm000336j. [DOI] [PubMed] [Google Scholar]; (b) Lee D, Long SA, Murray JH, Adams JL, Nuttall ME, Nadeau DP, Kikly K, Winkler JD, Sung CM, Ryan MD, Levy MA, Keller PM, DeWolf WE. J Med Chem. 2001;44:2015. doi: 10.1021/jm0100537. [DOI] [PubMed] [Google Scholar]; (c) Cole DC, Lennox WJ, Lombardi S, Ellingboe JW, Bernotas RC, Tawa GJ, Mazandarani H, Smith DL, Zhang G, Coupet J, Schechter LE. J Med Chem. 2005;48:353. doi: 10.1021/jm049243i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.