

Abstract
We have investigated the dynamic properties of the switch I region of the GTP-binding protein Ras by using mutants of Thr-35, an invariant residue necessary for the switch function. Here we show that these mutants, previously used as partial loss-of-function mutations in cell-based assays, have a reduced affinity to Ras effector proteins without Thr-35 being involved in any interaction. The structure of Ras(T35S)⋅GppNHp was determined by x-ray crystallography. Whereas the overall structure is very similar to wildtype, residues from switch I are completely invisible, indicating that the effector loop region is highly mobile. 31P-NMR data had indicated an equilibrium between two rapidly interconverting conformations, one of which (state 2) corresponds to the structure found in the complex with the effectors. 31P-NMR spectra of Ras mutants (T35S) and (T35A) in the GppNHp form show that the equilibrium is shifted such that they occur predominantly in the nonbinding conformation (state 1). On addition of Ras effectors, Ras(T35S) but not Ras(T35A) shift to positions corresponding to the binding conformation. The structural data were correlated with kinetic experiments that show two-step binding reaction of wild-type and (T35S)Ras with effectors requires the existence of a rate-limiting isomerization step, which is not observed with T35A. The results indicate that minor changes in the switch region, such as removing the side chain methyl group of Thr-35, drastically affect dynamic behavior and, in turn, interaction with effectors. The dynamics of the switch I region appear to be responsible for the conservation of this threonine residue in GTP-binding proteins.
An estimated 60–100 different GTP-binding proteins of the Ras superfamily belonging to different subfamilies have been identified and shown to regulate a diverse array of signal transduction reactions and/or transport processes. These small GTP-binding proteins, but also the larger Gα proteins or the protein synthesis elongation factors, contain conserved signature motifs in the primary sequence. Accordingly, the structures of many of these proteins show a common structural core called the G domain, an α/β fold consisting of six β strands and five α helices (1). The common structural core is modified by insertions and/or additional structural elements. The structures of several proteins have been solved in both the GDP- and GTP-bound form, which showed that the conformational change is mostly confined to the loop L2-β2 (by using the Ras nomenclature) and the β3/α2 regions, which have accordingly been called switches I and II (2). It turned out that the active site of the GDP-bound conformations shows large variations, whereas the triphosphate structures are very similar (3). It was also deduced from these studies that the conformational change is triggered when two hydrogen bonds to the γ-phosphate from switches I and II, involving invariant Thr and Gly residues, respectively, are released after GTP hydrolysis (3). NMR structural studies have shown that the switch regions show an inherent mobility much higher than the rest of the G domain (4–6). From temperature-dependence measurements and line-shape analysis of 31P-NMR spectra of Ras⋅GppNHp, it had been deduced earlier that in solution, the effector region interconverts between two main conformations that are characterized by different chemical shift values for the resonances of the α- and β-phosphate groups (7). Furthermore, EPR studies of the Mg2+-binding site of Ras suggested that the coordination of the Thr-35 to the γ-phosphate might be transient in solution, indicating a high flexibility of the side chain and/or the loop containing the Thr-35 (8, 9).
The switch I region contains only one invariant residue, Thr-35 in Ras (10), which is involved via its side chain hydroxyl in the coordination of the crucial metal ion and, via its main chain NH, in contacting the γ-phosphate. Thr-35 is most likely invariant in all Ras-related proteins (not counting pseudogene products) and is never substituted by a Ser, in contrast to the P-loop motif GxxxxGKS/T, where the interaction with the metal ion is performed by either Ser or Thr. Following the observation that the T35S mutation—frequently used in biological studies as a partial loss-of-function mutation—has a reduced affinity to RalGDS (see below) without Thr making a significant contribution to the interaction (11, 12), we have investigated the role of Thr-35 for the properties of H-Ras (subsequently referred to as Ras) and found its methyl group to be responsible for the dynamic behavior of the switch region. This observation, in turn, is most likely the reason for the complete invariance of this threonine residue.
Methods
Protein Purification.
Wild-type and mutants of H-Ras (1) were expressed in Escherichia coli and purified as described (13). Nucleotide exchange to GppNHp as well as N-methylanthraniloyl-GppNHp was done as described by John et al. (14). Free nucleotide and phosphates were removed by gel filtration. The final purity of the protein was >95%, as judged by SDS/PAGE. The concentration of the protein solution was determined by Bradford assay, with BSA as standard (15). The amount and nature of protein-bound nucleotide were analyzed by C18 reverse-phase HPLC and quantified with a calibrated detector (Beckman Coulter) and integrator (Shimadzu). The Ras-binding domains of human c-Raf-1 and RalGDS were expressed in E. coli and purified as described (16, 17).
NMR Spectroscopy.
31P NMR spectra were recorded with a Bruker DRX-500 NMR spectrometer operating at 202 MHz. Measurements were performed in a 10-mm probe by using 8-mm Shigemi sample tubes at various temperatures. Ras protein solutions (1 mM) in 40 mM Hepes, pH 7.4/10 mM MgCl2/150 mM NaCl/2 mM dithioerythritol contained 10% D2O to get a lock signal. 2,2-Dimethyl-2-silapentane-5-sulfonate (0.1 mM) was added to calibrate the spectra by indirect referencing. A Ξ-value of 0.4048073561 reported by Maurer and Kalbitzer (18) was used, which corresponds to 85% external phosphoric acid contained in a spherical bulb.
Stopped-Flow Measurements.
Stopped-flow experiments using N-methylanthraniloyl-GppNHp bound to Ras were used to measure the interaction of different Ras-binding domains (RBDs) with Ras in an Applied Photophysics (Surrey, U.K.) SX16MV apparatus, as described (19, 20). The reactions were carried out in 40 mM Hepes, pH 7.4/10 mM MgCl2/150 mM NaCl/2 mM dithioerythritol at 10°C by using an excitation wavelength of 360 nm and a cutoff filter (408 nm) in front of the detector. Exponential and hyperbolic fits to the data, respectively, were done by using the program grafit (Erithakus Software, Horley, U.K.).
Binding Affinity of Mg2+ to Ras.
The affinity of Mg2+ to Ras nucleotide complexes was estimated by measuring the dependence of Ras⋅mGppNHp dissociation rate constants on the Mg2+ concentration at 25°C. Different amounts of Mg2+ were added to 0.5 μM Ras⋅mGppNHp in 40 mM Hepes, pH 7.5/150 mM NaCl/2 mM dithioerythritol. Low Mg2+ concentrations were adjusted with EDTA by using an apparent dissociation constant of 1.7 μM for the Mg2+/EDTA complex (21). Fluorescence decrease (excitation/emission at 360/450 nm) because of dissociation of N-methylanthraniloyl nucleotide was started by addition of 200 μM of nonlabeled GppNHp, and time traces were fitted exponentially.
X-Ray Crystallography.
Crystals of the T35S mutant of Ras in complex with GppNHp and magnesium were grown with a reservoir of 24% polyethylene glycol 1500/50 mM CaCl2/100 mM Tris, pH 7.5, by using 20 mg ml−1 protein stock solution. The drop was composed of 5 μl protein stock solution, 4 μl reservoir, and 1 μl Spermine-tetraHCl. Crystals grew to a final size of 0.4 × 0.1 × 0.05 mm in the space group P212121 with three molecules in the asymmetric unit. The crystals were very sensitive to changes in the mother liquor, therefore no ideal cryoprotectant was found, and the crystals were measured at room temperature. Data were collected on an FR591 (Nonius Delft, The Netherlands) rotating anode equipped with a MAR345 (Mar Research, Hamburg) image plate detector and processed with xds (22). The crystals diffracted to better than 2.5 Å, but because of the small size of the crystals, the data were usable only to 2.9 Å (Table 3). The structure was solved by using the program amore from the ccp4 suite (23) with Ras⋅GppNHp (Protein Data Bank ID code 5p21) as a search model and yielded unambiguous solutions for the three molecules in the asymmetric unit (R factor 38.4% and correlation 62.9%, next highest peak 44.8 and 48.3%). The model was built with o (24) and refined with cns (25) with noncrystallographic symmetry restraints to a final Rfree of 28.2% (Rcrys 24.2%) with good model geometries (Table 3). The final model comprises residues 1–29,38–60,70–166 for molecule A, 1–29,39–61,68–166 for molecule B, and 1–31,38–63,71–166 for molecule C.
Table 3.
Crystallographic data
| Space group: P212121 | |
| Unit cell: a = 63.89 Å, b = 79.06 Å, c = 94.04 Å, α = β = γ = 90.0 | |
| Resolution: 50.0–2.9 Å | |
| Unique reflections: 10,369; observed reflections, 60,892 | |
| Completeness: 95.2% to 2.9 Å (last shell 3.0–2.9 Å: 91.6%) | |
| I/σ: 7.13 (last shell 3.0–2.9 Å: 1.8) | |
| Rmrgd-F*: 20.4% (last shell 3.0–2.9 Å: 38.7%) | |
| Model: Protein atoms | 3,595 |
| Nucleotide atoms | 96 |
| Magnesium atoms | 3 |
| Refinement: Resolution | 50.0–2.9 Å |
| Reflections | 9,016 |
| Rcrys† | 24.2% |
| Rfree | 28.2% |
| rms deviations from expected geometry: | |
| Bond lengths, Å | 0.008 |
| Bond angles, deg | 1.2 |
| Overall B value, Å2 | 58.0 |
| B value Mg atoms, Å2 | 30.85 |
Quality of amplitudes (F) in the scaled data set; for definition, see ref. 50.
Rcrys = (hkl∥Fobs| − |Fcalc∥/(hkl∥Fobs|, where Fobs denotes the observed structure factor amplitude, and Fcalc denotes the structure factor amplitude calculated from the model. Ten percent of reflections were used to calculate Rfree.
Results and Discussion
Affinity of Wild-Type Ras, Ras(T35S), and Ras(T35A) to Different Effectors.
As is generally accepted, Ras and other Ras-like proteins use different effectors to relay signals into different cellular responses (26). The T35S mutation has originally been identified in a screen for mutations that selectively disrupt specific signaling pathways of the Ras protein (27). It has been proposed, by using either the two-hybrid method or pull-down assays, that the mutation affects only the interaction with RalGDS and is thus used as a tool to prove or disprove the participation of RalGDS in a particular pathway (28–35). However, quantitative measurements by the GDI method (16, 17) of the interaction of Ras(T35S) with effectors show that the mutation also drastically effects the interaction with other effectors. Table 1 shows that the affinity to Raf-RBD is decreased 60-fold, at least 29-fold to RalGDS-RBD, and effects binding to AF6-RBD and Byr2-RBD (36) as well. Because the absolute affinity of wild-type Ras to Raf-RBD is much higher than to the other effectors, this would be a possible explanation for the finding that in vivo T35S appears to selectively activate the Raf pathway. Replacing Thr-35 in Ras by an alanine residue leads to a further reduction of the affinity to Raf-RBD by a factor of three (16). For measuring binding of Ras(T35A) to the RBDs of RalGDS and AF6, the affinities were too low to be determined by the GDI method.
Table 1.
Interaction of Ras(wt) and Ras(T35S) with different effectors
| H-Ras-variant | Effector | KD/μM | Relative
affinity K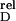 (mutant/wt) (mutant/wt)
|
|---|---|---|---|
| Ras(wt)⋅mGppNHp | Raf-RBD | 0.02 | |
| RalGDS-RBD | 1.0 | ||
| AF6-RBD | 3.0 | ||
| Byr2-RBD | 0.3 | ||
| Ras(T35S)⋅mGppNHp | Raf-RBD | 1.2 | 60 |
| RalGDS-RBD | >100 | >29 | |
| AF6-RBD | >100 | >25 | |
| Byr2-RBD | 3.7 | 12 |
The apparent dissociation constants KD were determined by the GDI method at 37°C, according to Herrmann et al. (17).
That the affinity between Ras and the effectors is substantially weakened by the T35S mutation is surprising at first glance, considering that in the three-dimensional structures of the Raps⋅Raf-RBD complex, which mimics the Ras⋅Raf-RBD complex, no direct interaction between T35 and the effector can be detected (37). Furthermore, the interaction of a Ras mutant with RalGDS-RBD showed only a weak water-mediated contact of Thr-35 (11) and none for wild-type Ras (12).
Conformational Transitions in Ras Proteins.
We have shown earlier that the switch I region of Ras in the GppNHp-bound state adopts at least two conformations, which can be detected by different chemical shifts in the 31P-NMR spectrum. From the two conformational states, conformation 2 is slightly preferred. From temperature-dependence measurements and line-shape analysis, the interconversion between the two conformations was found to be strongly temperature dependent, with a rate of 130 s−1 at 5°C and 1,900 s−1 at 25°C (7). Fig. 1 shows the 31P-NMR spectra of wild-type Ras and selected mutants with substitutions in the effector loop. Whereas wild-type Ras shows two resonance lines each for the α- and β-phosphate, as described earlier (7, 38), T35S shows only single peaks for the α-phosphate (−11.10 ppm) and β-phosphate (−2.57 ppm) (Table 2). The T35A mutation, also originally described as a partial loss-of-function mutation (27), again shows only single resonance lines for the α- and β-phosphate. This result indicates that in the time average, the effector loops of Ras(T35S) and Ras(T35A) adopt a similar conformation as in state 1 of wild-type Ras, and state 2 is not populated significantly. Because effector binding induces state 2 (7, 38, 39), it most likely corresponds to a rather well-defined conformation (Fig. 2), as found in the complexes with effectors (40), where the OH of Thr-35 coordinates the Mg2+ ion (distance 2.2 Å) and its methyl packs against the effector loop, and where Tyr-32 is close to the phosphates (closest approach to β-P 5.2 Å). State 1, however, may represent either a fixed conformation or an equilibrium of conformational substates in fast exchange on the NMR time scale. Both models would result in only one resonance each for the α- and β-phosphates. Such a spectrum would thus also be expected for an effector loop that has become more flexible (see below), because of the loss of the methyl group of Thr-35, an effect that would be even more pronounced for Ras(T35A), which has lost its hydroxyl group as well.
Figure 1.

31P-NMR spectra of wild-type or mutant Ras⋅GppNHp (in the presence of Mg2+). Spectra of different Ras proteins (1 mM) as indicated were recorded at 5°C, as described in Methods. The chemical shifts of the α- and β-phosphates corresponding to states 1 and 2 are summarized in Table 2.
Table 2.
31P chemical shifts and conformational states of Ras and Ras mutants in the presence and absence of different RBDs
| α
phosphate
|
β phosphate
|
γ
phosphate
|
K* | |||
|---|---|---|---|---|---|---|
| δ1/ppm | δ2/ppm | δ1/ppm | δ2/ppm | δ1,2/ppm | ||
| Ras(wt)⋅GppNHp | −11.20 | −11.70 | −2.56 | −3.31 | −0.25 | 1.09 |
| + Raf-RBD* | −11.7 | −3.6 | −0.3 | |||
| + RalGDS-RBD† | −11.6 | −3.4 | −0.4 | |||
| Ras(T35S)⋅GppNHp | −11.10 | −2.57 | −0.27 | <0.05 | ||
| + Raf-RBD | −11.60 | −3.41 | −0.29 | |||
| + RalGDS-RBD | −11.53 | −3.26 | −0.25 | |||
| Ras(T35A)⋅GppNHp | −11.09 | −2.49 | −0.33 | <0.05 | ||
| + Raf-RBD | −10.52 | −2.69 | −0.35 | |||
| + RalGDS-RBD | −11.09 | −2.49 | −0.32 | |||
Figure 2.

Structure of the nucleotide and effector region of Ras taken from the structure of Raps⋅GppNHp in complex with Raf-RBD (37). Ras is shown with van der Waals surface, highlighting the switch I region (red worm), with the indicated side chains and the guanine nucleotide in ball-and-stick representation. Mg2+ is shown as a yellow sphere. The image was prepared by using grasp (49).
The notion that “state 1” is not a well-defined arrangement of atoms fixed in space is supported by results from three glycine mutants of Ras, Ras(V29G), Ras(I36G), and the double mutant Ras(V29G/I36G). The mutated residues are sitting at positions where they are supposed to function as “hinges” for the effector loop, as their mutation to glycine was shown to increase the flexibility of the loop (41). Indeed, the 31P-NMR spectra of those mutants show single peaks for the phosphates at positions corresponding to the proposed flexible state 1 (Fig. 1; the spectrum of the double mutant is shown). In principle, the internal mobility of switches I and II could be studied directly in isotope-enriched Ras protein by relaxation time measurements. However, the relevant residues are not visible in wild-type Ras⋅Mg2+⋅GppNHp (5, 42), probably because they are exchange broadened beyond detection. It would be interesting to study the behavior of the T35 mutants by heteronuclear NMR spectroscopy. From our data, one would predict that for the mutants, the residues of the effector loop should now be visible in the multidimensional spectra because they occur only in state 1. Relaxation time measurements now feasible with these mutants should indicate that there is a fast internal mobility with a low-order parameter S (2) describing the conformational substates of the effector loop.
Crystal Structure of Ras(T35S)⋅Mg2+⋅GppNHp.
To get more information about the conformation of the effector loop, we solved the structure of Ras(T35S) by x-ray crystallography. The mutant crystallizes in a space group different from wild-type Ras (orthorhombic in contrast to the trigonal wild-type crystals), which indicates a rather drastic change in the structure/dynamics (Table 3). The overall fold is very similar, as expected (rms 1–29,39–60,71–166 for molA, 0.73 Å; molB, 0.47 Å; and molC, 0.5 Å) and contains three molecules in the asymmetric unit. The nucleotide is located in a similar position and makes basically the same contacts as in the wild-type protein (Fig. 3A). Although Ser-35 is not visible (see below), the position of the magnesium ion can be identified, which is somewhat (0.3 Å) further away from the β- and γ-phosphate oxygens as compared with wild-type Ras⋅GppNHp, where the hydroxyl group of Thr-35 contacts OH of Ser-17 (3.0 Å), the γ-phosphate oxygens (3.2 Å), the magnesium ion (2.3 Å), and two water molecules. However, there are significant differences in the switch regions. In all three T35S molecules, the switch I and II regions are disordered (Fig. 3A). In detail, the invisible regions comprise residues 30–37 and 61–69 (molA), 30–38 and 62–67 (molB), and 31–37 and 64–70 in molC, respectively, indicating that the effector loop is extremely mobile in the crystals (Fig. 3 A and B). The remainder of the structure is essentially unaltered. It can be shown that the differences are not because of (missing) crystal contacts by comparison with the structure of Ras⋅GppCH2p [Protein Data Bank ID code 6q21 (43)], which was crystallized in space group P21 with four molecules per asymmetric unit. In this space group, two molecules have no crystal contacts at all in the effector loop region, and still the effector loop shows density. The temperature of data collection, the crystallization conditions, and pH values were similar and thus cannot account for the observed differences in flexibility. In the T35S structure, one molecule (B) has no crystal contacts in the effector loop region (the effector loop was modeled according to the structure in 5p21 because no density was visible; see Fig. 3B), the other molecule could possibly have contacts between either Pro-34 and Arg-135 (A) or Tyr-32, and the symmetry-related Arg-41 and Asp-54 as well as Pro-34 with Glu-3 and Glu-31 with Ser-39 (C). Here, the putative effector loop taken from 5p21 would clash with the symmetry-related molecules so that the effector loop must be somewhere else (and flexible, because no density is visible).
Figure 3.

X-ray structural analysis of Ras(T355S) in the GppNHp-state. (A) Superimposition of the structures (in worm plot) of wild-type Ras (Protein Data Bank ID code 5p21) in blue and molA of Ras(T35S) in red. Neither switch I nor II of the mutant is visible. (B) Part of the final 2Fo − Fc map at 1.2 σ with the Ras(T35S) model (molecule C in the asymmetric unit) as a stick molecule and with the three phosphates of GppNHp in purple and Mg2+ as a ball in magenta, as indicated. The effector region from Tyr-32 to Glu-37, including Ser-35, is not visible. The corresponding region from the wild-type Ras⋅GppNHp structure (45) is shown in green, with corresponding residues as indicated.
Conformational State of the Effector Loop of Ras Bound to Effectors.
We have shown earlier that the two conformations represented by the 31P-NMR spectrum can be correlated to the biological function of Ras, as only one conformation (state 2) is found in the complex of Ras with either Raf-RBD (7), RalGDS-RBD (38), or AF6-RBD (39). The dynamic equilibrium of the switch region is thus of prime importance for the function of Ras in its interactions with effectors, probably also with regulators (7). In the spectrum shown in Fig. 1, Ras(T35S) can be seen exclusively in the first nonbonding conformation. Fig. 4 shows that increasing amounts of Raf-RBD or RalGDS-RBD induce a complete shift of the α and β resonances in the 31P-NMR spectrum to state 2. The chemical shift values are summarized in Table 2. Thus, although the mutant has a lower affinity and higher flexibility, it most likely adopts a conformation similar to that seen in the wild-type complex. The binding affinity of RalGDS-RBD, not measurable by the GDI or stopped-flow method, can be qualitatively determined with NMR titration, because the relation of unbound and complexed Ras protein can be taken from the integrals of the separated β lines. Using these, we get a dissociation constant between Ras(T35S)⋅GppNHp and RalGDS-RBD of about 360 μM, very much higher than the wild-type constant of 1 μM (Table 1).
Figure 4.

31P-NMR spectroscopic analysis of complex formation between Ras(T35S) (A and C) and Ras(T35A) (B and D) in the GppNHp form and cRaf-RBD (A and B) and RalGDS-RBD (C and D). Increasing amounts of the effectors were added to 1–2 mM Ras protein, and spectra were recorded as described in Fig. 1 and Methods. The ratio of RBDs to Ras proteins is indicated. The chemical shift values are summarized in Table 2.
In contrast to the above, the T35A mutant shows only a broadening of the resonance lines and a slight chemical shift on addition of the Raf-RBD (Table 2) but no shift to positions characteristic for state 2. Addition of RalGDS-RBD produces neither a shift of the β phosphate nor a pronounced line broadening, even at millimolar concentrations. When no special effects have to be considered such as line broadening by chemical exchange, the transverse relaxation rate 1/T2 and hence the line width should increase after binding of the effector because of an increase in the rotational correlation time of the complex. In a first approximation, the increase of 1/T2 is expected to be proportional to the increase of the molecular mass. Using the line broadening of the relatively narrow γ-phosphate line observed as function of the RalGDS-RBD concentration, we can estimate a dissociation constant of about 10 mM for the interaction with Ras(T35A).
Binding of Ras(V29G/I36G) to Raf-RBD again causes a shift toward state 2 (data not shown). However, much higher concentrations of RBD are needed compared with wild-type protein to shift the equilibrium. A dissociation constant of about 1 mM can be estimated. These results support the idea that the observed “state 1” in Ras(T35S) indeed corresponds to a highly flexible effector loop.
Kinetics of the Ras–Effector Interaction.
Kinetic stopped-flow experiments by using the fluorescent analogue
mGppNHp bound to Ras showed that the description of the binding of
effectors to Ras requires (at least) a two-step model. The data could
be fitted satisfactorily by assuming that an initial low-affinity
encounter complex isomerizes to the final high-affinity complex (19,
39), and that the Ras effector complex is highly dynamic, showing both
fast association and dissociation (19, 44). The binding reaction has
been described by Eq. 1. 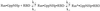
[1]
For wild-type Ras, plotting the observed pseudofirst-order association rate constants against the concentration of Raf-RBD (Fig. 5) and fitting the data according to Eq. 1 gives a dissociation constant K1 for the first step of 12 μM and 415 sec−1 for the rate-limiting isomerization reaction at 10°C. For Ras(T35S), the equilibrium constant K1 of the initial complex formation is higher by a factor of 5.9, and the isomerization rate constant drops down to 211 s−1. The overall affinity KD = K1 × K2 drops 42-fold to 2 μM, very close to what has been found by the equilibrium method (Table 1). The T35S mutant thus interacts with Raf-RBD qualitatively similar to the wild-type protein, although both the kinetic and equilibrium parameters of the binding are affected by the mutation. Ras(T35A) behaves quite differently: not only does it show a fluorescence increase on Raf binding (not shown), as opposed to a decrease observed for wild-type and T35S, but it also shows no saturation of the observed rate constants (Fig. 5, Table 4). This seems to indicate that alanine in position 35, which cannot coordinate to the Mg2+ ion as shown for the threonine residue by the three-dimensional structure (2, 45) and expected for serine, forms a different effector-binding conformation, and the association does not involve a rate-limiting conformational change. The ratio of dissociation and association rate constants gives an equilibrium dissociation constant for Raf-RBD of 7.2 μM, 3.5-fold higher than Ras(T35S). This is close to the results found by the GDI method, where Ras(T35A) shows a dissociation constant of 3.4 μM (16), 3-fold higher than the serine mutant (Table 1). Stopped-flow measurements of the association kinetics between both T35 mutants and RalGDS-RBD show no change in fluorescence signal.
Figure 5.

Kinetics of binding of Raf-RBD to Ras and Ras-mutants. 0.5 μM Ras complexed with mGppNHp was mixed with increasing concentrations of Raf-RBD in a stopped-flow apparatus, and the resulting pseudo-first-order reactions were measured (not shown). The observed pseudo-first-order rate constants are plotted against the concentrations of effector, as indicated, and the points are fitted to a two-step binding equation shown in the text. The resulting parameters are shown in Table 4.
Table 4.
Kinetics of interaction between wild-type and mutant Ras⋅mGppNHp with Raf-RBD measured at 10°C by stopped flow, as described in Methods
| Raf-RBD+ | K1 = k−1/k1, μM | k2, s−1 | kon = k2/K1, μM−1s−1 | koff = k−2, s−1 | KD = (K1K2), μM |
|---|---|---|---|---|---|
| Ras(wt) | 11.7 | 415 | 35.5 | 1.7 | 0.05 |
| Ras(T35S) | 68.8 | 211 | 3.1 | 6.5 | 2.1 |
| Ras(T35A) | — | — | 0.7 | 5.1 | 7.2 |
Because the x-ray structure shows the hydroxyl group of Thr-35 to be involved in the coordination of the metal ion, the differences of RBD-binding affinities between wild-type and Thr-35 mutants of Ras could be caused by the impaired Mg2+ of the T35A mutant. For the complex of Ras with the fluorescent nucleoside diphosphate mGDP, no significant change in the Mg2+ dissociation constants were previously found between the wild-type and the T35A-mutant (46). Here, we determined the affinity of Ras⋅mGppNHp for Mg2+ by measuring the dissociation rate of mGppNHp with varying Mg2+ concentrations and found only 3- and 4-fold decreases in Mg2+ affinity for the T35S and T35A mutant, respectively, as compared with wild type (data not shown). Under the experimental conditions used for 31P NMR experiments, the state of the protein can be directly observed. The protein is always complexed by Mg2+⋅GppNHp, because removal of the Mg2+ ion leads to characteristic chemical shift changes (38).
Conclusion
Our structural and kinetic data have shown that minor changes in the structure of Ras in and around the switch I region, such as replacement of GTP by GppNHp (7) or of Thr-35 by Ser or Ala, perturb the equilibrium of the conformational states and possibly also the rate of interconversion of these conformations, and thereby also effect interactions with target proteins. Thr-35, being invariant in all Ras-like (and other) GTP-binding proteins, is thus apparently conserved not for structural but rather for the dynamic properties of these switch molecules. Whereas a Thr→Ser mutation should be tolerated from structural considerations, the results presented here indicate that the methyl group of this residue is important for the capability of Ras to dynamically switch between different conformations. Because this residue has been mutated not only in Ras but also in other Ras-like proteins (i.e., refs. 47 and 48) and found to be important for function, dynamic flexibility in the switch regions of these proteins, a prerequisite for their function, might be commonly regulated by the Thr residue. Our results also show that the molecular switch function of GTP-binding proteins is not just because of whether the γ-phosphate is present on the guanine nucleotide, but that the dynamic interchange between different conformations is a structure-based inherent property of these molecules, and determined by a single methyl group in the effector region.
Acknowledgments
We thank Astrid Krämer for assistance with the stopped-flow measurements and Dorothee Kühlmann for cloning the glycine mutants. This work was supported by a Deutsche Forschungsgemeinschaft Grant KA647/10–2 (to H.R.K.) and by European Union Grants Bio4-CT96–1110 and QLK3-CT-1999–00875 (to A.W. and H.R.K.).
Abbreviation
- RBD
Ras-binding domain
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates reported in this paper have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1iaq).
References
- 1.Wittinghofer A, Pai E F. Trends Biochem Sci. 1991;16:382–387. doi: 10.1016/0968-0004(91)90156-p. [DOI] [PubMed] [Google Scholar]
- 2.Milburn M V, Tong L, deVos A M, Brunger A, Yamaizumi Z, Nishimura S, Kim S H. Science. 1990;247:939–945. doi: 10.1126/science.2406906. [DOI] [PubMed] [Google Scholar]
- 3.Wittinghofer A. In: The Functioning of Molecular Switches in Three Dimensions. Hall A, editor. Oxford: Oxford Univ. Press; 2000. pp. 244–310. [Google Scholar]
- 4.Redfield A G, Papastavros M Z. Biochemistry. 1990;29:3509–3514. doi: 10.1021/bi00466a013. [DOI] [PubMed] [Google Scholar]
- 5.Ito Y, Yamasaki K, Iwahara J, Terada T, Kamiya A, Shirouzu M, Muto Y, Kawai G, Yokoyama S, Laue E D, et al. Biochemistry. 1997;36:9109–9119. doi: 10.1021/bi970296u. [DOI] [PubMed] [Google Scholar]
- 6.Loh A P, Guo W, Nicholson L K, Oswald R E. Biochemistry. 1999;38:12547–12557. doi: 10.1021/bi9913707. [DOI] [PubMed] [Google Scholar]
- 7.Geyer M, Schweins T, Herrmann C, Prisner T, Wittinghofer A, Kalbitzer H R. Biochemistry. 1996;35:10308–10320. doi: 10.1021/bi952858k. [DOI] [PubMed] [Google Scholar]
- 8.Halkides C J, Bellew B F, Gerfen G J, Farrar C T, Carter P H, Ruo B, Evans D A, Griffin R G, Singel D J. Biochemistry. 1996;35:12194–12200. doi: 10.1021/bi9605954. [DOI] [PubMed] [Google Scholar]
- 9.Bellew B F, Halkides C J, Gerfen G J, Griffin R G, Singel D J. Biochemistry. 1996;35:12186–12193. doi: 10.1021/bi960594b. [DOI] [PubMed] [Google Scholar]
- 10.Valencia A, Chardin P, Wittinghofer A, Sander C. Biochemistry. 1991;30:4637–4648. doi: 10.1021/bi00233a001. [DOI] [PubMed] [Google Scholar]
- 11.Huang L, Hofer F, Martin G S, Kim S H. Nat Struct Biol. 1998;5:422–426. doi: 10.1038/nsb0698-422. [DOI] [PubMed] [Google Scholar]
- 12.Vetter I R, Linnemann T, Wohlgemuth S, Geyer M, Kalbitzer H R, Herrmann C, Wittinghofer A. FEBS Lett. 1999;451:175–180. doi: 10.1016/s0014-5793(99)00555-4. [DOI] [PubMed] [Google Scholar]
- 13.Tucker J, Sczakiel G, Feuerstein J, John J, Goody R S, Wittinghofer A. EMBO J. 1986;5:1351–1358. doi: 10.1002/j.1460-2075.1986.tb04366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.John J, Sohmen R, Feuerstein J, Linke R, Wittinghofer A, Goody R S. Biochemistry. 1990;29:6058–6065. doi: 10.1021/bi00477a025. [DOI] [PubMed] [Google Scholar]
- 15.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 16.Herrmann C, Martin G A, Wittinghofer A. J Biol Chem. 1995;270:2901–2905. doi: 10.1074/jbc.270.7.2901. [DOI] [PubMed] [Google Scholar]
- 17.Herrmann C, Horn G, Spaargaren M, Wittinghofer A. J Biol Chem. 1996;271:6794–6800. doi: 10.1074/jbc.271.12.6794. [DOI] [PubMed] [Google Scholar]
- 18.Maurer T, Kalbitzer H R. J Magn Reson. 1996;113:177–178. doi: 10.1006/jmrb.1996.0172. [DOI] [PubMed] [Google Scholar]
- 19.Sydor J R, Engelhard M, Wittinghofer A, Goody R S, Herrmann C. Biochemistry. 1998;37:14292–14299. doi: 10.1021/bi980764f. [DOI] [PubMed] [Google Scholar]
- 20.Ahmadian M R, Hoffmann U, Goody R S, Wittinghofer A. Biochemistry. 1997;36:4535–4541. doi: 10.1021/bi962556y. [DOI] [PubMed] [Google Scholar]
- 21.Reinstein J, Schlichting I, Frech M, Goody R S, Wittinghofer A. J Biol Chem. 1991;266:17700–17706. [PubMed] [Google Scholar]
- 22.Kabsch W. J Appl Crystallogr. 1993;26:795–800. [Google Scholar]
- 23.Collaborative Computational Projekt, N.4. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 24.Jones T A, Kjeldgaard M. Methods Enzymol. 1997;277:173–208. doi: 10.1016/s0076-6879(97)77012-5. [DOI] [PubMed] [Google Scholar]
- 25.Brunger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 26.McCormick F, Wittinghofer A. Curr Opin Biotechnol. 1996;7:449–456. doi: 10.1016/s0958-1669(96)80123-6. [DOI] [PubMed] [Google Scholar]
- 27.White M A, Nicolette C, Minden A, Polverino A, Van Aelst L, Karin M, Wigler M H. Cell. 1995;80:533–541. doi: 10.1016/0092-8674(95)90507-3. [DOI] [PubMed] [Google Scholar]
- 28.Akasaka K, Tamada M, Wang F, Kariya K, Shima F, Kikuchi A, Yamamoto M, Shirouzu M, Yokoyama S, et al. J Biol Chem. 1996;271:5353–5360. doi: 10.1074/jbc.271.10.5353. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez-Viciana P, Warne P H, Khwaja A, Marte B M, Pappin D, Das P, Waterfield M D, Ridley A, Downward J. Cell. 1997;89:457–467. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez-Viciana P, Warne P H, Vanhaesebroeck B, Waterfield M D, Downward J. EMBO J. 1996;15:2442–2451. [PMC free article] [PubMed] [Google Scholar]
- 31.Khosravi-Far R, White M A, Westwick J K, Solski P A, Chrzanowska W, Van Aelst L, Wigler M H, Der C J. Mol Cell Biol. 1996;16:3923–3933. doi: 10.1128/mcb.16.7.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.White M A, Vale T, Camonis J H, Schaefer E, Wigler M H. J Biol Chem. 1996;271:16439–16442. doi: 10.1074/jbc.271.28.16439. [DOI] [PubMed] [Google Scholar]
- 33.Joneson T, McDonough M, Bar-Sagi D, Van Aelst L. Science. 1996;274:1374–1376. doi: 10.1126/science.274.5291.1374. [DOI] [PubMed] [Google Scholar]
- 34.Joneson T, White M A, Wigler M H, Bar-Sagi D. Science. 1996;271:810–812. doi: 10.1126/science.271.5250.810. [DOI] [PubMed] [Google Scholar]
- 35.Wolthuis R M, Bauer B, van't Veer L J, Vries-Smits A M, Cool R H, Spaargaren M, Wittinghofer A, Burgering B M, Bos J L. Oncogene. 1996;13:353–362. [PubMed] [Google Scholar]
- 36.Tu H, Barr M, Dong D L, Wigler M. Mol Cell Biol. 1997;17:5876–5887. doi: 10.1128/mcb.17.10.5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nassar N, Horn G, Herrmann C, Scherer A, McCormick F, Wittinghofer A. Nature (London) 1995;375:554–560. doi: 10.1038/375554a0. [DOI] [PubMed] [Google Scholar]
- 38.Geyer M, Herrmann C, Wohlgemuth S, Wittinghofer A, Kalbitzer H R. Nat Struct Biol. 1997;4:694–699. doi: 10.1038/nsb0997-694. [DOI] [PubMed] [Google Scholar]
- 39.Linnemann T, Geyer M, Jaitner B K, Block C, Kalbitzer H R, Wittinghofer A, Herrmann C. J Biol Chem. 1999;274:13556–13562. doi: 10.1074/jbc.274.19.13556. [DOI] [PubMed] [Google Scholar]
- 40.Nassar N, Horn G, Herrmann C, Block C, Janknecht R, Wittinghofer A. Nat Struct Biol. 1996;3:723–729. doi: 10.1038/nsb0896-723. [DOI] [PubMed] [Google Scholar]
- 41.Kuppens S, Diaz J F, Engelborghs Y. Protein Sci. 1999;8:1860–1866. doi: 10.1110/ps.8.9.1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terada T, Ito Y, Shirouzu M, Tateno M, Hashimoto K, Kigawa T. J Mol Biol. 1999;286:219–232. doi: 10.1006/jmbi.1998.2472. [DOI] [PubMed] [Google Scholar]
- 43.Brunger A T, Milburn M V, Tong L, deVos A M, Jancarik J, Yamaizumi Z, Nishimura S, Ohtsuka E, Kim S H. Proc Natl Acad Sci USA. 1990;87:4849–4853. doi: 10.1073/pnas.87.12.4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gorman C, Skinner R H, Skelly J V, Neidle S, Lowe P N. J Biol Chem. 1996;271:6713–6719. doi: 10.1074/jbc.271.12.6713. [DOI] [PubMed] [Google Scholar]
- 45.Pai E F, Krengel U, Petsko G A, Goody R S, Kabsch W, Wittinghofer A. EMBO J. 1990;9:2351–2359. doi: 10.1002/j.1460-2075.1990.tb07409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.John J, Rensland H, Schlichting I, Vetter I, Borasio G D, Goody R S, Wittinghofer A. J Biol Chem. 1993;268:923–929. [PubMed] [Google Scholar]
- 47.Bae C D, Min D S, Fleming I N, Exton J H. J Biol Chem. 1998;273:11596–11604. doi: 10.1074/jbc.273.19.11596. [DOI] [PubMed] [Google Scholar]
- 48.Mott H R, Owen D, Nietlispach D, Lowe P N, Manser E, Lim L, Laue E D. Nature (London) 1999;399:384–388. doi: 10.1038/20732. [DOI] [PubMed] [Google Scholar]
- 49.Nicholls A, Sharp K A, Honig B. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 50.Diederichs K, Karplus P A. Nat Struct Biol. 19997;4:269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]